

Abstract
Aims
This research was conducted to evaluate the hypothesis that gastric ulcers caused by the NSAID diclofenac sodium (DCF) can be prevented by the soluble epoxide hydrolase inhibitor TPPU.
Main methods
Mice were administered a single dose of 10, 30 or 100 mg/kg of DCF. Once an ulcerative dose of DCF was chosen, mice were pretreated with TPPU for 7 days at 0.1 mg/kg to evaluate anti-ulcer effects of the sEH inhibitor on anatomy, histopathology, pH, inflammatory markers and epithelial apoptosis of stomachs.
Key findings
Diclofenac caused ulceration of the stomach at a dose of 100 mg/kg and a time post dose of 6 hours. Ulcers generated under these conditions were associated with a significant increase in the levels of TNF-α and IL-6 in serum and increased apoptosis compared to control mice. Pretreatment with TPPU resulted in a decrease of ulceration in mice treated with DCF with a significant decrease in the level of apoptosis, TNF-α and IL-6 in the serum in comparison to diclofenac-treated mice. TPPU did not affect the pH of the stomach, whereas omeprazole elevated the pH of the stomach as expected. A similar anti-ulcer effect was observed in sEH gene knockout mice treated with DCF.
Significance
The sEH inhibitor TPPU decreases NSAID-induced stomach ulcers.
Keywords: Diclofenac, soluble epoxide hydrolase inhibitor TPPU, stomach ulcer, apoptosis, TNF-α, IL-6
1. Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) have a long history of use worldwide for the treatment of pain, inflammation and fever. However, the gastrointestinal injury is an important use-limiting side effect of NSAIDs [1]. Approximately 50% of the patients who regularly use NSAIDs display gastric erosion and it is estimated that 15–30% have ulcers. Clinical treatment of this gastrointestinal toxicity is required for 3–4.5% of these patients [2]. Over 30 million people use NSAIDs daily [3], making gastrointestinal toxicity a major health issue. Though many drugs are available to alleviate NSAID-induced ulceration, inhibitors of soluble epoxide hydrolase (sEH), which decrease pain [4] and inflammation [5] hold promise to decrease such ulcers [1, 6].
The prostanoid PGE2 is an important potent lipid mediator of pain and inflammation. However, in the gastrointestinal system, its key function is to protect the stomach from acid secreted by parietal glands through stimulating the production of bicarbonate and mucus. The bicarbonates neutralize acid while the mucus covers the stomach mucosa and prevents contact of acid with mucosal cells. NSAIDs decrease the production of prostanoids, in particular, PGE2, but this lack of protective actions of PGE2 then leads to gastric lesion i.e. erosion and/ ulcers of gastric cells. Clinical treatment of gastric ulcers typically involves the use of agents which either decrease the secretion of acid or mimic the actions of PGE2 (prostaglandin analogs). Omeprazole (OME), the widely used inhibitor of proton pump (H+K+-ATPase) decreases the secretion of acid in the stomach [1]. Despite the wealth of knowledge on the effects of NSAIDs, the mechanistic aspects of NSAID-induced gastric ulcers are still not fully understood. In addition to PGE2, multiple other biological pathways seem responsible for the formation of NSAID-induced ulcers [7–11]. While NSAIDs suppress inflammation, NSAID-induced ulcers are associated with increases in the levels of inflammatory markers such as TNF-α and IL-6 [8, 12]. Although a downstream consequence of COX-2 activity, TNF-α is proposed to play an important role in ulcer pathways activated by prolonged use of NSAID. Anti-TNF-α antibodies for example significantly decrease necrotic and apoptotic lesions associated with ulcers in rats [13, 14] and TNF-α knockout mouse is more resistant to NSAID-induced ulceration [8].
In addition to its pro-inflammatory functions, TNF-α is known to be a strong inducer of apoptosis through at least several mechanisms [14]. Furthermore, a role for apoptosis in ulceration is supported in numerous studies [15–17]. Consequently, agents which decrease NSAID-induced apoptosis also decrease stomach ulcers [17]. NSAID-induced apoptosis of gastric cells seems to be driven by not only TNF-α but also by activated endoplasmic reticulum (ER) stress responses to an increase in intracellular Ca2+ concentrations [18].
The FitzGerald hypothesis suggests adverse effects of NSAIDs are due, in part, to an alteration in the ratio of thromboxane B2 (TxB2) and prostaglandin E2 (PGE2) [19]. Inhibitors of sEH not only synergize the anti-inflammatory and analgesic effects of NSAIDs but also balance the TxB2/PGE2 altered by treatment with some COX inhibitors [20]. Similarly, genetic deletion of sEH enzyme also leads to alterations in the level of TxB2 [21]. Previously published data suggest sEH inhibitors should protect animals from NSAID driven gastric ulcers [6, 22]. Several sEH inhibitors are in human clinical trials [23, 24] and these molecules display synergistic effects with NSAIDs [5]. Earlier, we demonstrated decreased NSAID-induced intestinal ulcers in mice treated with diclofenac and an inhibitor of sEH [6, 22]. NSAID-induced intestinal ulcer [6] is a serious problem in man. It is more likely that sEH inhibitor would decrease the dose of NSAID and mitigate adverse effect such as ulceration. Therefore, understanding the interactions of inhibition of sEH with NSAID-induced ulceration is important. Here, we evaluated the effects of inhibition of sEH on NSAID-induced gastric ulcers using a mouse model.
The sEH inhibitor trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB) was shown to decrease piroxicam (a COX inhibitor)-induced inflammatory bowel disease [22]. The sEH inhibitors also decrease apoptosis of cells [25] and ER stress [26, 27]. Therefore, we evaluated the preventative effects of N-[1-(1-oxopropyl)-4-piperidinyl]-N′-[4-(trifluoromethoxy)phenyl)-urea (TPPU) on diclofenac-induced gastric ulcers and the effect of this treatment on markers of inflammation (TNF-α and IL-6), and apoptosis. We selected the most commonly used sEH inhibitor in the field, TPPU, which is highly potent, has high oral availability and a longer half-life than the previously used t-AUCB in mice [28, 29]. Commercial availability of TPPU from several vendors should enable other laboratories to follow up on our results. Ulu et al. [30] recently demonstrated that when given in drinking water TPPU had high oral availability and quickly reached a steady state. Blood levels linearly correlated with the dose in drinking water. Here the effects of TPPU on diclofenac-induced gastric ulcers are reported. In addition, the effect of treatment was studied on pH of the stomach to find out whether sEH inhibitors influence ulceration through modulating pH. Omeprazole, a proton pump inhibitor (PPI) was used as a standard for comparison.
2. Materials and methods
2.1. Materials
Diclofenac (Sigma-Aldrich Co., St. Louis, MO), Mouse TNF-α and IL-6 ELISA kits (Thermo Scientific, Rockford, IL), Pierce® BCA protein assay reagent (Thermo Scientific, Rockford, IL), TACS® 2 TdT-DAB in situ apoptosis detection kit (Trevigen Inc., Gaithersburg, MD) were purchased. TPPU was synthesized previously in the laboratory and is available from commercial sources [28]. All other reagents were of analytical grade.
2.2. Animals
Male Swiss Webster mice, C57BL6 and sEH knockout mice in a C57BL6 background weighing 34–41 g were used for the research. Animals were 4 months old. The Institutional Animal Care and Use Committee (IACUC) of University of California, Davis reviewed and approved the animal protocol. The National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978) guidelines has been followed to conduct animal experiments. The animals were maintained in 12 h dark and 12 h light cycle with unlimited access to mouse chow and water. However, animals were kept fasting for 12 h before sacrifice to assess effects of treatment on stomach ulcers. In the cage, animals were kept 2.5 cm above bedding, by use of a wire mesh, to prevent them from eating their feces and bedding during fasting.
