

Abstract
Antibiotic effectiveness often changes when two or more such drugs are administered simultaneously, and unearthing antibiotic combinations with enhanced efficacy (synergy) has been a longstanding clinical goal. However, antibiotic resistance, which undermines individual drugs, threatens such combined treatments. Remarkably, it has emerged that antibiotic combinations whose combined effect is lower than that of at least one of the individual drugs can slow or even reverse the evolution of resistance. We synthesize and review studies of such so-called “suppressive interactions” in the literature. We examine why these interactions have been largely disregarded in the past, the strategies used to identify them, their mechanistic basis, demonstrations of their potential to reverse the evolution of resistance, and arguments for and against using them in clinical treatment. We suggest future directions for research on these interactions, aiming to expand the basic body of knowledge on suppression and to determine their applicability in the clinic.
Keywords: Antagonism, Antibiotic Resistance, Drug Interactions
Since the clinical introduction of penicillin in the 1940s, bacterial resistance to nearly all antibiotics has evolved and spread, presenting a serious threat to human health.1–3 Clinicians have treated resistant strains with ever higher doses of antibiotics,2, 3 often resulting in the evolution of bacteria capable of tolerating antibiotic levels exceeding those safe for the patient. In certain cases, major pathogens such as extensively drug resistant tuberculosis (XDR-TB), have developed resistance to virtually every effective antibiotic regime.4 To combat this impending crisis, researchers have focused on identifying antibiotic classes with novel cellular and metabolic targets,5 modifications to existing drugs that restore effectiveness against resistant bacteria,6 and co-administered chemical agents that disable specific drug resistance mechanisms,7, 8 increase antibiotic bioavailability,9 or decrease toxicity of high dosages to the patient.10 Researchers and clinicians have additionally taken into account physiological changes in patients that affect antibiotic concentrations and bacterial susceptibility (sensitivity) in order to maximize the effectiveness of antibiotics through individualized antibiotic dosing.11
These methods are designed to restore our capacity to treat resistant bacterial infections, but they often rely on fighting a traditional evolutionary “arms race” against the pathogens, where new antibiotic compounds are discovered and introduced as quickly as those in service are made obsolete by the spread of resistance. Given the decreasing discovery rate of novel antibiotics,12 the proven bacterial capacity to develop and disseminate novel resistance mechanisms,13 and the widespread exposure of bacteria to antibiotics in clinical and agricultural contexts,14 we do not expect to win this arms race.
Preserving the future efficacy of antibiotics therefore requires active reduction or even reversal of the selection that favors antibiotic resistance in the (necessary) presence of antibiotics. Indeed, various approaches to this goal are being explored.15 For instance, policy controls on antibiotic use in agriculture and their periodic removal from clinical use are aimed at reducing unnecessary selection for resistance,16 while screens for non-antibiotic compounds that target particular resistance mechanisms aim to eliminate them by increasing the cost of their carriage.8, 17
Strategies to combat resistance that employ drugs in combination have emerged as effective therapies since at least the 1940s18 and include important combinations used to combat HIV, tuberculosis, and to some degree, cancer.19–21 The range of organisms targeted by different antimicrobial compounds, as well as the considerable differences between the combined and individual effects of these drugs, have resulted in much research and clinical effort directed at identifying drug combinations that most effectively kill the widest range of pathogens.22, 23
Research on identifying and classifying drug combinations has been conducted both in the form of large screens20, 24, 25 and in specific studies focused mainly on synergistic combinations.26 While certain such combinations of compounds are more powerful together than predicted from their individual effects (termed synergistic interactions), the combined effects of others do not differ substantially from predicted effects (additive drug interactions). Yet others exhibit less inhibition than expected based upon their individual effects (antagonistic interactions). Amongst the set of antagonistic interactions, there exists a curious subset of drug combinations in which the joint inhibitory effect of two drugs is even less than the effect of one or both of the compounds alone. So-called “suppressive” interactions can be directional (the joint effect is reduced below that of one of the drugs) or reciprocal (the combination is less inhibitory than either individual component) (Figure 1). In order to understand, classify, and screen for different interactions, much effort has gone into establishing formal classification schemes (summarized in 27). These schemes typically capture many but not all aspects of such interactions and have usually not highlighted suppressive interactions.28
Figure 1. Schematic representation of suppressive drug interactions: Single concentrations and gradients.

Here we show suppressive drug interactions in two-drug and three-drug combinations, in both single concentrations per drug and gradients of drugs. The two most widely used systems for classifying drug interactions are Loewe additivity108 and Bliss independence.28 Loewe additivity relies on entire gradients of each drug, while Bliss independence categorizes drug interactions based on single-point concentrations per drug. Two-drug combinations: (a, b) Loewe and Bliss differ in how they define additivity. Loewe defines additive drugs as those whose inhibition is constant along lines of equal effective dosage in a two-drug concentration graph. In this case, isoboles (lines of constant inhibition) can be plotted based on data for two drugs across a range of dosages. Following previous papers,29, 35, 44 in (a), the non-monotonic concave isobole (red) defines synergy, the linear isobole (orange) defines additivity, the convex isobole (green) defines antagonism, and the isobole with even greater convexity (blue) defines suppression. (Note that this is a simplified depiction, since isoboles indicating suppression are often non-monotonic.35) Bliss defines additivity as the result when the relative effect of a drug at a certain concentration is independent of the presence of the other drug. If Drug X and Drug Y are additive, then wXY = wXwY, where w is relative fitness. Suppression occurs when the combined effect of two drugs is less than the effect of one or both (b) of the drugs. In (b), any growth response caused by Drug X + Drug Y that is above the dotted line indicates that Drug X and Drug Y are suppressive. Three-drug combinations: (c, d) Recently these systems have been extended to classify higher-order drug interactions. Interactions are considered suppressive if the combined effect of the three drugs is less than the effect of any of the pairwise combinations or the single drugs.53 Loewe’s extension to three drugs is shown in (c), and is characterized by an increase in growth (fitness) when all three drugs are present in high concentration. Growth response (fitness) increases as the combination becomes more suppressive (where red is synergistic, orange is additive, green is antagonistic, and blue is suppressive), as shown by the increasing convexity on the surfaces. In (d), Bliss’s extension to three drugs is shown. Any growth rate caused by Drug X + Drug Y + Drug Z that is above either dotted line (one for the highest single drug growth response (black) and one for the highest pairwise drug growth response (green)) indicates reciprocal suppression.
Recently, certain suppressive interactions between antibiotics administered in combination have been observed to select for susceptibility over resistance to the individual component drugs.29 Intriguingly, the increased cost of resistance over susceptibility imposed by the drug combination appears largely independent of the allele or even exact mechanism of resistance. This selection reversal occurs in the presence of susceptible and resistant bacterial populations, suggesting that suppressive antibiotic combinations might provide unusual antibiotic treatments which simultaneously treat infections while guarding against evolution of resistance.
In this review, we examine the suppressive interaction, its characterization, and the mechanisms of its effects on selection. We consider why it has been largely unappreciated despite identification well over a century ago, debate its potential for clinical applications, and suggest next steps for the investigation of these unusual interactions.
A Bias against Searching for and Reporting Suppressive Interactions
A study published by Thomas Fraser in 1872 is the first account of suppression in the literature. Working at a time when powerful modern tools of screening detection were unavailable, Fraser noted that when rabbits were injected with physostigmine, a cholinesterase inhibitor produced by Physostigma venenosum, within 20–30 minutes, it became difficult to “distinguish any respiratory movement or cardiac impulse [in the rabbit], and they soon altogether cease on the occurrence of death.”30 However, when the rabbits were injected with atropine subsequent to the same dose of physostigmine, they survived. Fraser termed this interaction between the drugs a “physiological antidote,” to stress that the source of the effect is an interaction between the physiological responses of the rabbit to the drugs in combination and not a purely chemical interaction between the compounds alone: “When… among the different counteracting actions that occur in general antagonism, there are included any by which the fatal effect of one or other of the substances is usually produced, the one substance may act towards the other as a physiological antidote.”30, 31
Fraser anticipated that many more studies of suppressive drug interactions would follow his publications,31 but these were few and far between for the next 100 years. While Fraser’s study received contemporary attention and a few scientists followed up on his research,26, 32–34 investigations of the suppressive interaction have been largely absent until this past decade. Indeed, an exhaustive search of the works citing Fraser found that the overwhelming majority of citations were for his development of the isobole method of representing drug interactions, with remarkably few papers mentioning his work on suppressive interactions. An increase in acknowledgements of this work has occurred during the past decade,35–38 with renewed interest in suppressive interactions, particularly to treat antibiotic resistant bacteria. A recently increased focus on the effects of antibiotic combinations on bacteria and new high-throughput measurement technology have enabled a more accurate classification and screening of antibiotic suppression,24, 29, 35, 39–43 and an increasing number of researchers have been drawn to the question of suppression.25, 29, 44–47 Finally, the potential of suppressive combinations to counter the evolution of antibiotic resistance29, 39, 48 has spurred additional interest in this topic.
