

Abstract
Gap junctions and hemichannels comprised of connexins impact many cellular processes. Significant advances in our understanding of the functional role of these channels have been made by the identification of a host of genetic diseases caused by connexin mutations. Prominent features of connexin disorders are the inability of other connexins expressed in the same cell type to compensate for the mutated one, and the ability of connexin mutants to dominantly influence the activity of other wild-type connexins. Functional studies have begun to identify some of the underlying mechanisms whereby connexin channel mutation contributes to the disease state. Detailed mechanistic understanding of these functional differences will help to facilitate new pathophysiology driven therapies for the diverse array of connexin genetic disorders.
Keywords: Connexin, Mutation, Genetic Disease, Hemichannel, Gap Junction
1. Introduction
Most of the human congenital diseases were described clinically long before the underlying genetic causes were known. Patients with similar characteristics were identified and clinicians then developed diagnostic criteria based on disease features to classify different disorders. Sequencing of the human genome dramatically increased the identification of underlying causative mutations, particularly for rare disorders, and uncovered new roles for the members of many gene families in the function of multiple organ systems. This has proven true for the connexin gene family, where half of its members have currently been linked to a broad spectrum of genetic diseases. In some cases, the same phenotypic disease can be caused by mutations in three different connexin genes. In other cases, mutations within the same connexin gene can result in up to eight distinct clinical disorders. A major challenge for the field is to understand how the specific functional consequences of the gene mutations relate to the clinical similarities and differences between diseases. Here, we will summarize the currently known connexin genetic disorders and review some of the recent mechanistic advances in our understanding of how the underlying connexin mutations contribute to disease.
1.1. Connexins and intercellular communication
Gap junction channels are made from a family of proteins called connexins (Cx), and allow the direct passage of small molecules between adjacent cells, coupling them both metabolically and electrically [1, 2]. Connexins have four transmembrane domains, TM1–TM4, connected by two extracellular loops, E1 and E2, which mediate docking. The N- and C- termini, and a loop connecting TM2 and TM3 are on the cytoplasmic side of the plasma membrane [3, 4].
Connexins oligomerize in the ER-Golgi pathway into hemichannels (half a gap junction channel, also called a connexon) containing six connexin monomers [5, 6]. The hemichannels are transported to the plasma membrane, where they can act as functional channels by themselves, or move to regions of cell contact and find a partner hemichannel from an adjacent cell to form a complete gap junction channel [3]. Unopposed hemichannels become active, or modulate their activity, under conditions of mechanical, or ischemic stress, allowing the flux of small molecules like Ca2+, ATP, glutamate, or NAD+ across the plasma membrane, thereby eliciting signaling cascades and diverse physiological responses [7, 8].
Channels formed by different connexins are functionally distinct in terms of their gating, conductance and permeability characteristics [3, 9–13]. Genetic studies in mice have shown that these functional differences between connexins are important, since the loss of one isoform cannot be compensated for by replacement with another [14–16]. Virtually all cell types make more than one connexin at any given time, as illustrated by keratinocytes which express Cx26, Cx30, Cx30.3, Cx31, and Cx43 [17, 18]. The co-expression of multiple connexins within a single cell diversifies the composition of the channels that can assemble. Hemichannels can be made either from a single connexin isoform (homomeric), or from more than one type (heteromeric). The formation of heteromeric hemichannels depends on the abilities of different connexins to interact; not all connexins can co-oligomerize with each other [19–22]. Docking of hemichannels to form homotypic and heterotypic gap junction channels adds an additional level of complexity. Docking compatibility is regulated by specific sequences in the E2 domain [21, 23, 24]. Thus, genetic mutation of a single connexin gene may influence many types of channel, which may contribute to the diverse variety of disease that can result.
2. Human disorders caused by connexin mutations
Mutations in ten different human connexin genes have been linked to twenty-eight distinct genetic diseases (Table 1). Eight of these disorders result from mutations in Cx26, and an additional six diseases are caused by mutations in Cx43. The highest number of distinct mutations has been identified in Cx32. Consequently, a great deal of the mechanistic research has focused on these connexins (see section 3). As the sequencing technologies used to identify and link mutations to human disease continue to improve, additional connexinopathies may be identified [25].
