

Abstract
Neurobehavioral disorders comprised of neurodegenerative, neurodevelopmental, and psychiatric disorders together represent leading causes of morbidity and mortality. Despite significant academic research and industry efforts to elucidate the disease mechanisms operative in these disorders and to develop mechanism‐based therapies, our understanding remains incomplete and our access to tractable therapeutic interventions severely limited. The magnitude of these short‐comings can be measured by the growing list of disappointing clinical trials based on initially promising compounds identified in genetic animal models. This review and commentary will explore why this may be so, focusing on the central role that genetic models of neurobehavioral disorders have come to occupy in current efforts to identify disease mechanisms and therapies. In particular, we will highlight the unique pitfalls and challenges that have hampered success in these models as compared to genetic models of non‐neurological diseases as well as to symptom‐based models of the early 20th century that led to the discovery of all major classes of psychoactive pharmaceutical compounds still used today. Using examples from specific genetic rodent models of human neurobehavioral disorders, we will highlight issues of reproducibility, construct validity, and translational relevance in the hopes that these examples will be instructive toward greater success in future endeavors. Lastly, we will champion a two‐pronged approach toward identifying novel therapies for neurobehavioral disorders that makes greater use of the historically more successful symptom‐based approaches in addition to more mechanism‐based approaches.
Keywords: behavioral genetics, behavioral neuroscience, CNS disorders, preclinical, rodent models
1. INTRODUCTION
Advances in the areas of the human genetics, alongside genetic and genomic engineering technologies have been instrumental in developing the laboratory rodent as one of the primary tools in disease pathogenesis studies. From discovery‐based observations of the consequences of disrupting genetic elements of interest conserved across rodents and humans, hypothesis‐based studies to delineate the precise mechanisms that may contribute and possibly lead to disease, and preclinical‐based studies evaluating interventions of putative therapeutic benefit for their eventual suitability in human clinical trials, genetic rodent models continue to serve as key players in the landscape of many scientific endeavors.
By leveraging the ability to manipulate the genetics of rodents, model organism studies have profoundly shaped human disease research and treatment. In the most “ideal” experimental scenario, one would predict high concordance between the phenotypic consequences caused by disruption of a gene conferring risk in a human population, and the disruption of its homolog in the mouse. This scenario, however, is an exception. In fact, some rodent models of rare monogenic disorders do not always show a direct correspondence in phenotype, underscoring the challenges of experimental modeling even when the primary genetic etiology and degree of penetrance is well known in humans. Variable penetrance and expressivity due to a multitude of factors including genetic as well as gene‐by‐environment interactions are common in disease (Hunter, 2005; Zlotogora, 2003) and may offer some explanation of why direct correlations are not necessarily observed in the most ideal scenario of modeling seemingly simple, less complex genetic disorders in rodents. Moreover, a prevailing school of thought in the context of disease modeling advocates that studying the consequences of genetic alterations in rodents despite this potential shortcoming will lead to a better understanding of idiopathic, heterogeneous disease states that may share some degree of clinical features present in monogenic disorders, and may also possibly be polygenic in origin (Geschwind, 2008; Lim et al., 2013; Manolio et al., 2009; Plomin, Haworth, & Davis, 2009). Genetic modeling in lower order organisms such as rodents, however, is still among the most robust strategies in mechanism‐based efforts to gain a deeper understanding of human biology and disease despite potential, perceived, and genuine limitations to their utility.
2. MODELING NEUROBEHAVIORAL PHENOTYPES IN GENETIC MODELS
At this juncture, we focus our attention toward a critical evaluation of the historical and current perspectives on how genetic modeling in rodents has impacted the neurosciences—an intersection of science and medicine that may be considered among the last frontiers for human biology and related disease research. In the modern genetic era, we find that the elucidation of many complexities of brain function have been empowered by a shift in experimental paradigms from an historical reliance on lesion‐based and pharmacological manipulations to model neurological phenotypes (Geyer & Paulus, 1992; Norton, 1973; Stromgaard, Krogsgaard‐Larsen, & Madsen, 2009) toward use of rodent models with precise genetic manipulations. As a result, the “toolbox” to evaluate how the brain functions expanded, and provided a path forward to better understand the regulation of both normal behavior and conditions associated with a broad spectrum of neurodevelopmental, neuropsychiatric, and neurological disorders. Currently, the institutional infrastructure and expertise to test the consequences of genetic alterations in rodents has become well‐established and pervasive across all disciplines of neuroscience. Moreover, the field has advanced beyond its roots characterizing the effects of loss‐of‐function “knock‐out” alleles toward more cutting‐edge genetic techniques in mice and, more recently, rats (Ellenbroek and Youn, 2016; Geurts et al., 2009; Homberg, Wöhr, & Alenina, 2017; Hamilton et al., 2014; Veeraragavan et al., 2016; Till et al., 2015) that allow manipulation of the spatial, temporal, and state‐dependent factors that influence rodent CNS function and may prove relevant to the clinical manifestations of human CNS disorders (Gunaydin et al., 2014; McGraw, Samaco, & Zoghbi, 2011; Samaco et al., 2009; Tsai et al., 2012).
However, even as technological advances have led to unprecedented levels of control and precision in developing animal models of disease, the central question of how valuable these models are for understanding and treating neurobehavioral phenotypes that occur in people remains curiously unanswered. One reason for this uncertainty is related to the inherent difficulty of expressing neurobehavioral phenotypes in objective, reproducible ways that have clear clinical correlates with human disease. This is in contrast to animal models of non‐neurological conditions such as cancer, heart disease and skeletal defects, in which disease modeling in rodents corresponds more precisely with the human disorder. For example, if the genetic root cause of these human conditions are highly penetrant and lead to direct developmental or physiologic abnormalities (e.g., aberrant tissue histology such as bone morphology in skeletal dysplasias), the manifestation of these specific pathologic disease phenotypes may be readily apparent in rodent models sharing the corresponding genetic perturbation (Fuster et al., 2017; Grafe et al., 2014; Nagy, Sweet, & Eng, 2004). In the arena of non‐neurological conditions, the link between pathogenesis studies in genetic rodent models and efforts to identify actionable therapeutic intervention has been strengthened by several recent successes (Grafe et al., 2016, 2014; McClung, 2017) because they have been able to convincingly demonstrate a combination of face validity, construct validity and predictive validity—the three major subdivisions of validity (Anderzhanova, Kirmeier, & Wotjak, 2017; Boer & Sitsen, 1994). Face validity refers to similarities between the model and the clinical appearance of the disease either behaviorally or with respect to molecular pathology. Construct validity refers to the similarity of etiologies of the disease and model. Predictive validity refers to the ability of a model to predict outcomes of treatments for the disease.
However, neurobehavioral phenotypes—the hallmark of CNS disorders, comprised of abnormalities in a spectrum of neurological and behavioral functions including anxiety, social behavior, learning and memory, pain tolerance, and many others—remain challenging targets for rodent models. Experiences with neurobehavioral disorders have been less successful perhaps due to at least two over‐arching reasons. First, there is often no straight‐forward path toward ideal construct validity because the etiology of neurodegenerative and neurodevelopmental disorders is rarely monogenic and their pathogenesis is frequently unclear. Except in a handful of genetic conditions, single gene mutations do not account for these disorders, so simply disrupting a homologous rodent gene may not be sufficient to generate a model that recapitulates all of the relevant phenotypes of the human disorder. However, this strategy appears to be the first‐line and most intuitive approach. Alternatively, for example in Alzheimer's disease (AD), researchers have combined a number of disease‐associated alleles to generate models with synthetic phenotypes, each of which bears some resemblance to various aspects of the disorder but are, in total, non‐ideal constructs (Duyckaerts, Potier, & Delatour, 2008; Webster, Bachstetter, Nelson, Schmitt, & Van Eldik, 2014). Therefore, it is conceivable that some of the therapies that may be discovered in these AD models could be ones that reverse the synthetic potentiation induced by the genetic engineering itself without yielding any appreciable effect on the authentic disease process as it occurs in the human population. The lackluster results of recent human clinical trials for AD underscores the frustration that has accompanied attempts to leverage these models to identify novel AD therapeutics, and is reviewed in further detail below in section 2.3.
