

SUMMARY
Localized protein synthesis is a mechanism for developing axons to react acutely and in a spatially restricted manner to extracellular signals. As such it is important for many aspects of axonal development, but its role in the formation of presynapses remains poorly understood. We found that the induced assembly of presynaptic terminals required local protein synthesis. Newly synthesized proteins were detectable at nascent presynapses within 15 minutes of inducing synapse formation in isolated axons. The transcript for the t-SNARE protein SNAP25, which is required for the fusion of synaptic vesicles with the plasma membrane, was recruited to presynaptic sites and locally translated. Inhibition of intra-axonal SNAP25 synthesis affected the clustering of SNAP25 and other presynaptic proteins and interfered with the release of synaptic vesicles from presynaptic sites. This study reveals a critical role for the axonal synthesis of SNAP25 in the assembly of presynaptic terminals.
Keywords: presynapse, synapse formation, local protein synthesis, SNAP25, β-catenin, synaptic vesicle release, axon
eTOC Blurb
Batista et al. find that during the assembly of presynaptic terminals, mRNA translation is upregulated at the nascent presynapses and required for the clustering of presynaptic proteins. Inhibition of local SNAP25 synthesis prevents proper formation of presynaptic terminal and interferes with synaptic vesicle release.
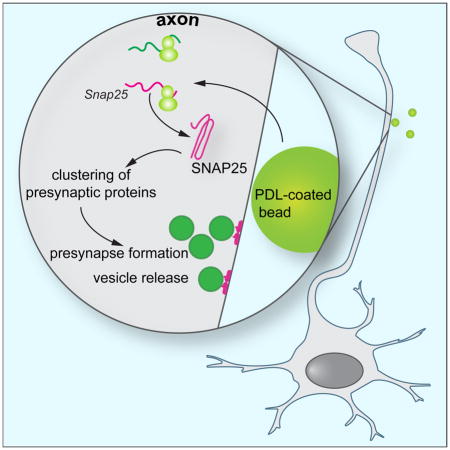
INTRODUCTION
During the development of the nervous system axons project over long distances to their cognate targets, until upon contact with target-derived adhesive or soluble factors the assembly of a presynaptic terminal is initiated (Chia et al., 2013; Jin and Garner, 2008). Application of these presynaptic organizing molecules to isolated axons is sufficient to induce presynapse formation from components that have been transported from the neuronal soma. An alternative source for at least some of the presynaptic proteins might be the axon itself through the process of local translation. Protein synthesis in axons is required for proper axon development (Campbell and Holt, 2001; Gracias et al., 2014; Hengst et al., 2009; Wu et al., 2005) by providing a spatially and temporally tightly restricted source of protein in response to extracellular signals (Batista and Hengst, 2016). Transcripts coding for several presynaptic proteins have been found in developing cortical axons (Taylor et al., 2009), and in Aplysia, protein synthesis is required for the formation of presynapses (Schacher and Wu, 2002). Recently, the importance of presynaptic protein synthesis in the control of neurotransmitter release was reported for the mature mammalian brain (Younts et al., 2016), but the role of local translation in the formation of presynapses remains poorly understood. Specifically, it is unknown whether axonal protein synthesis is required for the assembly of presynaptic terminals. So far only one locally synthesized protein has been described that accumulates at nascent presynapses, β-catenin, where it regulates the release of synaptic vesicles (Taylor et al., 2013). Here, we report the induced intra-axonal synthesis of the t-SNARE protein synaptosomal-associated protein 25 (SNAP25) as a necessary, early step for the clustering of presynaptic proteins and the formation and function of presynapses.
RESULTS
Presynaptic proteins cluster within 1 hour at contact sites with PDL-coated beads
To investigate whether the formation of presynapses requires local protein synthesis we cultured embryonic hippocampal neurons in tripartite microfluidic chambers that allow the fluidic isolation of axons from cell bodies and dendrites (Figure 1A) (Baleriola et al., 2014; Taylor et al., 2005). Poly-D-lysine (PDL)-coated latex beads were applied selectively to the axonal compartments to induce the clustering of presynaptic proteins (Lucido et al., 2009; Taylor et al., 2013). Immunostaining revealed significantly increased levels of several presynaptic proteins (β-catenin, synaptophysin, GAP-43, SNAP25) and tau at contact sites between axons and PDL-coated beads after 24 hours of incubation, while β-III tubulin levels were unchanged (Figure 1B). As local protein synthesis is frequently an acute reaction to an external stimulus we next investigated how early after addition of the PDL-coated beads we could detect clustering of presynaptic proteins. SNAP25 levels were significantly increased after 1 hour of contact with the PDL-coated beads (Figure 1C).
Figure 1. Clustering of presynaptic markers at PDL-coated beads in axons.

(A) Scheme of a tripartite microfluidic chamber used to selectively treat axons. Embryonic hippocampal neurons were seeded in the upper compartment of the chamber (teal) and axons cross through two microgroove barriers (500-μm-long) into the middle and lower axonal compartments (yellow). After DIV 10, PDL-coated or uncoated beads were added to the axonal compartments for 15 minutes to 1 hour, and clustering of presynaptic proteins (i.e. β-catenin, SNAP25) adjacent to the bead-axon contact sites was determined by IF.
(B) Axons were incubated with PDL-coated beads for 24 hours and immunostained for presynaptic and axonal proteins. Levels of β-catenin, synaptophysin, SNAP25, GAP43, and tau are significantly increased at axon-bead contact sites. IF and IF merged with DIC images are shown; yellow dashed circles outline beads. Quantifications are relative to off-bead fluorescence values. Mean ± SEM of 30–120 axonal fields (n = 3 biological replicates per condition). Unpaired t tests. n.s., not significant; **p < 0.01; ****p < 0.0001.
(C) Axons were incubated with PDL-coated and uncoated beads for the indicated times and immunostained for SNAP25. SNAP25 immunoreactivity is significantly increased at contact sites with PDL-coated beads at all time points. Fluorescence is normalized to off-bead values. Mean ± SEM of 30–50 axonal fields (n = 3–4 different biological replicates per condition). Two-way ANOVA with Bonferroni’s multiple comparison test. *p < 0.05; **p < 0.01.
Scale bars, 5 μm.
Inhibition of axonal protein synthesis prevents clustering of β-catenin and SNAP25 at 1 hour
Previously, it has been reported that the clustering of β-catenin at 3 hours after addition of PDL-coated beads requires local protein synthesis (Taylor et al., 2013). To determine whether the clustering of other presynaptic proteins is likewise dependent on axonal translation, we focused on SNAP25 and added the protein synthesis inhibitor emetine selectively to the axonal compartment during the treatment with uncoated or PDL-coated beads (Figure 2A). We quantified the fluorescent intensity within axons along a 30-μm-long line starting at the center of the beads (Figure 2B). Within 1 hour of treatment with PDL-coated beads, β-catenin and SNAP25 were significantly increased in the first 5 μm from the beads’ centers, i.e. at contact sites (Figures 2C–E). The clustering of β-catenin and SNAP25 did not show any bias for either the proximal (i.e. towards the cell body) or distal side of the beads (Figure S1); thus, for our analyses we did not distinguish between the proximal and distal sides. Uncoated beads did not induce clustering, and addition of emetine completely prevented the clustering of β-catenin and SNAP25. Neither tau nor β-III tubulin clustered at this early time point (Figures 2F and 2G). To determine whether the observed protein synthesis dependency of β-catenin and SNAP25 clustering at 1 hour was limited to this early time point we analyzed their clustering at 3, 6 and 12 hours after addition of beads and emetine to the axons. While SNAP25 clustering required protein synthesis at all time points tested, β-catenin clustering was significantly affected by emetine only at 3 hours (as previously reported by Taylor et al. (2013)) but not at the later time points (Figure 2H).