The sEH knockout mice were provided originally by Dr. Christopher J. Sinal from the Dr. Frank J. Gonzalez lab (Laboratory of Metabolism, Division of Basic Sciences, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA) and a colony of knockout mice has been maintained at the University of California, Davis under Mouse Biology Program with repeated back breeding into C57BL6 mice from Jackson Laboratories [31, 32, 33]. Therefore, C57BL6 mice were also used as controls for sEH (−/−) mice.
2.3. Induction and scoring of gastric ulcers
Diclofenac-induced ulceration was studied in mice using 3 doses i.e. 10, 30 and 100 mg/kg. Six hours after diclofenac dosing, mice were anesthetized with isoflurane. The abdominal cavity was opened to expose the stomach, which was opened along the greater curvature for evaluating the gastric damage. Ulcer indexes of stomachs exposed to different treatments were calculated as described by Naidu et al [34]. The images of stomachs were acquired with a digital camera (MA-1000, AmScope, Irvine, CA 92606, United States) mounted on SterioZoom 6 Plus Leica microscope (Leica Microsystems, Buffalo Grove, IL 60089, United States) at 7X magnification. Briefly, normal colored stomachs and pink colored stomachs were scored as 0 and 0.5 respectively. Spot ulcers were given a score of 1, whereas hemorrhagic streaks were given a score of 1.5. Numbers of ulcers exceeding 3 but less than 5 were scored as 2, and the number of ulcers ≥ 5 were given a score of 3. The final ulcer index was obtained by adding the scores for colored stomach, spot ulcer, hemorrhagic streak and full ulcers. The maximal score, warranted by the ulcer index, is 6. Ulcers were graded by a person not aware of details regarding the dosing. TPPU (0.1 mg/kg/day, 7 days in drinking water) and omeprazole (50 mg/kg/day, p.o., 5 days) were used to pre-treat the mice before dosing with DCF to test if these agents can decrease the ulcerative effect of the NSAID. The last doses of TPPU and omeprazole were administered 1 h before dosing with DCF. TPPU and OME were administered in 1% PEG400 solution in water. Since TPPU has high oral availability, and its efficacy in reducing intestinal ulcer as a function of oral dose in drinking water has been previously studied [6], this route of administration was selected as one minimizing trauma to the mice.
2.4. Sample collection and tissue processing
Prior to opening the stomach, it was washed with 0.5 mL of saline and the solution was stored in microcentrifuge tubes to measure the pH of gastric contents. Blood samples were collected via cardiac puncture to determine the levels of drugs, TxB2 and PGE2 in the plasma, and levels of cytokines in the serum. After scoring ulcers in stomachs, the tissues were placed in a neutral buffered formalin solution for 48 h and then transferred to 70% ethanol for further histopathological processing. The tissues were embedded in paraffin, cut into 5micron size sections and stained (with hematoxylin and eosin) for histopathological evaluation.
2.5. Quantification of pH in gastric content
The pHs of gastric contents were measured using pH indicator strips (Catalog number 9590, EMD Chemicals Inc., Gibbstown, NJ) in the range of 1–14 pH units [35].
2.6. Quantification of drugs in blood
Blood samples were collected in tubes containing K2EDTA solution (final concentration of 1.2 mg/mL EDTA in the blood) and subsequently mixed and stored on ice until centrifugation. Blood samples were centrifuged at 4000 rpm for 10 m at 4 °C. Plasma samples were then collected and stored at −80 °C until analyzed by LC-MS/MS [6].
2.7. Quantification of inflammatory markers by ELISA
Separately, blood samples collected from mice were centrifuged at 4000 rpm for 10 m at 4 °C without anticoagulant and the supernatant was collected. Serum samples of animals were analyzed to determine the levels of inflammatory markers, such as TNF-α and IL-6, according to instructions from the manufacturers [12, 36]. Mouse TNF-α and IL-6 enzyme-linked immunosorbent assay (ELISA) kits provided microtiter plates pre-coated with TNF-α and IL-6 antibodies respectively. These ELISAs were run in duplicate and values were similar.
2.8. Effect of treatment on apoptosis
The terminal deoxynucleotide transferase biotin-dUTP nick-end labeling (TUNEL) method was employed to detect DNA-strand breaks; a sign of gastric cell apoptosis [37, 38]. The effect of treatment on gastric cell apoptosis was determined using the TACS® 2 TdT-DAB in situ apoptosis detection kit of Trevigen Inc. The fixation conditions for the TUNEL assay were optimized before use. TUNEL (+) cells were counted in photos of gastric epithelial cells at 200X magnification.
2.9 The sEH activity assay
A 3H substrate labeled sEH activity assays were performed in vitro using stomachs of mice to evaluate the effect of treatment on the activity of sEH and to ensure the genetically knockout animals did not display any activity [39].
2.10 Quantification of PGE2 and TxB2
Stomach samples from the experimental mice were collected at the end of the experiment, frozen over dry ice and stored in −80 °C till further experiment. The level of PGE2 was quantified as a marker of COX activity. COX and cytochrome P450 mediated metabolites of arachidonic acid were extracted from tissue samples and quantified as described below [40].
Tissues (10–60 mg) were spiked with 400 μL + 100 μL methanol and 10 μL of a 400 nM solution containing deuterated oxylipins and subsequently homogenized with a bead beater. Samples were then centrifuged at 16,100g for 10 minutes; the supernatant was diluted with 1.5 mL 80:20 v/v water/methanol with 0.1% acetic acid, and then loaded onto Waters Oasis-HLB cartridges. The cartridges were washed twice with 1.5 mL 80:20 v/v water/methanol with 0.1% acetic acid, and dried under low vacuum for 20 min. SPE cartridges were eluted using 0.5 mL methanol and 1.5 mL ethyl acetate into 2 mL Eppendorf vials containing 6 μL 30% glycerol in methanol. Methanol and ethyl acetate were then evaporated from the samples using a Speed-Vac. The residues were reconstituted in 50 μL methanol, vortexed, and filtered prior to LC-MS/MS analysis. Chromatographic separation was performed using a Phenomenex Eclipse C18 column (150 × 4.6 mm; 5 μm). The analysis was carried out using an Agilent 1200 Series LC system coupled to an AB Sciex 4000 Qtrap-mass spectrometer in negative electrospray ionization mode. Samples were injected (5 μL) and separated using a gradient method over 21 min at 0.4 mL/min using acetonitrile/methanol (84:16) and water mobile phases, each containing 0.1% acetic acid. Quantification of oxylipins was calculated from constructed calibrations curves for each analyte, with respect to their deuterated internal standards [40].
The plasma samples were collected as discussed earlier. The levels of PGE2 and TxB2 were quantified in the plasma (approximately 250 μL) of animals as per established procedure [40].
2.11. Statistical analysis
Graphpad prism 4.03 (GraphPad Software, San Diego, CA, USA) was used for statistical calculation. Data are presented as the mean ± standard deviation or standard error of the mean of 3–5 readings. One-way ANOVA followed by Tukey’s multiple comparison test was used for calculating statistical significance. Differences between data sets were considered as significant when the p value was smaller than 0.05.
3. Results
3.1. Diclofenac causes ulcers in stomachs of mice
Ulceration was observed 6 hours post dosing of Swiss Webster and C57BL6 mice with DCF (Fig. 1). Since 100 mg/kg was the lowest in a series of ascending doses of diclofenac evaluated to give clear ulceration in the time frame investigated, this dose was selected to further investigate the effects of sEH inhibition on gastric erosion. The gastric ulcer score returned to normal levels 18 h after dosing mice with a single DCF (100 mg/kg) dose implying that gastric ulcers are reversible and will heal without intervention if the administration of the NSAID is stopped.
Fig. 1. Inhibition of sEH or genetic deletion decreases diclofenac-induced ulcers in stomachs of mice.
