

Abstract
Transthyretin (TTR) is a homotetrameric protein. Its dissociation into monomers leads to the formation of fibrils that underlie human amyloidogenic diseases. The binding of small molecules to the thyroxin-binding sites in TTR stabilizes the homotetramer and attenuates TTR amyloidosis. Herein, we report on boronic acid-substituted stilbenes that limit TTR amyloidosis in vitro. Assays of affinity for TTR and inhibition of its tendency to form fibrils were coupled with X-ray crystallographic analysis of nine TTR·ligand complexes. The ensuing structure–function data led to a symmetrical diboronic acid that forms a boronic ester reversibly with serine 117. This diboronic acid inhibits fibril formation by both wild-type TTR and a common disease-related variant, V30M TTR, as effectively as does tafamidis, a small-molecule drug used to treat TTR-related amyloidosis in the clinic. These findings establish a new modality for covalent inhibition of fibril formation and illuminate a path for future optimization.
Graphical abstract
INTRODUCTION
Amyloidosis is a disease caused by the aggregation of a normally soluble protein.1,2 Endogenous proteins can be causal for these diseases, which include Alzheimer’s, Huntington’s, and Parkinson’s.3 One such protein, transthyretin (TTR),4 is a homotetrameric protein comprised of four identical monomer units, each consisting of 127 amino-acid residues that fold into a β-sandwich (Figure 1).5,6 The dissociation of the TTR tetramer and aggregation of the ensuing monomers underlies familial amyloid polyneuropathy, familiar cardiomyopathy, and senile systemic amyloidosis.3,7
Figure 1.

Three-dimensional structure of the TTR·resveratrol complex. TTR monomers (tan, red, green, and purple ribbons) have a β-sandwich fold and assemble into a tetramer, which binds to two molecules of resveratrol (ball-and-stick). The rings of resveratrol (1) occupy inner and outer pockets of the two T4-binding sites at the dimer–dimer interfaces. The image was created with the program PyMOL and PDB entry 1dvs.5
TTR is present in both blood (0.25 g/L = 4 μM) and cerebrospinal fluid (0.1–0.4 μM).4,8 A primary role of TTR in vivo is to transport thyroxin (T4) and retinol, a hydrophobic hormone and fat-soluble vitamin A1, respectively. Due to the abundance of other lipid-binding proteins (e.g., thyroid-binding globulin and albumin), most of the T4-binding sites of TTR are empty in blood. In cerebrospinal fluid, TTR also binds to β-amyloid, attenuating the neurotoxicity that underlies Alzheimer’s disease.8–10
The binding of a ligand can stabilize the folded state of a protein.11–14 Evidence for the coupling of binding and stability appeared as early as 1890, when O’Sullivan and Thompson demonstrated that cane sugar increases markedly the thermostability of invertase, which is an enzyme that catalyzes the hydrolysis of sucrose.15 Since then, ligands have been used to enhance the conformational stability of countless proteins, including TTR. Many small molecules have been synthesized and tested as putative TTR ligands, and several have demonstrated efficacy in attenuating amyloidosis.16,17 Most efforts have focused on ligands that bind to the two identical T4-binding sites at a dimer–dimer interface (Figure 1), as such ligands discourage dissociation to the monomeric state.18 A few of these compounds have become viable treatment options, including diflunisal, which is an FDA-approved non-steroidal anti-inflammatory drug that has had limited adoption due to long-term gastrointestinal side-effects associated with cyclooxygenase inhibition,19,20 and tafamidis, which is used in the clinic to treat TTR-related amyloidosis.21–23
An attractive approach to increase the potency and pharmacokinetics of a ligand is to evoke the formation of a covalent bond.24–26 This strategy is well suited for TTR amyloidosis, not only because with an optimized dosage there might be no appreciable competition in serum with the natural ligand, T4, but also because sustained stabilization of the TTR tetramer deters the accumulation of monomers that leads to a cascade of aggregation.27 Previous work by Kelly and coworkers has shown that small molecules can modify TTR chemoselectively by targeting the ε amino group of Lys15/Lys15′ at the entry to the T4-binding binding site. This work employed irreversible reactions, including conjugate addition with activated esters and thioesters28,29 and vinyl sulfonamides,30 and sulfation with aryl fluorosulfates.31,32 Such ligands can, however, react irreversibly with other plasma proteins,30 leading to potential immunogenic responses to the protein–ligand adduct24 and the generation of potential toxic byproducts.29
To accrue the benefits of covalent binding without the liabilities, we sought ligands for TTR that bind in a covalent but reversible manner. Boronic acids interact with Lewis bases in aqueous media.33,34 Boronic acids (including the FDA-approved drug Bortezomib35) are well known as serine/threonine protease inhibitors,36,39 anti-microbial and anti-cancer agents,40,41 and delivery vehicles.42–45 Boronic acid-based fluorogenic probes have been developed for sensing both saccharides46 and reactive oxygen species,47,48 as well as for molecular recognition49 and protein conjugation.50,51 These applications arise from the ability of boronic acids to form a covalent bond with a Lewis base that is reversible under physiological conditions.52,53 Notably, boronic acids are benign, as their metabolic byproduct, boric acid, is present in a normal diet.54
Here, we report on the development of boronic acid-based ligands for the T4-binding site of the TTR tetramer. An iterative strategy involving chemical synthesis and structure–function analysis led us to covalent inhibitors of TTR aggregation. This strategy serves as a model for a new class of amyloidosis inhibitors.
RESULTS
We chose stilbene as a scaffold for the design of an initial series of boronic acid-containing TTR ligands (Chart 1). This scaffold is present in the natural product resveratrol (1), and has been employed in other TTR ligands.5,16,29,55–57 Moreover, stilbenes are readily accessible by a convergent synthetic route, as the two rings can be functionalized separately and then joined with a Wittig reaction (Schemes 1 and 2).
Chart 1.

Diphenol Ligands
Scheme 1.

Routes for the Synthesis of Stilbenes 2–6
Scheme 2.

Routes for the Synthesis of Stilbenes 7–11
Resveratrol occupies the T4-binding site with moderate affinity (Table 1 and Figure 1).5 We began by replacing the phenolic hydroxyl group, which is known to form a hydrogen bond with Ser117/117′, with a boronic acid group to generate stilbene 2 (Chart 1). Halogen substitution is known to provide additional van der Waals interactions within the inner pocket of the T4-binding site, enhancing the affinity of TTR ligands.58 Accordingly, we added a chloro group meta to the boronic acid moiety to generate stilbenes 3 and 4.
Table 1.
Interaction of Diphenols 1–4 with Wild-type TTR and its V30M Variant
| compound | Kd,2 (μM) | wild-type TTR | V30M TTR | binding modeb | ||
|---|---|---|---|---|---|---|
|
| ||||||
| %FF 1:1a | %FF 2:1a | %FF 1:1a | %FF 2:1a | |||
| 1 (resveratrol) | 0.47 ± 0.03 | 28 ± 8 | 9 ± 2 | 53 ± 12 | 26 ± 4 | forward |
| 2 | 0.47 ± 0.02 | 21 ± 4 | 9 ± 2 | 48 ± 11 | 23 ± 6 | reverse |
| 3 | 0.73 ± 0.02 | 22 ± 4 | 7 ± 3 | 44 ± 10 | 14 ± 4 | forward |
| 4 | 0.44 ± 0.01 | 19 ± 4 | 4 ± 2 | 36 ± 8 | 12 ± 3 | reverse |
We performed competitive binding assays to compare affinities among the diphenol series of ligands (Table 1 and Figure S1A). We observed no change in values of Kd,2 between stilbene 1 and 2. Interestingly, chlorinated stilbene 3 showed a higher value of Kd,2 relative to its non-halogenated counterpart, stilbene 1. This decrease in affinity contrasts with stilbene 4, which exhibited increased affinity as a result of chlorination and showed the strongest affinity for TTR of stilbenes 1–4, having a Kd,2 value of 441 nM.
Next, we assessed the ability of these molecules to inhibit fibril formation by both wild-type TTR and the common V30M variant, which is associated with familial amyloid polyneuropathy (Table 1 and Figure S2A). We found that all of the stilbenes in this series inhibited aggregation at 7.2 μM and at a 2:1 ligand/protein ratio. Stilbenes 1 and 2 showed 25% and 23% fibril formation for V30M TTR, whereas stilbenes 3 and 4 showed 14% and 11%, respectively. Herein, we consider compounds that limit aggregation to ≤10% over 96 h as potent inhibitors, and stilbenes 1–4 did not achieve this threshold.
Next, we solved co-crystal structures of TTR and resveratrol analogs 2–4 to discern if the boronated stilbenes formed a covalent bond with TTR. To our surprise, each phenylboronic acid moiety was observed in the “reverse” binding mode (Figure 2A and 2B) relative to its parent phenol, resveratrol (Figure 1 and 3A). In other words, the boronic acid group resided in the outer pocket of the T4-binding site No indication of boronic ester formation with amino acid residues was apparent (Figure S4 and S6).
Figure 2.

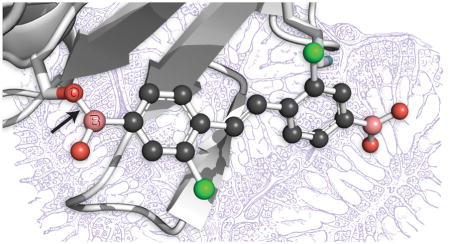
Three-dimensional structures of TTR·ligand complexes that contain a boronic acid group. One monomer (chain B) of the TTR tetramer is shown, and is in the same orientation in each panel. The main chain of TTR is rendered as a ribbon, and the side chains of Lys15 and Ser117 are shown explicitly. Ligands are depicted in a ball-and-stick rendition with CPK coloring and boron atoms labeled explicitly. Alternative conformations of Ser117 or the ligand are shown in some panels. Arrows indicate the O117γ–B bond in the TTR·10 and TTR·11 complexes. Images were created with the program PyMOL. (A) TTR·2 (PDB entry 5u48). (B) TTR·4 (5u4a). (C) TTR·6 (5u4c). (D) TTR·8 (5u4e). (E) TTR·10 (5u4f). (F) TTR·11 (5u4g).
Figure 3.

Three-dimensional structures of TTR·ligand complexes that do not contain a boronic acid group. One monomer of the TTR tetramer is shown, and is in the same orientation in each panel. The main chain of TTR is rendered as a ribbon, and the side chains of Lys15 and Ser117 are shown explicitly. Ligands are depicted in a ball-and-stick rendition with CPK coloring. Alternative conformations of Ser117 or the ligand are shown in panels B and C. Images were created with the program PyMOL. (A) TTR·3 (PDB entry 5u49, chain A). (B) TTR·5 (5u4b, chain B). (C) TTR·7 (5u4d, chain B).
To attempt to reconfigure this unexpected orientation, we designed a second series of stilbenes in which a carboxylic acid group was installed at the meta position of the ring not modified in the first series (Chart 2). Previous work had shown that incorporating an anionic substituent into ligands could introduce advantageous electrostatic interactions with Lys15/15′ of TTR.5,21,59 We hypothesized that this interaction would orient the boronic acid to the inner pocket of the T4-binding site, and perhaps promote boronate-ester formation.
Chart 2.

Carboxylic Acid Ligands
The first pair, stilbenes 5 and 6, exhibited Kd,2 values in the low micromolar range, 3- and 2-fold higher than those of stilbenes 1 and 2, respectively (Table 2 and Figure S1B). The installation of a chloro group in stilbenes 7 and 8 restored values of Kd,2 to the high nanomolar range. Consistent with a decreased affinity apparent in the value of Kd,2, the non-halogenated stilbenes 5 and 6 also showed a diminished ability to prevent fibril formation under acidic fibril-forming conditions (Table 2 and Figure S2B). Incubation of wild-type TTR with phenol 5 showed 77% fibril formation and 112% at 7.2 μM for the V30M variant, respectively. Boronic acid 6 performed better as an inhibitor than did phenol 5 for both wild-type TTR and the V30M variant (11% and 28%, respectively). The chlorinated pair 7 and 8 showed potent fibril inhibition (<10%) for both TTRs. These differences were, however, within experimental error. Again, co-crystallography revealed that boronic acids 6 and 8 were bound in the reverse mode, relative to their paired phenols. In other words, the carboxylic acid group was in the inner pocket, near Ser117/117′, and the boronic acid group was in the outer pocket, near Lys15/15′ (Figures 2C and 2D, and Table S11). As with boronic acids 2 and 4, boronic acids 6 and 8 did not exhibit covalent interactions with binding-pocket residues. The structure of 5 in complex with TTR may include alternative conformations for the phenolic group occupying the inner pocket but we were not confident placing those conformations (Figures S7A and S7B).
