

Abstract
The enoyl-ACP reductase (FabI) enzyme is a well validated target for anti-staphylococcal drug discovery and development. With the goal of finding alternate therapeutics for drug resistant strains of S. aureus, such as methicillin resistant S. aureus (MRSA), our previously published series of benzimidazole-based inhibitors of the FabI enzyme from F. tularensis (FtFabI) have been evaluated against FabI from S. aureus (SaFabI). We report here the preliminary structure-activity relationship of this series and the prioritization of compounds toward lead optimization. Mutational studies have identified key residues that contribute toward stabilizing the inhibitors in the active site of FabI. Mutations that do not significantly impact enzyme function but destabilize inhibitor binding are more likely to occur in nature as organisms evolve to evade the action of antibiotics leading to resistance. Identifying these residues provides guidance for minimizing susceptibility to resistance. Additionally, we have identified compounds that elicit antibacterial activity through off-target effects and observe that close analogs can display differing modes of action (on-target vs off-target) and need to be individually evaluated early on to prioritize compounds for lead optimization. Overall, our data suggest that the benzimidazole scaffold is a promising scaffold for anti-staphylococcal drug development.
Keywords: S. aureus, Fabl, benzimidazole inhibitor, enoyl-ACP reductase, mode of action
INTRODUCTION
The increased resistance of S. aureus strains to current antibiotics has been a cause for increasing alarm in the infectious diseases community.1 Methicillin resistant S. aureus (MRSA) has been a leading cause for hospital associated infections in the Unites States, with MRSA being responsible for a high fatality rate, bypassing that for HIV.1 To address MRSA, several new antibiotics have been introduced in the clinic or are undergoing clinical trials.2 With the exception of linezolid and daptomycin, these antibiotics are second or third generation compounds in their respective class. S. aureus has historically shown resistance to the parent compounds in each class, and hence second and third generation antibiotics could be easily rendered ineffective over time. Thus, there is an urgent need for novel therapeutics with different mechanisms of action to combat resistance.
The bacterial fatty acid biosynthesis pathway (FASII) is an attractive target for developing novel therapeutics. It differs from the human counterpart as each step in the bacterial FASII pathway is catalysed by separate enzymes while the human FASI system consists of one multifunctional enzyme complex. The enzyme FabI catalyses the rate-limiting step in this pathway, reducing enoyl-ACP to acyl-ACP with NADH or NADPH as a cofactor depending on the bacterial species.3–4 FabI is not a broad spectrum target as it is known that not all Gram-positive strains are susceptible to FabI inhibition.5 This is in part due to the fact that some Gram-positive organisms such as S. agalactiae can uptake required fatty acids from the host and suppress de novo fatty acid synthesis via feedback inhibition of acetyl-CoA carboxylase.6–8 Additionally, the presence of other FabI isoforms in some bacterial species, such as FabK, FabL and FabV, render the inhibition of FabI ineffective for such species. However, the essentiality of the FASII pathway and the FabI enzyme in S. aureus has been well established.7–11 As a result the FabI enzyme is of significant interest as a drug target for narrow spectrum antibiotics for S. aureus infections.12–18
We have previously reported the identification and characterization of a novel series of benzimidazole-based FabI inhibitors with potent nanomolar enzyme inhibitory activities against FabI from F. tularensis (FtFabI).19–22 In addition to F. tularensis, some of the compounds in this series also show promising antibacterial activities against S. aureus and MRSA.21 This work describes the evaluation of this benzimidazole-based series of FabI inhibitors to explore its potential for the development of novel anti-staphyloccocal drugs.
Structural and sequence comparison among the FabI enzymes from S. aureus and F. tularensis indicate that they possess a high level of structural identity in the catalytic site. As expected, our benzimidazole-based FtFabI inhibitors display excellent enzyme inhibitory activities against SaFabI. The co-crystal structure of SaFabI bound to one of our benzimidazole based FabI inhibitors confirms the binding mode and aids the structure based drug design process to help with iterative improvement of antibacterial activity while maintaining enzyme inhibitory activity.21 Additionally, mutational analysis has led to the identification of residues playing a role in stabilizing the inhibitors in the active site. We discuss the roles of these residues to both the catalytic function and inhibitor stabilization. We have also focused on the minimization of off-target activity, another important aspect that is often overlooked during initial stages of drug development. Compounds with off-target antibacterial effects showed no difference in antibacterial activities in S. aureus and S. aureus overexpressing our target, SaFabI. We observe that closely related analogs with excellent enzyme inhibitory activities can elicit antibacterial activity through differing modes of action with some being on-target and some off-target. Our work thus sets the stage for further lead generation by prioritizing compounds for the next stage of development.