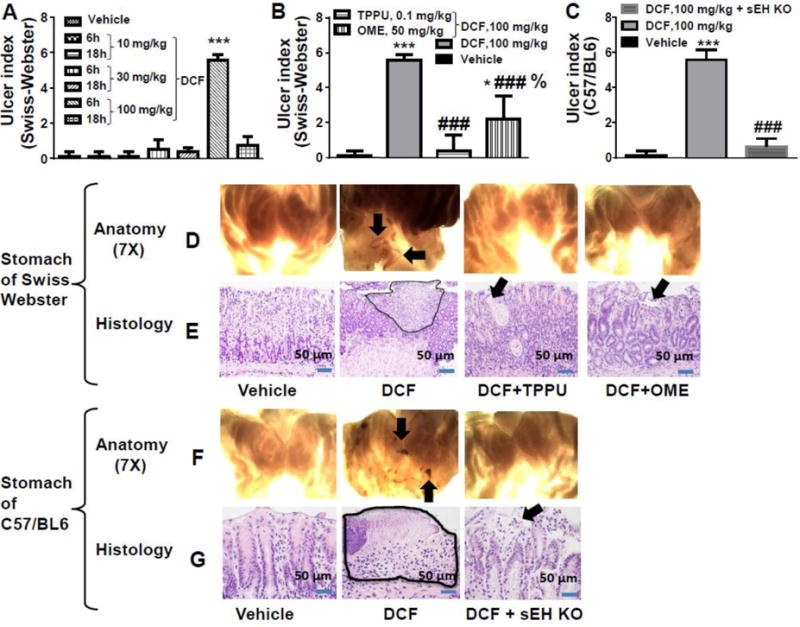
(A) Among the 3 doses tested for the ulcerative effect of diclofenac (DCF), the 100 mg/kg dose exhibited ulcers in Swiss Webster mice (*** p < 0.001 vs 10 mg/kg dose). The severity of the ulcers was greater at 6 h post diclofenac dose than at 18 h after diclofenac treatment. (B) 100 mg/kg of diclofenac-induced stomach ulcers significantly in Swiss Webster mice (*** p < 0.001, DCF treated animals vs vehicle control animals; * p < 0.01, DCF + OME vs vehicle-treated animals). Severity of diclofenac-induced ulcers decreased significantly with TPPU or OME treatment (### p < 0.001, DCF vs DCF + TPPU, DCF vs DCF + OME). OME-treated animals exposed to DCF had higher (% p < 0.01) incidence of ulceration than TPPU + DCF treated animals. (C) DCF significantly induced stomach ulcers in C57BL6 mice (*** p < 0.001, DCF vs vehicle). The sEH gene deletion (KO) also decreased ulcerative effects of diclofenac in stomach (### p < 0.001). The vehicle control animals used for this figure are C57BL6 mice. Values are presented as the mean ± standard deviation, n=4. (D) Anatomical observation at 7X under a dissecting microscope of stomachs of Swiss Webster mice administered with different treatments. Ulceration is pointed with an arrow. Stomachs from mice exposed to DCF exhibited ulcers and hemorrhagic streaks. Pre-treatment with the sEHI TPPU, omeprazole (OME), or (F) sEH knock out (KO) decreased stomach ulceration as shown at 7X under a dissecting microscope. (E) Histopathological observation of tissue section complemented anatomical observation. Morphology of gastric mucosa of vehicle treated Swiss Webster mice looked normal while ulceration was observed on stomachs of DCF treated mice. Luminal surface necrosis tissue debris is shown as highlighted area and it’s beneath granulation tissue formation is noticed. Ulceration was minimal in mice treated with DCF + TPPU. Minimal glandular cystic injury/erosion on surface of the mucosal layer in TPPU pre-treated mice is shown with an arrow. Omeprazole also minimized gastric erosion due to DCF. Minimal erosion on surface of mucosal layer is pointed by arrow. (G) Gastric mucosa of C57BL6 mice was normal but ulceration in mucosa (highlighted area) was observed in DCF treated mice. Mucosa with minimal injury/erosion and predominant reactive epithelial changes is seen in stomachs of sEH knockout mice. Doses of TPPU and OME were 0.1 mg/kg and 50 mg/kg respectively.
In general, NSAIDs/diclofenac-induced gastric lesion are categorized as 1) gastric mucosa erosion that is defined as acute mucosal inflammatory process and mucosa injury as loss of superficial epithelium above muscular mucosa, and 2) gastric acute ulcer that is defined as deep defect of mucosa, usually it is breach in muscularis mucosa of GI tract and begin as erosions. In our model, since the most of the diclofenac-induced mucosa injury is the deeper than muscular mucosa, we overall considered it as an acute gastric ulcer.
3.2. TPPU decreases diclofenac-induced gastric ulcer
Prophylactic treatment with TPPU significantly (p < 0.001) mitigated the ulcerative effects of DCF in Swiss Webster mice (Fig. 1). Genetic deletion of sEH also significantly (p < 0.001) prevented the formation of DCF-induced gastric ulcers (Fig. 1). Pre-treatment with omeprazole also decreased NSAID-induced stomach ulcers [7] (Fig. 1).
3.3. TPPU does not affect pH of gastric content
Parietal cells of epithelial gastric glands in the gastric mucosa produce gastric acid, which is secreted into the lumen of the stomach. Gastric acid helps in the digestion of food, but it can aggravate existing stomach ulcers. The pH of the gastric content of mice treated with the vehicle was found to be approximately 4. This is in accordance with the published literature [36, 41]. Treatment with DCF slightly decreased the pH of stomachs, but TPPU treatment normalized this trend. A similar gastric pH of 4 was observed in the sEH knockout mice treated with DCF. In this study, omeprazole administration increased the pH of stomachs as reported by published literature [42] (Fig. 2).
Fig. 2. Inhibition of sEH or gene deletion does not affect gastric pH.
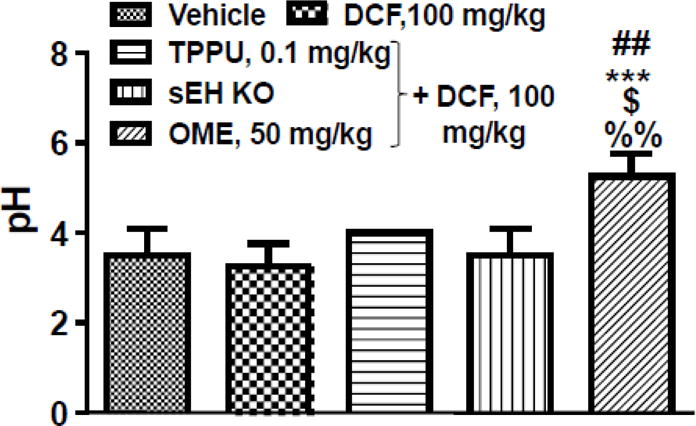
Diclofenac (DCF, 100 mg/kg, single dose) itself and diclofenac along with TPPU (0.1 mg/kg, 7 days) did not alter the pH of stomachs when compared to mice treated with vehicle. Similarly, DCF did not alter the pH in stomachs of sEH knockout mice. However, omeprazole treatment (50 mg/kg, 5 days) elevated the pH of stomachs and the difference was statistically significant when compared with vehicle (## p < 0.01), DCF (*** p < 0.001), DCF + TPPU ($ p < 0.05) and effect of DCF on gastric pH of sEH knockout animals (%% p < 0.01). One-way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis. Data are presented as the mean ± standard deviation of 4 readings.
3.4. TPPU does not alter concentration of diclofenac in plasma
The mean plasma concentration of DCF was 4 μg/ml in the mice those received DCF or DCF + TPPU. However, the concentration of DCF in plasma decreased in mice receiving OME, but the decrease was not statistically significant [43]. Diclofenac is known to be metabolized by uridine 5′-diphosphoglucuronosyl transferase 2B7, cytochrome P450s (CYP) 2C9 and 3A4 [44]. Omeprazole is known to increase activity of CYP3A4 [4]. We measured the concentration of DCF and omeprazole to evaluate if omeprazole treatment increased the metabolism of DCF. Though the mean concentration of diclofenac in plasma of mice treated with only diclofenac was approximately 2 times higher than the mice treated with DCF + OME the difference in concentration in both the groups was not significant which could be due to wide variation in DCF concentration and a low number of animals (n=4). Similarly, the mean plasma concentration of DCF in sEH knockout mice was not significantly different than other groups. Alterations in blood levels of TPPU and DCF was not observed when they were co-administered. Seven hours after TPPU administration, the concentration in plasma was still higher than its IC50 of 8 nM on the affinity purified recombinant mouse sEH [29] (Table 1).