Second, behavior is a challenging experimental readout for several reasons. One reason is that in neurodegenerative and neurodevelopmental disorders, where the primary clinical manifestations are related to abnormalities in a complex interaction between the affected organism and its environment and/or other organisms, the relevant phenotype is a functional one—not one that can be grossly ascertained or measured from a radiograph or a serum level, but rather is an expression of the animal's performance in one of a variety of neurobehavioral tests. As a result, these tests frequently have higher inter‐ and intra‐subject variability than disease‐relevant measures in other non‐neurological disorders, which in turn requires larger numbers of animals to discern significant differences using simple statistical methods (ANOVA) and can contribute to lower reproducibility if not adequately controlled.
A second reason that neurobehavioral disorders are challenging to model is that the behavioral assay or set of assays most well‐suited for mechanistic and/or translational applications is not necessarily known a priori. The methods for rodent behavioral phenotyping are well‐established and have indeed yielded a deeper understanding of the components of complex behavior (Gottesman & Gould, 2003; Gould & Gottesman, 2006; Rizzo & Crawley, 2017; Silverman, Yang, Lord, & Crawley, 2010). However, inferring relevance between the human disorder and the rodent model in either direction is not trivial. For example, the choice of tasks to examine as analogs or proxies for human impairments becomes daunting when the primary feature of the CNS disorder of interest is impairment of the individual's ability to meet the cognitive demands required to integrate seamlessly into society such as executive function, behavioral self‐regulation, autobiographical memory, and knowledge of social mores. Conversely, although social behavior assays (e.g., based on interaction time between rodents) may detect the abnormalities anticipated in models of human disorders characterized by impaired social behavior, it is not entirely clear whether studying rodent social behavior in these contexts adequately captures the complexities of normal human social behavior. Furthermore, avoiding anthropomorphizing bias (Sjoberg, 2017) is of utmost importance, the choice to select behavioral measures because they may “seem” relevant to humans without formal testing of their actual relevance also contributes to the artificiality of phenotypes. Even when the choice of the behavioral assay may appear intuitively obvious, factors frequently encountered in rodent models may also restrict the choice of assay or confound interpretations—such as background‐related predispositions to health problems including blindness, endocrinopathy, and premature lethality (Errijgers et al., 2007; Fontaine & Davis, 2016; Johnson, Zheng, & Noben‐Trauth, 2006; Ward et al., 2016). Finally, which among the repertoire of rodent behavioral assays is the most translationally relevant—that is, can be performed in a comparable manner in human patients in a way that allows testing results from animals to be predictive of results in humans—is an entirely separate but equally urgent question that must be answered with each disease model.
A third reason is the challenge of connecting the functional readout of behavior to an underlying neurophysiological abnormality despite what is frequently a non‐linear correspondence between these levels of disease expression. Behavior is a final culmination of many orchestrated events derived from neurophysiological properties of heterogeneous cell‐cell communications as a result of electrical‐chemical signals within interconnected brain regions. The brain as an organ is a complex machine, generating the final behavioral output as a non‐linear function on these complex neurophysiological interactions. Thus, unlike non‐neurological models of disease, there is the potential both for significant pathology to exist without a clinical correlate and for significant clinical dysfunction to exist without an identifiable pathological abnormality, at least not by conventional modes of testing. The former phenomenon has been observed in Parkinson's disease, where it is estimated that between 30% and 70% of dopaminergic neurons in the ventral tegmental area must be lost before the characteristic motor impairments manifest (Dauer & Przedborski, 2003; Fearnley & Lees, 1991; Lang and Lozano, 1998). This phenomenon is arguably a more pronounced problem in the field of neurobehavioral disease modeling than it is in non‐neurological diseases and likely relates to the importance of neurophysiological interactions (i.e., brain wave activity and neurotransmitter release) within and between brain regions for the proper regulation of normal behavior. For instance, long‐range interactions across brain regions may have a functional effect on behavior but would be difficult to evaluate without knowing, a priori, which brain regions to interrogate or without formal hypothesis‐based experimental designs (Harris & Gordon, 2015).
The final reason modeling behavior can be challenging is the skepticism on behalf of some investigators regarding whether any animal behavior has clinical relevance to that of humans, raising questions regarding the utility of model systems (Figure 1). Fortunately, however, there is strong evidence from the history of drug development using symptom‐based animal models of anxiety, acute mania, psychosis, and pain that animal behavior can be highly predictive of human responses to clinically relevant medications—despite the fact that the vast majority of these discoveries were made in animal models whose construct validity was arguably low. Many of the drugs in the World Health Organization essential drugs list arose from this “golden age” of reductionist models (WHO Expert Committee, 2015). For example, the antipsychotic haloperidol was originally identified by Janssen Pharmaceutica using a simple screen to identify compounds that provided analgesia during hot plate assay without causing pupil dilation in mice (Granger & Albu, 2005). Risperidone's potential was supported by its ability to counter interoception of LSD in rodents during drug discrimination testing (Meert, de Haes, & Janssen, 1989). Finally, among antiepileptic drugs (AEDs), acute inducible seizure models were sufficient to identify the vast majority of AEDs including phenytoin, carbamazepine, and valproic acid (Meunier, Carraz, Neunier, Eymard, & Aimard, 1963; Merritt & Putnam, 1984).
Figure 1.

General questions to evaluate a model's utility. The questions proposed serve as a guide to establish the limits of validity and utility for characterization of behavioral phenotypes present in brain disorders. While they are also more broadly applicable to molecular and cellular phenotypes, they seek to identify obstacles within a model that prevent successful translation from the research lab to the clinic
Despite the challenges discussed here, genetic approaches continue to hold unique promise for developing models of CNS disorders. In the following sub‐sections, we will illustrate how each of these challenges has manifest in the context of several specific disease models in the hope that these examples will be instructive toward greater success in future endeavors to model and develop treatments for neurobehavioral disorders.
2.1. Issues of Reproducibility
Reproducibility, especially with regard to models of CNS disorders, continues to be an ongoing issue. Several analyses, reviews, and commentaries have attempted to address the issue of reproducibility and have outlined a series of measures that the scientific community at large could adopt to counter systemic factors that impair reproducibility (Baker, 2016; Justice & Dhillon, 2016; Perrin, 2014; Voelkl & Würbel, 2016). This is also aggravated by a historical trend of conducting preclinical behavioral testing in statistically improper ways that increase the risk of type I errors (Ioannidis, 2005a, 2005b; Sjoberg, 2017). One example of reproducibility problems comes from the field of Amyotrophic Lateral Sclerosis (ALS). Reproduction studies have highlighted that additional rigorous testing would have tempered enthusiasm for drugs making their way from preclinical mouse work to clinical trials. These studies performed at the ALS‐Therapy Development Institute tested nine compounds proposed for the treatment of ALS. None reproduced the dramatic extension of lifespan indicated by their initial publications. Furthermore, eight of the nine outright failed in clinical trials (Perrin, 2014). While systematic reproduction studies have yet to be performed across many of the other models of neurological disease, there are indications that concerns with reproducibility are not specific to any particular field, but rather may be more widespread than previously appreciated. Even in cases of monogenic disorders, defined by a clear identifiable genetic mutation and clinical diagnostic criteria as is evident in Rett syndrome (RTT) and Fragile X syndrome (FXS), behavioral alterations may vary depending on factors such as genetic strain background and methodological approaches. In the field of RTT, treatments initially shown to improve symptomology such as bone marrow transplantation to restore wild‐type microglia function as well as stimulation of IGF‐1 receptors fail to replicate when attempted by other groups (Derecki et al., 2012; Pitcher et al., 2013; Tropea et al., 2009; Wang et al., 2015). Furthermore, characterization of Fmr1 knock‐out mice by multiple researchers has identified variation that may be driven by genetic strain, environment, or methodological differences (Bernardet & Crusio, 2006; Spencer et al., 2011). These limited examples from the literature are only a glimpse into the broader issue of reproducibility that can be resolved by implementing measures that include but are not limited to (i) providing incentives to publish rather than ignore negative data; (ii) improving the standards for the correct use of statistical analyses; (iii) incorporating statistical power analysis into experimental design before the onset of a study; (iv) increasing transparency of materials and methods such as strain backgrounds, husbandry practices, and testing protocols; and (v) funding systematic reproduction and validation studies across multiple sites. Reducing methodological and human biases that impair reproducibility will then allow fields to better distinguish signal from noise, and improve confidence in identifying biologically relevant factors that modify phenotypic penetrance and severity (Baker, 2016; Justice & Dhillon, 2016; Perrin, 2014).