Figure 2. Clustering of β-catenin and SNAP25 at PDL-coated beads requires local protein synthesis.

(A) Experimental design: on DIV 11 axons were treated with PDL-coated or uncoated beads in the presence of a protein synthesis inhibitor (emetine, 100 nM) or vehicle. Cultures were fixed at different time points after treatment.
(B) A 3-pixels-wide line was drawn along the axon, starting at the center of the bead. Fluorescence along this line was quantified for 30 μm and normalized against the average fluorescence in last 15 μm. Beads have a diameter of 5 μm.
(C–G) Axon were immunostained for β-catenin (D), SNAP25 (E), tau (F), and β-III tubulin (G) after 1 hour of treatment. β-catenin and SNAP25 increased in the direct vicinity of the beads in a protein synthesis inhibitor sensitive manner, while tau and β-III tubulin levels remained unchanged. IF and IF merged with DIC images are shown; yellow dashed circles outline beads. Fluorescence intensities obtained in the lines scans were averaged and normalized. The area under the curve (AUC) was then calculated for the first 5 μm and 5–15 μm from the bead center. Mean ± SEM of 30–120 beads (n = 3 biological replicates per condition).
(H) Axons were immunostained for β-catenin or SNAP25 after 3, 6 or 24 hours of treatment. SNAP25 levels were significantly increased at bead contact sites at all times tested. The effect for β-catenin was significant only at 3 hours and emetine did not affect β-catenin levels at 24 hours.
One-way ANOVA with Tukey’s multiple comparison tests and unpaired t tests. n.s., not significant; *p < 0.05; **p < 0.01; ***p < 0.001. Scale bars, 5 μm.
See also Figure S1.
PDL-coated beads induce protein synthesis at axonal contact sites within 15 minutes
The requirement for local translation for clustering after 1 hour of incubation with PDL-coated beads indicated that this treatment might trigger protein synthesis directly and acutely at contact sites with axons. We used puromycylation, also known as SUnSET (Figure 3A) (Schmidt et al., 2009), to detect protein synthesis events in axons. Puromycin is a tRNA analog that gets incorporated into the nascent protein chain (Yarmolinsky and Haba, 1959), allowing the detection of protein synthesis in situ with an anti-puromycin antibody. We detected a significant increase in the number of puromycin-positive puncta in axons at contact sites within 15 minutes of addition of PDL-coated beads (Figure 3B). The increase of the puromycin signal was accompanied by an increased presence of phospho-4EBP1, a marker for active translation, at contact sites with PDL-coated but not uncoated beads (Figure 3C). To investigate whether the immediate induction of protein synthesis was sustained over a longer time period, we added the beads for 1 hour and shifted the puromycylation time window to the last 10 minutes of the assay. As before, the addition of PDL-coated beads was associated with a significantly higher number of puromycylation-positive puncta in their immediate vicinity compared to uncoated beads, and addition of the protein synthesis inhibitor anisomycin completely abolished this effect (Figure 3D).
Figure 3. PDL-coated beads induce axonal protein synthesis.

(A) Puromycylation assay: ribosomes incorporate puromycin into nascent polypeptide chains, leading to elongation termination and premature chain release. An antibody against puromycin is used to label nascent proteins in situ.
(B) Axons were treated with PDL-coated beads and puromycin or puromycin alone for 15 minutes and immunostained for puromycin and β-III tubulin. The level of puromycylation, indicating de novo protein synthesis is significantly increased at contact sites with PDL-coated beads. Mean ± SEM of 30 axonal fields per condition (n = 3 biological replicates). Unpaired t test. *p < 0.05.
(C) Axons were treated with uncoated or PDL-coated beads and puromycin for 15 minutes and immunostained for phospho-4EBP1, β-III tubulin, and puromycin. The signal for p-4EBP1, indicating activation of translation, co-localizes with the puromycin signal at contact sites with PDL-coated beads.
(D) Uncoated beads, PDL-coated beads, or PDL-coated beads and the protein synthesis inhibitor anisomycin (10 μM) were added to the axonal compartments. Puromycin was added to axons during the last 10 minutes of the assay. The number of puromycin-positive puncta in a circle of 5 μm around each bead center and the percentage of all beads imaged in each condition with no puncta or at least one puncta in the 5 μm circle were plotted. Means ± SEM of 40–142 beads in 30 axonal fields (n = 3 different biological replicates). One-way ANOVA with Tukey’s multiple comparison test. *p < 0.05; ***p < 0.001.
Yellow dashed line represents bead location. Scale bars, 5 μm.
Detection of SNAP25 mRNA in axons by fluorescent in situ hybridization
mRNA encoding SNAP25 has previously been found in axons of cortical neurons (Taylor et al., 2009) but it was below the detection threshold in other axonal transcriptome datasets (Baleriola et al., 2014; Zivraj et al., 2010). Here, we used single molecule inexpensive fluorescent in situ hybridization (smiFISH; Tsanov et al., 2016) to directly visualize SNAP25 transcripts in axons of dissociated embryonic hippocampal neurons and to determine whether their intra-axonal localization changes in response to treatment with PDL-coated beads (Figure 4A). SNAP25 mRNA FISH-positive were readily detectable at contact sites with PDL-coated beads, indicating that contact with PDL-coated beads recruits SNAP25 transcripts. The axonal smiFISH signal for SNAP25 mRNA was specific as transfection of axons with a SNAP25 siRNA greatly reduced the number of positive puncta (Figure 4B).
Figure 4. SNAP25 is locally synthesized in axons.

(A) After DIV 11, PDL-coated beads were added to the axonal compartments, and 24 h later cells were fixed, processed for Snap25 or Gfp smiFISH, and Snap25 and Gfp positive puncta at bead contact sites were counted. SNAP25 transcripts accumulated at bead contact sites. Mean ± SEM of 10 axonal fields per condition (n=3 biological replicates). Unpaired t test. ****p < 0.0001. Scale bar, 5 μm.
(B) Hippocampal neurons were cultured in microfluidic chambers for 10 days. siRNA was applied only to the axonal compartments for 24 hours, and the neurons were processed for Snap25 smiFISH. Axonal transfection with SNAP25 siRNA greatly reduced the number of SNAP25 puncta, establishing the specificity of the axonal Snap25 smiFISH signal. Mean ± SEM of 10 axonal fields per condition. Unpaired t tests. *p < 0.05. Scale bar, 5 μm.