Table 1.
Concentration of different drugs in plasma of mice
| Group | Drug concentration in (ng/mL)
|
||
|---|---|---|---|
| Diclofenac | TPPU | Omeprazole | |
| Diclofenac | 4500 ± 2000 | – | – |
| Diclofenac + TPPU | 4900 ± 560 | 63 ± 5 | – |
| Diclofenac + omeprazole | 1400 ± 300 | – | 6 ± 2 |
| Diclofenac + sEH KO | 880 ± 520 | – | – |
Diclofenac (DCF) was administered to Swiss Webster and sEH knock out mice orally at a dose of 100 mg/kg and blood samples were collected after 6 h at the time of sacrifice. For prevention of ulcers, the sEH inhibitor TPPU (0.1 mg/kg/day, drinking water) was administered in drinking water for 7 days and administration was stopped 1 h before of DCF administration. Similarly, last dose of omeprazole (50 mg/kg, p.o., 5 days) was administered before 1 h of DCF administration. Values are presented as mean ± standard error of mean of 4 readings.
The concentration of omeprazole in plasma was lower than TPPU even though the dose of omeprazole was higher. This may be due to a very short half-life of omeprazole monitored in the blood i.e. 5–15 min. Though the concentration of omeprazole in the blood decreases significantly, the drug still binds to H+K+-ATPase in the stomach where it decreases acid secretion [45, 46]. The acidity of the stomach is maintained by histamine H2 receptors and proton pumps [47]. Helander et al demonstrated that 16 h after radioactive omeprazole administration, the radioactivity of the stomach was 100 times higher than blood [48]. The concentration of TPPU in plasma was similar to the previously published literature [4]. Like TPPU, omeprazole appears to show excellent target occupancy.
3.5. TPPU decreases levels of inflammatory markers in serum of diclofenac treated mice
DCF-induced gastrointestinal ulcers were associated with a significant increase (p < 0.05) in the levels of inflammatory markers TNF-α and IL-6 in serum when compared with mice receiving vehicle only. Treatment with TPPU, OME and sEH gene knockout normalized the level of these cytokines in serum of mice those received DCF (Fig. 3).
Fig. 3. Inhibition of sEH or knock out reverses diclofenac-induced increases in the levels of TNF-α and IL-6 in the serum of mice.
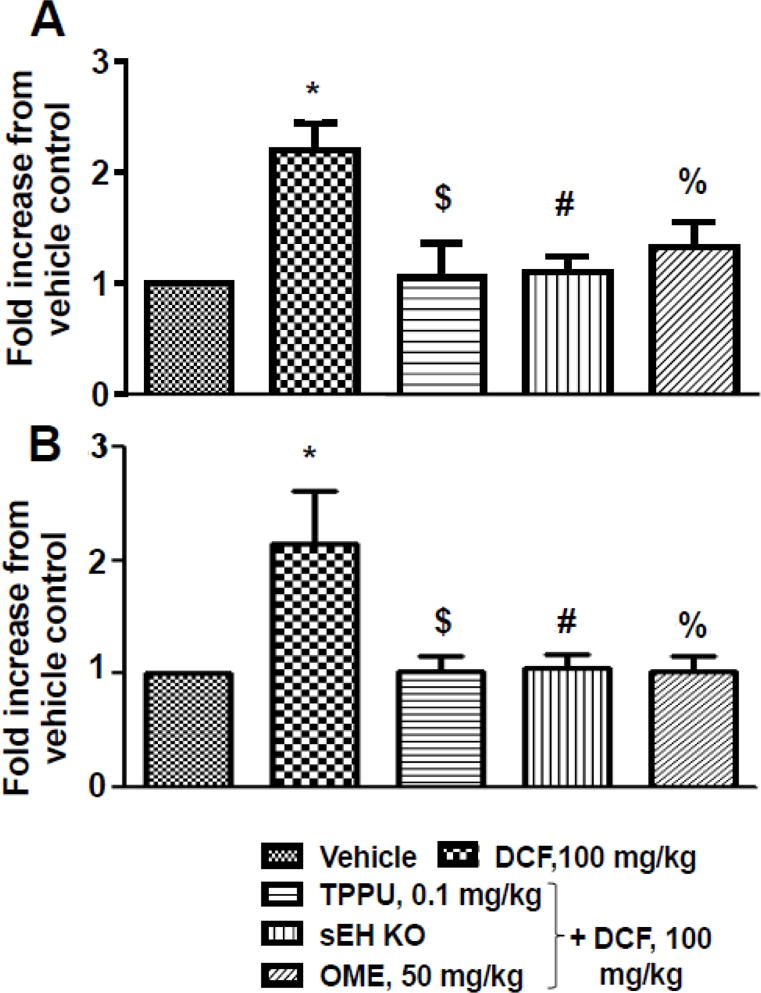
(A) Diclofenac (DCF) (100 mg/kg, p.o., single dose) treatment increased level of TNF-α significantly (* p < 0.01; DCF vs vehicle) while pretreatment of TPPU (0.1 mg/kg) ($ p < 0.01; DCF vs DCF+TPPU), omeprazole (OME) (50 mg/kg) (% p < 0.01; DCF vs DCF + OME) or sEH knock out mice (# p < 0.01; DCF vs DCF + sEH KO) decreased level of TNF-α significantly. (B) Diclofenac (DCF) treatment increased levels of IL-6 significantly (* p < 0.01; DCF vs vehicle) while pre-treatment of TPPU ($ p < 0.01; DCF vs DCF+TPPU), omeprazole (OME) (% p < 0.01; DCF vs DCF + OME) and sEH knocked out (# p < 0.01; DCF vs DCF + sEH KO) decreased level of IL-6 significantly. One-way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis. The assays were run in duplicate. Data are presented as mean ± standard deviation of 3–4 animals.
3.6. TPPU decreases diclofenac-induced gastric epithelial cell apoptosis
TUNEL positive cells are considered as markers of apoptosis. An increase in the number of TUNEL positive cells in the epithelial layer of the stomach was observed in the mice treated with DCF in comparison to vehicle-treated mice. Pre-treatment with TPPU and OME decreased DCF-induced apoptosis of gastric cells. Gene deletion of sEH also protected mice from apoptotic effects of DCF (Fig. 4).
Fig 4. Inhibition of sEH or gene knockout, and pre-treatment of Omeprazole decrease diclofenac-induced apoptosis in epithelial cells of stomachs. (A).