RESULTS AND DISCUSSION
Structure-Activity Relationship
Figure 1 presents the structures and IC50 values of the benzimidazole based FabI inhibitors tested in this study. Compound 1, our initial hit with FtFabI displays a promising IC50 of 370nM with SaFabI.19–20 The lack of methyl groups at positions 5 and 6 on the benzimidazole ring in compound 2 reduces inhibitory activity compared to compound 1. Also, the presence of hydrophobic substituents at positions 5 and 6 (as in compounds 4–12) enhances inhibitory activity compared to compound 2. Lack of a meta substituent on the phenyl ring, coupled with an unsubstituted benzimidazole ring at positions 5 and 6, as in compound 3, causes a complete loss in activity. A methyl substitution on the methylene linker in compound 4 is well tolerated compared to compound 1. However, a carbonyl group on the methylene linker (compound 5) causes a complete loss in activity, as the sp2 carbon renders the compound planar, thus changing its binding mode. Cyclopentane and cyclohexane rings are preferred over a dimethyl substitution at positions 5 and 6 as compounds 6 – 11 show an increase in enzyme inhibitory activities compared to compound 1. A dichloro substitution, methylenedioxy substitution, or m-methyl and p-methoxy at positions 12 and 13 on the phenyl ring (compounds 6–11) gave the most potent inhibitors in this series, with IC50 values in the 10 nM to 70 nM range. The cyclopentane in compounds 6 and 7, and cyclohexane rings in compounds 9 and 10 have similar effects on the inhibitory activity with no significant preference for one over the other. However, this trend is not observed with compounds 11 and 12 where the cyclopentane substitution has a 10-fold better inhibitory activity compared to the cyclohexane substitution. The reason for this discrepancy is not understood at this point. By comparing compound 13 with compound 6 we confirm that hydrophobic substituents are preferred at positions 5 and 6 as the cyclopentane ring is preferred over the tetrahydrofuran ring.
Figure 1.
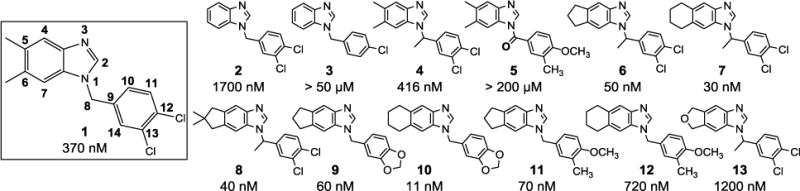
Structures and IC50 values of benzimidazole-based compounds against SaFabI. Atom numbering is highlighted in Compound 1.
Co-crystal structure
The co-crystal structure of SaFabI with compound 12 was solved to a resolution of 2.3 Å. The asymmetric unit consists of two chains as a homodimer. Clear electron density is visible in the active site in the omit map that allowed for unambiguous positioning of the ligand. This inhibitor binds to SaFabI in the presence of the cofactor NADPH (Fig. 2A), similar to FtFabI, which binds NADH (Fig. 2B, 2C). The benzimidazole nitrogen atom is well positioned to be within hydrogen bonding distance of the 2′-ribose hydroxyl group of NADPH and the hydroxyl group of Y157. The F204 residue in SaFabI also makes an edge-to-face interaction with the phenyl ring of compound 12, similar to FtFabI.20
Figure 2.
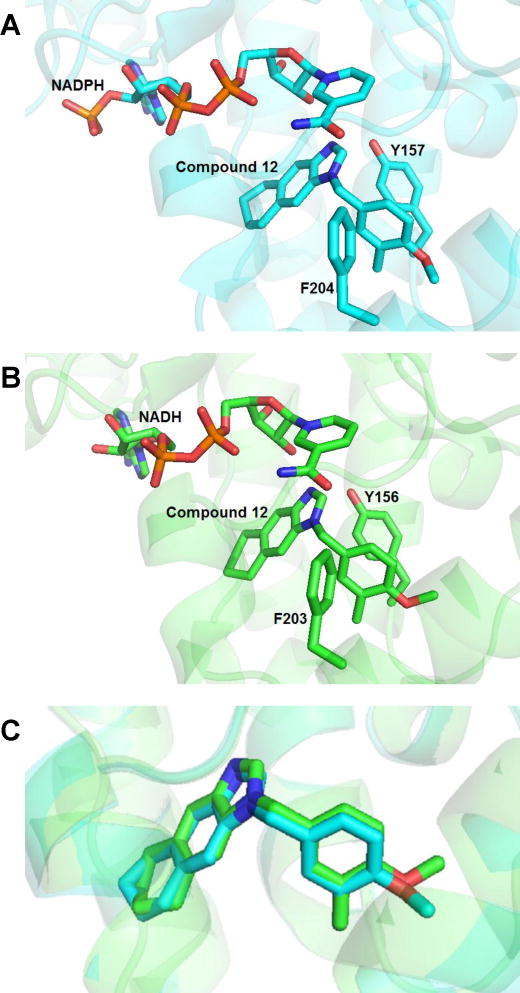
(A) Binding mode of compound 12 in the active site of SaFabI (PDB ID 4NZ9). (B) Binding mode of compound 12 in FtFabI (PDB ID 4J3F). (C) Overlay of compound 12 in the active sites of SaFabI (cyan backbone) and FtFabI (green backbone) indicates that the compound displays identical binding modes in both SaFabI and FtFabI.
Alignment of the co-crystal structures of FtFabI and SaFabI with compound 12 (PDB IDs 4J3F and 4NZ9) reveals the binding mode to be essentially identical in both structures (Fig. 2C).21 On comparison of the active site, we see that the active site opening around the phenyl ring is wider in SaFabI than in FtFabI (Fig. 3A, 3B), while they are similar in size around the cyclohexane ring (Fig. 3C, 3D). While the SAR of the benzimidazole-based FabI inhibitors in SaFabI is mostly similar to that for FtFabI, compounds 9 and 10 show more than an order of magnitude better activities in SaFabI (IC50 values of 60 nM and 10 nM, respectively) than FtFabI (IC50 values of 750 nM and 320 nM, respectively).21–22 We hypothesize this to be due to the differences in the environment surrounding the phenyl ring in the crystal structure of FtFabI and SaFabI as well as the size of the active site. The methylenedioxy substituted phenyl ring is better accommodated in SaFabI due to a more open active site compared with that of FtFabI.