Table 2.
Interactions of Carboxylic Acids 5–8 with Wild-type TTR and its V30M Variant
The final series of stilbenes contained a boronic acid moiety on each ring (Chart 3). We synthesized C2 symmetrical diboronic acid 10, which has two meta-chloro and para-boronic acid groups (relative to the stilbene olefin), as well as the analogous C2 symmetrical diphenol 9. Additionally, we synthesized stilbene 11, which has one boronic acid group meta to the linker, to investigate whether this position of the boronic acid enhances interactions with Lys15/15′ in the outer pocket. We also included tafamidis (12) in this series as a benchmark for our assays.
Chart 3.

Diboronic Acid and Related Ligands
In the competitive binding assay, diphenol 9 had a Kd,2 value of 819 nM (Table 3 and Figure S1C). The analogous diboronic acid, 10, had a Kd,2 value of 469 nM, which was the largest decrease in Kd,2 value that we observed between a boronic acid and its paired phenol. These two stilbenes comprise the only pair that can be compared directly, as their C2 symmetry precludes alternative binding orientations. The difference in affinity for TTR was amplified in the fibril formation assay, where diboronic acid 10 exhibited more potent inhibition of fibril formation than did diphenol 9, both for wild-type TTR (3% versus 12% at 7.2 μM) and for the V30M variant (8% versus 27% at 7.2 μM) (Table 1 and Figure S2C). The binding of diboronic acid 10 with TTR was reversible, as assessed with mass spectrometry (Figure S3). Asymmetric diboronic acid 11 had a value of Kd,2 similar to that of diphenol 9 (864 nM) as well as a similar ability to inhibit fibril formation, suggesting that the location of the boronic acid plays a role in optimizing interactions within the binding pocket. Gratifyingly, co-crystallographic data of TTR with either diboronic acid 10 or diboronic acid 11 showed the formation of a boronic ester with Ser117/117′ (Figures 2E and 2F).
Table 3.
Interactions of Compounds 9–12 with Wild-type TTR and its V30M Variant
| compound | Kd,2 (μM) | wild-type TTR | V30M TTR | binding modeb | ||
|---|---|---|---|---|---|---|
|
| ||||||
| %FF 1:1a | %FF 2:1a | %FF 1:1a | %FF 2:1a | |||
| 9 | 0.82 ± 0.01 | 30 ± 6 | 12 ± 2 | 62 ± 16 | 27 ± 10 | NDc |
| 10 | 0.47 ± 0.03 | 20 ± 3 | 3 ± 3 | 32 ± 9 | 8 ± 3 | covalent |
| 11 | 0.79 ± 0.03 | 26 ± 4 | 10 ± 3 | 54 ± 16 | 16 ± 5 | forward/covalent |
| 12 (tafamidis) | 0.37 ± 0.01 | 22 ± 5 | 0 ± 6 | 5 ± 13 | 8 ± 7 | forwardd |
DISCUSSION
We sought a new class of small-molecule ligands for TTR, which is a validated target for pharmacological intervention.7,21–23 Towards that goal, we investigated the effects of incorporating a boronic acid substituent on the well-known stilbene scaffold, embodied in resveratrol (1). The results enabled us to reach two conclusions. First, a boronic acid group can enhance the potency of a TTR ligand. Second, a boronate ester can form with a weakly nucleophilic amino-acid residue.
The installation of a boronic acid group tends to increase the affinity of a stilbene for TTR. The value of Kd,2 for each boronic acid ligand for TTR is lower than (or equivalent to) that of its analogous phenol (Tables 1–3). Moreover, a boronic acid group enhances the ability of a stilbene to deter the formation of TTR fibrils (Tables 1–3). Nonetheless, our structural studies revealed that in each complex between TTR and a stilbene containing a single boronic acid group, that group was in the outer pocket of the T4-binding site, exposed to solvent (Figures 2A–2D, S4, S6, S8, and S10).
The modest increase in affinity incurred by adding a single boronic acid group could be due to a weak noncovalent interaction between the ε-amino group of Lys15/15′ and the vacant p-orbital of the boron. Kelly and coworkers demonstrated that this amino group, which likely has a low pKa, can act as a nucleophile.28 In our complexes, the relevant B···N distances are 3.3–3.5 Å for stilbenes 2, 4, 6, 8, and 10 (Table S11). We did not, however, observe electron-densities or atomic geometries consistent with the formation of a dative N→B bond between these two functionalities, nor were any additional hydrogen bonds apparent between TTR residues and the boronic acid group (Figures 2A–2D, S4, S6, S8, and S10). Additional hydrogen bonds did, however, arise elsewhere in the complexes with a single boronic acid. For example, the two meta hydroxyl groups in stilbenes 2 and 4 (but not 1 and 3) interact closely (2.0–2.3 Å) with Ser117/117′ (Table S11).
Stilbenes and similar compounds bind to TTR in one of two modes.19,27,32,60–63 Although a consensus explanation is not apparent, the polarity of pendant functional groups can play a role in ligand orientation.56,64 In our stilbenes, however, the relevant logP values of phenol (1.46), benzoic acid (1.87), and phenylboronic acid (1.59) are similar,65 suggesting that hydrophilicity alone contributes little to their orientation in the T4-binding site. The installation of a chloro group on one ring, as in stilbenes 4 and 8, led to increased affinity and enhanced efficacy (Tables 1 and 2), but did not affect binding orientation (Figures 2B and 2D).
To negate binding orientation as a factor, we designed a class of molecules containing a boronic acid substituent on each stilbene ring. If such a ligand were to bind to TTR, then a boronic acid group would necessarily be in the inner pocket of the T4-binding site. We were gratified that this strategy was successful, as the boronic acid group of both stilbenes 10 and 11 that bound in the inner pocket formed an ester with Ser117/117′. Boronic esters have demonstrable utility as mimics of the high-energy tetrahedral intermediate in reactions catalyzed by serine/threonine proteases.36–39 In those covalent complexes, an active-site serine or threonine residue forms a tetrahedral, sp3-hybridized boronate ester with a boronic acid group.66
Despite the hydration of boronic acid groups in aqueous solution34 and in marked contrast to other known boronate esters with proteins, we observe planar, sp2-hybridized boronate esters with Ser117/117′ of TTR (Figures 2E and 2F). We are aware of only one other structure in which a planar boronate ester is formed with a hydroxyl residue of a protein (PDB entry 1p06).67 That other structure is, however, distinct because a lone pair of electrons from a proximal histidine residue appears to participate in a dative bond with the vacant p-orbital of the boron. The presence of a planar ester could indicate that formation of a tetrahedral adduct is hindered sterically, unlike in the active site of a serine/threonine protease that has evolved to bind tightly to a tetrahedral intermediate. Hence, our results could demarcate the lower limit of affinity enhancement that can be realized from boronic acid-based inhibitors.
Chloro groups have a variable contribution to the affinity of stilbenes for TTR. A chloro group can fill unoccupied cavities, which would otherwise compromise affinity.68,69 Second, the position of the chloro group in both of the ester-forming boronates enables the formation of a halogen bond.70 The relevant O···Cl distance is close to the sum of the van der Waals radii (rO + rCl = 3.27 Å; Figure 4).58 The geometries observed in the TTR complexes with stilbenes 10 and 11 (Table S12) suggest a halogen-bond energy of 0.7–0.9 kcal/mol.71 Still, the consequences of installing a chloro group on the stilbene scaffold are unlike those of installing a boronic acid group, which consistently increases affinity for TTR (vide supra). For example, the addition of a chloro group to stilbene 1 to form stilbene 3 decreases affinity, whereas the addition of a chloro group to stilbene 5 to form stilbene 7, increases affinity. Notably, the phenolic ring of stilbene 5 was found to occupy two conformations in the inner pocket of the T4-binding site, which could explain, in part, the lower affinity of stilbene 5 relative to stilbene 7 (Figures 3B, S7, and S9).
Figure 4.

Halogen-bonding interactions in the TTR·10 and TTR·11 complexes. One monomer (chain B) of the TTR tetramer is shown. Chloro groups in the two ester-forming boronates exhibit C–Cl···O108′ angles that are nearly linear and Cl··· O108′ distances (dashed yellow lines) that are 3.6–3.8 Å (Table S12). Images were created with the program PyMOL. (A) TTR·10 (PDB entry 5u4f). (B) TTR·11 (5u4g).
The judicious use of halogen substituents in boronated stilbenes merits further investigation. For example, a chloro group positioned meta to the boron atom has been found to decrease the pKa of phenyl boronic acid from 8.8 to 8.2.72 Such a more acidic boronic acid can form more stable boronate esters.73 An analogous difference in Lewis acidity could be responsible for some of the differences observed in the affinity of boronic acids for TTR.
Likewise, the esterification of TTR by boronic acid ligands warrants additional optimization. In particular, a mono-boronate analog of compound 10 that binds with its lone boronic acid group in the inner pocket could be used to reveal the precise contribution of a boronate ester to the thermodynamics and kinetics74 of binding. For proper orientation, such a ligand would likely require installation of a highly polar functional group that demands the solvation attainable in the outer pocket.
CONCLUSION
A series of paired stilbenes was designed, synthesized, and tested as ligands for TTR. Each ligand contained either a phenolic hydroxyl group or a phenylboronic acid group, which was intended to bind within the inner pocket of the T4-binding site in the dimer–dimer interface. We found that the functional groups on the stilbene can alter the binding mode, precluding a rigorous thermodynamic analysis. Nevertheless, our boronic acids are the first ligands observed to form a reversible covalent bond with TTR—one with a serine residue deep in the inner pocket. Crystal structures also revealed the first sp2-hybridized boronic esters observed with a protein. These stilbene boronic acids inhibit the TTR fibril-formation that leads to amyloidosis. Their efficacy extends to V30M TTR, which is a common disease-related variant. We envision that the unique attributes of boronic acid groups could find utility in pharmacological stabilizers of other proteins.
EXPERIMENTAL SECTION
Materials
Resveratrol (1), 8-anilino-1-naphthalenesulfonic acid (ANS), and other reagents for biochemical assays were from Sigma–Aldrich (St Louis, MO). Tafamidis (2-(3,5-dichlorophenyl)-6-benzoxazolecarboxylic acid) (12) was from Carbosynth Limited (Berkshire, UK). DNA oligonucleotides were from Integrated DNA Technologies (Coralville, IA).
Chemical Synthesis
Stilbenes 2–11 were synthesized by the routes shown in Schemes 1 and 2.
Materials
Reagents and solvents were from Sigma–Aldrich (Milwaukee, WI) and were used without further purification. All glassware was flame-dried, and all reactions were performed under an atmosphere of N2(g). Reagent-grade solvents: dichloromethane (DCM), tetrahydrofuran (THF), triethylamine (TEA), and dimethylformamide (DMF) were dried over a column of alumina and were removed from a dry still under an inert atmosphere. Flash column chromatography was performed with Silicycle 40–63 Å silica (230–400 mesh), and thin-layer chromatography (TLC) was performed with EMD 250-μm silica gel 60-F254 plates.
Conditions
All procedures were performed in air at ambient temperature (~22 °C) and pressure (1.0 atm) unless indicated otherwise.