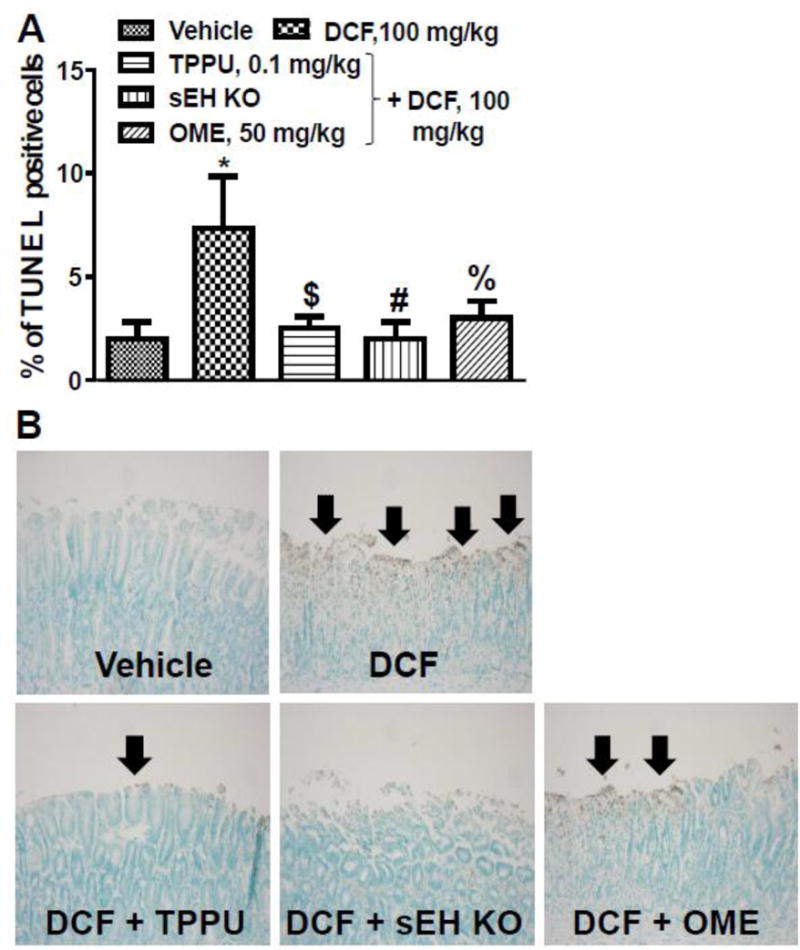
Apoptosis of stomach epithelial cells was detected by TUNEL positive cells with brown nuclei. DCF (100 mg/kg) treatment increased apoptosis significantly (* p < 0.001; DCF vs vehicle) while pre-treatment with TPPU (0.1 mg/kg) ($ p < 0.001; DCF vs DCF+TPPU), OME (50 mg/kg) (% p < 0.01; DCF vs DCF + OME) or sEH knockout (# p < 0.001; DCF vs DCF + sEH KO) decreased apoptosis significantly. One-way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis. Data are presented as the mean ± standard deviation of 3–4 observation (B) Stomach epithelial cells seen under a microscope at 200X magnification after TUNEL assay.
3.7 Diclofenac does not affect activity of the sEH
We did not observe any effect of DCF on the activity of sEH when assayed in vitro or in vivo. Inhibition of sEH by TPPU was not observed in vitro which was likely due to diffusion of the inhibitor from enzyme due to dilution of stomach homogenate during the enzyme assay. No sEH activity was observed in stomach homogenates of sEH KO mice (Fig. 5A).
Fig 5. Effect of treatment on activities of sEH and COX. (A).
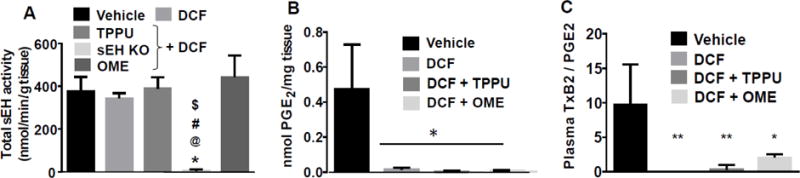
DCF (100 mg/kg) did not increase or decrease the activity of sEH in vitro. Negligible sEH activity was observed in the stomach of sEH knockout mice. The sEH activity in sEH KO mice was significantly less (p < 0.001) in comparison to vehicle (*), DCF (@), DCF + TPPU (#), DCF + OME ($). (B) Levels of PGE2 were quantified in stomachs of treated animals (n=4) as a marker of COX activity. Diclofenac alone and in combination with TPPU and OME inhibited COX as expected. (C) Levels of TxB2/PGE2 were quantified in the plasma of animals. Diclofenac treatment significantly decreased the levels of TxB2/PGE2. DCF + OME (* p < 0.05) decreased the level of TxB2/PGE2 to a lesser extent than DCF (** p < 0.01) and DCF + TPPU (** p < 0.01) in comparison to vehicle. One-way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis. The assay was run in triplicate. Data are presented as the mean ± standard deviation of 3–5 animals.
3.8 TPPU and OME do not affect activity of COX
Diclofenac significantly inhibited COX activity in the stomachs of treated mice which is evident from a significant decrease in the level of PGE2 in comparison to level of PGE2 in stomachs of mice treated with vehicle. Treatment with TPPU and/or OME did not increase the activity of COX, and these agents also did not further decrease the activity of COX (Fig. 5B). The accurate quantification of levels of TxB2, EETs and DHETs in stomach samples was not complete due to insufficient tissue samples (less than 50 mg).
3.9 Diclofenac and omeprazole alters level of PGE2/TxB2
Diclofenac significantly decreased the levels of TxB2/ PGE2 in plasma of mice in comparison to mice treated with vehicle. Levels of TxB2 was below the level of detection (20 pM) in mice treated with only DCF. TPPU and omeprazole increased the level of TxB2/PGE2 in DCF treated mice (Fig. 5C).
4. Discussion
Severe ulceration in the stomach of Swiss Webster mice was observed 6 h after a single administration of DCF. The resolution of this ulceration was time dependent with an apparent return to control levels at 18 h after DCF dose. Stomach ulcers were associated with an increase in the levels of TNF-α and IL-6 in serum and an increase in apoptosis in the epithelial mucosal layer. Pre-treatment of TPPU (0.1 mg/kg/day) for 7 days significantly decreased DCF-induced stomach ulcers and caused a significant decrease in levels of TNF-α and IL-6, as well as the degree of apoptosis. Unlike omeprazole, this anti-ulcer effect of TPPU was not associated with a significant elevation of the gastric pH. The ulcerative potential of DCF was also decreased by a 500-fold higher concentration of OME, a commonly used medicine for the management of NSAID-induced peptic ulcers by elevating gastric pH.
Elevating the pH of the stomach has been associated with alleviation and prevention of NSAID-induced peptic ulcers. Use of alkaline agents, histamine H2 blocker and PPI were found to decrease NSAID-induced ulcers. These pharmacological agents are a part of the management of NSAID-induced gastric ulcers [47, 48, 7]. A decrease in gastric juice volume and an increase in gastric pH and gastric intramucosal pH has been observed with administration of PPIs including OME [50]. However, the ulcer protective agent (sucralfate) and prostaglandin analogs including misoprostol, arbaprostil, enprostil and rioprostil [47] which do not alter pH of gastric acid also are reported to cure gastric ulcers. Sucralfate is shown to reduce ulcers by physically covering and protecting the ulcer area. Though gastric acid does not create ulcers in the stomach of healthy humans, it can aggravate previously formed ulcers. Prostaglandin is known to alleviate the harmful effects of acid by stimulating the release of bicarbonate and mucous which functions as a barrier for the epithelial cells of the stomach [47]. The anti-ulcer effect of the sEH inhibitor TPPU as shown here is not mediated by dramatically elevating the pH of the stomach.
The probable mechanism leading to healing of ulcers by TPPU is inhibition of the degradation of fatty acid epoxides by the sEH. Major anti-inflammatory fatty acid epoxides include epoxyeicosatrienoic acids (EETs) derived from arachidonic acid through the action of cytochrome P450s enzymes in the stomach tissue. These EETs also block ER stress and the associated activation of the apoptotic pathways. In a similar study, it was observed that TPPU decreased DCF-induced intestinal ulcers and this anti-ulcer effect was correlated with significant increases in EETs in plasma of animals due to the prevention of its metabolism [6]. Anti-inflammatory properties of EETs have been reported earlier [51, 52, 53]. Anti-inflammatory effects of sEH inhibitors are associated with the decrease in the levels of TNF-α [22, 6] and IL-6 [26, 6].
In this study, DCF-induced gastric ulcers were associated with an increase in the serum levels of TNF-α. Prophylactic treatment of TPPU, OME and gene deletion of sEH decreased ulceration which correlated with normalization of levels of TNF-α and IL-6. Levels of these cytokines and NSAID-induced gastric ulcers have previously been associated [12]. Specifically, NSAID-induced small intestinal and gastric ulcers were reduced by a TNF-α synthesis inhibitor [54, 55]. Similarly, indomethacin-induced inflammatory bowel disease was alleviated by the antibody to TNF-α or gene deletion of TNF-α [56, 8]. TNF-α mediated ulceration involves apoptosis [57].