The surprisingly few references to suppression in the literature are not limited to works citing Fraser. There is a significant bias against reporting antagonistic interactions (and especially suppressive interactions, which are often classified more broadly as antagonistic interactions when reported).25, 49–52 When comparing a database of interactions between antibiotics generated by manually curating Pubmed literature on in vitro antibiotic interaction experiments35 to a complete pairwise interaction network for 21 antibiotics24 and a complete three-way interaction network for 14 antibiotics,53 the database of literature-curated interactions had significantly more synergy and less antagonism in both cases (p-value<0.001). In studies with smaller numbers of drug-drug combinations, no suppressive interactions were found out of 10 total interactions,54 and 2 suppressive interactions were found out of 30 total interactions.55 However, the discrepancy in proportions of antagonism and suppression between the literature-curated data and whole network analyses suggests an overall under-representation of the discovery and reporting of these interactions (Figure 2).
Figure 2. Bias towards synergy in a literature-curated database of two-antibiotic combinations.

In a database of interactions between antibiotics based on manually curated Pubmed literature on in vitro antibiotic interaction experiments,35 61% of interactions were synergistic and 39% were antagonistic or suppressive. When a complete two-drug interaction network was studied using 21 drugs, 42% of drug interactions were synergistic and 58% were antagonistic or suppressive (chi-square, p-value<0.001).24 When a complete three-drug interaction network was studied using 14 drugs, 23% of interactions were synergistic and 77% were antagonistic or suppressive (chi-square, p-value<0.001).53 In the three-drug interaction dataset, the interactions measured were “emergent” interactions, meaning that these interactions were categorized based on deviations from all two-drug components. Inconclusive and additive cases were excluded when calculating proportions for all datasets.
Other researchers have found similar biases against reporting antagonistic, and specifically suppressive, interactions.49–52, 56 The most obvious explanation for this bias is that screens for antibiotic interactions typically have clinical applications in mind, and have targeted synergistically-interacting drugs. From a traditional, clinical viewpoint, treatments that require higher drug concentrations to achieve the same or lower degree of pathogen inhibition would appear to make little sense,57 since higher levels of antibiotics are more likely to adversely affect patients26, 58 and increase costs. Therefore, although suppressive combinations have the potential to reverse evolution of antibiotic resistance, to the best of our knowledge, no in vivo studies on suppressive drug combinations have been conducted.
Other reasons may also factor into the underrepresentation of antagonistic and suppressive combinations in the literature. The many different experimental methods for determining drug interactions and interaction classification schemes (see review in 27) can be confusing and result in varying levels of rigor,56 leading authors to overlook antagonistic results.49 For example, Bliss independence assumes that non-interacting toxins are completely independent of each other in their effects, so a drug can “interact” with itself, making classifications harder to interpret. Furthermore, since Bliss independence only requires four measurements of bacterial growth (in the absence of either drug, in the presence of drug A alone, drug B alone, and drug A and B together), the classification method may not adequately represent the different types of interactions drugs can demonstrate when measured under different sets of concentrations. While Loewe additivity rectifies the concentration issue by requiring measurements of growth rates over a 2-D field of pairwise dosages of each of the individual drugs, this can be extremely logistically challenging (Figure 1). However, mathematical solutions to Loewe additivity exist, allowing approximations to be obtained from fewer measurements.59
Variability in specific thresholds used for classification of an interaction and in the precision of the technology used to measure the interaction can also contribute to biases against reporting antagonistic results. Other factors such as genetic variations, environmental factors, host behavior,56 and isolate-specific interactions60, 61 may further affect the classification of drug interactions and could lead to the misreporting of antagonistic interactions. In particular, a drug combination can be antagonistic over one concentration range and synergistic over another. When interactions are not measured in 2-D concentration gradients, it becomes possible to report a combination as synergistic when it has the potential to be antagonistic or even suppressive.62
The underreporting of suppressive drug interactions is likely to diminish with the emergence of new technologies that allow for larger and less biased screens.24, 29, 35, 39–43 Ideally, more concentrations should be tested to detect concentration dependencies and enable researchers and clinicians to better understand the magnitude of interactions being reported. We suggest that it is important to search for and correctly identify suppressive drug combinations from both academic and clinical perspectives, since suppression is a distinct type of interaction with unique properties and consequences, including the potential to slow and even reverse the evolution of resistance.
Suppressive Drug Combinations and their Potential to Slow or Reverse Evolution of Resistance
Although it has been shown that antagonistic interactions can reduce the selective advantage of antibiotic resistant strains over susceptible ones,29, 39 only suppressive interactions can fully reverse this selective advantage.15 Suppressive antibiotic combinations can allow antibiotic-susceptible bacteria to out-compete bacteria that are resistant to one of the drugs in a two-drug treatment (Figure 3), as illustrated in reference 29.
Figure 3. How suppressive drug combinations can leave drug-space for wild-type bacteria to out-compete resistant mutants.
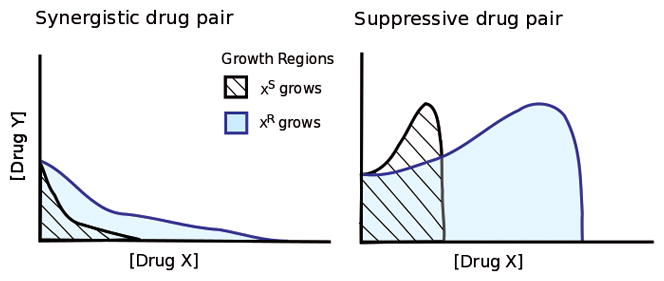
Synergistically interacting drugs amplify each other’s effects (left panel) while a drug in a suppressive pair cancels the effect of the other (right panel). Black lines outline minimum inhibitory isoboles for antibiotic X-susceptible (XS) bacteria in combinations of drugs X and Y. X-resistance (XR) reduces effective concentrations of the antibiotic X, producing isoboles and a growth region that are stretched along the X axis (blue lines). In both synergistic and suppressive drug combinations, this geometric scaling generates a region between the susceptible and resistant isoboles where only resistant mutants grow. In contrast, the suppressive combination also contains a region where the reduced level of drug X experienced by X-resistant strains is less inhibitory, but also less protective from even greater inhibition by drug Y. In such a region, the drugs, administered together, only support growth of the X-susceptible strain while preventing growth of the resistant strain, and selection for resistance is reversed.29
To demonstrate this potential, drug-susceptible bacteria and doxycycline-resistant mutants were each grown under two different drug pairings: a synergistic combination of doxycycline and erythromycin or a suppressive combination of doxycycline and ciprofloxacin. In the synergistic drug combination, the doxycycline-resistant bacteria appeared to gain relief both from doxycycline itself and from the additional inhibitory effect of the synergy with erythromycin; it handily out-competed the susceptible bacteria. However, at various concentrations of the antibiotics in the suppressive drug combination, the benefit of resistance to doxycycline was outweighed by the associated loss of its protection from ciprofloxacin. In these regimes, the doxycycline-susceptible bacteria out-competed the doxycycline-resistant strain, despite the presence of the resisted drug.29
Herein there is a potential concept for clinical translation: over time, resistant bacteria can be out-competed by more susceptible strains, leaving a population of treatable bacteria that would be more susceptible to classical antibiotics or synergistic combinations. Particular drug environments can generate local fitness peaks where greater relative fitness actually requires drug-susceptibility instead of resistance. While under synergistic combinations, bacteria always increase in fitness if they can gain resistance to one or more drugs; in the case of suppressive drug environments, if a bacterium gains resistance to one drug, its fitness may decrease. Selection could therefore constrain populations to a local fitness peak dominated by bacteria that are susceptible to certain antibiotics.29
Resistance mutations often impose fitness costs that favor susceptible over resistant bacteria at very low or no drug concentrations. Secondary, compensatory mutations that reduce these costs frequently follow. In the schematics in Figures 3 and 4, these more complex effects of resistance mutations are not displayed. While such factors should be included in a treatment strategy, this simplified two-drug environment presents an exciting potential for a dual treatment strategy using combination chemotherapy to treat an infection while simultaneously selecting for more treatable, antibiotic-susceptible bacteria.