Solvent Removal
The phrase “concentrated under reduced pressure” refers to the removal of solvents and other volatile materials using a rotary evaporator at water aspirator pressure (<20 torr) while maintaining a water bath below 40 °C. Residual solvent was removed from samples at high vacuum (<0.1 torr).
NMR Spectroscopy
1H and 13C NMR spectra were acquired with a Bruker Avance III 500i spectrometer at the National Magnetic Resonance Facility at Madison (NMRFAM). Chemical shift data are reported in units of δ (ppm) relative to residual solvent.
Mass Spectrometry
Mass spectra of small molecules were acquired with an LCT electrospray ionization (ESI) instrument from Waters. Mass spectra of proteins were acquired with a microflex LRF desorption ionization–time-of-flight (MALDI–TOF) instrument from Bruker (Billerica, MA). Both instruments were in the Paul Bender Chemical Instrumentation Center at the University of Wisconsin–Madison.
Melting Points
Melting points were determined with an OptiMelt MPA100 instrument from Stanford Research Systems (Sunnyvale, CA) over a range of 100–400 °C with a 0.5 °C/min heating rate. Melting points values are given as the single meniscus point. Compounds 2, 3, and 5 had melting points >400 °C.
Compound Purity
The purity of all compounds was judged to be ≥95%, as assessed by 1H and 13C NMR spectroscopy, mass spectrometry, melting-point determination, and reversed-phase high-performance liquid chromatography (HPLC) using a C18 column and 515/717/996 analytical instrument from Waters (Milford, MA) with a gradient of 10–80% v/v acetonitrile in water over 40 min.
5-[(1E)-2-(4-Bromo)ethenyl]-1,3-dimethoxybenzene (2a)
3,5-Dimethoxybenzyl bromide (1.2 g, 5.5 mmol) was dissolved in neat triethylphosphite (1.2 mL, 6.6 mmol) and heated to 150 °C for 4 h. The reaction mixture was cooled to 0 °C and diluted with DMF (40 mL). NaH (60% w/v in mineral oil, 0.28 g, 7.12 mmol) was added to the resulting solution, and the reaction mixture was stirred at 0 °C for 20 min. A solution of 4-bromo-benzaldehyde (1.0 g, 5.5 mmol) in DMF (15 mL) was then added dropwise. The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction mixture was then diluted with EtOAc (20 mL), and washed with 10% w/v citric acid (30 mL), followed by brine (30 mL). The organic layer was separated, dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (10% v/v EtOAc in hexanes) to afford compound 2a as a white solid (1.56 g, 89%). 1H NMR (500 MHz, CDCl3, δ): 3.83 (s, 6H), 6.41 (s, 1H), 6.65 (s, 2H), 6.98–7.01 (d, J = 16.28 Hz, 1H), 7.01–7.04 (d, J = 16.34 Hz, 1H), 7.35–7.37 (d, J = 8.48 Hz, 2H), 7.46–7.48 (d, J = 8.51 Hz, 2H); 13C NMR (100 MHz, CDCl3, δ): 55.42, 100.31, 104.74, 121.57, 128.03, 128.16, 129.50, 131.92, 136.19, 139.07, 161.12; ASAP–MS (m/z): [M + H]+ calcd for C16H15BrO2, 319.0329; found, 319.0331.
5-[(1E)-2-(4-Bromo)ethenyl]-1,3-benzenediol (2b)
Compound 2a (0.5 g, 1.6 mmol) was dissolved in DCM (7 mL), and the resulting solution was cooled to 0 °C. A solution of 1.0 M BBr3 in DCM (7.8 mL) was added dropwise at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred for 4 h. The reaction mixture was then poured carefully into a separation funnel containing ice water (~15 mL). The mixture was extracted with DCM (3 × 15 mL). The organic layers were combined and washed with brine (15 mL), dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was suspended in ice-cold DCM. The resulting precipitate was isolated by filtration to afford compound 2b as a white solid (0.326 g, 70%). 1H NMR (500 MHz, CD3OD, δ): 6.22 (s, 1H), 6.51 (s, 2H), 7.00–7.03 (d, J = 16.33 Hz, 1H), 7.04–7.07 (d, J = 16.33 Hz, 1H), 7.45–7.47 (d, J = 8.5 Hz, 2H), 7.50–7.51 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CD3OD, δ): 103.43, 106.15, 121.97, 128.05, 129.17, 130.97, 132.78, 138.04, 140.42, 159.80; HRMS–ESI (m/z): [M − H]− calcd for C14H11BrO2, 288.9870; found, 288.9869.
5-[(1E)-2-(4-Boronic Acid Pinacol Ester)ethenyl]-1,3-benzenediol (2c)
Compound 2b (0.250 g, 0.858 mmol), KOAc (0.245 g, 2.57 mmol), bis(pinacolato)diboron (0.652 g, 2.57 mmol), and Pd(dppf)Cl2 (0.062 g, 0.0858 mmol) were added to a Schlenk flask, which was then evacuated and backfilled with N2(g). Dioxane (8.5 mL) was deoxygenated by sonication under high vacuum and backfilled with N2(g). The deoxygenated dioxane was then added by cannula into the reaction flask, and the reaction mixture was heated to 80 °C and stirred overnight. The reaction mixture was then filtered, and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (20% v/v EtOAc in hexanes) to afford compound 2c as a white solid (0.245 g, 85%). 1H NMR (500 MHz, CD3OD, δ): 1.34 (s, 12H), 6.22 (s, 1H), 6.51 (s, 2H), 7.02–7.06 (d, J = 16.27 Hz, 1H), 7.06–7.10 (d, J = 16.36 Hz, 1H), 7.50–7.52 (d, J = 7.93 Hz, 2H), 7.71–7.72 (d, J = 7.79 Hz, 2H); 13C NMR (125 MHz, CD3OD, δ): 25.19, 85.04, 103.42, 160.21, 126.77, 129.23, 131.16, 136.08, 140.54, 141.63, 159.75; HRMS–ESI (m/z): [M − H]+ calcd for C20H23BO4, 336.1653; found, 336.1653.
5-[(1E)-2-(4-Boronic acid)ethenyl]-1,3-benzenediol (2)
Compound 2c (0.050 g, 0.147 mmol) was dissolved in 4:1 THF/H2O (1.5 mL). NaIO4 (0.157 g, 0.739 mmol) was added to the resulting solution, followed by 1.0 M HCl (36 μL, 0.036 mmol). The reaction mixture was allowed to stir overnight. The reaction mixture was then diluted with H2O (2 mL) and extracted with EtOAc (3 × 4 mL). The organic layers were combined and washed with brine (10 mL), dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (3% v/v CH3OH in DCM) to afford compound 2 as a white crystalline solid (0.027 g, 67%) with mp >400 °C. 1H NMR (500 MHz, CD3OD, δ): 6.20 (s, 1H), 6.50 (s, 2H), 7.95 (bs, 2H), 7.51–7.52 (d, J = 8.17 Hz, 2H), 7.60–7.62 (d, J = 8.13 Hz, 2H); 13C NMR (125 MHz, CD3OD, δ): 103.32, 106.13, 126.69, 129.26, 130.67, 135.09, 140.06, 140.64, 159.78; HRMS–ESI (m/z): [M − H]− calcd for the single methyl boronic ester C15H15BO4, 268.1027; found, 268.1027.
5-[(1E)-2-(2-Chloro-4-bromo)ethenyl]-1,3-dimethoxybenzene (3a)
3,5-Dimethoxybenzyl bromide (0.74 g, 3.4 mmol) was dissolved in neat triethylphosphite (0.7 mL, 4.0 mmol), and the resulting solution was heated to 150 °C for 4 h. The reaction mixture was cooled to 0 °C and added to DMF (20 mL). NaH (60% w/v in mineral oil, 0.17 g, 4.36 mmol) was added to the resulting solution, and the reaction mixture was stirred at 0 °C for 20 min. A solution of 4-bromo-2-chloro benzaldehyde (0.74 g, 3.36 mmol) in DMF (10 mL) was then added dropwise. The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction mixture was then diluted with EtOAc (15 mL), and washed with 10% w/v citric acid (20 mL), followed by brine (20 mL). The organic layer was separated, dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (5% v/v EtOAc in hexanes) to afford compound 3a as a white solid (1.10 g, 91% over 2 steps). 1H NMR (400 MHz, CDCl3, δ): 3.84 (s, 6H), 6.44 (s, 1H), 6.69 (s, 2H), 6.97–7.01 (d, J = 16.24 Hz, 1H), 7.36–7.40 (m, 2H), 7.50–7.52 (d, J = 8.42 Hz, 1H), 7.55 (s, 1H); 13C NMR (100 MHz, CDCl3, δ): 55.51, 100.60, 105.11, 121.37, 124.27, 127.60, 130.27, 131.89, 132.43, 134.20, 134.41, 138.81, 161.12; ASAP–MS (m/z): [M + H]+ calcd for C16H14BrClO2, 352.9939; found, 352.9939.
5-[(1E)-2-(2-Chloro-4-bromo)ethenyl]-1,3-benzenediol (3b)
Compound 3a (0.500 g, 1.42 mmol) was dissolved in DCM (14 mL), and the reaction mixture was cooled to 0 °C. A solution of 1.0 M BBr3 (7.10 mmol) in DCM (7.1 mL) was then added dropwise at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred for 4 h. The reaction mixture was then poured carefully into a separation funnel containing ice water (~10 mL) and extracted with DCM (3 × 10 mL). The organic layers were combined and washed with brine (15 mL), dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was suspended in cold DCM (5 mL). The resulting precipitate was filtered to afford compound 3b as a white solid (0.335 g, 72%). 1H NMR (500 MHz, CD3OD, δ): 6.23, (s, 1H), 6.51 (s, 2H), 7.03–7.07 (d, J = 16.23 Hz, 1H), 7.31–7.35 (d, J = 16.23 Hz, 1H), 7.45–7.47 (d, J = 8.31 Hz, 1H), 7.60 (s, 1H), 7.67–7.69 (d, J = 8.47 Hz, 1H); 13C NMR (125 MHz, CD3OD, δ): 103.79, 106.34, 124.89, 126.71, 133.26, 133.34, 133.90, 135.91, 137.59, 140.29, 159.89; HRMS–ESI (m/z): [M − H]− calcd for C14H10BrClO2, 322.9480; found, 322.9480.
5-[(1E)-2-(2-Chloro-4-boronic Acid Pinacol Ester)ethenyl]-1,3-benzenediol (3c)
Compound 3b (0.100 g, 0.308 mmol), KOAc (0.088 g, 0.926 mmol), bis(pinacolato)diboron (0.235 g, 0.926 mmol), and Pd(dppf )Cl2 (0.0225 g, 0.0308 mmol) were added to a flame-dried Schlenk flask, which was then evacuated and backfilled with N2(g). Dioxane (3.5 mL) was deoxygenated by sonication under high vacuum and backfilled with N2(g). The deoxygenated dioxane was then added by cannula into the reaction flask, and the reaction mixture was heated to 80 °C and stirred overnight. The reaction mixture was filtered and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (2% v/v CH3OH in DCM) to afford compound 3c as a white solid (0.080 g, 69%). 1H NMR (500 MHz, CD3OD, δ): 1.33 (s, 12H), 6.23 (s, 1H), 6.53 (s, 2H), 7.05–7.09 (d, J = 16.29 Hz, 1H), 7.40–7.44 (d, J = 16.28 Hz, 1H), 7.60–7.62 (d, J = 7.98 Hz, 1H), 7.70 (s, 1H), 7.72–7.74 (d, J = 7.83 Hz, 1H); 13C NMR (125 MHz, CD3OD, δ): 25.18, 75.82, 85.42, 103.90, 106.42, 124.82, 126.88, 133.83, 133.90, 134.11, 136.74, 139.11, 140.17, 159.84; HRMS–ESI (m/z): [M − H]− calcd for C20H22BClO4, 370.1263; found, 370.1263.