Table 1.
Genetic disorders caused by human connexin mutations
| Gene | Chromosome | Protein | Disorder(s) | OMIM |
|---|---|---|---|---|
| GJA1 | 6q22.31 | Cx43 | Craniometaphyseal dysplasia, autosomal recessive Erythrokeratodermia variabilis et progressiva Oculodentodigital dysplasia Oculodentodigital dysplasia, autosomal recessive Palmoplantar keratoderma with congenital alopecia Syndactyly, type III |
218400 133200 164200 257850 104100 186100 |
| GJA3 | 13q12.11 | Cx46 | Cataract | 601885 |
| GJA4 | 1p34.3 | Cx37 | ||
| GJA5 | 1q21.2 | Cx40 | Atrial fibrillation, familial, 11 Atrial standstill, digenic (GJA5/SCN5A) |
614049 108770 |
| GJA8 | 1q21.2 | Cx50 | Cataract | 116200 |
| GJA9 | 1p34.3 | Cx59 | ||
| GJA10 | 6q15 | Cx62 | ||
| GJB1 | Xq13.1 | Cx32 | Charcot-Marie-Tooth neuropathy, X-linked 1 | 302800 |
| GJB2 | 13q12.11 | Cx26 | Bart-Pumphrey syndrome Deafness, autosomal dominant 3A Deafness, autosomal recessive 1A Hystrix-like ichthyosis with deafness Keratitis-ichthyosis-deafness syndrome Keratoderma, palmoplantar, with deafness Vohwinkel syndrome Porokeratotic eccrine ostial and dermal duct nevus |
149200 601544 220290 602540 148210 148350 124500 |
| GJB3 | 1p34.3 | Cx31 | Deafness, autosomal dominant 2B Deafness, digenic, (GJB2/GJB3) Erythrokeratodermia variabilis et progressiva |
612644 220290 133200 |
| GJB4 | 1p34.3 | Cx30.3 | Erythrokeratodermia variabilis et progressiva | 133200 |
| GJB5 | 1p34.3 | Cx31.1 | ||
| GJB6 | 13q12.11 | Cx30 | Deafness, autosomal dominant 3B Deafness, autosomal recessive 1B Deafness, digenic (GJB2/GJB6) Ectodermal dysplasia 2, Clouston type |
612643 612645 220290 129500 |
| GJB7 | 6q14.3–q15 | Cx25 | ||
| GJC1 | 17q21.31 | Cx45 | ||
| GJC2 | 1q42.13 | Cx47 | Leukodystrophy, hypomyelinating, 2 Spastic paraplegia 44, autosomal recessive Lymphedema, hereditary, IC |
608804 613206 613480 |
| GJC3 | 7q22.1 | Cx30.2 | ||
| GJD2 | 15q14 | Cx36 | ||
| GJD3 | 17q21.2 | Cx31.9 | ||
| GJD4 | 10p11.21 | Cx40.1 | ||
| GJE1 | 6q24.1 | Cx23 |
2.1. Cx26 mutations cause non-syndromic deafness, syndromic deafness with skin disease, or skin disease
Mutations in the GJB2 gene encoding Cx26 are common in humans, with carrier frequencies in unaffected individuals reaching 2–4% in several populations [26–29]. Most of the Cx26 mutations result in non-syndromic deafness [30, 31] which can be autosomal recessive (DFNB1A) or autosomal dominant (DFNA3A). Non-syndromic deafness is a partial or total loss of hearing that is not associated with other pathologies, and occurs in ~1 in 1000 newborn children. Cx26 mutations contribute to ~50% of genetic deafness and hearing loss can range from mild to profound, be stable or progressive, and impact different sound frequencies [32, 33]. An abundance of data suggests that either partial or total loss of function mutations in Cx26 contribute to non-syndromic deafness [34, 35]. In addition to non-syndromic hearing loss, five syndromic forms of deafness that include skin disease are caused to mutations in Cx26.