An increase in apoptosis, evident from TUNEL staining, was noticed in gastric ulcers in mice treated with DCF. NSAID-induced gastric apoptosis could be due to an increase in the levels of TNF-α and its action via TNF receptor 1 (TNFR1) [16, 58, 59]. A baseline level of 2–3 % of apoptotic cells in the epithelial layer of the stomach is normal and the presence of more apoptotic cells than this is considered an indication of mucosal injury by stimuli such as NSAIDs [16]. Toxicity-induced cleavage of the DNA into fragments and production of DNA strand breaks are detected by nick end labeling [60]. A decrease in apoptosis of gastric epithelial cells was observed in the mice pre-treated with TPPU and OME followed by DCF. Omeprazole was reported to decrease NSAID (indomethacin)-induced gastropathy and cold stress-induced apoptosis of gastric epithelial cells [61]. A similar effect was also noticed with gene deletion of sEH.
We observed a wide difference in the concentration of DCF in plasma of mice. This could be due to inter-animal variation in metabolizing DCF and uneven blood loss at the site of ulceration. Pretreatment of OME and TPPU reduced variation in the concentration of DCF. Earlier, we reported that a significant loss of hemoglobin in animals treated with DCF using intestinal ulcer model [6]. However, these data are important because they indicate the range of concentration of diclofenac that is ulcerogenic in mice. The concentration of TPPU in plasma is important because it will help researchers to design further experiments accordingly. In this study, we administered TPPU chronically, but evaluation of the anti-ulcer effect of TPPU after a single dose, much like omeprazole is of interest. It is expected that plasma concentration of 60 ng/mL of TPPU is required to observe possible anti-ulcer effects.
Ulcerative effect of DCF was associated with a significant decrease (*p < 0.05, vs vehicle) in the levels of TxB2/PGE2 in the plasma of animals. Pretreatment of the sEH inhibitor and OME minimized the decrease in the levels of TxB2/PGE2 in DCF treated mice (Fig. 5c). Levels of PGE2 in the plasma of animals treated with vehicle, DCF, DCF + TPPU and DCF + OME were not statistically (data not shown) different whereas the levels of PGE2 in gastric samples were significantly decreased in the animals treated with DCF, DCF + TPPU and DCF + OME (Fig. 5B) in comparison to vehicle. Quantifying the levels of TxB2/PGE2 in stomach samples could have been better, but we were limited by the amount of stomach samples. In the DCF + OME group, the decrease of TxB2/PGE2 was least in comparison to other DCF treated groups. This could be due to the decrease in the levels of DCF in plasma of animals administered with OME. There is also an immunoassay-based method to quantify the activity of COX in vitro which uses supplemental arachidonic acid and measures the level of TxB2 and PGE2 produced [62–64]. The activity of COX in this assay may vary depending on the amount of arachidonic acid used unlike the assay performed by us which uses arachidonic acid available to the COX in the cells of the stomach.
5. Conclusion
Small molecule sEH inhibitors are in clinical trials to treat inflammation and are in development for managing pain [65, 66]. Since sEH inhibitors are likely to be used clinically with the standard of care, it is likely that they will be used with NSAIDs to control inflammation synergistically. Gastrointestinal erosion often is associated with NSAID use, an added benefit of co-administration with sEH inhibitors is reduction in ulceration. This anti-ulcer effect was supported by results from sEH knockout mice in diclofenac-induced gastric ulcer model. Therefore, co-administration of sEH inhibitors with NSAIDs can decrease dose and ulcerative potential of NSAIDs. Clinical studies are required to test these hypotheses in man and companion animals.
Acknowledgments
This study was partially funded by the National Institute of Environmental Health Sciences (NIEHS) [Grant R01 ES002710], NIEHS Superfund Research Program [Grant P42 ES004699], West Coast Central Comprehensive Metabolomics Center [Grant U24 DK097154] to BDH; National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) [grant R21 AR062866] to BI; and National Cancer Institute Grant [R01 CA172431 and R01 CA164041] to G-YY. We thank Sean D. Kodani for the valuable critique of the manuscript and Joseph Jilek for assistance during the animal study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The University of California holds numerous patents on the use of sEH inhibitors for the management of pain and inflammation. Dr. Bruce D. Hammock and Dr. Bora Inceoglu founded a company for the advancing research on the use of sEH inhibitors in companion animals and humans.
References
- 1.Sinha M, Gautam L, Shukla PK, Kaur P, Sharma S, Singh P. Current perspectives in NSAID-induced gastropathy. Mediators Inflamm. 2013;2013:258209. doi: 10.1155/2013/258209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laine L. Approaches to nonsteroidal anti-inflammatory drug use in the high-risk patient. Gastroenterology. 2001;120:594–606. doi: 10.1053/gast.2001.21907. [DOI] [PubMed] [Google Scholar]
- 3.Singh G. Gastrointestinal complications of prescription and over-the-counter nonsteroidal anti-inflammatory drugs: a view from the ARAMIS database. Arthritis, Rheumatism, and Aging Medical Information System. Am J Ther. 2000;7:115–121. doi: 10.1097/00045391-200007020-00008. [DOI] [PubMed] [Google Scholar]
- 4.Goswami SK, Inceoglu B, Yang J, Wan D, Kodani SD, da Silva CA, Morisseau C, Hammock BD. Omeprazole increases the efficacy of a soluble epoxide hydrolase inhibitor in a PGE₂ induced pain model. Toxicol Appl Pharmacol. 2015;289:419–427. doi: 10.1016/j.taap.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu JY, Yang J, Inceoglu B, Qiu H, Ulu A, Hwang SH, Chiamvimonvat N, Hammock BD. Inhibition of soluble epoxide hydrolase enhances the anti-inflammatory effects of aspirin and 5-lipoxygenase activation protein inhibitor in a murine model. Biochem Pharmacol. 2010;79:880–887. doi: 10.1016/j.bcp.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goswami SK, Wan D, Yang J, da Silva CAT, Morisseau C, Kodani SD, Yang GY, Inceoglu B, Hammock BD. Anti-ulcer efficacy of soluble epoxide hydrolase inhibitor TPPU on diclofenac-induced intestinal ulcers. J Pharmacol Exp Ther. 2016;357:529–536. doi: 10.1124/jpet.116.232108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatterjee A, Chatterjee S, Biswas A, Bhattacharya S, Chattopadhyay S, Bandyopadhyay SK. Gallic acid enriched fraction of Phyllanthus emblica potentiates indomethacin-induced gastric ulcer healing via e-nos-dependent pathway. Evid Based Complement Alternat Med. 2012;2012:487380. doi: 10.1155/2012/487380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukumoto K, Naito Y, Takagi T, Yamada S, Horie R, Inoue K, Harusato A, Hirata I, Omatsu T, Mizushima K, Hirai Y, Yoshida N, Uchiyama K, Ishikawa T, Handa O, Konishi H, Wakabayashi N, Yagi N, Kokura S, Ichikawa H, Kita M, Yoshikawa T. Role of tumor necrosis factor-α in the pathogenesis of indomethacin-induced small intestinal injury in mice. Int J Mol Med. 2011;27:353–359. doi: 10.3892/ijmm.2011.602. [DOI] [PubMed] [Google Scholar]