5-[(1E)-2-(2-chloro-4-hydroxyphenyl)ethenyl]-1,3-benzenediol (3)
Compound 3 was derived from the oxidation of compound 3c by an aryl N-oxide in one step as described previously.75 Here, compound 3c (0.020 g, 0.053 mmol) was dissolved in DCM (0.6 mL). N,N-Dimethyl-p-toluidine-N-oxide (0.012 g, 0.081 mmol) was added to the resulting solution, and the reaction mixture was stirred for 1 h. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (15% v/v EtOAc in hexanes) to afford compound 3 as a pale yellow solid (0.010 g, 70%) with mp >400 °C. 1H NMR (500 MHz, CD3OD, δ): 6.18–6.19 (t, J = 2.15 Hz, 1H), 6.47 (s, 2H), 6.74–6.76 (d, J = 8.66 Hz, 1H), 6.82 (s, 1H), 6.82–6.85 (d, J = 14.57 Hz, 1H), 7.31–7.34 (d, J = 16.16 Hz, 1H), 7.58–7.59 (d, J = 8.64 Hz, 1H); 13C NMR (125 MHz, CD3OD, δ): 103.10, 105.96, 115.92, 117.03, 124.95, 127.75, 128.32, 129.78, 134.77, 140.91, 159.08, 159.77; HRMS–ESI (m/z): [M − H]− calcd for C14H11ClO3, 261.0324; found, 261.0325.
5-[(1E)-2-(2-Chloro-4-boronic Acid)ethenyl]-1,3-benzenediol (4)
Compound 3c (0.050 g, 0.134 mmol) was dissolved in 4:1 THF/H2O (1.3 mL). NaIO4 (0.143 g, 0.670 mmol) was added to the resulting solution, followed by 1.0 M HCl (33 μL, 0.033 mmol). The reaction mixture was then stirred overnight. The reaction mixture was diluted with H2O (1.0 mL) and extracted with EtOAc (3 × 2 mL). The organic layers were combined and washed with brine (6 mL), dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude mixture was purified by flash column chromatography (4% v/v CH3OH in DCM) to afford compound 4 as a white crystalline solid (0.022 g, 88%) with mp 186.0 °C. 1H NMR (500 MHz, CD3OD, δ): 6.23 (s, 1H), 6.52 (s, 2H), 7.05–7.09 (d, J = 16.20 Hz, 1H), 7.41–7.45 (d, J = 16.23 Hz, 1H), 7.54–7.56 (d, J = 7.85 Hz, 1H), 7.63 (s, 1H), 7.75–7.76 (d, J = 7.81 Hz, 1H); 13C NMR (125 MHz, CD3OD, δ): 103.77, 106.32, 124.88, 126.71, 133.26, 133.34, 133.90, 135.91, 137.59, 140.29, 159.89; HRMS–ESI (m/z): [M − H]− calcd for C14H12BClO4, 288.0481; found, 288.0482.
3-[(1E)-2-(4-Bromophenyl)ethenyl]-benzoic Acid Methyl Ester (5a)
Ethyl 3-(bromomethyl)benzoate (2.0 g, 8.7 mmol) was dissolved in neat triethylphosphite (1.78 mL, 10.4 mmol) and heated to 150 °C for 4 h. The reaction mixture was cooled to 0 °C and diluted with DMF (87 mL). NaH (60% w/v in mineral oil, 0.69 g, 17.46 mmol) was added to the resulting solution, and the reaction mixture stirred at 0 °C for 20 min. A solution of 4-bromobenzaldehyde (1.62 g, 8.73 mmol) in DMF (87 mL) was then added dropwise. The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction mixture was then diluted with EtOAc (50 mL), and washed with 10% w/v citric acid (20 mL), followed by brine (20 mL). The organic layer was separated, dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (10 % v/v EtOAc in hexanes) to afford the 5a as a white solid (1.75 g, 63% over 2 steps). 1H NMR (500 MHz, CD3OD, δ): 3.84 (s, 3H), 6.94–6.96 (d, J = 8.70 Hz, 2H), 7.09–7.12 (d, J = 16.28, 1H), 7.20–7.25 (d, J = 16.41 Hz, 1H), 7.45–7.48 (t, J = 7.67, 1H), 7.53–7.55 (d, J = 8.62 Hz, 2H), 7.76–7.78 (d, J = 7.85 Hz, 1H), 7.88–7.90 (d, J = 7.72, 1H), 8.18 (s, 1H); 13C NMR (125 MHz, CD3OD, δ): 54.31, 113.73, 124.97, 126.93, 127.58, 127.80, 128.40, 129.13, 130.01, 138.21, 159.72, 168.62; HRMS–ESI (m/z): M+• calcd for C16H13BrO2, 316.0094; found, 316.0081.
3-[(1E)-2-(4-Bromophenyl)ethenyl]-benzoic Acid (5b)
Compound 5a (1.5 g, 4.7 mmol) was dissolved in 3:1 THF/EtOH (47 mL). A solution of 2 M NaOH (4.7 mL, 9.4 mmol) was added to the resulting solution, and the reaction mixture was stirred overnight. The reaction mixture was then diluted with EtOAc (20 mL), and washed with 10% w/v citric acid (20 mL), followed by brine (20 mL). The organic layer was separated, dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (20% v/v EtOAc in hexanes) to afford compound 5b as a white solid (1.36 g, 96%). 1H NMR (500 MHz, DMSO, δ): 7.33–7.36 (d, J = 16.52 Hz, 1H), 7.40–7.43 (d, J = 16.44 Hz, 1H), 7.50–7.54 (t, J = 7.68 Hz, 1H), 7.58–7.60 (d, J = 8.64 Hz, 2H), 7.63–7.61 (d, J = 8.66 Hz, 2H), 7.84–7.88 (t, J = 8.63 Hz, 2H), 8.16 (s, 1H), 13.10 (s, 1H); 13C NMR (125 MHz, DMSO, δ): 121.27, 127.81, 128.68, 128.92, 129.00, 129.10, 129.51, 131.04, 132.09, 136.61, 137.66, 167.69; HRMS–ESI (m/z): M+• calcd for C15H11BrO2, 301.9937; found, 301.9944.
3-[(1E)-2-(4-Boronic acid Pinacol Ester)ethenyl]-benzoic Acid (5c)
Compound 5b (1.0 g, 3.3 mmol), KOAc (0.971 g, 9.9 mmol), bis(pinacolato)diboron (2.5 g, 9.9 mmol), and Pd(dppf)Cl2 (0.24 g, 0.33 mmol) were added to a flame-dried Schlenk flask, which was then evacuated and backfilled with N2(g). Dioxane (33 mL) was deoxygenated by sonication under high vacuum and backfilled with N2(g). The deoxygenated dioxane was then added by cannula into the reaction flask, and the reaction mixture was stirred overnight at 80 °C. The reaction mixture was filtered, and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (20% v/v EtOAc in hexanes) to afford compound 5c as a white solid (0.850 g, 73%). 1H NMR (500 MHz, CDCl3, δ): 1.36, (s, 12H), 7.22 (s, 2H), 7.46–7.50 (t, J = 7.44 Hz, 1H), 7.54–7.55 (d, J = 7.72 Hz, 1H), 7.76–7.77 (d, J = 8.12 Hz, 2H), 7.81–7.83 (d, J = 8.10 Hz, 2H), 8.00–8.01 (d, J = 8.11 Hz, 1H), 8.27–8.28 (d, J = 7.72 Hz, 1H), 8.28 (s, 1H); 13C NMR (125 MHz, CDCl3, δ): 24.90, 83.87, 125.97, 128.19, 128.32, 128.94, 129.28, 129.70, 130.06, 131.67, 135.23, 137.70, 139.50; HRMS–ESI (m/z): [M + NH4]+ calcd for C21H23BO4, 367.2064; found, 367.2062.
3-[(1E)-2-(4-Hydroxyphenyl)ethenyl]-benzoic Acid (5)
Compound 5c (0.050 g, 0.142 mmol) was dissolved in DCM (1.4 mL). N,N-Dimethyl-p-toluidine-N-oxide (0.032 g, 0.213 mmol) was added to the resulting solution, and the reaction mixture was allowed to stir for 1 h. The reaction mixture was filtered, and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (2% v/v MeOH in DCM) to afford compound 5 as a white solid (0.030 g, 88%) with mp >400 °C. 1H NMR (500 MHz, CD3OD, δ): 6.78–6.80 (d, J = 8.61 Hz, 2H), 7.02–7.05 (d, J = 16.32 Hz, 1H), 7.15–7.19 (d, J = 16.34 Hz, 1H), 7.42–7.44 (d, J = 8.26 Hz, 2H), 7.42–7.45 (t, J = 7.28 Hz, 1H), 7.73–7.75 (d, J = 7.82 Hz, 1H), 7.85–7.87 (d, J = 7.79 Hz, 1H), 8.15 (s, 1H); 13C NMR (125 MHz, CD3OD, δ): 116.52, 125.56, 128.25, 129.04, 129.10, 129.80, 130.04, 130.90, 131.42, 132.30, 139.80; HRMS–ESI (m/z): [M − H]− calcd for C15H12O3, 239.0714; found, 239.0716.
3-[(1E)-2-(4-Boronic acid)ethenyl]-benzoic Acid (6)
Compound 5c (0.5 g, 1.48 mmol) was dissolved in 4:1 THF/H2O (15 mL). NaIO4 (0.405 g, 1.89 mmol) was added to the resulting solution, followed by 1.0 M HCl (0.15 mL, 0.158 mmol). The reaction mixture was then stirred overnight. The reaction mixture was diluted with H2O (10 mL) and extracted with EtOAc (3 × 15 mL). The organic layers were combined and washed with brine (20 mL), dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (10% v/v CH3OH in DCM) to afford compound 6 as a white crystalline solid (0.345 g, 90%) with mp 174.2 °C. 1H NMR (500 MHz, CDCl3, δ): 7.24–7.27 (d, J = 16.43 Hz, 1H), 7.28–7.32 (d, J = 16.45 Hz, 1H), 7.49–7.76 (t, J = 7.70 Hz, 1H), 7.58–7.59 (d, J = 7.94 Hz, 2H), 7.62–7.64 (d, J = 7.92 Hz, 2H), 7.80–7.82 (d, J = 7.87 Hz, 1H), 7.90–7.92 (d, J = 7.81 Hz, 1H), 8.21 (s, 1H); 13C NMR (125 MHz, CDCl3, δ): 125.51, 127.32, 127.86, 128.37, 128.51, 129.41, 130.43, 131.07, 133.70, 137.77, 138.27, 168.32; HRMS–ESI (m/z): [M − H]− calcd for single methyl boronic acid C16H15BO4, 280.1026; found, 280.1033.
Methyl 4-Bromo-2-chlorobenzoate (7a)
4-Bromo-2-chlorotoluene (1.0 g, 4.8 mmol) was dissolved in 1:1 water/tert-butanol (20 mL). KMnO4 (1.53 g, 9.7 mmol) was added to the resulting solution, and the reaction mixture was heated to 70 °C with a reflux condenser for 2 h. The reaction mixture was then allowed to cool to room temperature, and more KMnO4 (1.53 g, 9.7 mmol) was added. The reaction mixture was then reheated to 70 °C in a flask with a reflux condenser and stirred overnight at 70 °C. The warm reaction mixture was filtered, and the resulting KMnO4 cake was rinsed with water (~10 mL). The filtrate was acidified to pH 3 with concentrated HCl and extracted with EtOAc (3 × 20 mL). The organic layers were combined, dried with NaSO4(s), and filtered. The solvent was removed under reduced pressure to afford the carboxylic acid precursor as a white solid. This precursor was dissolved in 3 M HCl in MeOH (15 mL) and heated to reflux for 12 h. The reaction mixture was allowed to cool to room temperature, and N2(g) was bubbled through the solution for 20 min to remove excess HCl(g). The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (20% v/v EtOAc in hexanes) to afford compound 7a as a colorless oil (1.15 g, 96% yield over 2 steps). 1H NMR (500 MHz, CD3OD, δ): 3.93 (s, 3H), 7.59–7.61 (d, J = 8.47 Hz, 1H), 7.76–7.77 (m, 2H); 13C NMR (125 MHz, CD3OD, δ): 53.07, 127.34, 130.51, 131.41, 133.71, 134.63, 135.53, 166.72; HRMS–ESI (m/z): M+• calcd for C8H6BrClO2, 247.9235; found, 247.9237.