(C) Scheme of a circular microfluidic chamber used for selective axonal treatments and protein isolation. Embryonic hippocampal neurons are seeded in the inner compartment and axons cross through a microgroove barrier (500-μm-long) into an open circular (6-mm-diameter) compartment. After DIV 11, uncoated beads or PDL-coated beads with or without emetine (100 nM) were added to the axonal compartment only, and 24 hours later proteins were obtained from both somatic and axonal compartments and analyzed for immunoblot blot. Selective treatment of axons with PDL-coated beads resulted in an increase of SNAP25 protein only in axons but not in cell bodies. This increase was blocked with local application of the protein synthesis inhibitor emetine.
(D) Principle of puro-PLA: puromycin is incorporated by active ribosomes into nascent polypeptide chains. A proximity ligation assay with antibodies against puromycin and the protein of interest –here: SNAP25– is used to detect synthesis of this protein.
(E) Axons of hippocampal neurons cultured in microfluidic chambers for 11 days were treated with PDL-coated beads and puromycin or puromycin alone for 15 minutes and processed for puro-PLA against SNAP25. The number of SNAP25 puro-PLA puncta is significantly increased in axons incubated with PDL-coated beads. Mean ± SEM of 10 axonal fields per condition (n = 3 biological replicates). Unpaired t test. **p < 0.01.
(F) PDL-coated beads or PDL-coated beads and the protein synthesis inhibitor anisomycin (10 μM) were added to the axonal compartments for 1 hour. Puromycin was added to axons during the last 10 minutes of the assay. The number of SNAP25 puro-PLA-positive puncta in a circle of 5 μm around each bead center and the percentage of all beads imaged in each condition with no puncta or at least one puncta in the 5 μm circle were plotted. Means ± SEM of 76–84 beads in 5–10 axonal fields per condition (n = 4 different biological replicates). Scale bars, 5μm.
(G) Dissociated hippocampal neurons were cultured for 5, 10, or 15 days and incubated with puromycin for 10 minutes before fixation. Closed arrowheads indicate puro-PLA puncta and opened arrowheads PSD-95 puncta. Scale bars, 10 μm.
(H) Dissociated hippocampal neurons were cultured for 5, 10, or 15 days and processed for SNAP25 puro-PLA without puromycin incubation or after incubation with puromycin and anisomycin for 10 minutes before fixation. Closed arrowheads indicate puro-PLA puncta and opened arrowheads PSD-95 puncta. Scale bars, 10 μm.
See also Table S1.
SNAP25 is synthesized locally at contact sites of axons with PDL-coated beads
The protein synthesis-dependent clustering of SNAP25 and the presence of its mRNA in axons at contact sites with PDL-coated beads indicated that SNAP25 itself might be locally produced. To test this hypothesis, we adopted a different design of microfluidic chambers (Figure 4C) (Park et al., 2009). In these circular chambers, axons grow into the central open compartment, allowing the collection of axonal material in quantities required for biochemistry. SNAP25 protein was detectable by immunoblot in lysates of axons treated with uncoated beads. Application of PDL-coated beads for 24 hours greatly increased the presence of SNAP25 in axons and treatment of axons with emetine reduced SNAP25 below detection limit without affecting SNAP25 expression in the cell body compartment (Figure 4C). To directly visualize SNAP25 synthesis at contact sites with PDL-coated beads, we combined puromycylation with a proximity ligation assay (puro-PLA) (Figure 4D) (tom Dieck et al., 2015). Axons that were not treated with PDL-coated beads showed very few SNAP25 puro-PLA puncta while treatment with PDL-coated beads induced the local synthesis of SNAP25 (Figure 4E). As before, we tested how persistent the induction of SNAP25 synthesis was by incubating the axons for 1 hour with PDL-coated beads and adding puromycin for the last 10 minutes. SNAP25 puro-PLA puncta were readily detectable and their presence was abolished by the presence of the protein synthesis inhibitor anisomycin (Figure 4F). Together, these results establish that SNAP25 is locally synthesized upon contact of axons with PDL-coated beads.
SNAP25 is locally synthesized at synapses
So far, our approach has been to induce formation of presynapses by applying PDL-coated beads. This approach allows us to study presynaptic events required for the clustering of presynaptic proteins in the absence of a postsynaptic cell but it is necessarily artificial. To test whether SNAP25 is locally synthesized at synapses rather than at induced presynaptic specializations, we cultured embryonic hippocampal neurons in regular dissociated cultures and performed SNAP25 puro-PLA assays on DIV 5, 10, and 15 (Figure 4G). SNAP25 puro-PLA puncta were visible in axons and dendrites at all developmental stages and their frequency increased with the age of the cultures (Figure 4G). In DIV15 cultures, some SNAP25 puro-PLA puncta were found juxtapositioned to the postsynaptic protein PSD-95, indicating SNAP25 synthesis at established synapses. The appearance of SNAP25 puro-PLA puncta was prevented at all developmental stages if the cells were incubated with vehicle instead of puromycin or if the protein synthesis inhibitor anisomycin was added during the puromycylation (Figure 4H).
Axon-specific knockdown of SNAP25 mRNA reduces SNAP25 synthesis at contact sites with PDL-coated beads
To investigate the requirement of localized SNAP25 or β-catenin mRNAs, we selectively transfected the axons of hippocampal neurons grown in microfluidic chambers with siRNA (Figure 5A). Previously, we have found that RNAi is functional in axons (Hengst et al., 2006) and that localized mRNAs can be selectively knocked down using locally applied siRNAs without affecting protein expression in dendrites or cell bodies (Baleriola et al., 2014; Gracias et al., 2014; Hengst et al., 2009; Villarin et al., 2016). When we transfected axons with a SNAP25 siRNA, the effect was restricted to axons (Figure 5B). The knockdown of SNAP25 mRNA in axons was only partial, likely because of the tight packaging of axonal mRNAs in granules (Buxbaum et al., 2014), which makes them inaccessible for the RNAi machinery while silenced but susceptible to siRNA under conditions that activate their translation, as we have observed previously (Baleriola et al., 2014; Villarin et al., 2016). Knockdown of axonal SNAP25 transcripts significantly reduced the appearance of SNAP25 puro-PLA puncta at contact sites with PDL-coated beads and increased that percentage of beads that had no puncta in their vicinity (Figure 5C). Together, these results establish that axonal transfection of SNAP25 siRNA is an efficient method to prevent the local translation of SNAP25 at contact sites with PDL-coated beads.
Figure 5. Effect of local siRNA on β-catenin and SNAP25 clustering.

(A) Experimental design for C: hippocampal neurons were cultured in microfluidic chambers for 10 days. siRNA was applied only to the axonal compartments for 24 hours before PDL-coated beads were added for 1 hour. Puromycin was added to the axons for the last 10 minutes before fixation and processing for SNAP25 puro-PLA.