2.2. Selection of Appropriate Outcome Measures
An often underappreciated point is the importance of translationally valid outcome measures for studies. While many phenotypes provide reproducible and biologically meaningful data, they are often assumed to translate to clinical outcomes without sufficient vetting of their validity. A prime example is the use of survival as an outcome measure. Again the field of ALS can provide us with one example, mice overexpressing TDP43, an ALS‐associated gene. It was initially assumed that the mortality observed in TDP43 mice would be attributed to progressive muscle atrophy associated with ALS. However, the true cause of death in the TDP43 model was due to neurodegeneration in the myenteric plexus of the colon leading to diminished smooth muscle function and eventual bowel obstruction (Hatzipetros et al., 2014). A similar finding was also made for mouse models of RTT where decreased survival of male mice was used as a surrogate for the slightly increased mortality observed in female patients and early mortality in MeCp2‐related severe congenital encephalopathy. Unfortunately, kidney failure due to urethral obstruction by plugs of coagulated mouse semen was identified as a potential cause of death in the absence of Mecp2 function and suggests survival may be a poor outcome measure for preclinical studies of MeCp2‐related disorders (Ward et al., 2016).
The use of questionable outcome measures can be further subdivided into multiple categories: (i) measures that are phenotypically similar but mechanistically distinct; (ii) measures that do not have a clear clinical analog, or that are not feasible to test within a patient population; and (iii) measures that are subject to sources of variation that are poorly characterized, either in the animal models or the clinical population.
Although the above vignettes from the ALS and RTT fields illustrate two examples of phenotypically similar but mechanistically distinct outcome measures, the pursuit of mGluR5 as a target for FXS provides an additional case study highlighting how the sub‐optimal selection of outcome measures may have impeded progress in the area of therapy development. In recent years, mGluR5 (mG5), a group 1 metabotropic glutamate receptor expressed post‐synaptically throughout the brain (Shigemoto et al., 1997), has become an increasingly popular target for the treatment of neurological disorders. In the presence of glutamate, mG5 acts as a G‐protein coupled receptor, triggering release of intracellular calcium and stimulating a cascade of events to regulate excitability and promote translation at the synapse (Dhami & Ferguson, 2006). The role of mG5 in regulating translation at the synapse suggested a potential mechanism underlying the pathogenesis of FXS (Bear, Huber, & Warren, 2004). Conceptually, the model proposed by Bear et al. posited that in the normal state, FMRP inhibits translation of a pool of mRNA transcripts located at the synapse, with these transcripts among the distal effectors of mG5 activity; in contrast, in FXS, this translation is unchecked and pathology arises from excessive output from metabotropic glutamate signaling. This theory generated enormous excitement as a potential success of leveraging genetic animal models to generate and translate a therapy to treat people with FXS.
Despite the excitement, mavoglurant, one of the first mGluR5 negative allosteric modulating drugs tested for the treatment of FXS was unable to demonstrate efficacy for the improvement of patient outcome (Bailey et al., 2016; Berry‐Kravis et al., 2016). While the reported unsuccessful attempt to translate mavoglurant as a treatment FXS could be seen as an unfortunate aggregation of missteps in selecting the right druggable target, molecule, patient population, end points and so on (Clinical Trial Success Probabilities—some back of the envelope calculations • Fragile X Research—FRAXA Research Foundation, 2014), there is also clear indication that the selection of endpoints to evaluate progressing mavoglurant through the drug discovery pipeline were non‐ideal.
In preclinical trials with mouse models of FXS, there were multiple studies that probed whether genetic or pharmacological manipulation of mG5 modulates phenotypes associated with loss of Fmr1 function. Phenotypes apparently reversed by modulation of mG5 included alterations in social behavior, open field exploration, sensory motor gating, learning and memory, audiogenic seizures, ocular dominance plasticity, dendritic spine morphology, and aberrant protein synthesis at the synapse (de Vrij et al., 2008; Dölen et al., 2007; Gantois et al., 2013; Levenga et al., 2011; Pop et al., 2014; Yan, Rammal, Tranfaglia, & Bauchwitz, 2005). While each of these endpoints may serve to reveal biologically relevant information, the translational validity is less clear for some of the selected outcome measures. Two new categories of questionable endpoints become apparent from the preclinical data in support pharmacological manipulation of mG5 in FXS: endpoints for which the clinical analog is uncertain or untestable, and endpoints with variable penetrance within the mouse model.
Phenotypes such as abnormal ocular dominance plasticity, dendritic spine morphology, and aberrant protein synthesis at the synapse are inherently untestable in a clinical trial setting. In the case of inhibition of mG5 signaling via treatment with MPEP, exploration of the center of the open field was normalized which was argued as a normalization of anxiety (Yan et al., 2005). However, while open field locomotor activity is sometimes used as a test for hyperactivity, and rates of exploration of the center of the open field have previously been used as a surrogate measure for anxiety‐related behavior, more specialized tests for this behavioral domain such as the elevated plus maze or light‐dark box exploration tests would better distinguish the behavior that Yan and coworkers were actually measuring (Archer, 1973; File, 1985, 2001; File, Lippa, Beer, & Lippa, 2004; Spencer et al., 2011).
Furthermore, the phenotype described by Yan and coworkers is among several in the Fmr1 KO mice that are variable in penetrance. Fmr1 KO mice have demonstrated locomotor activity and exploration of the center of the open field at increased or comparable levels compared to their wild‐type littermates depending on the study (Mineur, Sluyter, de Wit, Oostra, & Crusio, 2002; Peier et al., 2000; Spencer, Alekseyenko, Serysheva, Yuva‐Paylor, & Paylor, 2005; Spencer et al., 2011). Similar variability in penetrance exists for assays of learning and memory in Fmr1 KO mice. To overcome this problem, Dölen et al. (2007) selected an assay for inhibitory avoidance extinction, a learning and memory task dependent on hippocampal protein synthesis, an alteration that is predicted to be affected in the mGluR theory of FXS (Power, Berlau, McGaugh, & Steward, 2006). While this is a fair logical progression based on the molecular model and the known regulation of the behavioral phenotype, it ignores the general absence of more penetrant and severe learning and memory deficits among Fmr1 KO mice. This would suggest that the model is insufficient as a predictor of learning and memory outcomes in humans (Bernardet & Crusio, 2006; Dobkin et al., 2000; Van Dam et al., 2000).
Additionally, the effect of mavoglurant on social behavior in Fmr1 KO mice was similarly confounded by genetic variation (Gantois et al., 2013). Mavoglurant was demonstrated to correct social behavior to wild‐type levels in the three chamber test for sociability. However, the Fmr1 KO mice tested by Gantois et al. (2013) were hypersocial which is in direct opposition to the social avoidance described in human patients. Furthermore, while it may be possible that assays of social behavior in the mouse may generally have validity with respect to human social behavior, the deviations from WT social behavior induced by genetic lesions in Fmr1 are especially sensitive to genetic strain background further suggesting poor validity when crossing the even wider genetic variation between mice and humans (Spencer et al., 2011). The genetic modification of symptom severity within the Fmr1 KO mice also suggests human genetic variation could be a large confound to clinical trials. Additional work leveraging differences in symptom severity across strains and to identify modifiers and biomarkers would improve patient selection and stratification in future trials, presumably improving the likelihood of selecting the correct patients for a treatment.