Figure 3.
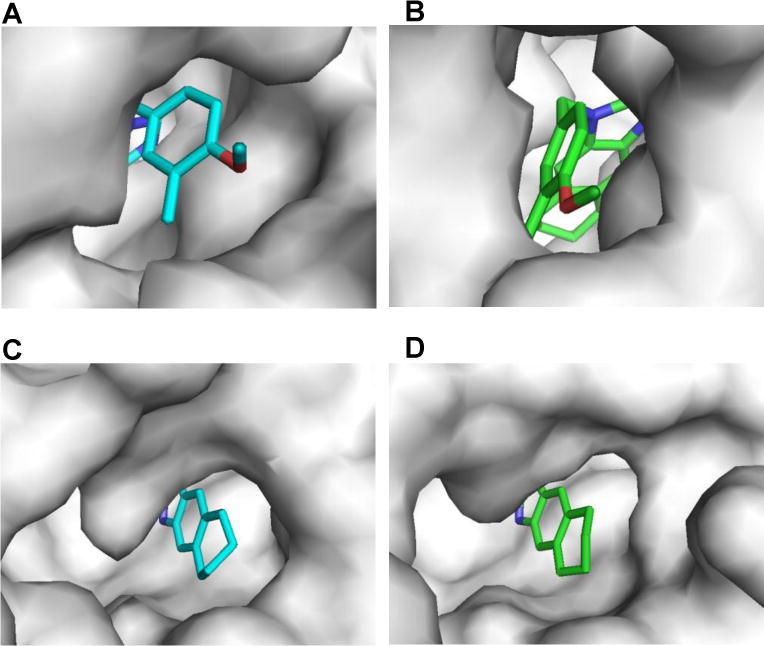
Comparison of the active sites in SaFabI and FtFabI. The active site around the phenyl ring of compound 12 is more open in SaFabI (A) than FtFabI (B). On the other hand, the active site opening around the cyclohexane ring of compound 12 is similar in both enzymes (C and D).
Mutational Studies
We used mutations to understand the contribution of key residues to the inhibitory activity of our compounds. Key residues identified for these studies include Y157, Y147 and F204. Y157 is a catalytic residue that is within H-bonding distance of the N-atom in the benzimidazole scaffold. Y147 is a residue known to be part of the SaFabI catalytic triad (Y147 (X)9 Y157 (X)6 K164), although its contribution to catalysis in SaFabI has long been questioned.23–24 We speculate that it could play an important role in stabilizing our inhibitors in the active site as it is in close proximity. The F204 residue is a part of the NADPH binding loop, but is not directly involved in an interaction with NADPH.25 As F204 contributes to the hydrophobic environment around the phenyl ring of our benzimidazole-based FabI inhibitors, the contribution of this residue to the stabilization of the inhibitors was also explored through mutation.
Table 1 highlights the kinetic parameters for the wild type and mutant forms of SaFabI. As expected the Y157 mutation drastically affected the enzyme activity. The mutation of this residue to a serine or alanine produced a complete loss of activity, while the Y157F mutant only retained ~ 4 % of the catalytic activity of the native SaFabI enzyme. Although the Km of the substrate (crotonyl-CoA) did not change for this mutant, the catalytic efficiency for this mutant was ~ 40-fold less than that for native SaFabI enzyme. This data confirms the previous speculation that Y157 is the catalytic residue.23 On the other hand, contrary to previous reports, we found the Y147F mutant to be stable, with catalytic activity similar to that for the native enzyme.24 The catalytic efficiency (kcat/Km) of the Y147F mutant is only 2-fold lower than the native enzyme (Table 1). In the case of the F204L mutant, as expected, the kinetic parameters did not change relative to the native enzyme.
Table 1. Kinetic parameters of SaFabI mutants relative to the wild type enzyme.
Saturating concentrations of 200 μM NADPH were used and Crotonyl-CoA concentrations were varied from 2000 μM to 31 μM.
| Enzyme |
kcat (min-1) |
Crot-CoA Km (μM) |
kcat/Km (min-1 μM-1) |
|---|---|---|---|
| WT SaFabI | 65.7 ± 2.2 | 236 ± 158.4 | 0.36 ± 0.25 |
| SaFabI Y157A | No activitya | ||
| SaFabI Y157S | No activitya | ||
| SaFabI Y157F | 2.9 ± 0.4 | 242 ± 17 | 0.01 ± 0.002 |
| SaFabI Y147F | 79.5 ± 12 | 454.5 ± 10.6 | 0.17 ± 0.02 |
| SaFabI F204L | 59.5 ± 3.6 | 258.5 ± 195.9 | 0.33 ± 0.26 |
No activity for mutant observed at 2 μM enzyme concentration.