(B) SNAP25 mRNA levels were quantified by RT-PCR in lysates obtained from the axonal and the cell body compartment 48 hours after transfection of the axons with siRNA. SNAP25 levels were reduced significantly reduced in axons but not in the cell bodies, demonstrating that the axonally applied siRNAs act exclusively locally. Mean ± SEM (n = 3 biological replicates).
(C) Transfection of axons with SNAP25 siRNA greatly reduced the synthesis of SNAP25 at contact sites with PDL-coated beads as measured with SNAP25 puro-PLA. Mean ± SEM of 80–103 beads in 10 axonal fields per condition (n = 3 biological replicates).
(D) Experimental design for E and F: Hippocampal neurons were cultured and their axons selectively transfected with siRNAs and incubated with PDL-coated beads as in A. The neurons were fixed after 1 hour and immunostained for SNAP25 (E) or β-catenin (F).
(E) SNAP25 immunostaining in the direct vicinity of PDL-coated beads is significantly reduced in axons transfected with SNAP25 siRNA. Average fluorescence values for 30 μm from the bead center, and the mean AUC for the 0–5 μm and 5–15 μm regions are plotted.
(F) β-catenin immunostaining in the 0–5 μm region is not significantly decreased in axons transfected with β-catenin siRNA. The signal intensity is increased in the 5–15 μm region. Mean ± SEM of 80–100 beads (n = 3 biological replicates).
(G) Dissociated hippocampal neurons were cultured for 11 days, transfected with siRNA and β-catenin mRNA levels were determined by RT-PCR after 48 hours.
Unpaired t tests. n.s., not significant; *p < 0.05; ***p < 0.001; ****p < 0.0001. Yellow dashed lines represent bead outline. Scale bars, 5 μm.
Clustering of SNAP25 and β-catenin requires the presence of their transcripts in axons
Next, we used this approach to investigate whether the local translation of SNAP25 or β-catenin was required for the clustering of these proteins. As before, we selectively knockdown SNAP25 or β-catenin transcripts in axons by locally applied siRNA and measured protein clustering 1 hour after the application of PDL-coated beads (Figure 5D). Knockdown of axonal SNAP25 mRNA significantly prevented the clustering of SNAP25 at contact sites with PDL-coated beads (Figure 5E). Knockdown of β-catenin mRNA did not prevent clustering of β-catenin directly at the contact sites (0–5 μm) but led to broadening of the peak with increased β-catenin levels at 5–10 μm (Figure 5F). This result indicates that the vast majority of β-catenin protein clustering at PDL-coated beads is not derived from acutely triggered local synthesis but rather is of somatic origin. However, a small amount of β-catenin whose local synthesis is prevented by the siRNA appears to be required to induce the clustering of β-catenin directly at contact sites. The β-catenin siRNA used here efficiently knocks down its target mRNA when applied to dissociated hippocampal neurons (Figure 5G). Together, these results suggest that most of the SNAP25 required during the early stages of presynapse formation is derived from local protein synthesis.
Clustering of SNAP25 and β-catenin require each other’s local synthesis
Next, we used the same experimental approach to test the reciprocal requirement of SNAP25 and β-catenin synthesis for the clustering of these proteins. Clustering of β-catenin protein was significantly reduced in axons depleted of SNAP25 mRNA (Figure 6A). In axons with β-catenin mRNA knockdown, clustering of SNAP25 protein was reduced as well, and the peak for SNAP25 was broadened as we had seen before for β-catenin protein itself (Figure 6B).
Figure 6. Reciprocal effects of local β-catenin and SNAP25 knockdown on clustering.

Hippocampal neurons were cultured and treated as in Figure 5D.
(A) Axons were transfected with scrambled or SNAP25 siRNA. 24 hours later, PDL-coated beads were added for 1 hour and β-catenin clustering was determined by IF. Fluorescence values were quantified for 30 μm starting at the bead center and mean AUC for the 0–5 μm and 5–15 μm regions are plotted.
(B) Axons were transfected with scrambled or β-catenin siRNA. 24 hours later, PDL-coated beads were added for 1 hour and SNAP25 clustering was determined by IF. Fluorescence values were quantified for 30 μm starting at the bead center and mean AUC for the 0–5 μm and 5–15 μm regions are plotted.
Mean ± SEM of 80–100 beads (n = of 3 different biological replicates). Unpaired t test. *p < 0.05 ****p < 0.0001. Yellow dashed lines represent bead outline. Scale bars, 5 μm.
Knockdown of axonal SNAP25 mRNA interferes with vesicle release from induced presynaptic sites
Lastly, we performed functional assays on induced presynaptic sites in axons transfected with siRNA similar to a previously described approach (Taylor et al., 2013). To study the release of synaptic vesicles at induced presynaptic sites we used FM 4–64. FM dyes are lipophilic dyes that can be endocytosed and incorporated into synaptic vesicles. We stimulated cells with a high potassium solution in the presence of FM 4–64 and let dye endocytosis occur. Cells were then imaged and stimulated a second time with high potassium. As vesicles are exocytosed, dye molecules are released and rapidly diffuse, resulting in nerve terminal destaining. In axons transfected with the siRNA targeting SNAP25, release of synaptic vesicles as measured by the disappearance of fluorescence was significantly slower than in scrambled siRNA transfected axons while the loading was not different (Figure 7A and 7B). Again, this effect was limited to the siRNA treated axons, as the unloading dynamic was unchanged at synapses in the cell body compartment (Figure 7C). As an additional control, we transfected axons with an siRNA targeting another presynaptic protein, piccolo (Fenster et al., 2000), whose transcript is absent from axonal transcriptome or translatome data sets (Shigeoka et al., 2016; Taylor et al., 2009). Neuron-wide knockdown of piccolo causes enhanced synaptic vesicles exocytosis rates (Leal-Ortiz et al., 2008), but axon-specific delivery of piccolo siRNA had no effect on synaptic vesicle release, again demonstrating that the siRNAs act only locally in axons and do not interfere with protein expression and transport from the cell body (Figure 7D).
Figure 7. Knockdown of axonal SNAP25 mRNA affects vesicle release of newly formed synaptic terminals.

(A) Experimental design: hippocampal neurons were cultured in microfluidic chambers for 10 days. Axons were transfected with siRNAs 24 hours before adding PDL-coated beads. 24 hours after bead incubation, cells were loaded with FM dyes and imaged.
(B) Sequential imaging and stimulation of FM4-64 puncta in scrambled or SNAP25 siRNA treated-axons at contact sites with PDL-coated beads. SNAP25 siRNA slows vesicle release after stimulation with potassium but does not impede FM4–64 loading. Pictures were taken every 15 seconds, high potassium was added after 30 seconds (black arrow), and axons were imaged for three and a half minutes more. Fluorescence at PDL-coated beads (yellow dashed circle) was quantified over time, and mean fluorescence, normalized to initial FM4-64 intensity, is shown with fitted exponential decay curves. The exponential destaining time constant (τ) for each bead and initial fluorescence values are plotted.