The syndromic deafness disorders are very rare, and can be divided into two broad groups. The first group includes Bart-Pumphrey syndrome (BPS) [36], Vohwinkel syndrome (VS) [37], and palmoplantar keratoderma with deafness (PPK) [38]. In addition to hearing loss and palmoplantar keratoderma (thickening of the skin on the palms and soles), BPS patients typically have leukonychia (white discoloration of the nails) and knuckle pads (verrucous growths on the knuckles) Skin abnormalities generally become noticeable during childhood, while hearing loss is present from birth [39]. Individuals affected with VS have hearing loss and thick, honeycomb-like calluses on the palms and soles that appear in infancy or early childhood. They may also show starfish-shaped patches of thickened skin on the tops of their digits or knees. VS patients develop constrictive bands of abnormal fibrous tissue around their fingers and toes that cut off the circulation to the digits leading to pseudoainhum (auto-amputation) [40]. Individuals with PPK have hearing loss and develop unusually thick skin on the palms of the hands and soles of the feet beginning in childhood with no other ectodermal abnormalities [41]. These three disorders have overlapping spectrums of both clinical similarities and specific pathogenic Cx26 mutations, and may represent phenotypic heterogeneity within a single broader genetic disorder [42].
Hystrix-like ichthyosis deafness syndrome (HID) [43] and Keratitis-ichthyosis-deafness (KID) syndrome [44, 45] make up the second group. HID is characterized by ichthyosis (dry, scaly skin) that can appear thick and spiky, resembling porcupine (hystrix) quills and profound hearing loss. Skin abnormalities are present at birth, become progressively worse, and eventually cover much of the body. HID patients may also have scarring alopecia (hair loss) [46, 47]. KID syndrome patients have hearing loss, keratitis (inflammation of the cornea) that can cause photophobia, scarring, and vison loss. They also have palmoplantar keratoderma, in addition to erythrokeratoderma (reddened skin) and ichthyosis (dry scaly skin). Partial alopecia is a common feature, frequently affecting the eyebrows and eyelashes. Squamous cell carcinoma occurs in ~15% of patients [48, 49]. Interestingly, only one causative mutation for HID syndrome has been identified, Cx26-D50N [43], which is also the most frequent Cx26 mutation found in KID patients [50]. Thus, HID may be a phenotypic variant of KID syndrome, with the most prominent difference being the absence of keratitis [42, 51]. Among KID patients, a strong genotype-phenotype correlation has emerged [52, 53]. Cx26-D50N patients live into adulthood, although they have vison loss from keratitis and are at high risk for developing squamous cell carcinomas [50, 54]. In contrast, ~10% of all KID patients die from septic complications in early childhood, and the most frequent mutations in this cohort are Cx26-G45E or Cx26-A88V [55–61].
The final Cx26 disorder is Porokeratotic eccrine ostial and dermal duct nevus (PEODDN), an epidermal nevus (an overgrowth of cells in a patch of skin) that follows the developmental path of ectodermal precursor cells, known as the lines of Blaschko [62]. PEODDN is caused by somatic mutations in Cx26 within these precursors, producing linear bands of diseased keratinocytes. The somatic mutations identified are the same as the germline Cx26 mutations causing KID syndrome [63, 64]. This suggested that PEODDN is a mosaic form of the epidermal features of KID syndrome caused by somatic mutations affecting only a subset of keratinocytes [65].
2.2. Cx30 mutations cause non-syndromic hearing loss, or skin disease
Mutations in the human GJB6 gene encoding Cx30 are less common than those in GJB2, but also contribute to non-syndromic deafness and skin disease. The first Cx30 mutation was found in a family with autosomal dominant (DFNA3B) middle-to-high frequency hearing loss [66]. This finding was consistent with the fact that the Cx26 and Cx30 genes share extensive sequence identity, are located close to each other on Chromosome 13, and both lie within the DFNA3/DFNB1 locus. It had been observed that a sizable fraction of deaf DFNB1 patients had only 1 mutant Cx26 allele at the GJB2 locus, and that additional DFNB1-linked familial cases with no Cx26 mutations had also been reported. It was then demonstrated that many of these patients were compound heterozygous for a mutation in one copy of Cx26 and a deletion in one copy of the GJB6 gene encoding Cx30. Two patients were homozygous for the Cx30 mutation, which was the second most frequent DFNB1 mutation in the Spanish population, and had no Cx26 mutations. Thus, mutations in Cx30 could contribute to autosomal recessive deafness (DFNB1B) by themselves [67], or cause deafness with a digenic pattern of inheritance in association with Cx26 [68, 69].