However, even if the phenotypes that were corrected in mouse showed similar restoration in the human population, the study endpoints chosen for the clinical trials were unlikely to reveal them. The outcome measures in these trials included several subjective rater scales, some of which were completed by the caregiver of the enrolled patients. Moreover, the main questionnaire used as the primary endpoint was a modified version of the aberrant behavior questionnaire tailored to the Fragile X community, ABC‐CFX, which generates a symptom score across dimensions of irritability, lethargy/withdrawal, stereotypic behavior, hyperactivity, inappropriate speech, and social avoidance (Sansone et al., 2012). The ABC‐CFX scores were also shown to be highly susceptible to placebo effect over the course of the clinical trials (Berry‐Kravis et al., 2016). Generally, the study did not leverage molecular biomarkers, was largely dependent on subjective metrics and was hindered by lacking objective quantifiable scores of symptom severity that could be generated for all of the enrolled patients.
In the end, the pursuit of the mG5 theory of FXS has left several questions unanswered, yet has been highly instructive. The “teachable moments” derived from this case study are generalizable across many genetic rodent models of brain disorders that have yet to yield a breakthrough in therapy. Successful integration of the lessons learned becomes essential to evaluating any model's role in any future attempts to treat disease (Figure 2). Thus, it becomes important to use models and compounds that have not, to date, successfully shown translational validity to inform us on the limits of our current approaches and so that the next iteration of models and methods can avoid similar pitfalls (Garner, 2014).
Figure 2.

Integration of “safeguards” in rodent model studies of brain disorders. In order to address barriers to translation such as those described in this review, we propose a series of suggestions in the development and use of rodent models, the design of preclinical studies that use them, and the clinical studies that are based upon successful preclinical work. Each of these “safeguards” helps to promote valid findings. Furthermore, if followed they will help to ensure that negative findings can still provide meaningful contributions to our understandings of the biology of the modeled disorders and the limits of our abilities to model them. While these safeguards were written with behavior in mind, they are also worthwhile considerations for models and diseases that involve molecular and histopathological components
2.3. Selection of Model, Guided by Purpose
Delineation of the experimental motivations for a rodent model is key to understanding the limits of its validity. Without understanding these limits, it becomes nearly impossible to distinguish between a biological artifact, a true cause of pathogenesis, or a component of a constellation of causes. This concept is perhaps underappreciated in modeling CNS disorders.
In the case of genetic modeling, disruptions of genes implicated in the human disorder are thought to result in high construct validity. Among monogenic and highly penetrant disorders (Amir et al., 1999; European Chromosome 16 Tuberous Sclerosis Consortium, 1993; van Slegtenhorst et al., 1997; Verkerk et al., 1991), the genetic rodent models do indeed recapitulate the chronic disease state that occurs in humans; there is an ongoing abnormal physiologic baseline, either due to developmental or progressive abnormalities. However, the nature of such genetic models cuts both ways, with the limitation applying to disease genes with low penetrant contributions in complex genetic disorders such as autism and neurodegenerative disease in which no single gene mutation confers 100% risk. With some of these genetic models, there is also a risk of failing to capture the essence of the disorders which in reality may be the consequence of a poorly understood combination of multiple genetic and environmental factors. Thus, the models become, at best, inadequate simulacra; that is, poor simulations or substitutes.
Furthermore, there are fundamental differences in the nature of the disease states in genetic models compared to the early acute reductionist models of decades past. With respect to the category of diseases that are directly attributable to specific genetic lesions, correcting chronic disease states may represent too great a challenge to overcome. New therapeutic strategies that are capable of eliciting long duration changes within critical windows of disease progression may be needed in the next stages of disease and therapy modeling.
In the era of mechanism‐based approaches for therapy discovery, the focus on molecular mechanism in models of brain disorders may also be leading us on unproductive detours when limitations are not clearly recognized. The logical trap that disease pathology is associated with the largest signal or a perturbation in a pathway overlooks potentially novel contributions to disease that may be more translationally relevant. For example, in the field of AD, potential mechanisms of disease are still hotly debated with the relative contributions of amyloid plaques and neurofibrillary tangles or other pathways still uncertain (Ballatore, Lee, & Trojanowski, 2007; Selkoe & Hardy, 2016). Preclinical proof‐of‐concept testing for antibody‐based therapies largely addressed the plaque burden and molecular load of amyloid‐β within the brains of AD‐related mouse models (Bard et al., 2000), with some studies also showing therapeutic improvement in learning and memory performance tasks (Antonios et al., 2015; Dodart et al., 2002; Wang, Yu, Liu, Zhao, & Xu, 2016). However, some of the animal model data demonstrated that acute intervention led to changes in learning and memory performance without an appreciable effect on amyloid‐β burden (Dodart et al., 2002), thereby dissociating the relationship between a pathological hallmark implicated in AD and the expected behavioral improvement. These discrepant findings may have signaled a pitfall in the mechanistic premise of antibody‐based therapies targeting amyloid‐β. Indeed, two antibody‐based therapies, solanuzumab and bapineuzumab, did not meet their primary study endpoints of cognitive improvement in clinical trials (Doody et al., 2014; Salloway et al., 2014; The Lancet Neurology, 2017). The problem of knowing whether the “correct target” is being manipulated is also an ongoing and difficult to resolve question. To the best of our knowledge, this mechanism‐based target identification approach has only infrequently produced a clinically successful treatment (e.g., in multiple sclerosis; Havrdova et al., 2017), and perhaps has yet to produce a truly groundbreaking or revolutionary treatment for a neurological disorder.
Although reliance on genetic models and mechanism‐based approaches may contribute to the challenges mentioned in this section, the rigorous and thoughtful use of these strategies does allow improved understanding of the basic biology of disease. Technological limitations of available tools are certainly roadblocks to the creation of truly polygenic multifactorial models of CNS disorder, the identification of authentic disease‐causing targets, and the development of ideal therapies for chronic conditions. Thus, it is important to make the best use of the tools we currently possess so that knowledge gained can be immediately leveraged as new innovative solutions are developed.
3. COMPLEMENTARY APPROACHES OF PATHOLOGY/MECHANISM‐ AND SYMPTOM‐BASED STRATEGIES
In the foregoing discussions, we have touched on several of the shortcomings of the leading modern approach to understanding genetic disorders of CNS, which is chiefly concerned with the identification of pathological molecular and cellular abnormalities to define each disorder. Here, we would like to take the time to formally discuss what may be perceived as the limits or blind spots in this approach, and to highlight the advantages of complementary symptom‐based screening strategies to understand CNS disorders.
As a motivating example, we would draw the readers’ attention to the fact that many clinically defined CNS disorders have more clinical similarity than genetic homogeneity, as evidenced by the fact that genome wide association and sequencing studies continue to identify genetic loci that can only account for a fraction of the genetic risk associated with each of these disorders (Manolio et al., 2009; Visscher, Brown, McCarthy, & Yang, 2012). This holds true of epilepsy (Epi4K Consortium et al., 2013), autism spectrum disorder (Chaste et al., 2015), and schizophrenia (Gejman, Sanders, & Duan, 2010; Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium, 2015). These observations imply that the abnormal phenotype in each disorder may be a common expression of dysfunction in a complex biological mechanism with several distinct genetic and developmental vulnerabilities. Various disruptions at different parts of this mechanism, therefore, represent necessary elements of different sufficient sets for producing the disease state, each of which is capable of giving rise to the symptoms associated with a disorder. At a certain level, this interpretation would imply that the upstream molecular profiles in these patients could be extremely different or perhaps partially overlapping, and in the worst case scenario a molecular‐based treatment approach may entail the discovery of a unique molecular mechanism for each patient. Yet, given the clinical similarity, there must be a common downstream endophenotype that explains each or all of these disorders, perhaps at the neurophysiological level which remains elusive. Application of the Research Domain Criteria to analyze the component constituents that may be modeled in rodents may likely help better resolve some of these questions (Anderzhanova et al., 2017; Carcone & Ruocco, 2017; Insel et al., 2010).