(C) FM 4–64 puncta (white arrows) of neurites in the cell body compartment of chambers where siRNA was applied only to axons were also stimulated and imaged. Knockdown of SNAP25 mRNA in axons does not affect vesicle dynamics in the cell body compartment. Exponential decay time constant (τ) and initial fluorescence are shown.
(D) Neurons were treated as in (A) but their axons were transfected with a piccolo siRNA. The piccolo siRNA does not change axonal vesicle dynamics. Mean fluorescence at beads over time with fitted exponential decay curves, exponential decay constant (τ) and initial fluorescence are represented.
Mean ± SEM of 45 beads per condition (n = 3 different biological replicates). Decay plots: Two-way ANOVA with multiple comparison test. Bar graphs: Unpaired t tests. n.s., not significant; **p < 0.01; ***p < 0.001****; p < 0.001. Scale bars, 5 μm.
(E) Dissociated hippocampal neurons were cultured for 11 days, transfected with siRNA and piccolo mRNA levels were determined by RT-PCR after 48 hours.
Mean ± SEM (n= 3 different biological replicates). Unpaired t test. *p<0.05.
DISCUSSION
Our results establish that SNAP25 synthesis is locally activated by a presynaptic organizing signal and required for presynaptic terminal assembly. Previously, it has been reported that β-catenin is locally synthesized at nascent presynaptic sites and that the locally produced protein regulates the release dynamics of synaptic vesicles (Taylor et al., 2013). The question of whether local protein synthesis was required for the assembly of presynaptic terminal was not directly addressed. Here we demonstrate that localized protein synthesis is a required step in the formation of presynaptic sites.
In accordance with Taylor et al. (2013) we also detect that β-catenin is locally synthesized. We find however differences in the requirement for local SNAP25 and β-catenin synthesis. While inhibition of protein synthesis prevents the accumulation of SNAP25 protein at contact sites with PDL-coated beads at all time points tested (1–12 hours), the accumulation of β-catenin is only affected until the three hours time point, indicating that at this time anterograde transport sufficiently meets the demand for β-catenin at presynapses. This difference is not easily explained by differences in the stability of SNAP25 and β-catenin: the half-life of SNAP25 during synaptogenesis in cerebellar granule neurons was reported to be 16 hours (Sanders et al., 1998), slightly longer than the half-life of β-catenin in PC12 cells of 12 hours (Bareiss et al., 2010). Instead, the role of the locally synthesized proteins appears to differ. While axonal specific siRNA treatment prevents the clustering of SNAP25 directly at contact sites with PDL-coated beads by around 50%, the analogous treatment leads to a broadening of the profile for β-catenin: the amount of protein found directly at beads is not significantly reduced but instead more β-catenin accumulates in the vicinity of the beads (5–15 μm distance from the bead center). These findings indicate that 1 hour after contact with the beads nearly half of presynaptic SNAP25 protein is derived from local synthesis. In contrast, nearly all β-catenin accumulates at or near the beads independently of local protein synthesis, i.e. by anterograde transport from the cell body. The small amount of locally produced β-catenin is required to cluster the anterogradely transported protein directly at presynaptic sites. Thus, while local production of β-catenin is required only during the first steps of presynapse assembly and does not generate the bulk of presynaptic β-catenin protein, SNAP25 synthesis persist at least until 12 hours after initiation of presynapse formation, it generates a substantial amount of synaptic SNAP25 proteins, and it continues to be required even in established synapses, as demonstrated by the effect of axonal SNAP25 mRNA knockdown on synaptic vesicle release. A recent translatome analysis of retinal ganglion cells axons identified SNAP25 as highly expressed not only in developing but also mature axons (Shigeoka et al., 2016), and inhibition of protein synthesis at established presynaptic terminals deregulates GABA release (Younts et al., 2016). The stability of SNAP25 at presynapses is controlled by activity induced ubiquitination and proteasome-dependent degradation (Sheehan et al., 2016). SNAP25 synthesis at established synapses might therefore be an important mechanism for the control of synaptic SNAP25 levels and synapse function.
mRNA localization to axons is generally understood to be controlled by RNA-binding proteins that associated with mRNAs through sequence elements nearly always found in the 3′ untranslated regions (3′UTRs) of the transcripts. Interestingly, several single-nucleotide polymorphisms located in the 3′UTR of SNAP25 are linked to adult attention deficit disorder (ADHD) (Barr et al., 2000; Brophy et al., 2002; Kustanovich et al., 2003; Mill et al., 2004). In the context of our findings it is tempting to speculate that these mutations might interfere with the local synthesis of SNAP25 at presynaptic sites. Future investigation of the potentially changes in subcellular localization and synaptic translation of SNAP25 mRNA might provide a mechanistic understanding of how these mutations are linked to hyperactive disorders.
In conclusion, we describe an alternative source for presynaptic proteins, intra-axonal synthesis, that is required the formation presynaptic terminals.
EXPERIMENTAL PROCEDURES
Compartmentalized culture of embryonic hippocampal neurons
All work involving animals was performed in accordance with NIH guidelines for the care and use of laboratory animals and was approved by the IACUC of Columbia University. All reagents were purchased from ThermoFisher Scientific unless otherwise noted. Hippocampal neurons were harvested from Sprague-Dawley embryonic day 17/18 rat embryos (Kaech and Banker, 2006). Embryonic rat neurons were grown in tripartite microfluidic chambers with 500-μm-long microgrooves connecting the three fluidically isolated compartments. Microfluidic chambers were produced according to published protocols (Gracias et al., 2014; Park et al., 2006). Primary hippocampal neurons (50,000 to 60,000 cells per chamber) were cultured in one of the side compartments and axons were allowed to grow into the other two compartments. Chambers were coated with 0.1 mg ml−1 poly- D-lysine (Sigma-Aldrich) and 2 μg ml−1 laminin (Trevigen). After 24 hours plating medium (neurobasal, 10% fetal bovine serum, 100 mM glutamine, 1mM sodium pyruvate, 100 IU ml−1 penicillin, 100 μg ml−1 streptomycin) was completely exchanged for growth medium (neurobasal, 1x B27, 100 mM glutamine). Half of this growth medium was replaced with fresh growth medium on DIV 4 and DIV 8. All experiments were performed at DIV 9–11. Whenever stated, axonal compartments were treated with emetine (100 nM, EMD Millipore) or anisomycin (10 μM, Sigma-Aldrich).
Presynaptic clustering with PDL-coated beads
Bead preparation and treatments were performed as described (Lucido et al., 2009; Taylor et al., 2013). Surfactant-free aliphatic amine latex microspheres 4–5 μm in diameter (Invitrogen) were coated in 50 μg ml−1 poly-D-lysine (Sigma-Aldrich) at 37°C for at least 2 hours, rinsed twice with water and diluted in growth medium. Uncoated beads were incubated in water. Around 150,000 PDL-coated beads were added to each axonal compartment through the side access ports. As adhesion of uncoated beads to axons is much lower for uncoated than for PDL-coated beads they were added at 5–10x excess.