Figure 4A shows the inhibitory activities of triclosan, and benzimidazole-based inhibitors 1, 6 and 7 on the native SaFabI enzyme and the Y157F, Y147F, and F204L mutants. Triclosan was found to be inactive against the SaFabI Y157F mutant at concentrations as high as 200 μM. Compounds 1, 6, and 7, on the other hand, display similar inhibitory activities with this mutant (<10 fold change). A possible explanation for this modest change in enzyme inhibitory activities is that the benzimidazole N-atom is still within H-bonding distance of NADPH and the loss of the H-bonding interaction with Y157 residue in the Y157F mutant does not significantly impact the binding mode (Fig. 4B). More importantly, these results suggest that mutation of this catalytic Y157 residue will not lead to the development of resistance unlike that seen with triclosan. All three compounds (1, 6, and 7) exhibit significant increase in IC50 values with both Y147F and F204L mutants (Fig. 4A). The activity of compound 1 decreased ~13-fold from 370 nM with the native enzyme to 5 μM with the Y147F mutant. Activity of compound 6 against the Y147F mutant decreased 24-fold compared to native SaFabI, with IC50 values increasing from 50 nM with the native enzyme to 1.2 μM with the mutant. Compound 7 also showed a similar trend with the IC50 increasing 60-fold from 30 nM with SaFabI to 1.8 μM with Y147F SaFabI. For the F204L mutant, the IC50 values for compounds 1, 6, and 7 increased ~11-fold, 38-fold, and 93-fold, respectively, compared to the native enzyme (IC50 values for compounds 1,6 and 7 increased from 370 nM, 50 nM and 30 nM, respectively, against native SaFabI to 4μM, 1.9μM, and 2.8μM against SaFabI F204L).
Figure 4.
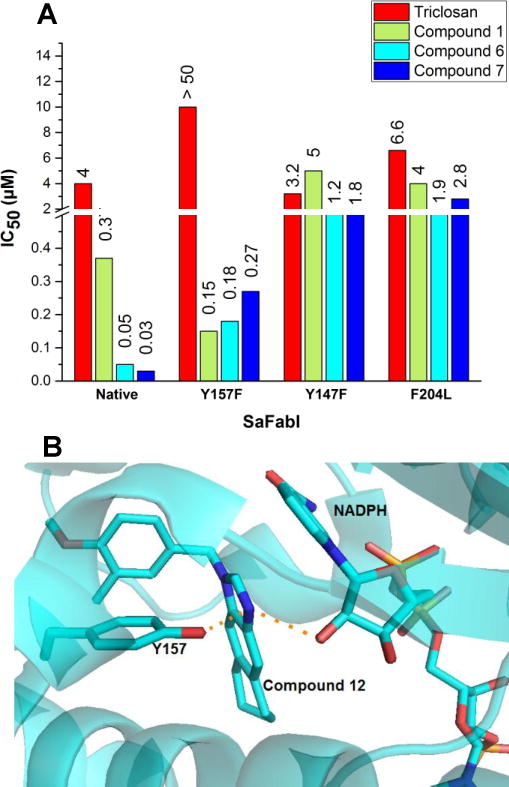
(A) Comparison of inhibitory activities of benzimidazole based inhibitors against native SaFabI and SaFabI Y157F, F204L and Y147F. Triclosan was used as a control.
(B) The benzimidazole N of compound 12 is within hydrogen-bonding distance from –OH of Y157 and the cofactor NADPH in the SaFabI active site. (PDB ID: 4NZ9)
Based on these studies we conclude that the inhibitory activities of the benzimidazole-based inhibitors against SaFabI are dependent on the contribution of the Y147 and F204 residues towards stabilizing these inhibitors in the active site. This provides key insight for the design of the next generation of inhibitors, with the goal of mitigating the dependence of activity on the F204 and Y147 residues, as these residues can easily mutate to destabilize inhibitor binding without significantly impacting enzyme catalysis.
Evaluation of on-target antibacterial activity
FabI overexpression in S. aureus has been previously used to confirm the mechanism of antibacterial activity of triclosan in S. aureus.26 An increase in MIC with the FabI overexpression strain is indicative of antibacterial activity being the result of on-target inhibition. The FabI inhibitors in this study were tested against both strains – the native S. aureus strain and S. aureus overexpressing FabI. We have not determined the total concentration of the FabI protein in the overexpression strain. However, from the plasmid copy number we estimate it to be >4 fold. Hence a four-fold or higher increase in MIC with the FabI overexpression strain is indicative of on-target FabI inhibition. Triclosan gave an 8-fold increase in MIC with the FabI overexpression strain and hence provides a good control.
Figure 5 sums up the change in individual compound MICs against the wild type S. aureus and S. aureus overexpressing FabI. Compounds with promising IC50 values of <1μM and low μg/mL MIC values with the native strain are reported here. Compound 1 shows a >4 fold increase in MIC with the SaFabI overexpressing strain indicating that the antibacterial effect is through FabI inhibition. However, compound 4, which has an IC50 and MIC in a range similar to those for compound 1, does not show a similar increase in MIC with the SaFabI overexpression strain. Similarly, compounds 6, 7, 8, 11, and 12, with excellent IC50s and promising MICs with the native S. aureus strain do not show an increase in MIC with the SaFabI overexpression strain. This indicates that the antibacterial effect for these compounds includes off-target effects. We suggest that this off-target activity could arise from the presence of another primary target within the bacterial cell. While there is no conclusive evidence supporting this, we have consequently de-prioritized compounds 6, 7, 8, 11, and 12. Compounds 9 and 10, on the other hand, display a 4-fold increase in MIC with the overexpression strain. These compounds were further confirmed to be on-target by testing with S. aureus carrying the empty plasmid (plasmid without the SaFabI insert) (data not shown).