Symptom‐based screening operates at this common downstream level to identify agents that reverse a behavior or symptom associated with the disorder irrespective of any particular molecular mechanism. Symptom‐based screening harkens back to the “golden age” of pharmaceutical discovery during which animal‐based screens led to the discovery of nearly all of today's major classes of pharmaceuticals including antipsychotics, antidepressants, anticonvulsants, and anxiolytics (Stromgaard et al., 2009). In general, these efforts identified novel compounds using animal models in which normal animals were treated with chemical, electrical, mechanical, or behavioral manipulations in order to induce an acutely abnormal physiological state that approximated the clinical condition in question, such as acute psychosis, depression, or seizure (Figure 3a). The success of this approach continues to astound many observers and there have been various explanations put forth to explain why these approaches appear to have been more productive than our modern efforts (Scannell & Bosley, 2016). Recently, there has been dwindling interest in using primarily symptom‐based screening in favor of developing more “authentic” genetic models of disease (for example, as championed by Wendland & Ehlers, 2016); however, we (and others [Pangalos, Schecter, & Hurko, 2007]) believe that there are certain advantages to symptom‐based screening that may help to overcome some of the shortcomings of the alternative “pathological” or “mechanism‐based” approach which is predicated on the assumption that identifying key molecular abnormalities and reversing them is the most actionable path toward identifying clinically effective therapies.
Figure 3.
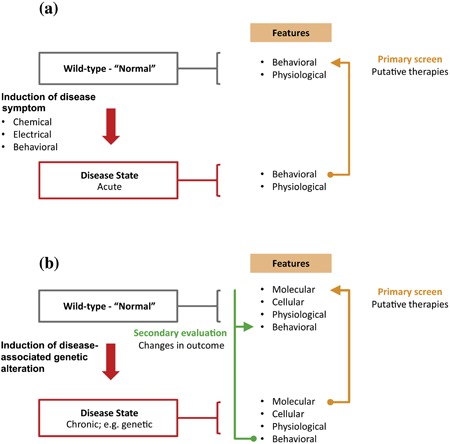
Comparison of perspectives on modeling and intervention strategies for CNS disorders. (a) The “golden age” of animal‐based modeling and therapeutic screens emphasized interventions in normal animals that resulted in changes in behavior based on acute interventions. (b) Broadly speaking all genetic CNS disorders can be modeled using the paradigm shown. An underlying genetic deficit (possibly in combination with other genetic susceptibility loci and environmental exposures) gives rise to a pathological sequence of molecular changes (gene expression, protein expression) leading to changes in cellular function, neurotransmitter release, neurophysiological function, and ultimately affecting the highest levels of organismal function mediated by the CNS, which may include motor control, learning/memory, executive processing, emotional regulation, and social behavior
One advantage to symptom‐based approaches is that they involve screening instead for agents that have effects on a functional read‐out such as behavior or neurophysiological measurements, similar to prior “golden age” assays (Figure 3b). The key element of this approach is that it prioritizes the discovery of treatments with high functional value regardless of any specific molecular mechanism. In fact, viewed from this direction, each positive hit teaches us precisely which molecular pathways are the most critical for treating the disorder, and may reveal novel mechanisms of action. For example, the antiepileptic properties of levetiracetam (Keppra) could only have been identified by screening, as its mechanism of action was later shown to be novel and distinct from other AEDs, being related not to ion channel function but rather synaptic vesicle glycoprotein 2A peptide, a molecule with no previously known role in seizure control (Bialer & White, 2010).
Overall, the two approaches remain complementary and investigations of brain disorders would benefit from utilizing the two in tandem. The symptom‐oriented approach would serve as a screen to identify all relevant molecular targets, and the subsequent molecular approach would aim to optimize the ability to target those pathways revealed by the more agnostic approach. In particular, the symptom‐oriented approach would be well‐suited for disease states lacking any effective treatments to date or well‐defined central basic science principles (e.g., social and cognitive impairment), are primarily neurophysiologic in nature (e.g., epilepsy), and/or are genetically heterogeneous among patients. A major barrier to the symptom‐oriented approach has been the logistically intense nature of behavioral screening compared to molecular studies. In addition, the enthusiasm for primarily (molecular) hypothesis‐testing research among funding agencies has overshadowed support for more mechanism‐naïve approaches. However, given the ongoing, less than favorable outcomes of clinical trials for CNS disorders, we suspect that this approach may be precisely what is needed.
4. CONCLUSION
Genetic rodent models of brain disorders are a necessary component to the dissection of neurobiology. They have offered a wealth of resources to explore genetic contributions to behavior and pathology, as well as explore mechanisms that link gene to function. However, the end goal of modeling human disorders and developing mechanistically derived therapies has yet to be reliably attained. The barriers to translation may involve limits to the validity of modeling human behavior and pathology in other species. Understanding these limits will be critical to determining the best applications for animal models. Unfortunately, problems across fields with reproducibility, discordant selection of outcome measures, and sub‐optimal models create a veil that masks and blurs the boundary delineating where these genetic models have potential to be successful and where they are no longer meaningful. Addressing these issues is paramount to the success of genetic neurobiology and development of novel therapies. In sum, taking lessons from earlier eras of drug discovery using clinical endpoint‐focused outcome measures may help bridge the gap to identify new targets which are missed by a narrow emphasis on established pathways and mechanisms.
ACKNOWLEDGMENTS
All authors contributed to the preparation and review of the manuscript. This work is supported in part by the U.S. National Institutes of Health Grants DP5OD009134, R01HD083181, the Stedman West Foundation, Texas Children's Hospital, and the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center (U54HD083092). The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
CONFLICTS OF INTEREST
The authors have no financial interests or conflicts of interest relevant to this manuscript to disclose.
Biographies
C. M. McGraw, MD, PhD, is a Resident Physician in the Department of Neurology at the University of California, San Francisco.

C. S. Ward, PhD, is a Research Associate in the Department of Molecular and Human Genetics at Baylor College of Medicine, and the Jan and Dan Duncan Neurological Research Institute at Texas Children's Hospital. He is also the Preclinical Studies Manager of the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center and Jan and Dan Duncan Neurological Research Institute Rodent Neurobehavioral Core Facilities.

R. C. Samaco, PhD, is an Assistant Professor in the Department of Molecular and Human Genetics at Baylor College of Medicine, the Program in Translational Biology and Molecular Medicine at Baylor College of Medicine, and the Jan and Dan Duncan Neurological Research Institute at Texas Children's Hospital. He is also the Associate Director of the Intellectual and Developmental Disabilities Research Center, and Director of the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center and Jan and Dan Duncan Neurological Research Institute Rodent Neurobehavioral Core Facilities.

McGraw CM, Ward CS, Samaco RC. Genetic rodent models of brain disorders: Perspectives on experimental approaches and therapeutic strategies. Am J Med Genet Part C Semin Med Genet. 2017;175C: 368–379. https://doi.org/10.1002/ajmg.c.31570
Christopher M. McGraw and Christopher S. Ward contributed equally to this work.