Immunofluorescence and line scans
Neurons grown in microfluidic chambers were treated on DIV9-11 and fixed for 20 minutes at room temperature in 4% paraformaldehyde (PFA) in cytoskeleton buffer (10 mM MES, 3 mM MgCl2, 138 mM KCl, 2 mM EGTA, 0.32 M sucrose, pH 6.1). Neurons were washed with PBS, permeabilized and blocked for 30 minutes with 3 mg ml−1 BSA, 100 mM glycine and 0.25% Triton X-100. Coverslips were incubated overnight at 4°C with primary antibodies: rabbit anti-β-catenin (1:500, Invitrogen, 71-2700), mouse anti-synaptophysin (1:500, BioLegend, SY38), mouse anti-GAP-43 (1:500, Invitrogen, 7B10), mouse anti-SNAP25 (1:1,000, BioLegend, SMI 81), rabbit anti-tau (1:500, GenScript, phospho-Ser235), rabbit anti-βIII-tubulin (1:1,000, BioLegend, Poly18020), mouse anti-βIII-tubulin (1:500, Abcam, TU-20). Neurons were washed with PBS and incubated with fluorophore-conjugated Alexa secondary antibodies (1:200) for 1 hour at room temperature. Samples were mounted with ProLong Diamond Antifade (Invitrogen) and images were acquired in Z-stacks using a 63x/1.3 oil objective on an Axio-Observer.Z1 microscope equipped with an AxioCam MRm Rev. 3 camera (Zeiss). Acquisition settings were kept the same for all samples in any given experiment. 5 random axonal fields containing beads were imaged per coverslip. To quantify average fluorescence intensity at axon-beads contact sites, a 5-μm-diameter circle around the bead center was draw in AxioVision and average pixel intensity was determined inside that circle. For off-bead values, average pixel intensity was determined in a 5 μm circle that encompassed axons not in proximity with beads. For each image, background fluorescence intensity was determined in an area with no axons and subtracted from all bead and off-bead values. To quantify fluorescence along the axons, starting at the center of the bead a 3-pixels-wide line was draw along the axon using ImageJ. Average pixel intensity in that line was determined for 30 μm. The intensity along the last 15 μm of each segment was averaged and the resulting off-bead mean axonal fluorescence intensity was subtracted from all values.
Immunoblot analysis
Hippocampal neurons were culture in circular microfluidic chambers modified after Park et al. (2009), in which the axon grow across a 500-μm-long microgroove barrier into the inner open compartment (6 mm diameter). For protein isolation, medium was carefully removed from axonal compartment and axons were collected in 50 μl of RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1%SDS, 1mM EDTA, supplemented with protease and phosphatase inhibitors (Pierce)), the chamber was then removed and somatic material was collected in 100 μl of RIPA buffer. The material from 6 different chambers was collected this way using the same buffer to increase the amount of protein. 20 μl of lysate from axons and 2 μl of lysate from cell bodies was used for Western blotting. Nitrocellulose membrane was incubated with anti-SNAP-25 (1:1,000, BioLegend, 836304) or anti-β-III tubulin (1:10,000, BioLegend, 802001) at 4°C overnight in TBS-T with 4% milk. For detection, blots were incubated with respective secondary antibodies (1:10,000, anti-Ms-HRP or anti-Rb-HRP, Invitrogen) and developed with 1-Shot Digital-ECL (KindleBio, R1003) and images were taken with the KwikQuant Imager (KindleBio, D1001).
Puromycylation and puromycylation-proximity ligation assays
To detect newly synthesized proteins, puromycin (1.8 μM, Sigma-Aldrich) or growth medium was added to axons in compartmentalized cultures or to dissociated neurons in regular cultures for 10–15 minutes, depending on the experiment, in the absence or presence of the protein synthesis inhibitor anisomycin (10 μM for axons, 40 μM for dissociated cultures). After incubation, cells were washed twice with warm PBS and fixed for 20 minutes at room temperature in 4% PFA in cytoskeleton buffer. Cells were washed with PBS, blocked and permeabilized in 3 mg ml−1 BSA, 100 mM glycine and 0.25% Triton X-100, and incubated overnight at 4°C with mouse anti-puromycin antibody (1:250, Millipore, MABE343) and rabbit anti-SNAP25 antibody (1:250, Sigma-Aldrich, S9684) for the puro-PLA assay. Detection of newly synthesized SNAP25 through PLA was performed according to the manufacturer’s recommendations, using rabbit PLAplus and mouse PLAminus probes, and red Duolink detection reagents (Sigma-Aldrich). Before mounting with Duolink In Situ Mounting Medium with DAPI (Sigma-Aldrich), coverslips were incubated with anti-βIII-tubulin (1:100, Abcam, 2G10; conjugated to Alexa Fluor 488) or anti- βIII-tubulin (1:100, Abcam, EP1569Y; Alexa Fluor 647) and anti-PSD-95 (1:50, Abcam, EP2652Y; Alexa Fluor 488) for 1 hour at room temperature. Images were acquired as described previously and quantified by counting the number of puromycin-positive or PLA puncta. SNAP25 puro-PLA experiments in dissociated cultures were imaged in a Zeiss LSM 800 confocal microscope using a 40x oil objective and Zen Blue 2.1 software.
Single molecule inexpensive fluorescence in situ hybridization (smiFISH)
Oligonucleotide probes were designed using Oligostan software (Tsanov et al., 2016). For Snap25, we obtained 30 probes while for Egfp we designed 15 probes due to the smaller coding sequence (Table S1). The probes were hybridized to a digoxigenin-labeled FLAP oligonucleotide to create FLAP-structured duplex probes (Tsanov et al., 2016). smiFISH was performed as described (Tsanov et al., 2016), with minor changes. On DIV10, beads were added to the axonal compartments of hippocampal neurons grown in microfluidic devices. Cells were fixed after 24 hours for 20 minutes in 4% PFA in cytoskeleton buffer. Coverslips were washed in PBS, permeabilized with 0.3% triton X-100 in PBS for 5 minutes, washed again with PBS. Coverslips were equilibrated at 37°C in 15% formamide, 1x SSC. Samples were incubated with 10 pmol of FLAP-structured duplex probes in 50 μl of hybridization buffer (15% formamide, 1X SSC, 10% dextran, 350 ng μl−1 yeast tRNA, 0.2 mg ml−1 BSA, 2 mM vanadyl ribonucleoside complex) overnight at 37°C. Coverslips were washed twice with 15% formamide in 1x SSC for 30 minutes at 37°C. Afterwards samples were washed three times with PBS-T (0.1% Tween-20) for 5 minutes, blocked with 3% BSA in PBS-T for 30 minutes and incubated with goat anti-digoxigenin (1:500, Vector Laboratories) overnight at 4°C. Samples were washed three times with PBS-T for 5 minutes and incubated with Alexa-488 anti-goat secondary (1:1,000), and anti-βIII-tubulin conjugated to Alexa-594 (Abcam, ab201740), for 1 hour at room temperature. They were then washed with PBS and mounted with ProLong Diamond Antifade Mountant (Invitrogen). Egfp fluorescence was used as a control for nonspecific hybridization and subtracted from all Snap25 values. The specificity of the Egfp probes was verified by performing smiFISH on cells transfected by a Gfp plasmid or control (data not shown). The specificity of the Snap25 probe was verified by RNAi in axons (Figure 4B).