Mutations in Cx30 also cause the autosomal dominant disease ectodermal dysplasia 2, Clouston type, also known as Clouston syndrome [70, 71]. The main features of Clouston syndrome are thickened dystrophic nails, hair defects that include brittleness, short wiry hair, or total alopecia, and moderate to severe palmoplantar keratoderma [72]. In rare cases, the backs of the hands and feet can develop enlarged eccrine syringofibroadenomas, a benign eccrine tumor [73].
2.3. Cx30.3 mutations cause skin disease
Mutations in the GJB4 gene encoding Cx30.3 cause erythrokeratodermia variabilis et progressiva (EKVP), a skin disorder that presents at birth, or appears during infancy [74, 75]. EKVP is characterized by areas of hyperkeratosis (rough, thickened skin) that can be widespread over the body or limited to a small area, but are generally symmetric and fixed. Transient erythematous patches (red areas with increased blood flow in superficial capillaries) are a second major feature of EKVP. Approximately half of the patients also have palmoplantar keratoderma [74, 76]. In addition to being caused by Cx30.3 mutations, EKVP can also result from mutations in the GJB3 and GJA1 connexin genes encoding Cx31 and Cx43, respectively [77, 78]. Thus, in the case of EKVP, mutations in any one of three independent connexin genes can result in the same clinically defined disease [79].
2.4. Cx31 mutations cause nonsyndromic hearing loss, or skin disease
Like many of the other B-group connexins described thus far, mutations in the human GJB3 gene encoding Cx31 also contribute to non-syndromic deafness and skin disease. Cx31 mutations were initially found in two families with autosomal dominant (DFNA2B) progressive high frequency hearing loss [80]. As described above for Cx30, digenic inheritance of nonsyndromic deafness caused by mutations in the GJB2 and GJB3 genes was also found in 3 out of 108 Chinese patients with autosomal recessive deafness and only 1 mutant copy of Cx26. The unaffected parents were heterozygous for 1 of the mutant Cx26 or Cx31 alleles, consistent with a digenic mode of inheritance [81]. One of the heterozygous Cx31 mutations has also been found in a deaf Korean patient with a heterozygous Cx26 mutation [82]. Thus, mutations in Cx31 can contribute to autosomal dominant deafness (DFNA2B) by themselves, or cause deafness with a digenic pattern of inheritance in association with Cx26.
In addition to autosomal dominant or digeneic hearing loss, mutations in Cx31 can also cause the same skin disorder, erythrokeratodermia variabilis et progressive (EKVP), as described above for Cx30.3 [78].
2.5. Cx32 mutations cause peripheral neuropathy
X-linked Charcot-Marie-Tooth disease (CMTX1) was the first human genetic disorder linked to a connexin, and is caused by mutations in the GJB1 gene encoding Cx32 [83, 84]. Charcot-Marie-Tooth (CMT) disease is a clinically and genetically heterogeneous group of hereditary motor and sensory peripheral neuropathies which affects ~1 in 2500 people. CMTX1 is the second most common form of CMT, comprising ~5% of all CMT cases and more than 400 distinct Cx32 mutations have been identified to date [85, 86]. CMTX1 is a demyelinating neuropathy characterized by severely reduced motor nerve conduction velocity, and is considered to be a X-linked dominant, or X-linked intermediate disorder, as hemizygous males are usually more severely affected than heterozygous females. The symptoms of CMTX1 include progressive weakness and atrophy in distal leg muscles with onset in early childhood in affected males. Over time, patients develop foot abnormalities, abnormal gait, diminished or absent reflexes, sensory loss in distal limbs, and the thenar muscles of the hands become atrophied [86]. Studies of large cohorts of patients have suggested that clear genotype-phenotype correlations for Cx32 and CMTX1 remain enigmatic, despite the large number of mutations identified, the relatively frequent occurrence of the disease and the 24 year time span that has elapsed since the first mutations were identified [87, 88].