REFERENCES
- Amir, R. E. , Van den Veyver, I. B. , Wan, M. , Tran, C. Q. , Francke, U. , & Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X‐linked MECP2, encoding methyl‐CpG‐binding protein 2. Nature Genetics, 23, 185–188. [DOI] [PubMed] [Google Scholar]
- Anderzhanova, E. , Kirmeier, T. , & Wotjak, C. T. (2017). Animal models in psychiatric research: The RDoC system as a new framework for endophenotype‐oriented translational neuroscience. Neurobiology of Stress, 7, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonios, G. , Borgers, H. , Richard, B. C. , Brauß, A. , Meißner, J. , Weggen, S. , … Bayer, T. A. (2015). Alzheimer therapy with an antibody against N‐terminal Abeta 4‐X and pyroglutamate Abeta 3‐X. Scientific Reports, 5, 17338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer, J. (1973). Tests for emotionality in rats and mice: A review. Animal Behavior, 21, 205–235. [DOI] [PubMed] [Google Scholar]
- Bailey, D. B. , Berry‐Kravis, E. , Wheeler, A. , Raspa, M. , Merrien, F. , Ricart, J. , … Apostol, G. (2016). Mavoglurant in adolescents with fragile X syndrome: Analysis of clinical global impression‐improvement source data from a double‐blind therapeutic study followed by an open‐label, long‐term extension study. Journal of Neurodevelopmental Disorders, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, M. (2016). 1,500 scientists lift the lid on reproducibility. Nature, 533, 452. [DOI] [PubMed] [Google Scholar]
- Ballatore, C. , Lee, V. M.‐Y. , & Trojanowski, J. Q. (2007). Tau‐mediated neurodegeneration in Alzheimer's disease and related disorders. Nature Reviews Neuroscience, 8, 663–672. [DOI] [PubMed] [Google Scholar]
- Bard, F. , Cannon, C. , Barbour, R. , Burke, R. L. , Games, D. , Grajeda, H. , … Yednock, T. (2000). Peripherally administered antibodies against amyloid beta‐peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nature Medicine, 6, 916–919. [DOI] [PubMed] [Google Scholar]
- Bear, M. F. , Huber, K. M. , & Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends in Neurosciences, 27, 370–377. [DOI] [PubMed] [Google Scholar]
- Bernardet, M. , & Crusio, W. E. (2006). Fmr1 KO mice as a possible model of autistic features. Scientific World Journal, 6, 1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry‐Kravis, E. , Des Portes, V. , Hagerman, R. , Jacquemont, S. , Charles, P. , Visootsak, J. , … von Raison, F. (2016). Mavoglurant in fragile X syndrome: Results of two randomized, double‐blind, placebo‐controlled trials. Science Translational Medicine, 8, 321ra5. [DOI] [PubMed] [Google Scholar]
- Bialer, M. , & White, H. S. (2010). Key factors in the discovery and development of new antiepileptic drugs. Nature Reviews. Drug Discovery, 9, 68–82. [DOI] [PubMed] [Google Scholar]
- Boer den, J. A. , & Sitsen, J. M. A. (Eds.). (1994). Handbook of depression and anxiety: A biological approach. New York: M. Dekker; (p. 691). [Google Scholar]
- Carcone, D. , & Ruocco, A. C. (2017). Six years of research on the National Institute of Mental Health's Research domain criteria (RDoC) initiative: A systematic review. Frontiers in Cellular Neuroscience, 11, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaste, P. , Klei, L. , Sanders, S. J. , Hus, V. , Murtha, M. T. , Lowe, J. K. , … Devlin, B. (2015). A genome‐wide association study of autism using the Simons Simplex Collection: Does reducing phenotypic heterogeneity in autism increase genetic homogeneity? Biological Psychiatry, 77, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinical Trial Success Probabilities—some back of the envelope calculations • Fragile X Research—FRAXA Research Foundation. (2014). https://www.fraxa.org/trialprobabilities/
- Dauer, W. , & Przedborski, S. (2003). Parkinson's disease: Mechanisms and models. Neuron, 39, 889–909. [DOI] [PubMed] [Google Scholar]
- Derecki, N. C. , Cronk, J. C. , Lu, Z. , Xu, E. , Abbott, S. B. G. , Guyenet, P. G. , & Kipnis, J. (2012). Wild‐type microglia arrest pathology in a mouse model of Rett syndrome. Nature, 484, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vrij, F. M. S. , Levenga, J. , van der Linde, H. C. , Koekkoek, S. K. , De Zeeuw, C. I. , Nelson, D. L. , … Willemsen, R. (2008). Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiology of Disease, 31, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhami, G. K. , & Ferguson, S. S. G. (2006). Regulation of metabotropic glutamate receptor signaling, desensitization and endocytosis. Pharmacology & Therapeutics, 111, 260–271. [DOI] [PubMed] [Google Scholar]
- Dobkin, C. , Rabe, A. , Dumas, R. , El Idrissi, A. , Haubenstock, H. , & Brown, W. T. (2000). Fmr1 knockout mouse has a distinctive strain‐specific learning impairment. Neuroscience, 100, 423–429. [DOI] [PubMed] [Google Scholar]
- Dodart, J.‐C. , Bales, K. R. , Gannon, K. S. , Greene, S. J. , DeMattos, R. B. , Mathis, C. , … Paul, S. M. (2002). Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nature Neuroscience, 5, 452–457. [DOI] [PubMed] [Google Scholar]
- Dölen, G. , Osterweil, E. , Rao, B. S. S. , Smith, G. B. , Auerbach, B. D. , Chattarji, S. , & Bear, M. F. (2007). Correction of fragile X syndrome in mice. Neuron, 56, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody, R. S. , Thomas, R. G. , Farlow, M. , Iwatsubo, T. , Vellas, B. , Joffe, S. , … Alzheimer's Disease Cooperative Study Steering Committee, Solanezumab Study Group. (2014). Phase 3 trials of solanezumab for mild‐to‐moderate Alzheimer's disease. New England Journal of Medicine, 370, 311–321. [DOI] [PubMed] [Google Scholar]
- Duyckaerts, C. , Potier, M.‐C. , & Delatour, B. (2008). Alzheimer disease models and human neuropathology: Similarities and differences. Acta Neuropathologica, 115, 5–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenbroek, B. , & Youn, J. (2016). Rodent models in neuroscience research: Is it a rat race? Disease Models & Mechanisms, 9, 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen, A. S. , Berkovic, S. F. , Cossette, P. , Delanty, N. , … Winawer, M. R. (2013). De novo mutations in epileptic encephalopathies. Nature, 501, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errijgers, V. , Van Dam, D. , Gantois, I. , Van Ginneken, C. J. , Grossman, A. W. , D'Hooge, R. , … Kooy, R. F. (2007). FVB.129P2‐Pde6b(+) Tyr(c‐ch)/Ant, a sighted variant of the FVB/N mouse strain suitable for behavioral analysis. Genes, Brain, and Behavior, 6, 552–557. [DOI] [PubMed] [Google Scholar]
- European Chromosome 16 Tuberous Sclerosis Consortium. (1993). Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell, 75, 1305–1315. [DOI] [PubMed] [Google Scholar]
- Fearnley, J. M. , & Lees, A. J. (1991). Ageing and Parkinson's disease: Substantia nigra regional selectivity. Brain: A Journal of Neurology, 114(Pt 5), 2283–2301. [DOI] [PubMed] [Google Scholar]
- File, S. E. (1985). What can be learned from the effects of benzodiazepines on exploratory behavior? Neuroscience and Biobehavioral Reviews, 9, 45–54. [DOI] [PubMed] [Google Scholar]
- File, S. E. (2001). Factors controlling measures of anxiety and responses to novelty in the mouse. Behavioural Brain Research, 125, 151–157. [DOI] [PubMed] [Google Scholar]
- File, S. E. , Lippa, A. S. , Beer, B. , & Lippa, M. T. (2004). Animal tests of anxiety. Current Protocols in Neuroscience, Chapter 8, Unit 8.3. [DOI] [PubMed] [Google Scholar]
- Fontaine, D. A. , & Davis, D. B. (2016). Attention to background strain is essential for metabolic research: C57BL/6 and the international knockout mouse consortium. Diabetes, 65, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster, J. J. , MacLauchlan, S. , Zuriaga, M. A. , Polackal, M. N. , Ostriker, A. C. , Chakraborty, R. , … Walsh, K. (2017). Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science, 355, 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantois, I. , Pop, A. S. , de Esch, C. E. F. , Buijsen, R. A. M. , Pooters, T. , Gomez‐Mancilla, B. , … Willemsen, R. (2013). Chronic administration of AFQ056/Mavoglurant restores social behaviour in Fmr1 knockout mice. Behavioural Brain Research, 239, 72–79. [DOI] [PubMed] [Google Scholar]