siRNA transfections
Axon-specific silencing of Snap25, ctnnb1 and pclo mRNAs was achieved using the following siRNAs: Snap25 (NM_001270575.1) 5′-CGUGUCGAAGAAGGGAUGAACCAUA-3′ and 5′-UAUGGUUCAUGCCUUCUUCGACACG-3′; ctnnb1 (NM_053357.2) 5′-UCUGCAUGCCCUCAUCUAGUGUCUC-3′ and 5′-GAGGUCGAAGAAGGCAUGAACCAUA-3′; pclo (NM_020098.1) 5′-CACCUUGCUGGUCUCUCACAUUAUU-3′ and 5′-AAUAAUGUGAGAGACCAGCAAGGUG-3′. Negative control siRNA was purchased from ThermoFisher Scientific (Stealth RNAi siRNA Negative Control Med GC Duplex #3). siRNAs were transfected into axons of DIV 10 neurons grown in microfluidic chambers using NeuroPORTER transfection reagent (Genlantis). Final siRNA concentration was 50 nM. Beads were added 24h after transfection.
Real Time RT-PCR
Total RNA from cell bodies (4 chambers or 150,000 cells) and axonal compartments (from a minimum of 6 chambers) was extracted with TRIzol, and RNA was purified using the Direct-zol RNA MiniPrep kit (Zymo Research). Axonal RNA was eluted in 10 μl of nuclease-free water and reverse transcribed to cDNA using the Superscript VILO cDNA synthesis kit. For cell body RNA, a total of 100 ng was reverse transcribed to cDNA. Axonal cDNA was preamplified using the TaqMan PreAmp kit, following manufacturer’s instructions for the 14 cycles preamplification. Real Time RT-PCR was performed with TaqMan Fast Advanced master mix in a StepOnePlus Real-Time PCR instrument, using pre-designed TaqMan probes. Amplification conditions were as follow: initial denaturing step at 95°C for 10 minutes, 40 cycles of denaturation at 95°C for 15 seconds and extension at 60°C for 1 minute. Snap25, ctnnb1 and pclo levels were normalized to gapdh.
FM 4–64 release assay
After DIV 11 the chamber medium was exchanged with tyrode’s solution (125 mM NaCl, 2 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 30 mM glucose, 25 mM HEPES, pH 7.4) and cells were let to equilibrate for at least 30 minutes. Then FM 4–64 (15 μM, N-(3-Triethylammoniumpropyl)-4-(6-(4-(Diethylamino) Phenyl) Hexatrienyl) Pyridinium Dibromide, Invitrogen), was loaded for 90 seconds in a high K+ solution (37 mM NaCl, 90 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 30 mM glucose, 25 mM HEPES, pH 7.4) with 10 μM CNQX (6-Cyano-7-nitroquinoxaline-2,3-dione disodium, Santa Cruz Biotechnology) and 50 μM DL-AP5 (DL-2-Amino-5-phosphonovaleric acid, Santa Cruz Biotechnology). Cells were loaded for 10 more minutes in tyrode’s solution with FM 4–64, CNQX and DL-AP5. Chambers were then briefly washed twice in low Ca2+ solution (112 mM NaCl, 2 mM KCl, 0.5 mM CaCl2, 10 mM MgCl2, 30 mM glucose, 25 mM HEPES, pH 7.4) with 0.5 mM advasep-7 (Sigma-Aldrich), CNQX and DL-AP5, and three times in tyrode’s solution with CNQX and DL-AP5. Unloading was then evoked with high K+ solution. During imaging neurons were kept in a CO2 and humidity controlled incubation chamber at 37°C. Images were acquired every 15 seconds, for a total of 4 minutes. High K+ was added after 30 seconds. The intensity of FM 4–64 puncta around beads was quantified over time. Relative fluorescence values were plotted and entered in SPSS (IBM). A monoexponential fit was calculated for each individual bead data with the equation, F = Fmax × e−time/τ + Ffinal, with Fmax being the fluorescence value right before K+ stimulation, τ the exponential decay constant, and Ffinal the final plateau. τ values were plotted and compared between conditions.
Statistical Methods
Statistical analysis was performed using Prism 7 (GraphPad Software; La Jolla, CA). The means of two groups were compared with unpaired t-tests, the means of three groups were compared using one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. The means of multiple groups with two independent variables were compared using two-way ANOVA with Bonferroni’s multiple comparisons tests.
Supplementary Material
Highlights.
Protein synthesis is rapidly induced at contact sites with presynaptic organizers
The presynaptic protein SNAP25 is locally synthesized at induced presynaptic sites
Local SNAP25 synthesis is required for formation of presynapses
Inhibition of local SNAP25 synthesis interferes with vesicle release
Acknowledgments
A.F.R.B. was supported by the Portuguese Foundation for Science and Technology (FCT) in the context of the FCT funded University of Minho MD/PhD Program (SFRH/BD/52322/2013). J.C.M. was supported by the National Institutes of Health (F31GM116617). U.H. was supported by the NIH (R01MH096702), the BrightFocus Foundation, and the Irma T. Hirschl Trust.