4-Bromo-2-chlorobenzyl Alcohol (7b)
Compound 7a (1.00 g, 4.01 mmol) was dissolved in THF (47 mL), and the resulting solution was cooled to 0 °C. 2 M LiBH4 in THF (12 mL, 23.5 mmol) was then added dropwise, followed by methanol (4 mL). The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction mixture was quenched by adding EtOAc (20 mL) dropwise, followed by water (15 mL), and then acidification to pH 5 with 1.0 M HCl. The resulting lithium salts were removed by filtration, and the filtrate was extracted with EtOAc (3 × 15 mL). The organic layers were combined, dried with NaSO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (20% v/v EtOAc in hexanes) to afford compound 7b as a white solid (0.843 g, 95%). 1H NMR (500 MHz, CDCl3, δ): 1.91 (bs, 1H), 4.74 (s, 2H), 7.38–7.39 (d, J = 8.22 Hz, 1H), 7.42–7.44 (d, J = 8.23, 1H) 7.53 (s, 1H); 13C NMR (500 MHz, CDCl3, δ): 62.48, 121.68, 129.93, 130.42, 132.05, 133.54, 137.43; HRMS–ESI (m/z): M+• calcd for C7H6OBrCl, 219.9286; found, 219.9282.
4-Bromo-2-chlorobenzaldehyde (7c)
Compound 7b (0.5 g, 2.28 mmol) was dissolved in DCM (22 mL). Pyridinium dichromate (PDC, 2.57 g, 6.84 mmol) was added to the resulting solution, and the reaction mixture was allowed to stir overnight. The reaction mixture was then filtered through a pad of Celite®, and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (10% v/v EtOAc in hexanes) to afford compound 7c as a white solid (0.440 g, 88%). 1H NMR (500MHz, CDCl3, δ): 7.54–7.55 (d, J = 8.25 Hz, 1H), 7.66 (s, 1H), 7.78–7.80 (d, J = 8.32 Hz, 1H), 10.42 (s, 1H); 13C NMR (125 MHz, CDCl3, δ) 129.78, 130.55, 131.09, 131.45, 133.49, 138.71, 188.95; ASAP–MS (m/z): [M+H]+ calcd for C7H4BrClO, 218.9207; found, 218.9216.
3-[(1E)-2-(2-Chloro-4-bromophenyl)ethenyl]-benzoic Acid Methyl Ester (7d)
Ethyl 3-(bromomethyl)benzoate (1.04 g, 4.55 mmol) was dissolved in neat triethylphosphite (0.9 mL, 5.46 mmol), and the resulting solution was heated to 150 °C for 4 h. The reaction mixture was cooled to 0 °C and diluted with DMF (40 mL). NaH (60% w/v in mineral oil, 0.23 g, 5.91 mmol) was added to the resulting solution, and the reaction mixture was stirred at 0 °C for 20 min. A solution of 4-bromo-2-chlorobenzaldehyde (7c; 1.0 g, 4.6 mmol) in DMF (5 mL) was added dropwise. The reaction mixture was then allowed to warm to room temperature and stirred overnight. The reaction mixture was diluted with EtOAc (5 mL), and washed with 10% w/v citric acid (5 mL), followed by brine (5 mL). The organic layer was separated, dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (10% v/v EtOAc in hexanes) to afford compound 7d as a white solid (1.3 g, 83% over 2 steps). 1H NMR (500MHz, CDCl3, δ): 3.95 (s, 3H), 7.08–7.11 (d, J = 16.31 Hz, 1H), 7.39–7.41 (d, J = 8.53 Hz, 1H), 7.44–7.47 (t, J = 8.02 Hz, 1H), 7.45–7.49 (d, J = 16.56 Hz, 1H), 7.53–7.55 (d, J = 8.45 Hz, 1H), 7.56 (s, 1H), 7.71–7.73 (d, J = 7.73 Hz, 1H), 7.96–7.97 (d, J = 7.70 Hz, 1H), 8.19 (s, 1H); 13C NMR (125 MHz, CDCl3, δ): 52.43, 121.70, 125.02, 127.66, 128.15, 129.02, 129.35, 130.39, 130.88, 131.12, 132.55, 134.26, 134.35, 137.19, 167.02; HRMS–ESI (m/z): [M + NH4]+ calcd for C16H12BrClO2, 368.0048; found, 368.0053.
3-[(1E)-2-(2-Chloro-4-bromophenyl)ethenyl]-benzoic Acid (7e)
Compound 7d (1.34 g, 3.83 mmol) was dissolved in 3:1 THF/EtOH (40 mL). 2.0 M NaOH (3.3 mL, 7.7 mmol) was added to the resulting solution, and the reaction mixture was stirred overnight. The reaction mixture was diluted with EtOAc (20 mL), and washed with 10% w/v citric acid (30 mL), followed by brine (30 mL). The organic layer was separated, dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (20% v/v EtOAc in hexanes) to afford compound 7e as a white solid (1.22 g, 95%). 1H NMR (500 MHz, DMSO, δ): 7.42–7.45 (d, J = 16.39 Hz, 1H), 7.47–7.50 (d, J = 16.41 Hz, 1H), 7.53–7.56 (t, J = 7.71 Hz, 1H), 7.60–7.62 (d, J = 8.54, 1H), 7.79 (s, 1H), 7.86–7.88 (d, J = 8.63 Hz, 1H), 7.88–7.92 (t, J = 7.86, 2H), 8.15 (s, 1H), 13.01 (bs, 1H); 13C NMR (125 MHz, DMSO, δ): 121.99, 124.85, 128.89, 129.10, 129.94, 130.17, 131.41, 131.73, 132.08, 132.38, 132.87, 134.61, 135.22, 138.07, 167.31; HRMS–ESI (m/z): [M − H]− calcd for C15H10BrClO2, 334.9479; found, 334.9476.
3-[(1E)-2-(2-Chloro-4-boronic Acid Pinacol Ester)ethenyl]-benzoic Acid (7f)
Compound 7e (0.200 g, 0.580 mmol), KOAc (0.171 g, 1.791 mmol), bis(pinacolato)diboron (0.45 g, 1.79 mmol), and Pd(dppf)Cl2 (0.043 g, 0.059 mmol) were added to a flame-dried Schlenk flask, which was then evacuated and backfilled with N2(g). Dioxane (6 mL) was deoxygenated by sonication under high vacuum and backfilled with N2(g). The deoxygenated dioxane was then added by cannula into the reaction flask, and the reaction mixture was stirred overnight at 80 °C. The reaction mixture was filtered, and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (1% v/v CH3OH in DCM) to afford compound 7f as a white solid (0.183 g, 82%). 1H NMR (500 MHz, CD3OD, δ): 1.36 (s, 12H), 7.33–7.36 (d, J = 16.30 Hz, 1H), 7.49–7.52 (t, J = 8.41 Hz, 1H), 7.61–7.64 (d, J = 16.47 Hz, 1H), 7.66–7.67 (d, J = 7.46, 1H), 7.73 (s, 1H), 7.83–7.85 (d, J = 7.75 Hz, 2H), 7.95–7.97 (d, J = 7.73, 1H), 8.24 (s, 1H); 13C NMR (125 MHz, CD3OD, δ): 25.19, 85.51, 126.33, 127.18, 129.00, 130.07, 130.41, 132.16, 132.61, 134.18, 136.79, 138.74, 138.86, 169.56; HRMS–ESI (m/z): [M + NH4]+ calcd for C21H22BClO4, 401.1675; found, 401.1666.
3-[(1E)-2-(2-Chloro-4-hydroxyphenyl)ethenyl]-benzoic Acid (7)
Compound 7f (0.100 g, 0.259 mmol) was dissolved in DCM (2.6 mL). N,N-Dimethyl-p-toluidine-N-oxide (0.060 g, 0.389 mmol) was added to the resulting solution, and the reaction mixture was then stirred overnight. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (2% v/v MeOH in DCM) to afford compound 7 as a white solid (0.051 g, 72%) with mp 213.6 °C. 1H NMR (500 MHz, CD3OD, δ): 6.77–6.68 (d, J = 8.63 Hz, 1H), 6.84 (s, 1H), 7.06–7.10 (d, J = 16.34 Hz, 1H), 7.45–7.48 (t, J = 7.72 Hz, 1H), 7.49–7.52 (d, J = 16.31 Hz, 1H), 7.65–7.67 (d, J = 8.63 Hz, 1H), 7.75–7.77 (d, J = 7.91 Hz, 1H), 7.89–7.91 (d, J = 7.78 Hz, 1H), 8.17 (s, 1H); 13C NMR (125 MHz, CD3OD, δ): 115.98, 117.09, 126.42, 127.43, 128.47, 128.48, 128.58, 129.60, 129.92, 131.70, 132.43, 135.06, 139.41, 159.45, 169.74; HRMS–ESI (m/z): [M − H]− calcd for C15H11ClO3, 273.0324; found, 273.0327.
3-[(1E)-2-(2-Chloro-4-boronic Acid)ethenyl]-benzoic (8)
Compound 7f (0.100 g, 0.260 mmol) was dissolved in 4:1 THF/H2O (2.6 mL). NaIO4 (0.28 g, 1.30 mmol) was added to the resulting solution, followed by 0.02 mL of 1.0 M HCl (0.02 mmol). The reaction mixture was then stirred overnight. The reaction mixture was diluted with H2O (2 mL) and extracted with EtOAc (3 × 3 mL). The organic layers were combined and washed with brine (5 mL), dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (1–3% v/v CH3OH in DCM) to afford compound 8 as a white crystalline solid (0.058 g, 75%) with mp 161.0 °C. 1H NMR (500 MHz, CD3OD, δ): 7.32–7.35 (d, J = 16.37 Hz, 1H), 7.51–7.54 (t, J = 7.73 Hz, 1H), 7.51–7.67 (m, 3H), 7.84–7.85 (d, J = 7.29 Hz, 2H), 7.97–7.98 (d, J = 7.76 Hz, 1H), 8.25 (s, 1H); 13C NMR (125 MHz, CD3OD, δ): 126.41, 126.94, 128.93, 130.04, 130.29, 132.07, 132.63, 133.31, 134.12, 135.95, 137.29, 138.82, 169.61; HRMS–ESI (m/z): [M − H]− calcd for the single methyl boronic ester C16H14BClO4, 314.0637; found, 314.0635.
1,1′-(1E)-(1,2-Ethenediyl)bis[2-chloro-4-bromo]-benzene (9a)
4-Bromo-1-(bromomethyl)-2-chloro-benzene (0.300 g, 1.05 mmol) was dissolved in neat triethylphosphite (0.217 mL, 1.26 mmol) and heated to 150 °C for 4 h. The reaction mixture was then cooled to 0 °C and diluted with DMF (8 mL). NaH (60% w/v in mineral oil, 0.054 g, 1.36 mmol) was added to the resulting solution, and the reaction mixture was stirred at 0 °C for 20 min. A solution of 4-bromo-2-chloro-benzaldehyde (7c; 0.230 g, 1.05 mmol) in DMF (2.5 mL) was then added dropwise. The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction mixture was diluted with EtOAc (8 mL), and washed with 10% w/v citric acid (10 mL), followed by brine (10 mL). The organic layer was then separated, dried with Na2SO4 (s), and filtered. The solvent was removed under reduced pressure, and the crude product was suspended in cold DCM (10 mL). The resulting precipitate was collected by filtration to afford compound 9a as a white solid (0.306 g, 72% yield over 2 steps). 1H NMR (500 MHz, CDCl3, δ): 7.39 (s, 2H), 7.41–7.43 (d, J = 8.42 Hz, 2H), 7.58 (s, 2H), 7.58–7.59 (d, J = 7.12 Hz, 2H); 13C NMR (125 MHz, CDCl3, δ): 121.99, 126.70, 127.83, 130.35, 132.45, 133.89, 134.30; ASAP–MS (m/z): M+• calcd for C14H8Br2Cl2, 403.8365; found, 403.8367.