- Garner, J. P. (2014). The significance of meaning: Why do over 90% of behavioral neuroscience results fail to translate to humans, and what can we do to fix it? ILAR Journal, 55, 438–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gejman, P. , Sanders, A. , & Duan, J. (2010). The role of genetics in the etiology of schizophrenia. Psychiatric Clinics of North America, 33, 35–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind, D. H. (2008). Autism: Many genes, common pathways? Cell, 135, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts, A. M. , Cost, G. J. , Freyvert, Y. , Zeitler, B. , Miller, J. C. , Choi, V. M. , … Buelow, R. (2009). Knockout rats via embryo microinjection of zinc‐finger nucleases. Science, 325, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer, M. A. , & Paulus, M. P. (1992). Multivariate and nonlinear approaches to characterizing drug effects on the locomotor and investigatory behavior of rats. NIDA Research Monograph, 124, 203–235. [PubMed] [Google Scholar]
- Gottesman, I. I. , & Gould, T. D. (2003). The endophenotype concept in psychiatry: Etymology and strategic intentions. American Journal of Psychiatry, 160, 636–645. [DOI] [PubMed] [Google Scholar]
- Gould, T. D. , & Gottesman, I. I. (2006). Psychiatric endophenotypes and the development of valid animal models. Genes, Brain, and Behavior, 5, 113–119. [DOI] [PubMed] [Google Scholar]
- Grafe, I. , Alexander, S. , Yang, T. , Lietman, C. , Homan, E. P. , Munivez, E. , … Lee, B. (2016). Sclerostin antibody treatment improves the bone phenotype of Crtap(‐/‐) mice, a model of recessive osteogenesis imperfecta. Journal of Bone and Mineral Research, 31, 1030–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafe, I. , Yang, T. , Alexander, S. , Homan, E. P. , Lietman, C. , Jiang, M. M. , … Lee, B. (2014). Excessive transforming growth factor‐β signaling is a common mechanism in osteogenesis imperfecta. Nature Medicine, 20, 670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger, B. , & Albu, S. (2005). The haloperidol story. Annals of Clinical Psychiatry, 17, 137–140. [DOI] [PubMed] [Google Scholar]
- Gunaydin, L. A. , Grosenick, L. , Finkelstein, J. C. , Kauvar, I. V. , Fenno, L. E. , Adhikari, A. , … Deisseroth, K. (2014). Natural neural projection dynamics underlying social behavior. Cell, 157, 1535–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, S. M. , Green, J. R. , Veeraragavan, S. , Yuva, L. , McCoy, A. , Wu, Y. , … Paylor, R. (2014). Fmr1 and Nlgn3 knockout rats: Novel tools for investigating autism spectrum disorders. Behavioral Neuroscience, 128, 103–109. [DOI] [PubMed] [Google Scholar]
- Harris, A. Z. , & Gordon, J. A. (2015). Long‐range neural synchrony in behavior. Annual Review of Neuroscience, 38, 171–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzipetros, T. , Bogdanik, L. P. , Tassinari, V. R. , Kidd, J. D. , Moreno, A. J. , Davis, C. , … Perrin, S. (2014). C57BL/6J congenic Prp‐TDP43A315T mice develop progressive neurodegeneration in the myenteric plexus of the colon without exhibiting key features of ALS. Brain Research, 1584, 59–72. [DOI] [PubMed] [Google Scholar]
- Havrdova, E. , Giovannoni, G. , Gold, R. , Fox, R. J. , Kappos, L. , Phillips, J. T. , … Marantz, J. L. (2017). Effect of delayed‐release dimethyl fumarate on no evidence of disease activity in relapsing‐remitting multiple sclerosis: Integrated analysis of the phase III DEFINE and CONFIRM studies. European Journal of Neurology, 24, 726–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homberg, J. R. , Wöhr, M. , & Alenina, N. (2017). Comeback of the rat in biomedical research. ACS Chemical Neuroscience, 8, 900 –903. [DOI] [PubMed] [Google Scholar]
- Hunter, D. J. (2005). Gene‐environment interactions in human diseases. Nature Reviews. Genetics, 6, 287–298. [DOI] [PubMed] [Google Scholar]
- Insel, T. , Cuthbert, B. , Garvey, M. , Heinssen, R. , Pine, D. S. , Quinn, K. , … Wang, P. (2010). Research domain criteria (RDoC): Toward a new classification framework for research on mental disorders. American Journal of Psychiatry, 167, 748–751. [DOI] [PubMed] [Google Scholar]
- Ioannidis, J. P. A. (2005a). Contradicted and initially stronger effects in highly cited clinical research. JAMA, 294, 218–228. [DOI] [PubMed] [Google Scholar]
- Ioannidis, J. P. A. (2005b). Why most published research findings are false. PLoS Medicine, 2, e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, K. R. , Zheng, Q. Y. , & Noben‐Trauth, K. (2006). Strain background effects and genetic modifiers of hearing in mice. Brain Research, 1091, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice, M. J. , & Dhillon, P. (2016). Using the mouse to model human disease: Increasing validity and reproducibility. Disease Models & Mechanisms, 9, 101–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, A. E. , & Lozano, A. M. (1998). Parkinson's disease. New England Journal of Medicine, 339, 1044–1053. [DOI] [PubMed] [Google Scholar]
- Levenga, J. , Hayashi, S. , de Vrij, F. M. S. , Koekkoek, S. K. , van der Linde, H. C. , Nieuwenhuizen, I. , … Oostra, B. A. (2011). AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiology of Disease, 42, 311–317. [DOI] [PubMed] [Google Scholar]
- Lim, E. T. , Raychaudhuri, S. , Sanders, S. J. , Stevens, C. , Sabo, A. , MacArthur, D. G. , … Daly, M. J. (2013). Rare complete knockouts in humans: Population distribution and significant role in autism spectrum disorders. Neuron, 77, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio, T. A. , Collins, F. S. , Cox, N. J. , Goldstein, D. B. , Hindorff, L. A. , Hunter, D. J. , … Visscher, P. M. (2009). Finding the missing heritability of complex diseases. Nature, 461, 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung, M. R. (2017). Clinical utility of anti‐sclerostin antibodies. Bone, 96, 3–7. [DOI] [PubMed] [Google Scholar]
- McGraw, C. M. , Samaco, R. C. , & Zoghbi, H. Y. (2011). Adult neural function requires MeCP2. Science, 333, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meert, T. F. , de Haes, P. , & Janssen, P. A. (1989). Risperidone (R 64 766), a potent and complete LSD antagonist in drug discrimination by rats. Psychopharmacology, 97, 206–212. [DOI] [PubMed] [Google Scholar]
- Merritt, H. H. , & Putnam, T. J. (1984). Landmark article Sept 17, 1938: Sodium diphenyl hydantoinate in the treatment of convulsive disorders. By H. Houston Merritt and Tracy J. Putnam. JAMA, 251, 1062–1067. [DOI] [PubMed] [Google Scholar]
- Meunier, H. , Carraz, G. , Neunier, Y. , Eymard, P. , & Aimard, M. (1963). Pharmacodynamic properties of N‐dipropylacetic acid. Therapie, 18, 435–438. [PubMed] [Google Scholar]
- Mineur, Y. S. , Sluyter, F. , de Wit, S. , Oostra, B. A. , & Crusio, W. E. (2002). Behavioral and neuroanatomical characterization of the Fmr1 knockout mouse. Hippocampus, 12, 39–46. [DOI] [PubMed] [Google Scholar]
- Nagy, R. , Sweet, K. , & Eng, C. (2004). Highly penetrant hereditary cancer syndromes. Oncogene, 23, 6445–6470. [DOI] [PubMed] [Google Scholar]
- Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium. (2015). Psychiatric genome‐wide association study analyses implicate neuronal, immune and histone pathways. Nature Neuroscience, 18, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton, S. (1973). Amphetamine as a model for hyperactivity in the rat. Physiology & Behavior, 11, 181–186. [DOI] [PubMed] [Google Scholar]
- Pangalos, M. N. , Schecter, L. E. , & Hurko, O . (2007) Drug development for CNS disorders: Strategies for balancing risk and reducing attrition. Nature Reviews. Drug Discovery, 6(7), 521 –532. [DOI] [PubMed] [Google Scholar]
- Peier, A. M. , McIlwain, K. L. , Kenneson, A. , Warren, S. T. , Paylor, R. , & Nelson, D. L. (2000). (Over)correction of FMR1 deficiency with YAC transgenics: Behavioral and physical features. Human Molecular Genetics, 9, 1145–1159. [DOI] [PubMed] [Google Scholar]
- Perrin, S. (2014). Preclinical research: Make mouse studies work. Nature, 507, 423–425. [DOI] [PubMed] [Google Scholar]