Figure 4. Suppressive drug combinations may have smaller mutant selection windows (MSWs) compared to synergistic combinations.
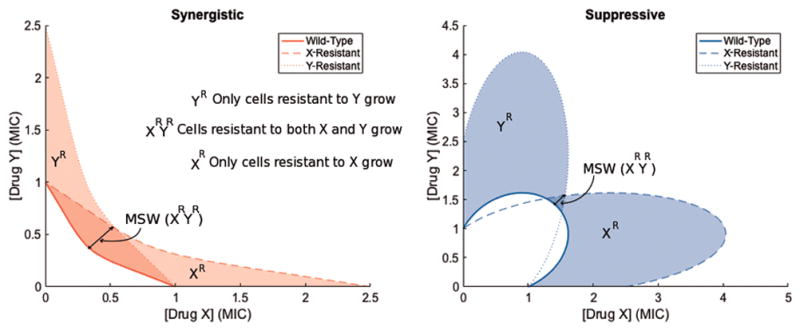
Consider a bacterial population with three subpopulations characterized by MIC isoboles: the wild-type (solid line), a mutant population resistant only to drug X (dashed line), and a mutant population resistant only to drug Y (dotted line). In this simple scenario, without double mutants or cross-resistance, one mutation renders resistance to only drug X and another mutation renders resistance to only drug Y. The mutant prevention concentration (MPC) line is shown by the outermost isobole, and the MSW is shown by the shaded area between the outermost and innermost isoboles. The MSW is larger in the synergistic combination than in the suppressive combination.
In addition to their potential to reverse evolution of resistance by imposing selective pressure, suppressive drug combinations can create lower rates of adaptation to antibiotics compared to adaptation rates under synergistic combination treatments. When populations of E. coli were subjected to the same synergistic and suppressive drug combinations used in reference 29, the rate of adaptation was observed to be greater under the synergistic pair than under the suppressive pair or indeed under either of the two drugs alone.45 In order to better understand this effect, the growth rates of E. coli under two drug pairs (ciprofloxacin + amikacin and doxycycline + amikacin) and multiple concentrations per drug pair, whose classifications range from antagonism to synergy depending on the dose level,45 were measured. Intriguingly, a significant positive correlation was found between the rate of adaptation and degree of synergy between the drugs,45 suggesting the enhanced effect of synergistic combinations comes also with an enhanced risk. This observation further underscores the importance of evaluating not only the direct inhibitory impact of a drug combination on a strain, but also the sign and intensity of selection it imposes on that strain relative to others with altered susceptibility.
Consideration of the above factors underscores the complexity in optimizing treatment strategies. The selective advantage of resistant cells under synergistic drug combinations may lead resistant mutants to out-compete susceptible cells and accumulate further resistance mutations more quickly than under antagonistic or suppressive combinations.45 While this suggests that using antagonistic or suppressive drug pairs can slow the rate at which bacteria increase their resistance to antibiotics, it is important to note that the drug comparisons in this study were conducted at roughly equal levels of inhibition.45 In other scenarios, more efficient killing by synergistic drug pairs could also provide a means to slow the evolution of resistant mutants.63 Practically, effectively using drug combinations to cure infections while also reducing the threat of resistance may necessitate monitoring factors besides selection, such as population size, ecological carrying capacity, and mutational potential.63
We can further understand the trade-off between efficacy of inhibition and likelihood to evolve resistance by examining the mutant selection window (MSW), which measures the range of drug concentrations above the minimum inhibitory concentration (MIC), where wild-type cells cease to measurably grow, and below the mutant prevention concentration (MPC), where no mutant survival is measured.64 As shown in Figure 4, synergistic drug pairs have larger MSWs than either of the individual constituents and antagonistic and suppressive drug pairs have smaller MSWs, since they have larger combined MICs than synergistic drug pairs.39 This simple model adds to the suggestion that there are trade-offs between efficacy of inhibition and likelihood of resistance evolution.
Increased efficacy can be a poor long-term decision if the initial dose of drugs does not kill the entire population. This is because resistant mutants are enriched as drugs decay over time, and ultimately, the most aggressive and potent treatments can end up yielding the highest density of bacteria.65 In a study by Pena-Miller et al., when bacteria were initially treated with the strongest synergistic drug combinations, remaining bacteria were resistant to the drug combination and thus the population subsequently grew at a faster rate in days two through five, compared to when the population was initially treated by just a single drug or a weaker synergistic combination. This finding is consistent with results on the rate of adaptation described earlier,45 in which synergy yields faster rates of adaptation.
Mechanisms of Suppression
Understanding how two-drug interactions occur mechanistically may yield valuable insights that could lead to new ways to manipulate cellular growth in clinical settings. This has been especially true of synergistic interactions, where the hope is to create rational combinations with maximum killing effect. The underlying mechanisms of these interactions are likely diverse, yet even for synergistic interactions, the number of discoveries has been limited.66 Much less is known about mechanisms behind suppressive and antagonistic interactions, likely due to the same reason there is a bias towards reporting synergy in the literature: thinking that suppressive/antagonistic mechanisms are either outliers or clinically irrelevant. Below we detail several types of mechanisms for suppression/antagonism:
1. Bacteriostatic-bactericidal
A prevalent mechanism of antibiotic antagonism is found in the interaction between bacteriostatic and bactericidal antibiotics, which halt cellular growth and kill growing cells, respectively.67 When resistance to the bacteriostatic agent restores growth, the resistant strain lies exposed to the second, bactericidal stress and is eliminated. The quiescent, susceptible strain experiences the reduced effect of the antagonistic combination and survives, thus creating an evolutionary advantage for susceptibility.67–72 While it would be useful to check bacteriostatic-bactericidal combinations for suppression, simply combining drugs based on their -static or -cidal effects does not necessarily yield similar phenotypic effects (e.g. trimethoprim (bacteriostatic) and streptomycin (bactericidal) are synergistic, not antagonistic24).
2. Cellular Physiology
Antibiotic interactions can be studied from Fraser’s perspective, in that they result from interfering effects of each of the individual substances on the cells’ physiology. Gene-expression patterns in bacteria subjected to the known suppressive combination of DNA synthesis inhibitors (e.g. ciprofloxacin, nalidixic acid) and protein synthesis inhibitors (e.g. spiramycin, tetracycline)24, 29 revealed insight into how bacteria regulate their behavior non-optimally under varying circumstances.44 In the absence of antibiotics, synthesis rates of the different compartments of the cell (e.g. DNA, protein, cell-wall) are balanced to produce rapid growth. In the presence of DNA replication inhibitors, such as ciprofloxacin, protein synthesis is not adequately down-regulated, causing an imbalance in the ratio of DNA to protein. The addition of a translation-inhibiting drug, spiramycin, can force down the rate of protein synthesis and thus restore the ratio of DNA to protein to a closer to optimal value, enabling the population to grow faster under the combination of both drugs than ciprofloxacin alone. Thus, it was observed that certain suppressive antibiotic interactions find their origins in the non-optimal cross-regulation of the cellular DNA and protein synthesis processes44 (Figure 5).
Figure 5. A mechanism of suppression between DNA-synthesis inhibitors and protein-synthesis inhibitors.