1,1′-(1E)-(1,2-Ethenediyl)bis[2-chloro-4-boronic Acid Pinacol Ester]-benzene (9b)
Compound 9a (0.050 g, 0.123 mmol), KOAc (0.071 g, 0.742 mmol), bis(pinacolato)diboron (0.187 g, 0.742 mmol), and Pd(dppf)Cl2 (9 mg, 0.012 mmol) were added to a flame-dried Schlenk flask, which was then evacuated and backfilled with N2(g). Dioxane (2 mL) was deoxygenated by sonication under high vacuum and backfilled with N2(g). The deoxygenated dioxane was then added by cannula into the reaction flask, and the reaction mixture was heated to 80 °C and stirred overnight (Note: higher yields of the diboronated product were found at more dilute reaction concentrations). The reaction mixture was filtered, and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (50% v/v DCM in hexanes) to afford 9b as a white solid (0.056 g, 92%). 1H NMR (500 MHz, CDCl3, δ): 1.35 (s, 24H), 7.56 (s, 2H), 7.67–7.69 (d, J = 7.76 Hz, 2H), 7.73–7.75 (d, J = 7.85 Hz, 2H), 7.83 (s, 2H); 13C NMR (125 MHz, CDCl3, δ): 24.88, 84.18, 126.17, 127.94, 133.02, 133.45, 136.06, 137.45; ASAP–MS (m/z): [M + H]+ calcd for C26H32B2Cl2O4, 499.2009; found, 499.2001.
1,1′-(1E)-(1,2-Ethenediyl)bis[2-chloro-4-hydroxy]-benzene (9)
Compound 9b (0.020 g, 0.040 mmol) was dissolved in DCM (0.5 mL). N,N-Dimethyl-p-toluidine-N-oxide (0.018 g, 0.120 mmol) was added to the resulting solution, and the reaction mixture was stirred for 1 h. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (2% v/v MeOH in DCM) to afford compound 9 as a white solid (9 mg, 80%) with mp 203.9 °C. 1H NMR (500 MHz, CD3OD, δ): 6.75–6.77 (d, J = 8.61 Hz, 2H), 6.83 (s, 2H), 7.24 (s, 2H), 7.55–7.57 (d, J = 8.59 Hz, 2H); 13C NMR (125 MHz, CD3OD, δ): 115.93, 117.07, 125.22, 128.00, 128.36, 134.73, 159.09; HRMS–ESI (m/z): [M − H]− calcd for C14H10Cl2O2, 278.9985; found, 278.9985.
1,1′-(1E)-(1,2-Ethenediyl)bis[2-chloro-4-boronic acid]-benzene (10)
Compound 9b (0.020 g, 0.040 mmol) was dissolved in 4:1 THF/H2O (0.6 mL). NaIO4 (0.042 g, 0.200 mmol) was added to the resulting solution, followed by a few drops of 1.0 M HCl. The reaction mixture was then stirred overnight. The reaction mixture was then diluted with H2O (1 mL) and extracted with EtOAc (3 × 2 mL). The organic layers were combined and washed with saturated brine (3 mL), dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (3% v/v CH3OH in DCM) to afford compound 10 as a white crystalline solid (0.010 g, 74%) with mp 234.2 °C. 1H NMR (500 MHz, CD3OD, δ): 7.59 (s, 1H), 7.59–7.60 (d, J = 8.42 Hz, 2H), 7.66 (s, 1H), 7.78–7.80 (d, J = 7.78 Hz, 2H); 13C NMR (125 MHz, CD3OD, δ): 127.14, 128.69, 133.38, 134.25, 135.98; MALDI–MS (m/z): M+• calcd for C14H12B2Cl2O4, 336.03; found, 336.00.
2-Bromo-1-[(1E)-2-(3-bromophenyl)ethenyl]-4-chloro-benzene (11a)
3-Bromobenzyl bromide (0.500 g, 2.00 mmol) was dissolved in neat triethylphosphite (0.411 mL, 2.4 mmol), and the resulting solution was heated to 150 °C for 4 h. The reaction mixture was cooled to 0 °C and then diluted with DMF (10 mL). NaH (60% w/v in mineral oil, 0.096 g, 2.4 mmol) was added to the resulting solution, and the reaction mixture stirred at 0 °C for 20 min. A solution of 4-bromo-2-chloro-benzaldehyde (7c; 0.447 g, 2.04 mmol) in DMF (10 mL) was then added drop-wise. The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction mixture was diluted with EtOAc (20 mL), and washed with 10% w/v citric acid (30 mL) followed by brine (30 mL). The organic layer was separated, dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was suspended in ice-cold DCM (5 mL). The resulting precipitate was collected by filtration to afford compound 11a as a white solid (0.514 g, 69% over 2 steps). 1H NMR (500 MHz, CD3OD, δ): 7.09–7.13 (d, J = 16.3 Hz, 1H), 7.26–7.29 (d, J = 12.53 Hz, 2H), 7.35–7.39 (t, J = 7.61 Hz, 1H), 7.53 (s, 3H), 7.61–7.63 (d, J = 7.74 Hz, 1H), 7.79 (s, 1H); 13C NMR (125 MHz, CD3OD, δ): 124.42, 125.32, 126.40, 127.54, 127.67, 131.61, 131.80, 131.80, 132.97, 134.94, 135.71, 141.09; ASAP–MS (m/z): M+• calcd for C14H9Br2Cl, 369.8754; found, 369.8740.
2-Boronic Acid Pinacol Ester-1-[(1E)-2-(3-Boronic acid pinacol ester)ethenyl]-4-chloro-benzene (11b)
Compound 11a (0.100 g, 0.270 mmol), KOAc (0.152 g, 1.62 mmol), bis(pinacolato)diboron (0.410 g, 1.62 mmol), and Pd(dppf)Cl2 (0.0270 g, 0.020 mmol) were added to a flame-dried Schlenk flask, which was then evacuated and backfilled with N2(g). Dioxane (3.6 mL) was deoxygenated by sonication under high vacuum and backfilled with N2(g). The deoxygenated dioxane was then added by cannula into the reaction flask, and the reaction mixture was heated to 80 °C and stirred overnight. The reaction mixture was filtered, and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (2% v/v DCM in hexanes) to afford 11b as a white solid (0.111 g, 87%). 1H NMR (500 MHz, CD3OD, δ): 1.36 (s, 12H), 1.38 (s, 12H), 7.27–7.30 (d, J = 16.29 Hz, 1H), 7.38–7.41 (t, J = 7.47 Hz, 1H), 7.55–7.58 (d, J = 16.30 Hz, 1H), 7.64–7.66 (d, J = 7.73 Hz, 1H), 7.67–7.70 (t, J = 7.88 Hz, 2H), 7.72 (s, 1H), 7.81–7.82 (d, J = 7.75 Hz, 1H), 7.96 (s, 1H); 13C NMR (125 MHz, CD3OD, δ): 26.45, 26.48, 50.43, 50.60, 86.55, 86.74, 126.43, 128.29, 130.59, 132.18, 134.79, 135.29, 135.31, 135.40, 136.90, 138.03, 138.91, 140.44; HRMS–ESI (m/z): [M + NH4]+ calcd for C26H33B2ClO4, 484.2607; found, 484.2600.
2-Boronic Acid-1-[(1E)-2-(3-boronic Acid)ethenyl]-4-chloro-benzene (11)
Compound 11b (0.100 g, 0.214 mmol) was dissolved in 4:1 THF/H2O (2.1 mL). NaIO4 (0.227 g, 1.07 mmol) was added to the resulting solution, followed by a few drops of 1.0 M HCl. The reaction mixture was then stirred overnight. The reaction mixture was diluted with H2O (2 mL) and extracted with EtOAc (3 × 2 mL). The organic layers were combined and washed with brine (4 mL), dried with Na2SO4(s), and filtered. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (2% v/v CH3OH in DCM) to afford compound 11 as a white crystalline solid (0.040 g, 63%) with mp 146.3 °C. 1H NMR (500 MHz, CD3OD, δ): 7.24–7.27 (d, J = 16.3 Hz, 1H), 7.37–7.40 (t, J = 7.57 Hz, 1H), 7.52–7.57 (m, 3H), 7.63–7.64 (d, J = 4.84 Hz, 2H), 7.79 (s, 2H); 13C NMR (125 MHz, CD3OD, δ): 125.12, 126.78, 128.85, 129.02, 129.16, 129.80, 133.24, 133.95, 134.31, 135.93, 137.64; HRMS–ESI (m/z): [M − H]− calcd for the single methyl boronate ester C15H14B2ClO4, 315.0772; found, 315.0772.
Protein Expression and Purification
Plasmids that direct the expression of wild-type TTR and its V30M variant were prepared in the pET32b vector from Merck KGaA (Darmstadt, Germany) by standard methods.76 To create the plasmid encoding V30M TTR, two double-stranded DNA fragments were prepared by PCR using complementary primers containing V30M-generating substitutions and gene-specific primers targeting opposite termini for assembly with the plasmid fragment. Wild-type TTR and its V30M variant were produced in Escherichia coli strain BL-21 from Merck KGaA cultured in Luria–Bertani medium containing ampicillin (200 μM) at 37 °C. Gene expression was induced when OD600 nm reached ~2.0, and cells were then grown for an additional 4 h at 37 °C. Cell pellets were resuspended in 20 mM Tris–HCl buffer, pH 7.4, containing EDTA (1.0 mM) and lysed with a high-pressure cell disruptor from Constant Systems (Kennesaw, GA). The soluble fraction was isolated by centrifugation for 10 min at 10,500g and for 1 h at 30,000g.
Wild-type TTR and its V30M variant were purified as described previously,77 with minor modifications. The lysate was fractionated with aqueous ammonium sulfate at 60–85% saturation. The precipitate was dissolved in 20 mM Tris–HCl buffer, pH 7.8, containing EDTA (1.0 mM) and dialyzed overnight against this same buffer. The isolate was clarified at 30,000g for 30 min, filtered, and applied to a Hitrap Q HP column from GE Healthcare Life Sciences (Pittsburgh, PA) that had been equilibrated with the dialysis buffer. TTR was eluted with the same buffer containing NaCl (1.0 M) and was subjected to gel-filtration chromatography on a Superdex 75 column from GE Healthcare Life Sciences (Pittsburgh, PA) that had been pre-equilibrated with 10 mM sodium phosphate buffer, pH 7.6, containing KCl (100 mM). Pure tetrameric TTR eluted at ~0.6 column volumes as monitored by SDS–PAGE. The concentration of wild-type TTR and its V30M variant was determined from the A280 nm by using ε = 18.5 × 103 M−1cm−1 and confirmed with a bicinchoninic acid (BCA) assay using a kit from Pierce Biotechnology (Rockford, IL).
Competitive Fluorescence Assay
Fluorescence measurements were performed with a Photon Technology International Quantamaster spectrofluorometer (Edison, NJ). Wild-type TTR was found to form a complex with ANS that has a Kd value of 3.2 μM (data not shown).78 To determine the affinity for ligands, wild-type TTR tetramers (500 nM) were incubated in 2.00 mL of 10 mM sodium phosphate buffer, pH 7.6, containing KCl (100 mM) and ANS (5.0 μM) until the fluorescence signal (excitation: 410 nm; emission: 460 nm) was stable (~30 min). A ligand (1 nM–10 μM) was then added in aliquots (5 μL) from a stock solution in dimethyl formamide (DMF). The fluorescence intensity at each ligand concentration was recorded before adding the next dose. Control experiments with only the solvent (DMF) revealed that the fluorescence intensity dropped proporationally with the dilution factor. Accordingly, average intensities were adjusted for the dilution incurred upon adding ligand and were expressed as a percent change from the initial measurement. The final DMF concentration was 6.3% by volume after 27 additions of ligand.