2.6. Cx40 mutations cause atrial fibrillation or standstill
Heterozygous somatic, or germline, mutations in the GJA5 gene encoding Cx40 cause familial atrial fibrillation-11 (ATFB11) [89–93]. ATFB11 is characterized by uncoordinated electrical activity in the atria of the heart, causing the heartbeat to become fast and irregular, in addition to increasing the risk of stroke and sudden death [94]. Gap junctions in the atrial myocardium contain predominately Cx40, with smaller amounts of Cx43 and Cx45 [95, 96].
Cx40 mutations can also contribute familial atrial standstill (ATRST1) with a digenic pattern of inheritance with mutations in the sodium voltage-gated channel alpha subunit 5 (SCN5A) gene [97]. ATRST1 is a rare condition characterized by the absence of electrical and mechanical activity in the atria that can be persistent or transient, as well as diffuse or partial. About half of the patients experience syncope (fainting) [98].
2.7. Cx43 mutations cause development disorders, or skin disease
Mutations in the GJA1 gene encoding Cx43 cause a variety of rare genetic diseases. Most mutations cause autosomal dominant oculodentodigital dysplasia (ODDD) [99, 100], which can also be inherited in an autosomal recessive pattern [101]. ODDD is a complex developmental disorder characterized by syndactaly (webbing of skin between fingers), microphthalmia (small eyes), craniofacial and dental abnormalities. In addition, some patients display microcephaly (small head), hypotrichosis (sparse hair growth), brittle nails, syndactyly of the toes, and a cleft palate [99, 102]. In exceptional cases, patients may also have with palmoplantar keratoderma [103]. In some patients with Cx43 mutations, syndactyly (type III) is the most prominent feature, and other typical ODDD manifestations are greatly reduced, or absent. Thus, it has been suggested that isolated syndactyly type III, which is also caused by Cx43 mutations, and ODDD may not be separate genetic conditions, but instead represent a broader spectrum of phenotypes within a single disease [104].
Cx43 mutations can also cause autosomal recessive craniometaphyseal dysplasia (CMDR) [105]. CMDR is a rare condition, evident at birth, which is characterized by progressive thickening of bones in the skull and abnormalities at the ends of long bones of the limbs. Patients display distinctive facial features including a prominent forehead, wide nasal bridge, wide-set eyes and a prominent jaw. X-rays reveal unusually shaped long bones, with wider metaphyses (ends), particularly in the legs. The typical ocular, digital, or dental features of ODDD are absent in CMDR patients [106, 107].
Cx43 mutations can also be linked to epidermal disorders such as palmoplantar keratoderma and congenital alopecia-1 (PPKCA1, also called keratoderma-hypotrichosis-leukonychia totalis) [108]. PPKCA1 is a rare autosomal dominant disorder characterized by severe hyperkeratosis, congenital alopecia and leukonychia [109].
Finally, mutations in Cx43 can also cause the same skin disorder, erythrokeratodermia variabilis et progressive (EKVP), as described above for Cx30.3 and Cx31 [77]. In Cx43-associated EKVP, patients had two additional features, prominent white lunulae and periorificial darkening, which had not been previously reported in the Cx30.3 and Cx31 linked cases.
2.8 Cx46 mutations cause cataract
Mutations in the GJA3 gene encoding Cx46 have been found to cause autosomal dominant cataract (CTRCT14), multiple types of which have been described as zonular pulverulent, posterior polar, nuclear coralliform, embryonal nuclear, and Coppock-like [110]. Bilateral opacities are diagnosed at a median age of 5 years, but can appear as early as 6 months. Opacities affect the lens nucleus and are often surrounded by dust-like precipitates in the lens cortex. Many patients require cataract surgery in childhood [111, 112]. No other ocular abnormalities have been reported. Mutations in connexins currently account for ~16% of all genetic cataract where the causative gene is known [113, 114].
2.9 Cx47 mutations cause leukodystrophy, spastic paraplegia, or lymphedema
Autosomal recessive mutations in the GJC2 gene encoding Cx47 cause hypomyelinating leukodystrophy-2 (HLD2, also called Pelizaeus-Merzbacher like disease) [115, 116]. HLD2 is an autosomal recessive leukodystrophy characterized by early-onset nystagmus (repetitive, uncontrolled eye movements), delayed motor development, progressive spasticity, ataxia (uncoordinated voluntary muscle movement), and diffuse leukodystrophy (degeneration of myelin) [117, 118].