Figure 5.
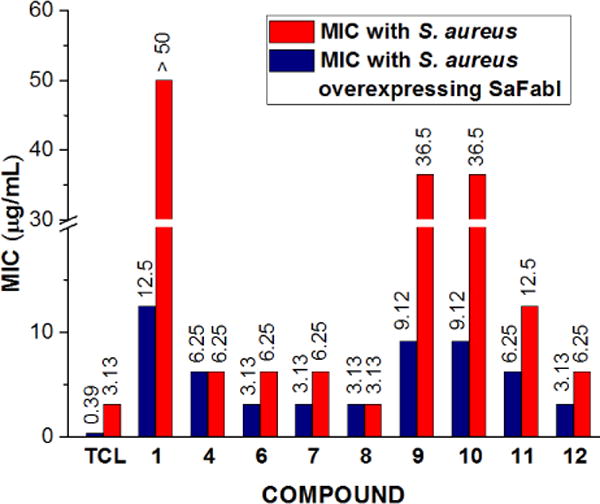
Evaluation of on-target antibacterial activity. Comparison of MIC of compounds with wild type S. aureus (red bars) and S. aureus strain overexpressing SaFabI (blue bars) to determine on-target antibacterial activity. Compounds with promising IC50s and MICs (with wild S. aureus) are shown here. The ≥4 fold increase in MIC with the SaFabI overexpression strain indicates antibacterial activity is the result of on-target inhibition as seen in the case of compounds 1, 9, and 10. Triclosan (TCL) is used as a control.
In conclusion, this work provides compelling evidence that the benzimidazole based FabI inhibitors are capable of eliciting antibacterial activity by inhibiting the intended target, FabI, in S. aureus as three of our compounds show increased MICs with SaFabI overexpression. We also show that close analogs can elicit differing mechanisms of antibacterial activity and this needs to be identified early on during the drug development process to correctly prioritize compounds for pharmacokinetic analysis. One drawback with the current generation of the benzimidazole-based SaFabI inhibitors is the high logD values (~4). In addition to improving the drug-like properties and reducing logD, our goal with the next generation of inhibitors will be to also minimize resistance development by mitigating the dependence of the observed enzyme inhibitory activities on the Y147 and F204 residues by monitoring on-target activity. The benzimidazole-based scaffold is thus a promising first in-class scaffold for future anti-staphylococcal drug-development efforts.
MATERIALS AND METHODS
SaFabI inhibitors
Compounds 1 and 2 were purchased from ChemBridge Corporation (ID# 7725253 and 5571325, respectively). Compound 3 was purchased from ASDI (ID# 100017167). Synthetic analogs 4–13 were previously synthesized by Dr. Arun Ghosh at Purdue University and their activity reported with FtFabI.21
Cloning and purification
The fabi gene from S. aureus strain Rosenbach (AATCC BAA-1556D-5) was PCR amplified using the following forward (SF-for) and reverse (SF-rev) primers: SF-for: 5′ TTATAA GGAGTTA CTCGAG ATGTTAAATC TTGAAAAC -3′ and SF-rev : 5′- CAA AGC TGT TGA GGA TCC TTA TTT AAT TGC G -3′ respectively. The fabI gene was inserted into the pET15b vector and ligation, transformation and protein purification were carried out using standard protocols.27 The fabI gene in the pET15b vector was used as the template for site-directed mutagenesis using the Q5 site directed mutagenesis kit from New England Biolabs with the vendor-recommended protocol for PCR and transformation into E. coli DH5 . The correct clones were then transformed into E. coli BL21 cells. Purification of mutant SaFabI was carried out using the same protocol as that for SaFabI.
Km determination
Reactions were carried out in 50 mM MES buffer at pH 5.5 with 100 mM NaCl, 0.1 mg/ml BSA and 0.01% triton and 200 nM enzyme in a final volume of 200 μL in a 96-well plate format. NADPH absorbance was measured at 340 nm. The Michaelis-Menten kinetic constant for crotonyl-CoA in the presence of NADPH was determined by varying the substrate concentration from 2000 to 31.25 μM, while maintaining NADPH at a fixed concentration of 200 μM and enzyme at 200 nM. Substrate Km determinations for all mutants were carried out using this same method.
IC50 determinations
The assay was conducted using the above-mentioned buffer conditions with 100 nM enzyme and 200 μM substrate measuring NADPH fluorescence at 340 nm/460 nm in a 50 μL assay volume to measure the reaction rate. Compound concentrations were varied from 200 μM to 0.4 nM. The compounds were incubated with the enzyme in the well for 20 minutes before addition of the substrate. Linear slopes for the first ten minutes were used to determine reaction rate.