The data did not fit well to the one- or two-site competitive binding models used previously to describe other systems.78 The asymmetric behavior is likely a consequence of two distinct binding events and a modest decrease in fluorescence intensity resulting from the first ANS dissociation event. The steepest inflection point at higher ligand concentrations can be attributed to the half-maximal concentration required to compete ANS from the second binding site. The Prusoff–Cheng relation was used to account for this competition, resulting in a logistic equation:79
| (1) |
where Kd,2 is the equilibrium dissociation constant of the second site, n is the Hill coefficient, and S is the symmetry parameter. Values of Kd,2 were determined with Prism 6 software from Graphpad (La Jolla, CA) by holding constant the ligand concentration, ANS concentration, and Kd,ANS = 3.2 μM and varying other parameters to maximize the value of R2, which was >0.99 for all datasets. The value of Kd,2 = (373 ± 10) nM for the TTR·tafamidis complex determined with this method is similar to the value of Kd,2 = 278 nM determined with isothermal titration calorimetry.21 Values of Kd,1 were not obtainable by this method, though modest inflection points were observed in the low nanomolar range (Figure S1). Reported values of Kd,1 for other ligands fall within this range, however, our efforts to improve kinetic stabilization properties focus on reducing the Kd,2 value.
Fibril-Formation Assay
Light-scattering at 400 nm was used to assess the formation of fibrils under acidic conditions, as described previously.80 In this assay, turbidity is assumed to be proportional to the mass of the protein converted to fibrils. Ligands (7.2 or 14.4 μM) were incubated with TTR tetramers (7.2 μM) for 30 min prior to twofold dilution with 50 mM sodium acetate buffer, pH 4.4, containing KCl (100 mM). Absorbance at 400 nm was measured ten times with four replicates in clear, flat-bottomed, 96-well plates at 0 and 96 h using an M1000 plate reader from Tecan (Maennedorf, Switzerland). Percent fibril formation was calculated from the difference in the light scattering that accumulated after 96 h in ligand-containing wells versus wells containing only vehicle (DMF), using the equation:
| (2) |
Standard deviations from the four replicates were propagated through this calculation.
Protein Crystallization and X-ray Structure Determination
Crystals were grown by vapor diffusion using a solution of wild-type TTR (~6.0 mg/mL in 10 mM sodium phosphate buffer, pH 7.6, containing 100 mM KCl) and stilbenes 2–8, 10, or 11 (7.2 mM, added from a stock solution in DMF). Hanging drops (2 μL of solution containing protein and ligand plus 2 μL of mother liquor) were equilibrated with a reservoir of 1.0–1.3 M sodium citrate buffer, pH 5.5, containing glycerol (1–3% v/v).5 Crystals appeared after 1–3 days. TTR precipitated in the presence of stilbene 9 (7.2 mM). Crystals were cryoprotected by brief transfer into a solution of 1.5 M sodium citrate buffer, pH 5.5, containing glycerol (10% v/v). Diffraction data were collected at Sector 21 of the Life Sciences Collaborative Access Team (LS-CAT) at the Advanced Photon Source of Argonne National Laboratory (Argonne, IL). Data were reduced using HKL2000 (Tables S2–S10).81 Boronate ester restraints were obtained by measuring the bond lengths and angles from ten CSD small-molecule structures (Table S1). Initial phases were obtained by molecular replacement using the protein atoms of Protein Data Bank (PDB) entry 2qgb as a model.64 Refinement and model building were conducted with the programs Phenix and Coot (Tables S2–S10 and Figures S4–S12).82,83 Ligand models in idealized geometries were prepared with the program WebMO, and restraints were prepared in Phenix with the program eLBOW. Restraints generated by eLBOW were modified to impose planarity on the four carbon atoms in the olefin of the stilbene and on the carbon, boron, and two oxygen atoms in a boronic acid group. Short interatomic distances in TTR·ligand complexes are listed in Table S11. Atomic coordinates and structure factors for all nine TTR·ligand complexes have been deposited in the PDB.
Supplementary Material
Acknowledgments
We are grateful to Drs. R. M. Murphy and C. L. Jenkins for contributive discussions. I.W.W. was supported by Biotechnology Training Grant T32 GM008349 (NIH) and a Genentech Predoctoral Fellowship. This work was supported by Grants R01 GM044783 (NIH) and MCB 1518160 (NSF), and made use of NMRFAM (University of Wisconsin–Madison), which is supported by Grant P41 GM103399 (NIH).
ABBREVIATIONS USED
- EDTA
ethylenediaminetetraacetic acid
- FDA
Food and Drug Administration USA
- PDB
protein data bank
- TTR
transthyretin
- T4
thyroxin
Footnotes
Notes
The authors declare no competing financial interest.
PDB ID CODES
5u48, TTR·2 complex; 5u49, TTR·3 complex; 5u4a, TTR·4 complex; 5u4b, TTR·5 complex; 5u4c, TTR·6 complex; 5u4d, TTR·7 complex; 5u4e, TTR·8 complex; 5u4f, TTR·10 complex; 5u4g, TTR·11 complex.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.xxxxxxx.
Assay data; crystallographic data; HPLC traces for compounds 2, 3, and 5; 1H and 13C NMR spectra of all synthetic compounds; molecular formula strings
References
- 1.Dobson CM. Protein folding and misfolding. Science. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 2.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 3.Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 2014;15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- 4.Richardson SJ. Cell and molecular biology of transthyretin and thyroid hormones. Int Rev Cytol. 2007;258:137–193. doi: 10.1016/S0074-7696(07)58003-4. [DOI] [PubMed] [Google Scholar]
- 5.Klabunde T, Petrassi HM, Oza VB, Raman P, Kelly JW, Sacchettini JC. Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol. 2000;7:312–321. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- 6.Hamilton JA, Benson MD. Transthyretin: A review from a structural perspective. Cell Mol Life Sci. 2001;58:1491–1521. doi: 10.1007/PL00000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hund E. Familial amyloidotic polyneuropathy: Current and emerging treatment options for transthyretin-mediated amyloidosis. Appl Clin Genet. 2012;5:37–41. doi: 10.2147/TACG.S19903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang DT, Joshi G, Cho PY, Johnson JA, Murphy RM. Transthyretin as both a sensor and a scavenger of β-amyloid oligomers. Biochemistry. 2013;52:2849–2861. doi: 10.1021/bi4001613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP, Johnson JA. Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APPSW mice resulting in tau phosphorylation and loss of hippocampal neurons: Support for the amyloid hypothesis. J Neurosci. 2004;24:7707–7717. doi: 10.1523/JNEUROSCI.2211-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X, Zhang X, Ladiwala ARA, Du D, Yadav JK, Tessier PM, Wright PE, Kelly JW, Buxbaum JN. Mechanisms of transthyretin inhibition of β-amyloid aggregation in vitro. J Neurosci. 2013;33:19423–19433. doi: 10.1523/JNEUROSCI.2561-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller DW, Dill KA. Ligand binding to proteins: The binding landscape model. Protein Sci. 1997;6:2166–2179. doi: 10.1002/pro.5560061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Celej MS. Protein stability induced by ligand binding correlates with changes in protein flexibility. Protein Sci. 2003;12:1496–1506. doi: 10.1110/ps.0240003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 14.Hingorani KS, Metcalf MC, Deming DT, Garman SC, Powers ET, Gierasch LM. Ligand-promoted protein folding by biased kinetic partitioning. Nat Chem Biol. 2017;13:369–371. doi: 10.1038/nchembio.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Sullivan C, Tompson FW. LX.—Invertase: A contribution to the history of an enzyme or unorganised ferment. J Chem Soc. 1890;57:834–931. [Google Scholar]
- 16.Nencetti S, Orlandini E. TTR fibril formation inhibition: Is there a SAR? Curr Med Chem. 2012;19:2356–2379. doi: 10.2174/092986712800269326. [DOI] [PubMed] [Google Scholar]
- 17.Obici L, Merlini G. An overview of drugs currently under investigation for the treatment of transthyretin-related hereditary amyloidosis. Expert Opin Invest Drugs. 2014;23:1239–1251. doi: 10.1517/13543784.2014.922541. [DOI] [PubMed] [Google Scholar]
- 18.Johnson SM, Wiseman RL, Sekijima Y, Green NS, Adamski-Werner SL, Kelly JW. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: A focus on the transthyretin amyloidoses. Acc Chem Res. 2005;38:911–921. doi: 10.1021/ar020073i. [DOI] [PubMed] [Google Scholar]
- 19.Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogs stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J Med Chem. 2004;47:355–374. doi: 10.1021/jm030347n. [DOI] [PubMed] [Google Scholar]
- 20.Castano A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congestive Heart Failure. 2012;18:315–319. doi: 10.1111/j.1751-7133.2012.00303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bulawa CE, Connelly S, DeVit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, Wilson IA, Kelly JW, Labaudinière R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109:9629–9634. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sekijima Y. Recent progress in the understanding and treatment of transthyretin amyloidosis. J Clin Pharm Ther. 2014;39:225–233. doi: 10.1111/jcpt.12145. [DOI] [PubMed] [Google Scholar]
- 23.Scott LJ. Tafamidis: A review of its use in familial amyloid polyneuropathy. Drugs. 2014;74:1371–1378. doi: 10.1007/s40265-014-0260-2. [DOI] [PubMed] [Google Scholar]
- 24.Johnson DS, Weerapana E, Cravatt BF. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med Chem. 2010;2:949–964. doi: 10.4155/fmc.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller RM, Taunton J. Targeting protein kinases with selective and semipromiscuous covalent inhibitors. Methods Enzymol. 2014;548:93–116. doi: 10.1016/B978-0-12-397918-6.00004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baillie TA. Targeted covalent inhibitors for drug design. Angew Chem, Int Ed. 2016;55:2–17. doi: 10.1002/anie.201601091. [DOI] [PubMed] [Google Scholar]
- 27.Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: From delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency–approved drug. J Mol Biol. 2012;421:185–203. doi: 10.1016/j.jmb.2011.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi S, Connelly S, Reixach N, Wilson IA, Kelly JW. Chemoselective small molecules that covalently modify one lysine in a non-enzyme protein in plasma. Nat Chem Biol. 2010;6:133–139. doi: 10.1038/nchembio.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi S, Ton Ong DS, Kelly JW. A stilbene that binds selectively to transthyretin in cells and remains dark until it undergoes a chemoselective reaction to create a bright blue fluorescent conjugate. J Am Chem Soc. 2010;132:16043–16051. doi: 10.1021/ja104999v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suh EH, Liu Y, Connelly S, Genereux JC, Wilson IA, Kelly JW. Stilbene vinyl sulfonamides as fluorogenic sensors of and traceless covalent kinetic stabilizers of transthyretin that prevent amyloidogenesis. J Am Chem Soc. 2013;135:17869–17880. doi: 10.1021/ja408230k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grimster NP, Connelly S, Baranczak A, Dong J, Krasnova LB, Sharpless KB, Powers ET, Wilson IA, Kelly JW. Aromatic sulfonyl fluorides covalently kinetically stabilize transthyretin to prevent amyloidogenesis while affording a fluorescent conjugate. J Am Chem Soc. 2013;135:5656–5668. doi: 10.1021/ja311729d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baranczak A, Liu Y, Connelly S, Han Du WG, Greiner ER, Genereux JC, Wiseman RL, Eisele YS, Bradbury NC, Dong J, Noodleman L, Sharpless KB, Wilson IA, Encalada SE, Kelly JW. A fluorogenic aryl fluorosulfate for intraorganellar transthyretin imaging in living cells and in Caenorhabditis elegans. J Am Chem Soc. 2015;137:7404–7414. doi: 10.1021/jacs.5b03042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lorand JP, Edwards JO. Polyol complexes and structure of the benzeneboronate ion. J Org Chem. 1958;24:769–774. [Google Scholar]