Autosomal recessive mutations in Cx47 can also cause spastic paraplegia type 44 (SPG44) [119]. SPG44 is an exceptionally rare form of hereditary spastic paraplegia characterized by a milder phenotype than that of HLD2. Nystagmus is not present, and patients display a late-onset, slowly progressive spastic paraplegia associated with mild ataxia, dysarthria (slurred speech), dysmetria (difficulty judging distance, or scale) and mild cognitive impairment [118].
In addition, hereditary lymphedema type IC (LMPH1C) can be caused by autosomal dominant mutations in Cx47 [120, 121]. LMPH1C is caused by anatomic or functional defects in the lymphatic system, resulting in chronic swelling. There may be accompanying nail and skin changes, such as nail dysplasia or papillomatosis. Onset is often at birth, or early childhood, but can also occur later. Swelling occurs most frequently in the legs, but may also be manifest in the arms, face, genitalia and torso [122].
2.10 Cx50 mutations cause cataract
Similar to the situation described above for Cx46, mutations in the GJA8 gene encoding Cx50 have been found to cause multiple types of autosomal dominant cataract (CTRCT1) [123, 124]. These can be congenital, zonular pulverulent, nuclear progressive, nuclear pulverulent, stellate nuclear, nuclear total, total, and posterior subcapsular. In some cases, cataract caused by Cx50 mutations is associated with microcornea [114, 125]. Bilateral lens opacities may be first seen at birth, or early childhood, and often progress. The embryonic and fetal lens nuclei are usually affected and diffuse cortical opacities may appear in some patients. Of historical note, the Cx50 cataract locus was the very first human genetic disease to be assigned to an autosome [126, 127].
3. Functional consequences of connexin mutations
As described in section 2, Cx26 and Cx43 have the greatest number of distinct disorders linked to them, while Cx32 was the first Cx disease gene identified and has the largest number of known mutations. As a consequence, much of the mechanistic research has emphasized mutations in these three connexins. In the case of Cx43, it has been determined that GJA1 gene mutations cause ODDD by at least ten distinct mechanisms [100, 128]. In contrast, for Cx32, most of the GJB2 mutations fail to form functional channels, suggesting a consistent loss of function mechanism [86, 129, 130]. For brevity, we will focus here on the functional consequences of mutations in Cx26. Other recent reviews have examined the various proposed mechanisms of disease caused by mutations in Cx32 and Cx43, and a similar spectrum of functional consequences are found across many of the different connexin disorders. Cx26 mutations occur with the highest frequently in humans, with a vast majority resulting in non-syndromic deafness without skin pathology [30, 31].
3.1. Loss of function mutations
The majority of recessive nonsyndromic deafness Cx26 mutations found in patients are deletions, truncations, or frameshifts suggesting that total loss of gap junction and/or hemichannel function produces hearing loss. In support of this idea, many of the single amino acid substitution mutations are also devoid of channel activity [34, 35]. In the cochlea, Cx26 is expressed in the supporting cells of the Organ of Corti surrounding the sensory hair cells, and in the lateral wall containing the stria vascularis [131–135]. The stria vascularis produces the endolymph and generates the endocochlear potential, which is critical for sound transduction by the hair cells. Gap junctions in the stria vascularis may contribute to K+ transport and the generation of the endocochlear potential. However, within the sensory epithelium, the contribution of Cx26 gap junctions to K+ transport does not appear to be critical [136–140]. Instead, Cx26 gap junctions are more likely to transfer nutrients and signaling molecules like glucose or IP3, which, in turn, help maintain sensory epithelial integrity and hair cell function [141–144]. There is also expression of Cx26 in basolateral and apical regions of supporting cells in regions devoid of cell-cell contact, suggesting that hemichannels may communicate with the perilymphatic and endolymphatic compartments [145]. The absence of skin disorders in patients with loss-of-function mutations suggests human skin does not need functional Cx26, either during development or for normal homeostasis, and that syndromic deafness is a Cx26 gain-of-function disease [51, 53, 146].