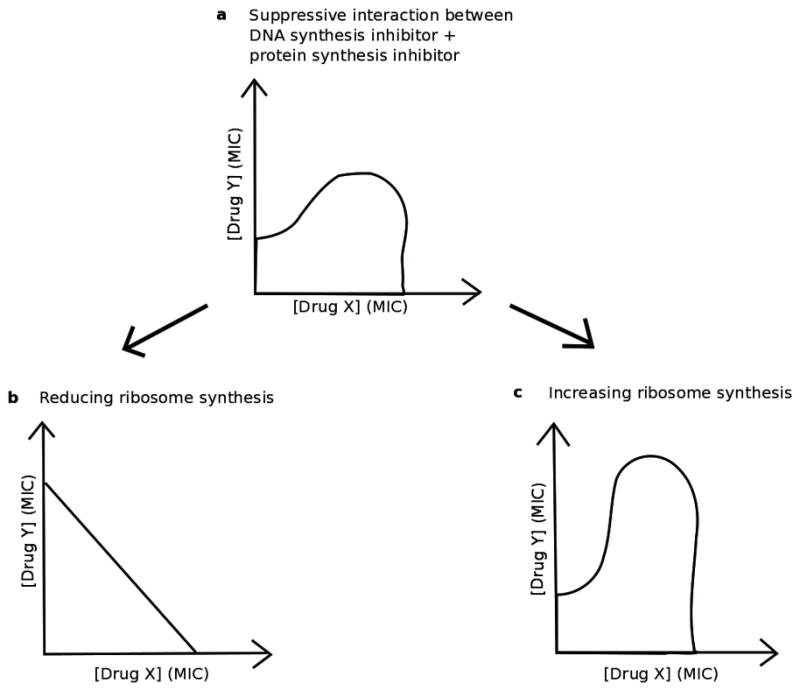
When a DNA-synthesis inhibitor and a protein-synthesis inhibitor are used in combination, the result can be a suppressive interaction.44 In (a), this interaction is shown by its outermost isobole, and the fitness of the bacteria greatly increases when both Drug X and Drug Y are in high concentration because the bacteria experience a combined effect less than one or both of the drugs in the combination (a). However, when this same drug combination is given to a population of bacteria with genetically reduced level of ribosome synthesis, the suppression disappears (b), as shown by an outermost isobole in which increasing the concentration of both Drug X and Drug Y swiftly decreases the bacterial fitness. In populations of genetically manipulated bacteria that overproduce ribosomes, the suppression intensifies (c), as shown by an outermost isobole in which increasing the concentration of both Drug X and Drug Y results in an even bigger increase in fitness than in (a). Bollenbach et al. show that a mismatch in the amount of DNA and ribosomes produced in a cell can more effectively hamper population growth when compared to an overall decrease in both DNA and ribosome production.44
Understanding the suppressive interaction in the context of physiological consequences of these two antibiotics’ activities has two notable results. First, suppression may be somewhat robust to the precise antibiotics used (and their particular target sites), provided that one mainly inhibits DNA synthesis and the other protein translation. Similarly, since diverse mechanisms of antibiotic resistance commonly reduce effective antibiotic concentrations upstream of their targets without altering downstream physiology, the interaction may also be somewhat robust to resistance mechanism. In contrast to mechanism-dependent selection against resistance, as seen with collateral sensitivity73 (susceptibility), resistance mechanism-independence has been seen in assays of selection against efflux-, drug degradation-, and target-site modification-mediated tetracycline resistance by suppressive ciprofloxacin-doxycycline combinations.29, 40
3. Unknown Mechanisms
Other instances of suppression have been found but the mechanisms are currently unclear. A recent study demonstrated that colistin can essentially suppress vancomycin by enabling MRSA to survive in vancomycin-enriched environments. While the exact mechanism is unknown, gene expression changes in MRSA in the presence of colistin resemble the changes seen in mutant strains that are resistant to vancomycin.46 Both cases involved the GraRS regulon, a two-component regulatory system that is involved in bacterial response to cationic antimicrobial peptides.46
Drug interactions may also be sensitive to changes in the genotype of the organism. A large screen of antibiotic interaction mechanisms based on systematic analysis of dose effects in E. coli knockout strains found that most genetic perturbations do not affect drug interactions.47 However, some specific instances of suppression could be amplified, altered, synthetically created, or removed based on certain genetic perturbations.47 For example, perturbing tRNA processing consistently removed chloramphenicol-nitrofurantoin suppression, ribosome production and assembly altered ciprofloxacin-tetracycline suppression, chaperone deletions consistently caused suppression for distinct drug pairs, and perturbing amino acid synthesis amplified chloramphenicol-nitrofurantoin suppression but removed trimethoprim-chloramphenicol antagonism. In particular, polysaccharide synthesis affected the majority of drug interactions, and inhibiting ATP synthase affected the way in which E. coli reacted to many different drugs. These findings suggest that only a few recurring cellular functions control multiple drug interactions.47 Further study is needed to understand why this is so.
Lipopolysaccharide (LPS) and ATP synthase inhibition are mechanistically interesting because although LPS and ATP synthesis are central cellular functions; they are not common antibiotic targets. LPS removal leads to regulated changes in cell composition, affecting drug uptake and proving to be an underlying mechanism of chloramphenicol-nitrofurantoin suppression and other interactions.47 ATP-dependent processes such as DNA repair and synthesis are likely the growth-limiting processes of bacteria, with decreased ATP concentrations in the presence of DNA synthesis inhibitors. Inhibiting more than one of these processes can lead to suppression, as discussed by Bollenbach and colleagues.44
Understanding these mechanisms of suppression and means of altering them could prove useful in clinical treatments. The removal of chloramphenicol-nitrofurantoin suppression by LPS synthesis inhibitors could increase the combination’s killing efficacy while preserving its advantages as an unused drug combination.47 Similarly, the use of thiamin synthesis inhibitors can make the chloramphenicol-trimethoprim combination reciprocally suppressive.47 This type of suppression, in which a drug combination is less inhibitory than either of its individual components, has hardly been found among antibiotics, but could potentially make the path to multidrug resistance even harder by selecting against resistance to both drugs.29
Suppression in Non-Antibacterial Contexts
Antagonism and suppression have been found in many non-antibacterial contexts, including genetic interactions,74 antifungal drugs,37, 75 HIV drugs,76 metal stress,77 biocontrol agents,78 enzyme modifiers,79 plant defense compounds,52 and antimalarial drugs.50 In a study that assayed drug interactions within a set of 175 anti-fungal drug pairs, a large number of suppressive combinations (17%) was found, indicating that suppression (at least in antifungal drug sets) is much more common than previously thought. Interestingly, there were suppression “hubs” where certain drugs were much more likely to suppress a large number of other drugs. The investigators also found 17 reciprocally suppressive interactions.25
A few potential mechanisms of suppression in non-antibiotic contexts have been suggested.25, 80, 81 In antifungal research, suppressive combinations have also been shown to select against resistance.82, 83 Mechanisms explaining the tendency of staurosporine to be suppressed and of bromopyruvate to act as a suppressor have been proposed.25 Staurosporine is an ATP-competitive kinase inhibitor and bromopyruvate acts by inhibiting the glycolysis pathway. Bromopyruvate may be a strong suppressor of staurosporine and other drugs because it causes media acidification, modifying the cellular concentration of suppressed drugs by affecting drug transporters.25 However, as with all explanations of the mechanisms dictating suppression, we must be careful about extrapolating to in vivo situations. Media acidification and similar effects may be phenomena only observed in vitro, thus further highlighting the many difficulties in translating in vitro results to in vivo situations.
There has been research on other aspects of suppressive interactions in non-antibacterial systems, such as cancer. In a tumor setting, cell cycle-dependent antagonistic effects, in which one drug prevents entry of cells into a phase in which the other drug is more effective, have been found between taxol and ϒ-radiation,84 cisplatin and the antimicrotubule agents taxol and vincristine,85 and novel anti-HER2 monoclonal antibodies and tumor necrosis factor-α.86 Cell cycle-dependent antagonisms in cancer treatment84, 86 suggest that cell cycle-dependent phenomena may be a rich avenue for investigating suppressive interactions. Selection against resistant tumor cells due to the suppressive drug combination of doxorubicin and taxol has been shown.87
Furthermore, the exploration of higher-order (more than two component) drug combinations in different systems may reveal different patterns of interactions and levels of antagonism and suppression. Wood and colleagues found that higher-order drug combinations yielded primarily additive interactions.88 However, re-scaling produced a multi-modal distribution from which more synergistic and antagonistic interactions could be straightforwardly identified.53 Furthermore, when examining emergent interactions in higher-order combinations, that is, interactions that rely on all three drugs being present rather than the interaction primarily being the result of interactions between only two of the three drugs, it appears that emergent interactions may be skewed towards increasing antagonism.53, 89 Identification and classification of higher-order interactions is an exciting current area of research due to the important distinction between net (overall) and emergent interactions,89 the multiple classification systems already existing at the pairwise level,27 and considerable debate about whether higher-order interactions need to be considered at all or whether pairwise interactions are sufficient to predict the overall behavior of complex systems.90, 91
Prevalence of suppressive interactions varies greatly depending on the elements tested, model used, and classification method. While we know that suppressive interactions are rarely reported in the antibiotic literature, they made up ~9% of interactions in a study of a complete pairwise interaction network between 21 drugs.24 Studies have found other systems have higher, yet also varied, rates of suppression.25, 37, 78, 92 Antagonistic interactions have been observed to be more common than synergistic interactions among biological control agents (living organisms used to control threats to agriculture)78 and herbicides,80 although reasons for this are unclear.