- 34.Peters JA. Interactions between boric acid derivatives and saccharides in aqueous media: Structures and stabilities of resulting esters. Coord Chem Rev. 2014;268:1–22. [Google Scholar]
- 35.Adams J. The development of proteasome inhibitiors as anticancer drugs. Cancer Cell. 2003;5:417–421. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 36.Antonov VK, Ivanina TV, Berezin IV, Martinek K. n-Alkylboronic acids as bifunctional reversible inhibitors of α-chymotrypsin. FEBS Lett. 1970;7:23–25. doi: 10.1016/0014-5793(70)80607-x. [DOI] [PubMed] [Google Scholar]
- 37.Koehler KA, Lienhard GE. 2-Phenylethaneboronic acid, a possible transition-state analog for chymotrypsin. Biochemistry. 1971;10:2477–2483. doi: 10.1021/bi00789a008. [DOI] [PubMed] [Google Scholar]
- 38.Kettner C, Shenvi A. Inhibition of the serine proteases leukocyte elastase, pancreatic elastase, cathepsin G, and chymotrypsin by peptide boronic acids. J Biol Chem. 1984;259:15106–15114. [PubMed] [Google Scholar]
- 39.Smoum R, Rubinstein A, Dembitsky VM, Srebnik M. Boron containing compounds as protease inhibitors. Chem Rev. 2012;112:4156–4220. doi: 10.1021/cr608202m. [DOI] [PubMed] [Google Scholar]
- 40.Trippier PC, McGuigan C. Boronic acids in medicinal chemistry: Anticancer, antibacterial and antiviral applications. MedChemComm. 2010;1:183–198. [Google Scholar]
- 41.Adamczyk-Woźniak A, Borys KM, Sporzyński A. Recent developments in the chemistry and biological applications of benzoxaboroles. Chem Rev. 2015;115:5224–5247. doi: 10.1021/cr500642d. [DOI] [PubMed] [Google Scholar]
- 42.Ellis GA, Palte MJ, Raines RT. Boronate-mediate biologic delivery. J Am Chem Soc. 2012;134:3631–3634. doi: 10.1021/ja210719s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma R, Shi L. Phenylboronic acid-based glucose-responsive polymeric nanoparticles: Synthesis and applications in drug delivery. Polym Chem. 2014;5:1503. [Google Scholar]
- 44.Wang J, Wu W, Jiang X. Nanoscaled boron-containing delivery systems and therapeutic agents for cancer treatment. Nanomedicine. 2015;10:1149–1163. doi: 10.2217/nnm.14.213. [DOI] [PubMed] [Google Scholar]
- 45.Andersen KA, Smith TP, Lomax JE, Raines RT. Boronic acid for the traceless delivery of proteins into cells. ACS Chem Biol. 2016;11:319–323. doi: 10.1021/acschembio.5b00966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu X, Li Z, Chen XX, Fossey JS, James TD, Jiang YB. Selective sensing of saccharides using simple boronic acids and their aggregates. Chem Soc Rev. 2013;42:8032–8048. doi: 10.1039/c3cs60148j. [DOI] [PubMed] [Google Scholar]
- 47.Lippert AR, Van de Bittner GC, Chang CJ. Boronate oxidation as a bioorthogonal reaction approach for studying the chemistry of hydrogen peroxide in living systems. Acc Chem Res. 2011;44:793–804. doi: 10.1021/ar200126t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoang TT, Smith TP, Raines RT. A boronic acid conjugate of angiogenin that shows ROS-responsive neuroprotective activity. Angew Chem, Int Ed. 2017;56:2619–2622. doi: 10.1002/anie.201611446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whyte GF, Vilar R, Woscholski R. Molecular recognition with boronic acids—applications in chemical biology. J Chem Biol. 2013;6:161–174. doi: 10.1007/s12154-013-0099-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cal PMSD, Frade RFM, Cordeiro C, Gois PMP. Reversible lysine modification on proteins using functionalized boronic acids. Chem Eur J. 2015;21:8182–8187. doi: 10.1002/chem.201500127. [DOI] [PubMed] [Google Scholar]
- 51.Akçay G, Belmonte MA, Aquila B, Chuaqui C, Hird AW, Lamb ML, Rawlins PB, Su N, Tentarelli S, Grimster NP, Su Q. Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain. Nat Chem Biol. 2016;12:931–936. doi: 10.1038/nchembio.2174. [DOI] [PubMed] [Google Scholar]
- 52.Hall DG. Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials. Wiley–VCH Verlag; Weinheim, Germany: 2011. [Google Scholar]
- 53.Bull SD, Davidson MG, van den Elsen JMH, Fossey JS, Jenkins ATA, Jiang YB, Kubo Y, Marken F, Sakurai K, Zhao J, James TD. Exploiting the reversible covalent bonding of boronic acids—recognition, sensing, and assembly. Acc Chem Res. 2012;46:312–326. doi: 10.1021/ar300130w. [DOI] [PubMed] [Google Scholar]
- 54.Toxicological profile for boron. US Agency for Toxic Substances and Disease Registry; [Accessed December 12: 2016]. http://www.atsdr.cdc.gov/toxprofiles/tp26-c6.pdf. [PubMed] [Google Scholar]
- 55.Baures PW, Peterson SA, Kelly JW. Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorg Med Chem. 1998;6:1389–1401. doi: 10.1016/s0968-0896(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 56.Johnson SM, Cohen SM, Wilson IA, Kelly JW. Toward optimization of the linker substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J Med Chem. 2008;51:6348–6358. doi: 10.1021/jm800435s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bourgault S, Choi S, Buxbaum JN, Kelly JW, Price JL, Reixach N. Mechanisms of transthyretin cardiomyocyte toxicity inhibition by resveratrol analogs. Biochem Biophys Res Commun. 2011;410:707–713. doi: 10.1016/j.bbrc.2011.04.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cotrina EY, Pinto M, Bosch L, Vila M, Blasi D, Quintana J, Centeno NB, Arsequell G, Planas A, Valencia G. Modulation of the fibrillogenesis inhibition properties of two transthyretin ligands by halogenation. J Med Chem. 2013;56:9110–9121. doi: 10.1021/jm401061w. [DOI] [PubMed] [Google Scholar]
- 59.Palaninathan SK. Nearly 200 X-ray crystal structures of transthyretin—what do they tell us about this protein and the design of drugs for TTR amyloidoses? Curr Med Chem. 2012;19:2324–2342. doi: 10.2174/092986712800269335. [DOI] [PubMed] [Google Scholar]
- 60.Muzio T, Cody V, Luft JR, Pangborn W, Wojtczak A. Complex of rat transthyretin with tetraiodothyroacetic acid refined at 2.1 and 1.8 Å resolution. Acta Biochim Polonica. 2001;48:877–884. [PubMed] [Google Scholar]
- 61.Oza VB, Smith C, Raman P, Koepf EK, Lashuel HA, Petrassi HM, Chiang KP, Powers ET, Sacchettini JC, Kelly JW. Synthesis, structure, and activity of diclofenac analogues as transthyretin amyloid fibril formation inhibitors. J Med Chem. 2002;45:321–332. doi: 10.1021/jm010257n. [DOI] [PubMed] [Google Scholar]
- 62.Neumann P, Cody V, Wojtczak A. Ligand binding at the transthyretin dimer–dimer interface: Structure of the transthyretin–T4Ac complex at 2.2 Å resolution. Acta Crystallogr Sect D, Biol Crystallogr. 2005;61:1313–1319. doi: 10.1107/S0907444905022523. [DOI] [PubMed] [Google Scholar]
- 63.Palaninathan SK, Mohamedmohaideen NN, Orlandini E, Ortore G, Nencetti S, Lapucci A, Rossello A, Freundlich JS, Sacchettini JC. Novel transthyretin amyloid fibril formation inhibitors: Synthesis, biological evaluation, and X-ray structural analysis. PLoS ONE. 2009;4:e6290. doi: 10.1371/journal.pone.0006290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnson SM, Connelly S, Wilson IA, Kelly JW. Biochemical and structural evaluation of highly selective 2-arylbenzoxazole-based transthyretin amyloidogenesis inhibitors. J Med Chem. 2008;51:260–270. doi: 10.1021/jm0708735. [DOI] [PubMed] [Google Scholar]
- 65.Society of Chemistry; London, UK: [accessed December 7: 2016]. www.chemspider.com. [Google Scholar]
- 66.Groll M, Berkers CR, Ploegh HL, Ovaa H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure. 2006;14:451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 67.Bone R, Frank D, Kettner CA, Agard DA. Structural analysis of specificity: α-Lytic protease complexes with analogues of reaction intermediates. Biochemistry. 1989;28:7600–7609. doi: 10.1021/bi00445a015. [DOI] [PubMed] [Google Scholar]
- 68.Eriksson AE, Baase WA, Zhang XJ, Heinz DW, Blaber M, Baldwin EP, Matthews BW. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science. 1992;255:178–183. doi: 10.1126/science.1553543. [DOI] [PubMed] [Google Scholar]
- 69.Kawasaki Y, Chufan EE, Lafont V, Hidaka K, Kiso Y, Mario Amzel L, Freire E. How much binding affinity can be gained by filling a cavity? Chem Biol Drug Des. 2010;75:143–151. doi: 10.1111/j.1747-0285.2009.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Voth AR, Khuu P, Oishi K, Ho PS. Halogen bonds as orthogonal molecular interactions to hydrogen bonds. Nat Chem. 2009;1:74–79. doi: 10.1038/nchem.112. [DOI] [PubMed] [Google Scholar]
- 71.Riley KE, Merz KM., Jr Insights into the strength and origin of halogen bonding: The halobenzene-formaldehyde dimer. J Phys Chem A. 2007;111:1688–1694. doi: 10.1021/jp066745u. [DOI] [PubMed] [Google Scholar]
- 72.Yamamoto Y, Matsumura T, Takao N, Yamagishi H, Takahashi M, Iwatsuki S, Ishihara K. Fast trigonal/tetragonal interconversion followed by slow chelate-ring closure in the complexation of boronic acids. Inorg Chim Acta. 2005;358:3355–3361. [Google Scholar]
- 73.Peters JA. Interactions between boric acid derivatives and saccharides in aqueous media: Structures and stabilities of resulting esters. Coord Chem Rev. 2015;268:1–22. [Google Scholar]
- 74.Alhamadsheh MM, Connelly S, Cho A, Reixach N, Powers ET, Pan DW, Wilson IA, Kelly JW, Graef IA. Potent kinetic stabilizers that prevent transthyretin-mediated cardiomyocyte proteotoxicity. Sci Transl Med. 2011;3:97ra81. doi: 10.1126/scitranslmed.3002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhu C, Wang R, Falck JR. Mild and rapid hydroxylation of aryl/heteroaryl boronic acids and boronate esters with N-oxides. Org Lett. 2012;14:3494–3497. doi: 10.1021/ol301463c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Windsor IW, Raines RT. Fluorogenic assay for inhibitors of HIV-1 protease with sub-picomolar affinity. Sci Rep. 2015;5:11286. doi: 10.1038/srep11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McCutchen SL, Colon W, Kelly JW. Transthyretin mutation Leu-55-Pro significantly alters tetramer stability and increases amyloidogenicity. Biochemistry. 1993;32:12119–12127. doi: 10.1021/bi00096a024. [DOI] [PubMed] [Google Scholar]
- 78.Grimm FA, Lehmler HJ, He X, Robertson LW, Duffel MW. Sulfated metabolites of polychlorinated biphenyls are high-affinity ligands for the thyroid hormone transport protein transthyretin. Environ Health Perspect. 2013;121:657–662. doi: 10.1289/ehp.1206198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Giraldo J, Vivas NM, Vila E, Badia A. Assessing the (a)symmetry of concentration-effect curves: Empirical versus mechanistic models. Pharmacol Ther. 2002;95:21–45. doi: 10.1016/s0163-7258(02)00223-1. [DOI] [PubMed] [Google Scholar]
- 80.Miroy GJ, Lai Z, Lashuel HA, Peterson SA, Strang C, Kelly JW. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc Natl Acad Sci U S A. 1996;93:15051–15056. doi: 10.1073/pnas.93.26.15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 82.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D, Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D, Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.