Crystallization, Data Collection, Structure solution and Refinement
SaFabI at a concentration of 11.6 mg/mL was incubated with 5 mM NADPH and 1.5 mM compound for 1 hour at room temperature prior to crystallization set up. Diffraction quality crystals were obtained by the hanging drop method with buffer containing 200 mM lithium sulfate, 40% PEG400 in 0.1M Tris-HCl pH 8.5. Crystals were cryoprotected with 15% glycerol and data was collected at the LS-CAT 21-ID-D beamline at the Advanced Photon Source, Argonne National Laboratory using a wavelength of 1.12 Ǻ, a sample-to-detector-distance of 250mm and an oscillation angle of 0.5°. Diffraction data was processed using XDS and the structures were solved by molecular replacement using Phaser in the CCP4 program suite and the coordinates of FtFabI complexed with compound 12 (PDB ID 4J3F).21, 28–29 Refinement was performed using Refmac5.5 and the models built using the program COOT.30–31 The models were validated by Molprobity.32 The data collection statistics, final refinement statistics, and the statistics for the final geometry of the models are presented in Table 2. Coordinates of the final structures have been deposited in the PDB under the accession code 4NZ9.
Table 2.
Data collection and Refinement Statistics
| Data Collection: | Compound 11 + SaFabI (PDB ID: 4NZ9) |
|---|---|
| Space group | P21 212 |
| Unit cell parameters: | |
| a,b,c (Å) |
a = 80.5, b = 61.0 c = 112.6 |
| Resolution (Å) | 2.18 |
| No. reflections | 232529 |
| No. averaged reflections (unique) | 29318 |
| Rmerge (%) | 14.7 |
| I/σI | 12.1 |
| Completeness % | 98.8 |
| Refinement | |
|
| |
| Resolution range (Å) | 20.0 – 2.30 |
| no. reflections in working set | 24060 |
| No. of free reflections | 1265 |
| Rcrys (%) | 22.6 |
| Rfree (%) | 27.8 |
| Figure of merit | 0.83 |
| average B-factor (Å2) (protein) | 26.6 |
| No. of protein molecules in asymmetric unit | 2 |
| RMSD from ideal geometry: | |
| Bond lengths (Å) | 0.01 |
| Bond angles (deg) | 1.17 |
| Ramachandran plot | |
| favored (%) | 93.5 |
| outliers (%) | 1.0 |
Generation of the S. aureus strain overexpressing SaFabI
We used the E.coli-B. thuringensis shuttle vector pHT370 for overexpression of FabI in S. aureus.33 This vector contains genes for ampicillin and erythromycin resistance, for selection of correct clones in E. coli and S. aureus respectively. The gene encoding the FabI protein, along with its putative promoter was PCR amplified from the S. aureus ATCC BAA-1556D5 genomic DNA. The sequences of the forward and reverse primers were as follows: Forward primer: 5′ - CAA ACA TTT ATC GCA TGC GTT GTA ATA CGT - 3′, Reverse primer: 5′ - CAA AGC TGT TGA GGA TCC TTA TTT AAT TGC G -3′. Initial selection of the correct clone was done in E. coli DH5α cells. The correct clone was extracted and then transformed into S. aureus. The S. aureus strain used for these studies was the RN4220 strain. Electro-competent S. aureus was prepared in the lab as follows. Wild Type Sa RN4220 was grown overnight in Tryptic Soy Broth (TSB) and the next day diluted to a final O.D. of 0.5 in fresh, pre-warmed media before growing for an additional 30 minutes at 37 °C. The culture was then chilled in an ice slurry for 10 minutes and then harvested (by centrifuging at 2500 rpm for 15 minutes at 4 °C). The pellet was re-suspended in an equal volume of sterile, chilled water and centrifuged again. This step was repeated twice, followed by sequential re-suspension and centrifugation of the pellet in 1/10th and 1/25th the original volume. The resulting cell pellet was finally resuspended in 1/200th of the original volume. To preserve the integrity of the cells and increase the efficiency of electroporation, electro-competent S. aureus cells was prepared fresh, on the day of the eletroporation. For electroporation, 100 μL of the cell suspension was centrifuged for 1 minute at 5000 × g. The pellet was re-suspended in 50 μL of a chilled, filter sterilized solution of 500 mM sucrose with 10% glycerol. Approximately 5 μg of either the empty pHT370 plasmid or the pHT370 plasmid with SaFabI insert was added to the re-suspended electro-competent cells. The mix was transfered to a chilled electroporation tube (2 mm, Bio-Rad). A Bio-Rad electroporator with a preset protocol for S. aureus transformation was used. Immediately after the electroporation, 900 μL of a sterile solution of 500 mM sucrose in TSB was added to the tube and mixed well before transferring the mix to a 15 mL culture tube in which it was incubated with shaking for 1 hour at 37 °C. The cells were plated on TSA plates with 2 μg/mL erythromycin and incubated overnight at 37 °C. Selection of the right clone with the SaFabI insert was done by following the MICs with triclosan and erythromycin. Colonies with no insert showed an increase in the MIC for erythromycin only when compared to the wild type S. aureus strain RN4220, while colonies with the SaFabI insert showed an increase in the MIC of both triclosan and erythromycin compared with the wild type S. aureus strain RN4220.
Acknowledgments
We thank Dr. Hyunwoo Lee for the pHT370 vector and his guidance in preparing electrocompetent S. aureus RN4220 and transformation of the pHT370 vector into S. aureus and Dr. Alexander Mankin for letting us use his electroporation apparatus. This work was supported in part by National Institutes of Health Grants AI077949 and AI110090.