Use of Suppressive Drug Combinations in the Clinic
Using suppressive drug combinations to combat drug resistance in the clinic presents significant challenges. Living systems are much more complex than in vitro systems due to factors such as differential degradation of drugs,11 the role of the immune system in selecting for resistant bacteria,93 and the increased efficacy of bacteriostatic agents.94 To our knowledge, all studies demonstrating the potential of suppressive drug combinations to reverse evolution of resistance have been performed in vitro or in silico.
Using suppressive drug combinations in vivo likely represents a trade-off between killing efficacy and likelihood of future evolution of resistance.15, 39 Even if suppressive regimens work perfectly, we must consider the fundamental issue that therapies are most often designed to treat the individual, but what is in the best interest of the individual may not always be good for the public at large.95 The increased dosages required in suppressive combinations could in some cases increase the risk of toxic side effects to the patient (especially for the case of bactericidal drugs), yet there may be benefits to the general population in terms of reduced risk of resistance evolution.
When strong selection for resistance is generated by a combined antibiotic treatment (e.g. for synergistic combinations) and resistance evolves, the cost of more effective individual treatment is borne by the public in the form of long-term loss of treatment options and more recalcitrant future infections. Suppressive combinations would present a cost to the individual (higher doses of drugs, with potentially more toxicity, to achieve the same rate of infection clearance), but would also benefit the public at large by reducing the level of resistance and maintaining more treatable infections for the future. Therefore, the use of both suppressive and synergistic drug combinations should take into account both the specific public and individual interests involved in a specific infection and environment. For instance, toxicity allowing, a suppressive drug combination may potentially be useful in the case of TB96 and other scenarios in which antibiotics are used in widespread, long-term therapy and the emergence of resistance is a major threat to both active patients and the public.97
A dynamic treatment protocol that uses both suppressive combinations (or other tactics to give susceptible bacteria a selective advantage) and regular antibiotic treatment may be a solution that could maximize killing efficacy while minimizing resistance.97 Recent mathematical work compares growth of susceptible and resistant bacteria under different conditions and demonstrates how a precisely timed combination of treatments can select for susceptibility. In the model used, a costly plasmid confers antibiotic resistance.97 The work suggests that the optimal amount of time to apply suppressive combinations to a resistant population until an antibiotic treatment is effective depends on three main factors: the pathogen division rate, the rate of plasmid loss, and the difference in growth rate between susceptible and resistant strains.97 Since the optimal strategy changes as the bacterial population changes, we must further understand under exactly which conditions to apply which treatment, and how to quickly and cheaply monitor these conditions to appropriately schedule such treatments in bacterial infections.15
We can better understand how the composition of a bacterial infection affects treatment strategies by considering a computational infection model that takes into account the overall strength of the drug-drug interaction and the relative and absolute numbers of susceptible and resistant subpopulations competing for a limited supply of nutrients.63 In the absence of any resource competition, highly effective synergistic drug pairs represent the optimal treatment, rapidly eliminating infections and thus best preventing the prolonged cycles of division and mutation that allow resistance to the combination to evolve.
However, when the bacteria exist in a milieu in which they must compete for resources, the picture changes dramatically. If a synergistic drug pair is applied, the resources freed by the rapid removal of the susceptible strain allow a strain that has become resistant to one of the antibiotics to grow and mutate to resist both drugs much more rapidly while it is still present in large numbers. On the other hand, if a suppressive drug combination is used, the susceptible bacteria are killed much more slowly than the single-drug resistant cells.63 While the single-drug resistant population is being killed, the overall infection remains sufficiently large such that nutrient limitation suppresses growth and thus slows generation of novel mutations in the singly resistant subpopulation. Once only susceptible bacteria are left in this (still relatively large) population, it can be cleared with a single antibiotic or synergistic drug combination. In this model, synergistic drug combinations rapidly eliminate the infection but can increase the risk of generating mutants that are entirely resistant to the drug combination. In contrast, antagonistic or suppressive drug pairs are less effective at removing the infection but are typically less likely to produce conditions favorable to the evolution of doubly resistant mutants.63 As noted above, certain cases also exist where a synergistic combination better prevents mutations and the emergence of resistance, contingent on the specific ecology of the infection.63
While some researchers have advocated for this more ecology-centric approach to the problem of drug resistance,97–100 there are many challenges to taking this approach. These include determining the extent to which resource competition plays a role in such a complex environment as the patient’s body98 and obtaining accurate parameters of traits that determine resource competition in the human body,98 including nutrient availability in various sites of infection.101
Another difficulty with using suppressive drug combinations is that physiological interactions can change across different environments and genetic backgrounds, or as pathogens evolve.15 In addition, there are only narrow windows of drug concentrations for which these strategies have been shown to work,29 and different factors such as time since last dose102 and the tissue being targeted103 can change the intended effects of drugs in the body. Of particular concern, it has been found that even a weak antagonistic interaction leads to a very dynamic optimal treatment protocol104 due to the changing resistance profile of the infection continuously redefining the efficacy of chosen treatment strategies.29 Tailoring treatments to take into account dynamic bacterial resistance profiles and factors that affect the strength and type of physiological interactions presents a major area for further study.
Future Directions
While suppressive drug combinations are one of only a few potential strategies to combat emergence of resistance, their effects on treatment efficacy and resistance prevention are likely to be affected by a number of factors, and there is a long way to go before they can be used clinically. We have identified five research directions as especially pressing: (1) technological advances and/or high-throughput screening methods to more efficiently identify suppression; (2) further identification of suppressive mechanisms of action; (3) examination of other factors (e.g. type of bacteria, dose-dependence) that can affect suppressive interactions; (4) investigation of the ecological role that resource competition could play in treatment; and (5) in vivo studies to better understand the effects of suppressive interactions in the body.
Even with new technologies, we cannot simply test all drug combinations for suppression—given the large numbers of drug combinations, this would be logistically difficult.105 We need to develop more efficient ways to identify suppression, such as conducting screens to discover if the types of suppression hubs found in antifungal drug screenings25 also exist in antibiotic networks. Suppression often cannot be predicted based on mechanisms of action,62 but continuing to identify suppressive mechanisms of action may lead to the discovery of generalized suppression between certain drug classes, such as DNA synthesis inhibitors and protein synthesis inhibitors.44
Other factors, such as how adding a third component to a pairwise suppressive combination can increase the magnitude of suppression53, 106 or create a reciprocally suppressive combination, should also be investigated. It is also of interest to understand the effect of type of bacteria on observed interaction type60, 61 and to conduct careful dose-dependent tests in vitro to establish a clearer idea of what concentrations are suppressive and how wide or narrow this range is.
In addition, there needs to be careful examination of whether and how different ecological components of resource competition are relevant to treatment using suppression. For example, it would be useful to understand the extent to which different environmental parameters affect resource competition, and whether resource competition could be exploited clinically through the use of dynamic treatment protocols or the introduction of drug-susceptible non-pathogenic strains.63, 97 These factors may affect the number and magnitude of suppressive combinations.
Ultimately, basic in vivo studies must be conducted to determine whether the results of in vitro and in silico experiments hold when treating infections, as well as to more explicitly identify the potential risks and complications of using suppressive drug combinations in treatment. After conducting careful dose-dependence tests in vitro, the next key step in translating suppressive combinations to the clinic would be testing suppressive combinations in animal models to establish better understanding of risks as well as necessary concentrations, pharmacodynamics, and pharmacokinetics.107 These tests would also help us understand how quickly resistance profiles of bacterial populations change in response to suppressive drugs in vivo. This would in turn inform the timing of switches from suppressive to synergistic or classic antibiotic treatment once most of the resistant bacteria have been killed.97
If suppressive drug combinations show in vivo potential in animal models, a first case in which to use them clinically may be in the treatment of diseases like tuberculosis (TB).96 Since TB requires long-term treatment, the patient receiving the suppressive drug treatment would be able to reap the benefits of reversal of resistance, rather than only the general public benefiting from the likely more toxic and higher dosage suppressive treatment regimen.