Footnotes
AUTHOR CONTRIBUTIONS
A.F.R.B. and U.H. designed the experiments. A.F.R.B. and J.C.M. performed the experiments and data analysis. A.F.R.B. and U.H. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baleriola J, Walker CA, Jean YY, Crary JF, Troy CM, Nagy PL, Hengst U. Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell. 2014;158:1159–1172. doi: 10.1016/j.cell.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareiss S, Kim K, Lu Q. Delta-catenin/NPRAP: A new member of the glycogen synthase kinase-3beta signaling complex that promotes beta-catenin turnover in neurons. J Neurosci Res. 2010;88:2350–2363. doi: 10.1002/jnr.22414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr CL, Feng Y, Wigg K, Bloom S, Roberts W, Malone M, Schachar R, Tannock R, Kennedy JL. Identification of DNA variants in the SNAP-25 gene and linkage study of these polymorphisms and attention-deficit hyperactivity disorder. Mol Psychiatry. 2000;5:405–409. doi: 10.1038/sj.mp.4000733. [DOI] [PubMed] [Google Scholar]
- Batista AF, Hengst U. Intra-axonal protein synthesis in development and beyond. Int J Dev Neurosci. 2016;55:140–149. doi: 10.1016/j.ijdevneu.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brophy K, Hawi Z, Kirley A, Fitzgerald M, Gill M. Synaptosomal-associated protein 25 (SNAP-25) and attention deficit hyperactivity disorder (ADHD): evidence of linkage and association in the Irish population. Mol Psychiatry. 2002;7:913–917. doi: 10.1038/sj.mp.4001092. [DOI] [PubMed] [Google Scholar]
- Buxbaum AR, Wu B, Singer RH. Single beta-actin mRNA detection in neurons reveals a mechanism for regulating its translatability. Science. 2014;343:419–422. doi: 10.1126/science.1242939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DS, Holt CE. Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron. 2001;32:1013–1026. doi: 10.1016/s0896-6273(01)00551-7. [DOI] [PubMed] [Google Scholar]
- Chia PH, Li P, Shen K. Cell biology in neuroscience: cellular and molecular mechanisms underlying presynapse formation. J Cell Biol. 2013;203:11–22. doi: 10.1083/jcb.201307020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenster SD, Chung WJ, Zhai R, Cases-Langhoff C, Voss B, Garner AM, Kaempf U, Kindler S, Gundelfinger ED, Garner CC. Piccolo, a presynaptic zinc finger protein structurally related to bassoon. Neuron. 2000;25:203–214. doi: 10.1016/s0896-6273(00)80883-1. [DOI] [PubMed] [Google Scholar]
- Gracias NG, Shirkey-Son NJ, Hengst U. Local translation of TC10 is required for membrane expansion during axon outgrowth. Nat Commun. 2014;5:3506. doi: 10.1038/ncomms4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengst U, Cox LJ, Macosko EZ, Jaffrey SR. Functional and selective RNA interference in developing axons and growth cones. J Neurosci. 2006;26:5727–5732. doi: 10.1523/JNEUROSCI.5229-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengst U, Deglincerti A, Kim HJ, Jeon NL, Jaffrey SR. Axonal elongation triggered by stimulus-induced local translation of a polarity complex protein. Nat Cell Biol. 2009;11:1024–1030. doi: 10.1038/ncb1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Garner CC. Molecular mechanisms of presynaptic differentiation. Annu Rev Cell Dev Biol. 2008;24:237–262. doi: 10.1146/annurev.cellbio.23.090506.123417. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kustanovich V, Merriman B, McGough J, McCracken JT, Smalley SL, Nelson SF. Biased paternal transmission of SNAP-25 risk alleles in attention-deficit hyperactivity disorder. Mol Psychiatry. 2003;8:309–315. doi: 10.1038/sj.mp.4001247. [DOI] [PubMed] [Google Scholar]
- Leal-Ortiz S, Waites CL, Terry-Lorenzo R, Zamorano P, Gundelfinger ED, Garner CC. Piccolo modulation of Synapsin1a dynamics regulates synaptic vesicle exocytosis. J Cell Biol. 2008;181:831–846. doi: 10.1083/jcb.200711167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucido AL, Suarez Sanchez F, Thostrup P, Kwiatkowski AV, Leal-Ortiz S, Gopalakrishnan G, Liazoghli D, Belkaid W, Lennox RB, Grutter P, et al. Rapid assembly of functional presynaptic boutons triggered by adhesive contacts. J Neurosci. 2009;29:12449–12466. doi: 10.1523/JNEUROSCI.1381-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mill J, Richards S, Knight J, Curran S, Taylor E, Asherson P. Haplotype analysis of SNAP-25 suggests a role in the aetiology of ADHD. Mol Psychiatry. 2004;9:801–810. doi: 10.1038/sj.mp.4001482. [DOI] [PubMed] [Google Scholar]
- Park J, Koito H, Li J, Han A. Microfluidic compartmentalized co-culture platform for CNS axon myelination research. Biomed Microdevices. 2009;11:1145–1153. doi: 10.1007/s10544-009-9331-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Vahidi B, Taylor AM, Rhee SW, Jeon NL. Microfluidic culture platform for neuroscience research. Nat Protoc. 2006;1:2128–2136. doi: 10.1038/nprot.2006.316. [DOI] [PubMed] [Google Scholar]
- Sanders JD, Yang Y, Liu Y. Differential turnover of syntaxin and SNAP-25 during synaptogenesis in cultured cerebellar granule neurons. J Neurosci Res. 1998;53:670–676. doi: 10.1002/(SICI)1097-4547(19980915)53:6<670::AID-JNR5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Schacher S, Wu F. Synapse formation in the absence of cell bodies requires protein synthesis. J Neurosci. 2002;22:1831–1839. doi: 10.1523/JNEUROSCI.22-05-01831.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- Sheehan P, Zhu M, Beskow A, Vollmer C, Waites CL. Activity-Dependent Degradation of Synaptic Vesicle Proteins Requires Rab35 and the ESCRT Pathway. J Neurosci. 2016;36:8668–8686. doi: 10.1523/JNEUROSCI.0725-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeoka T, Jung H, Jung J, Turner-Bridger B, Ohk J, Lin JQ, Amieux PS, Holt CE. Dynamic Axonal Translation in Developing and Mature Visual Circuits. Cell. 2016;166:181–192. doi: 10.1016/j.cell.2016.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Berchtold NC, Perreau VM, Tu CH, Li Jeon N, Cotman CW. Axonal mRNA in uninjured and regenerating cortical mammalian axons. J Neurosci. 2009;29:4697–4707. doi: 10.1523/JNEUROSCI.6130-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Wu J, Tai HC, Schuman EM. Axonal translation of beta-catenin regulates synaptic vesicle dynamics. J Neurosci. 2013;33:5584–5589. doi: 10.1523/JNEUROSCI.2944-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- tom Dieck S, Kochen L, Hanus C, Heumuller M, Bartnik I, Nassim-Assir B, Merk K, Mosler T, Garg S, Bunse S, et al. Direct visualization of newly synthesized target proteins in situ. Nat Methods. 2015;12:411–414. doi: 10.1038/nmeth.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsanov N, Samacoits A, Chouaib R, Traboulsi AM, Gostan T, Weber C, Zimmer C, Zibara K, Walter T, Peter M, et al. smiFISH and FISH-quant - a flexible single RNA detection approach with super-resolution capability. Nucleic Acids Res. 2016;44:e165. doi: 10.1093/nar/gkw784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarin JM, McCurdy EP, Martinez JC, Hengst U. Local synthesis of dynein cofactors matches retrograde transport to acutely changing demands. Nat Commun. 2016;7:13865. doi: 10.1038/ncomms13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KY, Hengst U, Cox LJ, Macosko EZ, Jeromin A, Urquhart ER, Jaffrey SR. Local translation of RhoA regulates growth cone collapse. Nature. 2005;436:1020–1024. doi: 10.1038/nature03885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarmolinsky MB, Haba GL. Inhibition by Puromycin of Amino Acid Incorporation into Protein. Proc Natl Acad Sci U S A. 1959;45:1721–1729. doi: 10.1073/pnas.45.12.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younts TJ, Monday HR, Dudok B, Klein ME, Jordan BA, Katona I, Castillo PE. Presynaptic Protein Synthesis Is Required for Long-Term Plasticity of GABA Release. Neuron. 2016;92:479–492. doi: 10.1016/j.neuron.2016.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivraj KH, Tung YC, Piper M, Gumy L, Fawcett JW, Yeo GS, Holt CE. Subcellular profiling reveals distinct and developmentally regulated repertoire of growth cone mRNAs. J Neurosci. 2010;30:15464–15478. doi: 10.1523/JNEUROSCI.1800-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.