ABBREVIATIONS
- MRSA
Methicillin resistant Staphylococcus aureus
- FAS
Fatty acid synthase
- Enoyl-ACP
Enoyl-Acyl Carrier Protein
- SaFabI
Staphylococcus aureus FabI
- FtFabI
Francisella tularensis FabI
- crotonyl-CoA
Crotonyl-coenzyme A
- NADPH
β-nicotinamide adenine dinucleotide phosphate
- NADH
β-Nicotinamide adenine dinucleotide, reduced
- MIC
Minimum Inhibitory Concentration
Footnotes
Author contributions:
T.L.M. performed all enzyme kinetics, inhibition assays and mode of antibacterial activity studies and wrote the manuscript. S.M. solved the crystal structure (PDB: 4NZ9). L.T. set up crystals. A.K.G. synthesized compounds 4 to 13. M.E.J. and S.M. supervised the project and helped write the manuscript.
References
- 1.Deleo FR, Chambers HF. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J Clin Invest. 2009;119:2464–2474. doi: 10.1172/jci38226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bassetti M, Righi E. Development of novel antibacterial drugs to combat multiple resistant organisms. Langenbecks Arch Surg. 2015;400:153–165. doi: 10.1007/s00423-015-1280-4. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Ma ST. Recent advances in inhibitors of bacterial fatty acid synthesis type II (FASII) system enzymes as potential antibacterial agents. ChemMedChem. 2013;8:1589–1608. doi: 10.1002/cmdc.201300209. [DOI] [PubMed] [Google Scholar]
- 4.Heath RJ, Rock CO. Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J Biol Chem. 1995;270:26538–26542. doi: 10.1074/jbc.270.44.26538. [DOI] [PubMed] [Google Scholar]
- 5.Kingry LC, Cummings JE, Brookman KW, Bommineni GR, Tonge PJ, Slayden RA. The Francisella tularensis FabI enoyl-acyl carrier protein reductase gene Is essential to bacterial viability and is expressed during infection. J Bacteriol. 2013;195:351–358. doi: 10.1128/jb.01957-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brinster S, Lamberet G, Staels B, Trieu-Cuot P, Gruss A, Poyart C. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature. 2009;458:83–U85. doi: 10.1038/nature07772. [DOI] [PubMed] [Google Scholar]
- 7.Balemans W, Lounis N, Gilissen R, Guillemont J, Simmen K, Andries K, Koul A. Essentiality of FASII pathway for Staphylococcus aureus. Nature. 2010;463:E3–E4. doi: 10.1038/nature08667. [DOI] [PubMed] [Google Scholar]
- 8.Parsons JB, Frank MW, Subramanian C, Saenkham P, Rock CO. Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc Natl Acad Sci U S A. 2011;108:15378–15383. doi: 10.1073/pnas.1109208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heath RJ, Su N, Murphy CK, Rock CO. The enoyl- acyl-carrier-protein reductases FabI and FabL from Bacillus subtilis. J Biol Chem. 2000;275:40128–40133. doi: 10.1074/jbc.M005611200. [DOI] [PubMed] [Google Scholar]
- 10.Heath RJ, Rock CO. Microbiology - A triclosan-resistant bacterial enzyme. Nature. 2000;406:145–146. doi: 10.1038/35018162. [DOI] [PubMed] [Google Scholar]
- 11.Massengo-Tiasse RP, Cronan JE. Vibrio cholerae FabV defines a new class of enoyl-acyl carrier protein reductase. J Biol Chem. 2008;283:1308–1316. doi: 10.1074/jbc.M708171200. [DOI] [PubMed] [Google Scholar]
- 12.Escaich S, Prouvensier L, Saccomani M, Durant L, Oxoby M, Gerusz V, Moreau F, Vongsouthi V, Maher K, Morrissey I, Soulama-Mouze C. The MUT056399 inhibitor of FabI is a new antistaphylococcal compound. Antimicrob Agents Chemother. 2011;55:4692–4697. doi: 10.1128/aac.01248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerusz V, Denis A, Faivre F, Bonvin Y, Oxoby M, Briet S, LeFralliec G, Oliveira C, Desroy N, Raymond C, Peltier L, Moreau F, Escaich S, Vongsouthi V, Floquet S, Drocourt E, Walton A, Prouvensier L, Saccomani M, Durant L, Genevard JM, Sam-Sambo V, Soulama-Mouze C. From triclosan toward the clinic: Discovery of nonbiocidal, potent FabI inhibitors for the treatment of resistant bacteria. J Med Chem. 2012;55:9914–9928. doi: 10.1021/jm301113w. [DOI] [PubMed] [Google Scholar]
- 14.Kaplan N, Albert M, Awrey D, Bardouniotis E, Berman J, Clarke T, Dorsey M, Hafkin B, Ramnauth J, Romanov V, Schmid MB, Thalakada R, Yethon J, Pauls HW. Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob Agents Chemother. 2012;56:5865–5874. doi: 10.1128/aac.01411-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller WH, Seefeld MA, Newlander KA, Uzinskas IN, Burgess WJ, Heerding DA, Yuan CCK, Head MS, Payne DJ, Rittenhouse SF, Moore TD, Pearson SC, Berry V, DeWolf WE, Keller PM, Polizzi BJ, Qiu XY, Janson CA, Huffman WF. Discovery of aminopyridine-based inhibitors of bacterial enoyl-ACP reductase (FabI) J Med Chem. 2002;45:3246–3256. doi: 10.1021/jm020050+. [DOI] [PubMed] [Google Scholar]