The properties of antagonistic and suppressive drug combinations to slow or even reverse the evolution of increasing drug resistance suggest intriguing clinical potential. However, as we have noted, there are a great many exigencies which desperately require more studies (particularly in vivo) to clarify whether cases for clinical use exist. Given our diminishing options to treat increasingly resistant pathogens and the critical need to protect available treatment options, we suggest that the long-known, yet underappreciated, suppressive interactions are a worthy research direction to pursue.
Acknowledgments
We thank Remy Chait, Jeffrey H. Miller, Lei Dai, Casey Beppler, Matthew Sit, and Roy Kishony for discussion and/or comments on the manuscript.
References
- 1.Taubes G. The bacteria fight back. Science. 2008;321(5887):356–361. doi: 10.1126/science.321.5887.356. [DOI] [PubMed] [Google Scholar]
- 2.Levy SB. The antibiotic paradox: how the misuse of antibiotics destroys their curative powers. Da Capo Press; 2002. [Google Scholar]
- 3.Lewis K, Salyers A, Taber H, Wax R. Bacterial resistance to antimicrobials: mechanisms, genetics, medical practice and public health. New York: Marcel Dekker; 2001. [Google Scholar]
- 4.Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. The challenge of new drug discovery for tuberculosis. Nature. 2011;469(7331):483–490. doi: 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- 5.Clardy J, Fischbach MA, Walsh CT. New antibiotics from bacterial natural products. Nature biotechnology. 2006;24(12):1541–1550. doi: 10.1038/nbt1266. [DOI] [PubMed] [Google Scholar]
- 6.Devasahayam G, Scheld WM, Hoffman PS. Newer antibacterial drugs for a new century. Expert opinion on investigational drugs. 2010;19(2):215–234. doi: 10.1517/13543780903505092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drawz SM, Bonomo RA. Three decades of β-lactamase inhibitors. Clinical microbiology reviews. 2010;23(1):160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stone LK, Baym M, Lieberman TD, Chait R, Clardy J, Kishony R. Compounds that select against the tetracycline-resistance efflux pump. Nature Chemical Biology. 2016;12:902–904. doi: 10.1038/nchembio.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Everitt E, Han E, Cherian S, Kempf D, Sham H, Ng S-C. Compositions and methods for enhancing the bioavailability of pharmaceutical agents. Google Patents. 2002 [Google Scholar]
- 10.Ali B. Agents ameliorating or augmenting experimental gentamicin nephrotoxicity: some recent research. Food and Chemical Toxicology. 2003;41(11):1447–1452. doi: 10.1016/s0278-6915(03)00186-8. [DOI] [PubMed] [Google Scholar]
- 11.Roberts JA, et al. Individualised antibiotic dosing for patients who are critically ill: challenges and potential solutions. The Lancet. Infectious diseases. 2014;14(6):498–509. doi: 10.1016/S1473-3099(14)70036-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boucher HW, et al. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clinical Infectious Diseases. 2009 doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 13.Perry JA. The antibiotic resistome: what’s new? 2014;21:45–50. doi: 10.1016/j.mib.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Boeckel TPV, et al. Global trends in antimicrobial use in food animals. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(18) doi: 10.1073/pnas.1503141112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baym M, Stone LK, Kishony R. Multidrug evolutionary strategies to reverse antibiotic resistance. Science. 2016;351(6268):aad3292. doi: 10.1126/science.aad3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith DL, Harris AD, Johnson JA, Silbergeld EK, Morris JG. Animal antibiotic use has an early but important impact on the emergence of antibiotic resistance in human commensal bacteria. Proceedings of the National Academy of Sciences. 2002;99(9):6434–6439. doi: 10.1073/pnas.082188899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stone GW, et al. Mechanism of Action of NB2001 and NB2030, Novel Antibacterial Agents Activated by β-Lactamases. Antimicrobial Agents and Chemotherapy. 2004;48(2):477–483. doi: 10.1128/AAC.48.2.477-483.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munck C, Gumpert HK, Wallin AIN, Wang HH, Sommer MO. Prediction of resistance development against drug combinations by collateral responses to component drugs. Science translational medicine. 2014;6(262):262ra156–262ra156. doi: 10.1126/scitranslmed.3009940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shafer R, Vuitton D. Highly active antiretroviral therapy (HAART) for the treatment of infection with human immunodeficiency virus type 1. Biomedicine & pharmacotherapy. 1999;53(2):73–86. doi: 10.1016/s0753-3322(99)80063-8. [DOI] [PubMed] [Google Scholar]
- 20.Ramón-García S, et al. Synergistic drug combinations for tuberculosis therapy identified by a novel high-throughput screen. Antimicrobial agents and chemotherapy. 2011;55(8):3861–3869. doi: 10.1128/AAC.00474-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bozic I, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. eLife. 2013:2. doi: 10.7554/eLife.00747. [DOI] [PMC free article] [PubMed]
- 22.Keith CT, Borisy AA, Stockwell BR. Multicomponent therapeutics for networked systems. Nature reviews Drug discovery. 2005;4(1):71–78. doi: 10.1038/nrd1609. [DOI] [PubMed] [Google Scholar]
- 23.Braberg H, et al. Quantitative analysis of triple-mutant genetic interactions. Nature protocols. 2014;9(8):1867–1881. doi: 10.1038/nprot.2014.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeh P, Tschumi AI, Kishony R. Functional classification of drugs by properties of their pairwise interactions. Nature genetics. 2006;38(4):489–494. doi: 10.1038/ng1755. [DOI] [PubMed] [Google Scholar]
- 25.Cokol M, et al. Large-scale identification and analysis of suppressive drug interactions. Chemistry & biology. 2014;21(4):541–551. doi: 10.1016/j.chembiol.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lepper MH, Dowling HF. Treatment of pneumococcic meningitis with penicillin compared with penicillin plus aureomycin: studies including observations on an apparent antagonism between penicillin and aureomycin. AMA archives of internal medicine. 1951;88(4):489–494. doi: 10.1001/archinte.1951.03810100073006. [DOI] [PubMed] [Google Scholar]
- 27.Foucquier J, Guedj M. Analysis of drug combinations: current methodological landscape. Pharmacology Research & Perspectives. 2015;3(3) doi: 10.1002/prp2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bliss C. The toxicity of poisons applied jointly1. Annals of applied biology. 1939;26(3):585–615. [Google Scholar]
- 29.Chait R, Craney A, Kishony R. Antibiotic interactions that select against resistance. Nature. 2007;446(7136):668–671. doi: 10.1038/nature05685. [DOI] [PubMed] [Google Scholar]
- 30.Fraser TR. Lecture on the antagonism between the actions of active substances. British medical journal. 1872;2(617):457. doi: 10.1136/bmj.2.617.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fraser TR. 5. An Experimental Research on the Antagonism between the Actions of Physostigma and Atropia. Proceedings of the Royal Society of Edinburgh. 1872;7:506–511. [Google Scholar]
- 32.Weiss S. Persistence of Action of Physostigmin and the Atropin-Physostigmin Antagonism in Animals and in Man. Journal of Pharmacology and Experimental Therapeutics. 1926;27(3):181–188. [Google Scholar]
- 33.Browning C, Gulbransen R. An interference phenomenon in the action of chemotherapeutic substances in experimental trypanosome infections. The Journal of Pathology and Bacteriology. 1922;25(3):395–397. [Google Scholar]
- 34.Proudfoot A. The Early Toxicology of Physostigmine | SpringerLink. Toxicological Reviews. 2006;25(2):99–138. doi: 10.2165/00139709-200625020-00004. [DOI] [PubMed] [Google Scholar]
- 35.Yeh PJ, Hegreness MJ, Aiden AP, Kishony R. Drug interactions and the evolution of antibiotic resistance. Nature Reviews Microbiology. 2009;7(6):460–466. doi: 10.1038/nrmicro2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeh P, Kishony R. Networks from drug–drug surfaces. Molecular systems biology. 2007;3(1):85. doi: 10.1038/msb4100133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Vos MG, Bollenbach T. Suppressive Drug Interactions between Antifungals. Chemistry & biology. 2014;21(4):439–440. doi: 10.1016/j.chembiol.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 38.Stechmann B, et al. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell. 2010;141(2):231–242. doi: 10.1016/j.cell.2010.01.043. [DOI] [PubMed] [Google Scholar]