- Pitcher, M. R. , Ward, C. S. , Arvide, E. M. , Chapleau, C. A. , Pozzo‐Miller, L. , Hoeflich, A. , … Neul, J. L. (2013). Insulinotropic treatments exacerbate metabolic syndrome in mice lacking MeCP2 function. Human Molecular Genetics, 22, 2626 –2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomin, R. , Haworth, C. M. A. , & Davis, O. S. P. (2009). Common disorders are quantitative traits. Nature Reviews. Genetics, 10, 872–878. [DOI] [PubMed] [Google Scholar]
- Pop, A. S. , Levenga, J. , de Esch, C. E. F. , Buijsen, R. A. M. , Nieuwenhuizen, I. M. , Li, T. , … Willemsen, R. (2014). Rescue of dendritic spine phenotype in Fmr1 KO mice with the mGluR5 antagonist AFQ056/Mavoglurant. Psychopharmacology, 231, 1227–1235. [DOI] [PubMed] [Google Scholar]
- Power, A. E. , Berlau, D. J. , McGaugh, J. L. , & Steward, O. (2006). Anisomycin infused into the hippocampus fails to block “reconsolidation” but impairs extinction: The role of re‐exposure duration. Learning and Memory (Cold Spring Harbor, NY), 13, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo, S. J. S. , & Crawley, J. N. (2017). Behavioral phenotyping assays for genetic mouse models of neurodevelopmental, neurodegenerative, and psychiatric disorders. Annual Review of Animal Biosciences, 5, 371–389. [DOI] [PubMed] [Google Scholar]
- Salloway, S. , Sperling, R. , Fox, N. C. , Blennow, K. , Klunk, W. , Raskind, M. , … Bapineuzumab 301 and 302 Clinical Trial Investigators. (2014). Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. New England Journal of Medicine, 370, 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaco, R. C. , Mandel‐Brehm, C. , Chao, H.‐T. , Ward, C. S. , Fyffe‐Maricich, S. L. , Ren, J. , … Neul, J. L. (2009). Loss of MeCP2 in aminergic neurons causes cell‐autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proceedings of the National Academy of Sciences of the United States of America, 106, 21966–21971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansone, S. M. , Widaman, K. F. , Hall, S. S. , Reiss, A. L. , Lightbody, A. , Kaufmann, W. E. , … Hessl, D. (2012). Psychometric study of the Aberrant Behavior Checklist in Fragile X Syndrome and implications for targeted treatment. Journal of Autism and Developmental Disorders, 42, 1377–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannell, J. W. , & Bosley, J. (2016). When quality beats quantity: Decision theory, drug discovery, and the reproducibility crisis. PLoS ONE, 11, e0147215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe, D. J. , & Hardy, J. (2016). The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Molecular Medicine, 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto, R. , Kinoshita, A. , Wada, E. , Nomura, S. , Ohishi, H. , Takada, M. , … Mizuno, N. (1997). Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 17, 7503–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman, J. L. , Yang, M. , Lord, C. , & Crawley, J. N. (2010). Behavioural phenotyping assays for mouse models of autism. Nature Reviews Neuroscience, 11, 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoberg, E. A. (2017). Logical fallacies in animal model research. Behavioral and Brain Functions, 13, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer, C. M. , Alekseyenko, O. , Hamilton, S. M. , Thomas, A. M. , Serysheva, E. , Yuva‐Paylor, L. A. , & Paylor, R. (2011). Modifying behavioral phenotypes in Fmr1KO mice: Genetic background differences reveal autistic‐like responses. Autism Research: Official Journal of the International Society, 4, 40–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer, C. M. , Alekseyenko, O. , Serysheva, E. , Yuva‐Paylor, L. A. , & Paylor, R. (2005). Altered anxiety‐related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes, Brain, and Behavior, 4, 420–430. [DOI] [PubMed] [Google Scholar]
- Stromgaard, K. , Krogsgaard‐Larsen, P. , & Madsen, U. (2009). Textbook of drug design and discovery, Fourth Edition. Florida: CRC Press. (p. 476). [Google Scholar]
- The Lancet Neurology Null. (2017). Solanezumab: Too late in mild Alzheimer's disease? Lancet Neurology, 16, 97. [DOI] [PubMed] [Google Scholar]
- Till, S. M. , Asiminas, A. , Jackson, A. D. , Katsanevaki, D. , Barnes, S. A. , Osterweil, E. K. , … Kind, P. C. (2015). Conserved hippocampal cellular pathophysiology but distinct behavioural deficits in a new rat model of FXS. Human Molecular Genetics, 24, 5977–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea, D. , Giacometti, E. , Wilson, N. R. , Beard, C. , McCurry, C. , Fu, D. D. , … Sur, M. (2009). Partial reversal of Rett syndrome‐like symptoms in MeCP2 mutant mice. Proceedings of the National Academy of Sciences of the United States of America, 106, 2029–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, P. T. , Hull, C. , Chu, Y. , Greene‐Colozzi, E. , Sadowski, A. R. , Leech, J. M. , … Sahin, M. (2012). Autistic‐like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature, 488, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam, D. , D'Hooge, R. , Hauben, E. , Reyniers, E. , Gantois, I. , Bakker, C. E. , … De Deyn, P. P. (2000). Spatial learning, contextual fear conditioning and conditioned emotional response in Fmr1 knockout mice. Behavioural Brain Research, 117, 127–136. [DOI] [PubMed] [Google Scholar]
- van Slegtenhorst, M. , de Hoogt, R. , Hermans, C. , Nellist, M. , Janssen, B. , Verhoef, S. , … Kwiatkowski, D. J. (1997). Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science, 277, 805–808. [DOI] [PubMed] [Google Scholar]
- Veeraragavan, S. , Wan, Y.‐W. , Connolly, D. R. , Hamilton, S. M. , Ward, C. S. , Soriano, S. , … Samaco, R. C. (2016). Loss of MeCP2 in the rat models regression, impaired sociability and transcriptional deficits of Rett syndrome. Human Molecular Genetics, 25, 3284 –3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk, A. J. , Pieretti, M. , Sutcliffe, J. S. , Fu, Y. H. , Kuhl, D. P. , Pizzuti, A. , … Zhang, F. P. (1991). Identification of a gene (FMR‐1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell, 65, 905–914. [DOI] [PubMed] [Google Scholar]
- Visscher, P. M. , Brown, M. A. , McCarthy, M. I. , & Yang, J. (2012). Five years of GWAS discovery. American Journal of Human Genetics, 90, 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelkl, B. , & Würbel, H. (2016). Reproducibility crisis: Are we ignoring reaction norms? Trends in Pharmacological Sciences, 37, 509–510. [DOI] [PubMed] [Google Scholar]
- Wang, H.‐C. , Yu, Y.‐Z. , Liu, S. , Zhao, M. , & Xu, Q. (2016). Peripherally administered sera antibodies recognizing amyloid‐β oligomers mitigate Alzheimer's disease‐like pathology and cognitive decline in aged 3× Tg‐AD mice. Vaccine, 34, 1758–1766. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Wegener, J. E. , Huang, T.‐W. , Sripathy, S. , De Jesus‐Cortes, H. , Xu, P. , … Pieper, A. A. (2015). Wild‐type microglia do not reverse pathology in mouse models of Rett syndrome. Nature, 521, E1–E4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, C. S. , Huang, T.‐W. , Herrera, J. A. , Samaco, R. C. , Pitcher, M. R. , Herron, A. , … Neul, J. L. (2016). Loss of MeCP2 causes urological dysfunction and contributes to death by kidney failure in mouse models of rett syndrome. PLoS ONE, 11, e0165550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster, S. J. , Bachstetter, A. D. , Nelson, P. T. , Schmitt, F. A. , & Van Eldik, L. J. (2014). Using mice to model Alzheimer's dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Frontiers in Genetics, 5, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland, J. R. , & Ehlers, M. D. (2016). Translating neurogenomics into new medicines. Biological Psychiatry, 79(8), 650 –656. [DOI] [PubMed] [Google Scholar]
- WHO Expert Committee. 2015. The selection and use of essential medicines Geneva: World Health Organization. (p. 568).
- Yan, Q. J. , Rammal, M. , Tranfaglia, M. , & Bauchwitz, R. P. (2005). Suppression of two major fragile X syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology, 49, 1053–1066. [DOI] [PubMed] [Google Scholar]
- Zlotogora, J. (2003). Penetrance and expressivity in the molecular age. Genetics in Medicine, 5, 347–352. [DOI] [PubMed] [Google Scholar]