3.2. Gain of function mutations
Pathogenesis in syndromic deafness and skin disease caused by dominant Cx26 mutations may be caused by several mechanisms. Two gain of function changes have emerged for Cx26 mutations that cause PPK and deafness, the formation of active heteromeric hemichannels with Cx43, and trans-dominant inhibition of gap junction channels formed by other keratinocyte connexins. For six different Cx26 PPK mutations, it has been shown that they all lacked intrinsic gap junction channel activity when expressed as homotypic channels. In addition, five of the six displayed dominant-negative inhibition of coupling when co-expressed with wild-type Cx26, suggesting a mechanism for their contribution to hearing loss. When co-expressed with wild-type Cx30 or Cx43, all of these mutations also resulted in trans-dominant inhibition of gap junction conductance [147–151]. Thus, one common gain of function mechanism for Cx26 skin disease mutations linked to PPK is trans-dominant inhibition of other keratinocyte connexins, i.e. where the non-functional mutated connexin is able to inhibit the function of another wild-type connexin. In addition, one recent study demonstrated the ability of two Cx26 PPK mutations to form functional heteromeric hemichannels, even though both mutations failed to form functional homomeric hemichannels when expressed alone. Co-expression of Cx43 with either of the Cx26 mutants showed a significant increase in hemichannel activity when compared to Cx43 alone. Co-immunoprecipitation confirmed that Cx43 could be pulled down with either Cx26 PPK mutation, confirming the formation of heteromeric hemichannels [150].
The cause of pathogenesis caused by dominant Cx26 mutations linked to KID syndrome is thought to be due to the formation of aberrantly functioning hemichannels. Work from several labs has confirmed that dysregulation of mutant Cx26 hemichannels is a common gain of function, seen for all known KID mutations, with one exception [52, 152–159]. The lone exception was the Cx26-S17F mutation, which a recent study has shown results in increased hemichannel activity in the presence of wild-type Cx43, as described for some of the PPK mutations above [160]. Unlike the PPK mutations, several of the Cx26 KID mutations can also form functional gap junction channels [153–155].
The majority of KID syndrome mutants cluster in protein domains that contribute to the aqueous pore of the hemichannel, and that form the structural elements that regulate gating [53]. Thus, many KID mutations show altered voltage gating and reduced inhibition by extracellular Ca2+, which results in increased hemichannel activity. KID mutant residues identified as pore-lining using cysteine scanning result in hemichannels with altered unitary conductance and permeability to Ca2+, and likely other signaling molecules such as ATP [155, 156, 161]. Purinergic signaling plays an important role in cochlear function [162] and hemichannel-mediated release of Ca2+ and/or ATP has been implicated in proper cochlear development and sustaining auditory function [163, 164]. Ca2+ is also an important regulator of keratinocyte differentiation and the epidermis maintains a Ca2+ gradient, with the highest levels in the granular layer and an abrupt reduction in the cornified layer [165]. The altered Ca2+ permeation/gating properties of KID hemichannels would almost certainly alter this gradient and affect keratinocyte differentiation (Figure 2). Studies of mouse models of KID syndrome (Figure 1) have supported the hypothesis that mutant hemichannels disrupt calcium homeostasis in vivo. Transgenic mice expressing Cx26 KID mutations develop skin disease and display increased hemichannel activity in their keratinocytes [166, 167]. Examination of the epidermal Ca2+ gradient in the skin of these mice showed that it was elevated in the cornified layer. This change in the Ca2+ gradient was correlated with altered lipid secretion and defects in the epidermal water barrier [168].
Figure 2.