- 39.Michel J-B, Yeh PJ, Chait R, Moellering RC, Kishony R. Drug interactions modulate the potential for evolution of resistance. Proceedings of the National Academy of Sciences. 2008;105(39):14918–14923. doi: 10.1073/pnas.0800944105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chait R, Shrestha S, Shah AK, Michel J-B, Kishony R. A differential drug screen for compounds that select against antibiotic resistance. PLoS One. 2010;5(12):e15179. doi: 10.1371/journal.pone.0015179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cokol M, et al. Systematic exploration of synergistic drug pairs. Molecular systems biology. 2011;7(1):544. doi: 10.1038/msb.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mitosch K, Bollenbach T. Bacterial responses to antibiotics and their combinations. Environmental microbiology reports. 2014;6(6):545–557. doi: 10.1111/1758-2229.12190. [DOI] [PubMed] [Google Scholar]
- 43.Palmer AC, Kishony R. Opposing effects of target overexpression reveal drug mechanisms. Nature communications. 2014:5. doi: 10.1038/ncomms5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bollenbach T, Quan S, Chait R, Kishony R. Nonoptimal microbial response to antibiotics underlies suppressive drug interactions. Cell. 2009;139(4):707–718. doi: 10.1016/j.cell.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hegreness M, Shoresh N, Damian D, Hartl D, Kishony R. Accelerated evolution of resistance in multidrug environments. Proceedings of the National Academy of Sciences. 2008;105(37):13977–13981. doi: 10.1073/pnas.0805965105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haaber J, Friberg C, McCreary M, Lin R, Cohen SN, Ingmer H. Reversible antibiotic tolerance induced in Staphylococcus aureus by concurrent drug exposure. Mbio. 2015;6(1):e02268–02214. doi: 10.1128/mBio.02268-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chevereau G, Bollenbach T. Systematic discovery of drug interaction mechanisms. Molecular systems biology. 2015;11(4):807. doi: 10.15252/msb.20156098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Motter AE. Improved network performance via antagonism: From synthetic rescues to multi - drug combinations. Bioessays. 2010;32(3):236–245. doi: 10.1002/bies.200900128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berenbaum M. Synergy, additivism and antagonism in immunosuppression. A critical review. Clinical and experimental immunology. 1977;28(1):1. [PMC free article] [PubMed] [Google Scholar]
- 50.Bell A. Antimalarial drug synergism and antagonism: mechanistic and clinical significance. FEMS microbiology letters. 2005;253(2):171–184. doi: 10.1016/j.femsle.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 51.Geary N. Understanding synergy. American Journal of Physiology-Endocrinology and Metabolism. 2013;304(3):E237–E253. doi: 10.1152/ajpendo.00308.2012. [DOI] [PubMed] [Google Scholar]
- 52.Nelson AC, Kursar TA. Interactions among plant defense compounds: a method for analysis. Chemoecology. 1999;9(2):81–92. [Google Scholar]
- 53.Tekin E, Beppler C, White C, Mao Z, Savage VM, Yeh PJ. Enhanced identification of synergistic and antagonistic emergent interactions among three or more drugs. Journal of The Royal Society Interface. 2016;13(119):20160332. doi: 10.1098/rsif.2016.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou A, et al. Synergistic Interactions of Vancomycin with Different Antibiotics against Escherichia coli: Trimethoprim and Nitrofurantoin Display Strong Synergies with Vancomycin against Wild-Type E. coli. Antimicrobial Agents and Chemotherapy. 2015;59:276–281. doi: 10.1128/AAC.03502-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nguyen C, Zhou A, Khan A, Miller JH, Yeh P. Pairwise antibiotic interactions in Escherichia coli: triclosan, rifampicin and aztreonam with nine other classes of antibiotics. The Journal of Antibiotics. 2016;69(11):791–797. doi: 10.1038/ja.2016.26. [DOI] [PubMed] [Google Scholar]
- 56.Jia J, Zhu F, Ma X, Cao ZW, Li YX, Chen YZ. Mechanisms of drug combinations: interaction and network perspectives. Nature reviews Drug discovery. 2009;8(2):111–128. doi: 10.1038/nrd2683. [DOI] [PubMed] [Google Scholar]
- 57.Osborne CK, Kitten L, Arteaga CL. Antagonism of chemotherapy-induced cytotoxicity for human breast cancer cells by antiestrogens. Journal of Clinical Oncology. 1989;7(6):710–717. doi: 10.1200/JCO.1989.7.6.710. [DOI] [PubMed] [Google Scholar]
- 58.French GL, Ling TK, Davies DP, Leung DT. Antagonism of ceftazidime by chloramphenicol in vitro and in vivo during treatment of gram negative meningitis. Br Med J (Clin Res Ed) 1985;291(6496):636–637. doi: 10.1136/bmj.291.6496.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jonker DM, Visser SAG, van der Graaf, Piet H, Voskuyl RA, Danhof M. Towards a mechanism-based analysis of pharmacodynamic drug-drug interactions in vivo. Pharmacology & Therapeutics. 2005;106(1):1–18. doi: 10.1016/j.pharmthera.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 60.Jacqueline C, et al. In Vitro and In Vivo Synergistic Activities of Linezolid Combined with Subinhibitory Concentrations of Imipenem against Methicillin-Resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2005;49(1):45–51. doi: 10.1128/AAC.49.1.45-51.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wicha SG, Kees MG, Kuss J, Kloft C. Pharmacodynamic and response surface analysis of linezolid or vancomycin combined with meropenem against Staphylococcus aureus | SpringerLink. Pharmaceutical Research. 2015;32(7):2410–2418. doi: 10.1007/s11095-015-1632-3. [DOI] [PubMed] [Google Scholar]
- 62.Ankomah P, Johnson PJ, Levin BR. The pharmaco–, population and evolutionary dynamics of multi-drug therapy: Experiments with S. aureus and E. coli and computer simulations. PLoS Pathog. 2013;9(4):e1003300. doi: 10.1371/journal.ppat.1003300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Torella JP, Chait R, Kishony R. Optimal drug synergy in antimicrobial treatments. PLoS Comput Biol. 2010;6(6):e1000796. doi: 10.1371/journal.pcbi.1000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dong Y, Zhao X, Domagala J, Drlica K. Effect of fluoroquinolone concentration on selection of resistant mutants of Mycobacterium bovis BCG and Staphylococcus aureus. Antimicrobial Agents and Chemotherapy. 1999;43(7):1756–1758. doi: 10.1128/aac.43.7.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pena-Miller R, et al. When the most potent combination of antibiotics selects for the greatest bacterial load: the smile-frown transition. PLoS Biol. 2013;11(4):e1001540. doi: 10.1371/journal.pbio.1001540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cottarel G, Wierzbowski J. Combination drugs, an emerging option for antibacterial therapy. Trends in Biotechnology. 2007;25(12):547–555. doi: 10.1016/j.tibtech.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 67.Ocampo PS, et al. Antagonism between bacteriostatic and bactericidal antibiotics is prevalent. Antimicrobial agents and chemotherapy. 2014;58(8):4573–4582. doi: 10.1128/AAC.02463-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dance D, Wuthiekanun V, Chaowagul W, White N. Interactions in vitro between agents used to treat melioidosis. Journal of Antimicrobial Chemotherapy. 1989;24(3):311–316. doi: 10.1093/jac/24.3.311. [DOI] [PubMed] [Google Scholar]
- 69.Davis BD. Isolation of biochemically deficient mutants of bacteria by penicillin. Journal of the American Chemical Society. 1948;70(12):4267–4267. doi: 10.1021/ja01192a520. [DOI] [PubMed] [Google Scholar]
- 70.Tuomanen E, Cozens R, Tosch W, Zak O, Tomasz A. The rate of killing of Escherichia coli by β-lactam antibiotics is strictly proportional to the rate of bacterial growth. Microbiology. 1986;132(5):1297–1304. doi: 10.1099/00221287-132-5-1297. [DOI] [PubMed] [Google Scholar]