- 9.LoGuidice A, Ramirez-Alcantara V, Proli A, Gavillet B, Boelsterli UA. Pharmacologic targeting or genetic deletion of mitochondrial cyclophilin D protects from NSAID-induced small intestinal ulceration in mice. Toxicol Sci. 2010;118:276–85. doi: 10.1093/toxsci/kfq226. [DOI] [PubMed] [Google Scholar]
- 10.Ramirez-Alcantara V, LoGuidice A, Boelsterli UA. Protection from diclofenac-induced small intestinal injury by the JNK inhibitor SP600125 in a mouse model of NSAID-associated enteropathy. Am J Physiol Gastrointest Liver Physiol. 2009;297:G990–G998. doi: 10.1152/ajpgi.00219.2009. [DOI] [PubMed] [Google Scholar]
- 11.Tsutsumi S, Gotoh T, Tomisato W, Mima S, Hoshino T, Hwang HJ, Takenaka H, Tsuchiya T, Mori M, Mizushima T. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ. 2004;11:1009–1016. doi: 10.1038/sj.cdd.4401436. [DOI] [PubMed] [Google Scholar]
- 12.Kinsey SG, Nomura DK, O’Neal ST, Long JZ, Mahadevan A, Cravatt BF, Grider JR, Lichtman AH. Inhibition of monoacylglycerol lipase attenuates nonsteroidal anti-inflammatory drug-induced gastric hemorrhages in mice. J Pharmacol Exp Ther. 2011;338:795–802. doi: 10.1124/jpet.110.175778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeuchi T, Miura S, Wang L, Uehara K, Mizumori M, Kishikawa H, Hokari R, Higuchi H, Adachi M, Nakamizo H, Ishii H. Nuclear factor-kappaB and TNF-alpha mediate gastric ulceration induced by phorbol myristate acetate. Dig Dis Sci. 2002;47:2070–2078. doi: 10.1023/a:1019633114854. [DOI] [PubMed] [Google Scholar]
- 14.Laster SN, Wood JG, Goding LR. Tumor necrosis factor can induce both apoptotic and necrotic form of cell lysis. J Immunol. 1988;141:2629–2634. [PubMed] [Google Scholar]
- 15.Sánchez-Fidalgo S, Martín-Lacave I, Illanes M, Motilva V. Angiogenesis, cell proliferation and apoptosis in gastric ulcer healing. Effect of a selective cox-2 inhibitor. Eur J Pharmacol. 2004;505:187–94. doi: 10.1016/j.ejphar.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 16.Szabó I, Tarnawski AS. Apoptosis in the gastric mucosa: molecular mechanisms, basic and clinical implications. J Physiol Pharmacol. 2000;51:3–15. [PubMed] [Google Scholar]
- 17.Pal C, Bindu S, Dey S, Alam A, Goyal M, Iqbal MS, Maity P, Adhikari SS, Bandyopadhyay U. Gallic acid prevents nonsteroidal anti-inflammatory drug-induced gastropathy in rat by blocking oxidative stress and apoptosis. Free Radic Biol Med. 2010;49:258–67. doi: 10.1016/j.freeradbiomed.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 18.Ishihara T, Tanaka K, Tashiro S, Yoshida K, Mizushima T. Protective effect of rebamipide against celecoxib-induced gastric mucosal cell apoptosis. Biochem Pharmacol. 2010;79:1622–1633. doi: 10.1016/j.bcp.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 19.McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A. 1999;96:272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci USA. 2005;102:9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oni-Orisan A, Deng Y, Schuck RN, Theken KN, Edin ML, Lih FB, Molnar K, DeGraff L, Tomer KB, Zeldin DC, Lee CR. Dual modulation of cyclooxygenase and CYP epoxygenase metabolism and acute vascular inflammation in mice. Prostaglandins Other Lipid Mediat. 2013;104–105:67–73. doi: 10.1016/j.prostaglandins.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang W, Yang AL, Liao J, Li H, Dong H, Chung YT, Bai H, Matkowskyj KA, Hammock BD, Yang GY. Soluble epoxide hydrolase gene deficiency or inhibition attenuates chronic active inflammatory bowel disease in IL-10(−/−) mice. Dig Dis Sci. 2012;57:2580–2591. doi: 10.1007/s10620-012-2217-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lazaar AL, Yang L, Boardley RL, Goyal NS, Robertson J, Baldwin SJ, Newby DE, Wilkinson IB, Tal-Singer R, Mayer RJ, Cheriyan J. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br J Clin Pharmacol. 2016;81:971–979. doi: 10.1111/bcp.12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen D, Whitcomb R, MacIntyre E, Tran V, Do ZN, Sabry J, Patel DV, Anandan SK, Gless R, Webb HK. Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J Clin Pharmacol. 2012;52:319–328. doi: 10.1177/0091270010397049. [DOI] [PubMed] [Google Scholar]
- 25.Luo P, Chang HH, Zhou Y, Zhang S, Hwang SH, Morisseau C, Wang CY, Inscho EW, Hammock BD, Wang MH. Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J Pharmacol Exp Ther. 2010;334:430–438. doi: 10.1124/jpet.110.167544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bettaieb A, Nagata N, AbouBechara D, Chahed S, Morisseau C, Hammock BD, Haj FG. Soluble epoxide hydrolase deficiency or inhibition attenuates diet-induced endoplasmic reticulum stress in liver and adipose tissue. J Biol Chem. 2013;288:14189–14199. doi: 10.1074/jbc.M113.458414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris TR, Bettaieb A, Kodani S, Dong H, Myers R, Chiamvimonvat N, Haj FG, Hammock BD. Inhibition of soluble epoxide hydrolase attenuates hepatic fibrosis and endoplasmic reticulum stress induced by carbon tetrachloride in mice. Toxicol Appl Pharmacol. 2015;286:102–111. doi: 10.1016/j.taap.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rose TE, Morisseau C, Liu JY, Inceoglu B, Jones PD, Sanborn JR, Hammock BD. 1-Aryl-3-(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: structure-activity relationships, pharmacokinetics, and reduction of inflammatory pain. J Med Chem. 2010;53:7067–7075. doi: 10.1021/jm100691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu JY, Lin YP, Qiu H, Morisseau C, Rose TE, Hwang SH, Chiamvimonvat N, Hammock BD. Substituted phenyl groups improve the pharmacokinetic profile and anti-inflammatory effect of urea-based soluble epoxide hydrolase inhibitors in murine models. Eur J Pharm Sci. 2013;48:619–627. doi: 10.1016/j.ejps.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ulu A, Stephen Lee KS, Miyabe C, Yang J, Hammock BG, Dong H, Hammock BD. An omega-3 epoxide of docosahexaenoic acid lowers blood pressure in angiotensin-II-dependent hypertension. J Cardiovasc Pharmacol. 2014;64:87–99. doi: 10.1097/FJC.0000000000000094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyata M, Kudo G, Lee YH, Yang TJ, Gelboin HV, Fernandez-Salguero P, Kimura S, Gonzalez FJ. Targeted disruption of the microsomal epoxide hydrolase gene. Microsomal epoxide hydrolase is required for the carcinogenic activity of 7, 12-dimethylbenz[a]anthracene. J Biol Chem. 1999;274:23963–23968. doi: 10.1074/jbc.274.34.23963. [DOI] [PubMed] [Google Scholar]
- 32.Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem. 2000;275:40504–40510. doi: 10.1074/jbc.M008106200. [DOI] [PubMed] [Google Scholar]
- 33.Luria A, Morisseau C, Tsai HJ, Yang J, Inceoglu B, De Taeye B, Watkins SM, Wiest MM, German JB, Hammock BD. Alteration in plasma testosterone levels in male mice lacking soluble epoxide hydrolase. Am J Physiol Endocrinol Metab. 2009;297:E375–E383. doi: 10.1152/ajpendo.00131.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naidu PS, Booker L, Cravatt BF, Lichtman AH. Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J Pharmacol Exp Ther. 2009;329:48–56. doi: 10.1124/jpet.108.143487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.More DG, Boutagy J, Shenfield GM. pH testing paper for measurement of intragastric acidity: an assessment. Anaesth Intensive Care. 1983;11:147–150. doi: 10.1177/0310057X8301100211. [DOI] [PubMed] [Google Scholar]