- 16.Park HS, Yoon YM, Jung SJ, Yun INR, Kim CM, Kim JM, Kwak JH. CG400462, a new bacterial enoyl-acyl carrier protein reductase (FabI) inhibitor. Int J Antimicrob Agents. 2007;30:446–451. doi: 10.1016/j.ijantimicag.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Park HS, Yoon YM, Jung SJ, Kim CM, Kim JM, Kwak JH. Antistaphylococcal activities of CG400549, a new bacterial enoyl-acyl carrier protein reductase (FabI) inhibitor. J Antimicrob Chemother. 2007;60:568–574. doi: 10.1093/jac/dkm236. [DOI] [PubMed] [Google Scholar]
- 18.Sampson PB, Picard C, Handerson S, McGrath TE, Domagala M, Leeson A, Romanov V, Awrey DE, Thambipillai D, Bardouniotis E, Kaplan N, Berman JM, Pauls HW. Spiro-naphthyridinone piperidines as inhibitors of S. aureus and E. coli enoyl-ACP reductase (FabI) Bioorg Med Chem Lett. 2009;19:5355–5358. doi: 10.1016/j.bmcl.2009.07.129. [DOI] [PubMed] [Google Scholar]
- 19.Hevener KE, Mehboob S, Su PC, Truong K, Boci T, Deng JP, Ghassemi M, Cook JL, Johnson ME. Discovery of a novel and potent class of F. tularensis enoyl-reductase (FabI) inhibitors by molecular shape and electrostatic matching. J Med Chem. 2012;55:268–279. doi: 10.1021/jm201168g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mehboob S, Hevener KE, Truong K, Boci T, Santarsiero BD, Johnson ME. Structural and enzymatic analyses reveal the binding mode of a novel series of Francisella tularensis enoyl reductase (FabI) inhibitors. J Med Chem. 2012;55:5933–5941. doi: 10.1021/jm300489v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mehboob S, Song JH, Hevener KE, Su PC, Boci T, Brubaker L, Truong L, Mistry T, Deng JP, Cook JL, Santarsiero BD, Ghosh AK, Johnson ME. Structural and biological evaluation of a novel series of benzimidazole inhibitors of Francisella tularensis enoyl-ACP reductase (FabI) Bioorg Med Chem Lett. 2015;25:1292–1296. doi: 10.1016/j.bmcl.2015.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su PC, Tsai CC, Mehboob S, Hevener KE, Johnson ME. Comparison of radii sets, entropy, QM methods, and sampling on MM-PBSA, MM-GBSA, and QM/MM-GBSA ligand binding energies of F-tularensis enoyl-ACP reductase (FabI) J Comput Chem. 2015;36:1859–1873. doi: 10.1002/jcc.24011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schiebel J, Chang A, Merget B, Bommineni GR, Yu WX, Spagnuolo LA, Baxter MV, Tareilus M, Tonge PJ, Kisker C, Sotriffer CA. An ordered water channel in Staphylococcus aureus FabI: Unraveling the mechanism of substrate recognition and reduction. Biochemistry. 2015;54:1943–1955. doi: 10.1021/bi5014358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao J, Maxwell JB, Rock CO. Resistance to AFN-1252 arises from missense mutations in Staphylococcus aureus enoyl-acyl carrier protein reductase (FabI) J Biol Chem. 2013;288:36261–36271. doi: 10.1074/jbc.M113.512905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Priyadarshi A, Kim EE, Hwang KY. Structural insights into Staphylococcus aureus enoyl-ACP reductase (FabI), in complex with NADP and triclosan Proteins: Structure, Function, and Bioinformatics. Proteins. 2010;78(2):480–486. doi: 10.1002/prot.22581. [DOI] [PubMed] [Google Scholar]
- 26.Slater-Radosti C, Van Aller G, Greenwood R, Nicholas R, Keller PM, DeWolf WE, Fan F, Payne DJ, Jaworski DD. Biochemical and genetic characterization of the action of triclosan on Staphylococcus aureus. J Antimicrob Chemother. 2001;48:1–6. doi: 10.1093/jac/48.1.1. [DOI] [PubMed] [Google Scholar]
- 27.Mehboob S, Truong K, Santarsiero BD, Johnson ME. Structure of the Francisella tularensis enoyl-acyl carrier protein reductase (FabI) in complex with NAD+ and triclosan. Acta Crystallogr., Sect F. 2010;66:1436–1440. doi: 10.1107/s1744309110039862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;26:795–800. doi: 10.1107/s0021889893005588. [DOI] [Google Scholar]
- 29.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/s0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr., Sect D. 2004;60:2184–2195. doi: 10.1107/s0907444904023510. [DOI] [PubMed] [Google Scholar]
- 31.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr., Sect D. 2010;66:486–501. doi: 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect D. 2010;66:12–21. doi: 10.1107/s0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arantes O, Lereclus D. Construction of cloning vectors for Bacillus thuringiensis. Gene. 1991;108:115–119. doi: 10.1016/0378-1119(91)90495-w. [DOI] [PubMed] [Google Scholar]