- 71.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305(5690):1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 72.Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nature reviews microbiology. 2004;2(2):95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 73.Li Q, et al. NB2001, a novel antibacterial agent with broad-spectrum activity and enhanced potency against β-lactamase-producing strains. Antimicrobial agents and chemotherapy. 2002;46(5):1262–1268. doi: 10.1128/AAC.46.5.1262-1268.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.MacLean RC, Hall AR, Perron GG, Buckling A. The population genetics of antibiotic resistance: integrating molecular mechanisms and treatment contexts. Nature Reviews Genetics. 2010;11(6):405–414. doi: 10.1038/nrg2778. [DOI] [PubMed] [Google Scholar]
- 75.Anderson JB, Ricker N, Sirjusingh C. Antagonism between two mechanisms of antifungal drug resistance. Eukaryotic cell. 2006;5(8):1243–1251. doi: 10.1128/EC.00048-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bretscher MT, Althaus CL, Müller V, Bonhoeffer S. Recombination in HIV and the evolution of drug resistance: for better or for worse? Bioessays. 2004;26(2):180–188. doi: 10.1002/bies.10386. [DOI] [PubMed] [Google Scholar]
- 77.Taylor GJ, Stadt KJ, Dale MR. Modelling the interactive effects of aluminum, cadmium, manganese, nickel and zinc stress using the Weibull frequency distribution. Environmental and experimental botany. 1992;32(3):281–293. [Google Scholar]
- 78.Xu X-M, Jeffries P, Pautasso M, Jeger MJ. Combined use of biocontrol agents to manage plant diseases in theory and practice. Phytopathology. 2011;101(9):1024–1031. doi: 10.1094/PHYTO-08-10-0216. [DOI] [PubMed] [Google Scholar]
- 79.Schenker P, Baici A. Simultaneous interaction of enzymes with two modifiers: reappraisal of kinetic models and new paradigms. Journal of theoretical biology. 2009;261(2):318–329. doi: 10.1016/j.jtbi.2009.07.033. [DOI] [PubMed] [Google Scholar]
- 80.Cedergreen N, Kudsk P, Mathiassen SK, Streibig JC. Combination effects of herbicides on plants and algae: do species and test systems matter? Pest management science. 2007;63(3):282–295. doi: 10.1002/ps.1353. [DOI] [PubMed] [Google Scholar]
- 81.Farha MA, et al. Antagonism screen for inhibitors of bacterial cell wall biogenesis uncovers an inhibitor of undecaprenyl diphosphate synthase. Proceedings of the National Academy of Sciences. 2015;112(35):11048–11053. doi: 10.1073/pnas.1511751112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cowen LE. The evolution of fungal drug resistance: modulating the trajectory from genotype to phenotype. Nature Reviews Microbiology. 2008;6(3):187–198. doi: 10.1038/nrmicro1835. [DOI] [PubMed] [Google Scholar]
- 83.Anderson JB. Evolution of antifungal-drug resistance: mechanisms and pathogen fitness. Nature Reviews Microbiology. 2005;3(7):547–556. doi: 10.1038/nrmicro1179. [DOI] [PubMed] [Google Scholar]
- 84.Sui M, Dziadyk JM, Zhu X, Fan W. Cell Cycle–Dependent Antagonistic Interactions between Paclitaxel and γ-Radiation in Combination Therapy. Clinical cancer research. 2004;10(14):4848–4857. doi: 10.1158/1078-0432.CCR-03-0707. [DOI] [PubMed] [Google Scholar]
- 85.Rowinsky EK, Citardi MJ, Noe DA, Donehower RC. Sequence-dependent cytotoxic effects due to combinations of cisplatin and the antimicrotubule agents taxol and vincristine. Journal of cancer research and clinical oncology. 1993;119(12):727–733. doi: 10.1007/BF01195344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ceran C, Cokol M, Cingoz S, Tasan I, Ozturk M, Yagci T. Novel anti-HER2 monoclonal antibodies: synergy and antagonism with tumor necrosis factor-α. BMC cancer. 2012;12(1):450. doi: 10.1186/1471-2407-12-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blagosklonny M. Drug-resistance enables selective killing of resistant leukemia cells: exploiting of drug resistance instead of reversal. Leukemia. 1999;13:2031–2035. doi: 10.1038/sj.leu.2401623. [DOI] [PubMed] [Google Scholar]
- 88.Wood K, Nishida S, Sontag ED, Cluzel P. Mechanism-independent method for predicting response to multidrug combinations in bacteria. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(30):12254–12259. doi: 10.1073/pnas.1201281109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beppler C, et al. Uncovering emergent interactions in three-way combinations of stressors. Journal of the Royal Society Interface. 2016;13(125) doi: 10.1098/rsif.2016.0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wood KB. Pairwise interactions and the battle against combinatorics in multidrug therapies. PNAS. 2016;113(37):10231–10233. doi: 10.1073/pnas.1612365113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tekin E, Savage VM, Yeh PJ. Measuring higher-order drug interactions: A review of recent approaches. Current Opinion in Systems Biology. 2017;4:16–23. [Google Scholar]
- 92.Steinbach WJ, Stevens DA, Denning DW. Combination and sequential antifungal therapy for invasive aspergillosis: review of published in vitro and in vivo interactions and 6281 clinical cases from 1966 to 2001. Clinical Infectious Diseases. 2003;37(Supplement 3):S188–S244. doi: 10.1086/376524. [DOI] [PubMed] [Google Scholar]
- 93.Brandl K, et al. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature. 2008;455(7214):804–807. doi: 10.1038/nature07250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pankey GA, Sabath LD. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2004;38(6):864–870. doi: 10.1086/381972. [DOI] [PubMed] [Google Scholar]
- 95.Foster KR, Grundmann H. Do we need to put society first? The potential for tragedy in antimicrobial resistance. PLoS Med. 2006;3(2):e29. doi: 10.1371/journal.pmed.0030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Connolly LE, Edelstein PH, Ramakrishnan L. Why is long-term therapy required to cure tuberculosis? PLoS Med. 2007;4(3):e120. doi: 10.1371/journal.pmed.0040120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gomes AL, Galagan JE, Segrè D. Resource competition may lead to effective treatment of antibiotic resistant infections. PloS one. 2013;8(12):e80775. doi: 10.1371/journal.pone.0080775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Day T, Huijben S, Read AF. Is selection relevant in the evolutionary emergence of drug resistance? Trends in microbiology. 2015;23(3):126–133. doi: 10.1016/j.tim.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Singer RS, Ward MP, Maldonado G. Can landscape ecology untangle the complexity of antibiotic resistance? Nature Reviews Microbiology. 2006;4(12):943–952. doi: 10.1038/nrmicro1553. [DOI] [PubMed] [Google Scholar]
- 100.Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nature medicine. 2004;10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 101.Smith VH, Holt RD. Resource competition and within-host disease dynamics. Trends in ecology & evolution. 1996;11(9):386–389. doi: 10.1016/0169-5347(96)20067-9. [DOI] [PubMed] [Google Scholar]
- 102.Kano Y, Ohnuma T, Okano T, Holland JF. Effects of vincristine in combination with methotrexate and other antitumor agents in human acute lymphoblastic leukemia cells in culture. Cancer research. 1988;48(2):351–356. [PubMed] [Google Scholar]
- 103.Torchilin VP. Drug targeting. European Journal of Pharmaceutical Sciences. 2000;11:S81–S91. doi: 10.1016/s0928-0987(00)00166-4. [DOI] [PubMed] [Google Scholar]
- 104.Orlando PA, Gatenby RA, Brown JS. Cancer treatment as a game: integrating evolutionary game theory into the optimal control of chemotherapy. Physical biology. 2012;9(6):065007. doi: 10.1088/1478-3975/9/6/065007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bollenbach T. Antimicrobial interactions: mechanisms and implications for drug discovery and resistance evolution. Current opinion in microbiology. 2015;27:1–9. doi: 10.1016/j.mib.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 106.Rodea-Palomares I, González-Pleiter M, Martín-Betancor K, Rosal R, Fernández-Piñas F. Additivity and Interactions in Ecotoxicity of Pollutant Mixtures: Some Patterns, Conclusions, and Open Questions. Toxics. 2015;3(4):342–369. doi: 10.3390/toxics3040342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Beppler C, et al. When more is less: Emergent suppressive interactions in three-drug combinations. BMC Microbiology. 2017;17(1):107. doi: 10.1186/s12866-017-1017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Loewe S. The problem of synergism and antagonism of combined drugs. Arzneimittel-Forschung. 1953;3(6):285. [PubMed] [Google Scholar]