- 36.Li WF, Hao DJ, Fan T, Huang HM, Yao H, Niu XF. Protective effect of chelerythrine against ethanol-induced gastric ulcer in mice. Chem Biol Interact. 2014;208:18–27. doi: 10.1016/j.cbi.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 37.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu CC, Lu YZ, Wu LL, Yu LC. Role of myosin light chain kinase in intestinal epithelial barrier defects in a rat model of bowel obstruction. BMC Gastroenterol. 2010;10:39. doi: 10.1186/1471-230X-10-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Borhan B, Mebrahtu T, Nazarian S, Kurth MJ, Hammock BD. Improved radio labeled substrates for soluble epoxide hydrolase. Anal Biochem. 1995;231:188–200. doi: 10.1006/abio.1995.1520. [DOI] [PubMed] [Google Scholar]
- 40.Yang J, Schmelzer K, Georgi K, Hammock BD. Quantitative profiling method for oxylipin metabolome by liquid chromatography electrospray ionization tandem mass spectrometry. Anal Chem. 2009;81:8085–8093. doi: 10.1021/ac901282n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jain NK, Kulkarni SK, Singh A. Modulation of NSAID-induced antinociceptive and anti-inflammatory effects by alpha2-adrenoceptor agonists with gastroprotective effects. Life Sci. 2002;70:2857–2869. doi: 10.1016/s0024-3205(02)01549-7. [DOI] [PubMed] [Google Scholar]
- 42.Bavishi C, Dupont HL. Systematic review: the use of proton pump inhibitors and increased susceptibility to enteric infection. Aliment Pharmacol Ther. 2011;34:1269–1281. doi: 10.1111/j.1365-2036.2011.04874.x. [DOI] [PubMed] [Google Scholar]
- 43.Andersson T, Bredberg E, Lagerström PO, Naesdal J, Wilson I. Lack of drug-drug interaction between three different non-steroidal anti-inflammatory drugs and omeprazole. Eur J Clin Pharmacol. 1998;54:399–404. doi: 10.1007/s002280050482. [DOI] [PubMed] [Google Scholar]
- 44.Tang W. The metabolism of diclofenac–enzymology and toxicology perspectives. Curr Drug Metab. 2003;4:319–329. doi: 10.2174/1389200033489398. [DOI] [PubMed] [Google Scholar]
- 45.Regårdh CG, Gabrielsson M, Hoffman KJ, Löfberg I, Skånberg I. Pharmacokinetics and metabolism of omeprazole in animals and man–an overview. Scand J Gastroenterol Suppl. 1985;108:79–94. doi: 10.3109/00365528509095821. [DOI] [PubMed] [Google Scholar]
- 46.Tallman MN, Ali SY, Smith PC. Altered pharmacokinetics of omeprazole in cystic fibrosis knockout mice relative to wild-type mice. Drug Metab Dispos. 2004;32:902–905. [PubMed] [Google Scholar]
- 47.Mejia A, Kraft WK. Acid peptic diseases: pharmacological approach to treatment. Expert Rev Clin Pharmacol. 2009;2:295–314. doi: 10.1586/ecp.09.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Helander HF, Ramsay CH, Regårdh CG. Localization of omeprazole and metabolites in the mouse. Scand J Gastroenterol Suppl. 1985;108:95–104. doi: 10.3109/00365528509095822. [DOI] [PubMed] [Google Scholar]
- 49.Maity P, Bindu S, Choubey V, Alam A, Mitra K, Goyal M, Dey S, Guha M, Pal C, Bandyopadhyay U. Lansoprazole protects and heals gastric mucosa from non-steroidal anti-inflammatory drug (NSAID)-induced gastropathy by inhibiting mitochondrial as well as Fas-mediated death pathways with concurrent induction of mucosal cell renewal. J Biol Chem. 2008;283:14391–14401. doi: 10.1074/jbc.M800414200. [DOI] [PubMed] [Google Scholar]
- 50.Gursoy O, Memiş D, Sut N. Effect of proton pump inhibitors on gastric juice volume, gastric pH and gastric intramucosal pH in critically ill patients: a randomized, double-blind, placebo-controlled study. Clin Drug Investig. 2008;28:777–782. doi: 10.2165/0044011-200828120-00005. [DOI] [PubMed] [Google Scholar]
- 51.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chiamvimonvat N, Ho CM, Tsai HJ, Hammock BD. The soluble epoxide hydrolase as a pharmaceutical target for hypertension. J Cardiovasc Pharmacol. 2007;50:225–237. doi: 10.1097/FJC.0b013e3181506445. [DOI] [PubMed] [Google Scholar]
- 53.Thomson SJ, Askari A, Bishop-Bailey D. Anti-inflammatory effects of epoxyeicosatrienoic acids. Int J Vasc Med. 2012;2012:605101. doi: 10.1155/2012/605101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bertrand V, Guimbaud R, Tulliez M, Mauprivez C, Sogni P, Couturier D, Giroud JP, Chaussade S, Chauvelot-Moachon L. Increase in tumor necrosis factor-alpha production linked to the toxicity of indomethacin for the rat small intestine. Br J Pharmacol. 1998;124:1385–1394. doi: 10.1038/sj.bjp.0701968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fiorucci S, Antonelli E, Migliorati G, Santucci L, Morelli O, Federici B, Morelli A. TNF-α processing enzyme inhibitors prevents aspirin-induced TNFalpha release and protection against mucosal injury in rats. Aliment Pharmacol Ther. 1998;12:1139–1153. doi: 10.1046/j.1365-2036.1998.00409.x. [DOI] [PubMed] [Google Scholar]
- 56.Cury DH, Costa JE, Irika K, Mijji L, Garcez A, Buchiguel C, Silva I, Sipahi A. Protective effect of octreotide and infliximab in an experimental model of indomethacin-induced inflammatory bowel disease. Dig Dis Sci. 2008;53:2516–2520. doi: 10.1007/s10620-007-0172-z. [DOI] [PubMed] [Google Scholar]
- 57.Fiorucci S, Santucci L, Federici B, Antonelli E, Distrutti E, Morelli O, Renzo GD, Coata G, Cirino G, Soldato PD, Morelli A. Nitric oxide-releasing NSAIDs inhibit interleukin-1beta converting enzyme-like cysteine proteases and protect endothelial cells from apoptosis induced by TNFalpha. Aliment Pharmacol Ther. 1999;13:421–435. doi: 10.1046/j.1365-2036.1999.00442.x. [DOI] [PubMed] [Google Scholar]
- 58.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 59.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 60.Kaur Saini M, Kaur J, Sharma P, Nath Sanyal S. Chemopreventive response of diclofenac, a non-steroidal anti-inflammatory drug in experimental carcinogenesis. Nutr Hosp. 2009;24:717–723. [PubMed] [Google Scholar]
- 61.Biswas K, Bandyopadhyay U, Chattopadhyay I, Varadaraj A, Ali E, Banerjee RK. A novel antioxidant and antiapoptotic role of omeprazole to block gastric ulcer through scavenging of hydroxyl radical. J Biol Chem. 2003;278:10993–11001. doi: 10.1074/jbc.M210328200. [DOI] [PubMed] [Google Scholar]
- 62.Guo JS, Cho CH, Wang WP, Shen XZ, Cheng CL, Koo MW. Expression and activities of three inducible enzymes in the healing of gastric ulcers in rats. World J Gastroenterol. 2003;9:1767–1771. doi: 10.3748/wjg.v9.i8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tomlinson A, Appleton I, Moore AR, Gilroy DW, Willis D, Mitchell JA, Willoughby DA. Cyclo-oxygenase and nitric oxide synthase isoforms in rat carrageenin-induced pleurisy. Br J Pharmacol. 1994;113:693–698. doi: 10.1111/j.1476-5381.1994.tb17048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vane JR, Mitchell JA, Appleton I, Tomlinson A, Bishop-Bailey D, Croxtall J, Willoughby DA. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc Natl Acad Sci USA. 1994;91:2046–2050. doi: 10.1073/pnas.91.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kodani SD, Hammock BD. The 2014 Bernard B. Brodie award lecture-epoxide hydrolases: drug metabolism to therapeutics for chronic pain. Drug Metab Dispos. 2015;43:788–802. doi: 10.1124/dmd.115.063339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. 2013;53:37–58. doi: 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]