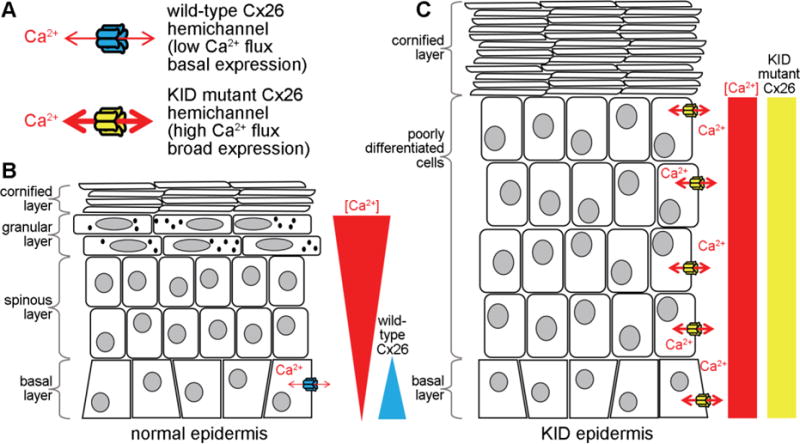
Disruption of the epidermal Ca2+ gradient by mutant Cx26 hemichannels in KID syndrome. (A). Cx26 KID mutant hemichannels have a higher net flux of Ca2+ and broader expression pattern than wild-type Cx26. (B). In normal epidermis, Cx26 is restricted to cells in the basal layer, and there is a gradient of Ca2+ with the highest levels in the granular layer, and low levels in the basal layer. In KID epidermis, mutant Cx26 expression expands across the epithelium, increasing Ca2+ flux and eliminating the normal Ca2+ gradient. The loss of the Ca2+ gradient disrupts the normal regulation of keratinocyte proliferation (favored in low Ca2+) and differentiation (favored in high Ca2+) resulting in a greatly thickened epidermis and a decrease in differentiated cells.
Figure 1.
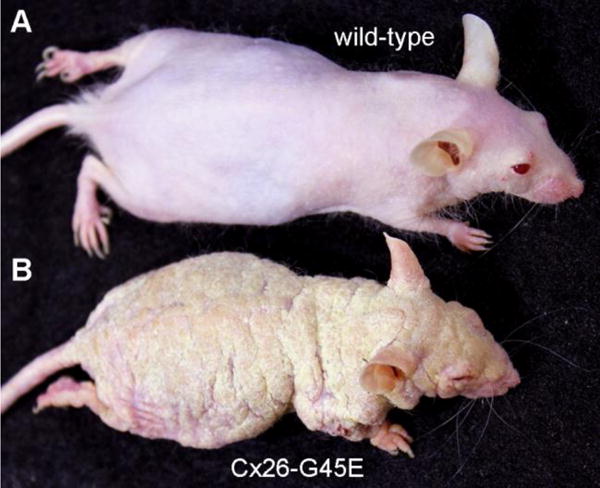
A mouse model of KID syndrome. (A). A wild-type mouse of the SKH1 hairless strain. (B). An inducible transgenic SKH1 mouse expressing the Cx26-G45E mutation in keratinocytes develops the epidermal features of KID syndrome. See [166] for details.
3.3. Unifying themes in connexin congenital disorders
A growing body of evidence suggests that many, if not a majority, of the dominant connexin mutations associated with human genetic diseases consistently acquire unique gain of function activities [94, 128]. Eleven of the twenty-eight connexinopathies are skin diseases that are caused by mutations in five different connexin genes [42]. In the case of Cx26, increased hemichannel activity has been compellingly linked to mutations causing both KID syndrome, and more recently PPK.
Accumulating evidence suggests that dysregulated hemichannels may also play a pathogenic role in some of the other connexin skin disorders. For example, studies of Cx30 mutations associated with Clouston syndrome have provided evidence that increased hemichannel function may contribute to this skin diseases. Expression of these mutants in cultured cells induced large voltage-activated currents and caused cell death, consistent with formation of active connexin hemichannels. In addition, higher rates of ATP leakage were measured in cells expressing the mutant connexins than controls, implying that hemichannel mediated ATP release may contribute to Clouston syndrome pathology [169]. In a similar fashion, Cx31 mutations linked to EKVP were shown to result in increased hemichannel activity, ATP leakage, and necrotic cell death when expressed in transfected cells [170].
Additional support for abberant hemichannels being a general pathological mechanism contributing to skin disease came from analysis of a Cx43 mutation (Cx43-G8V) linked to PPKCA1. Expression of the Cx43-G8V mutant in transfected cells resulted in greatly increased whole cell membrane current, Ca2+ influx, and cell death when compared to wild-type Cx43, all consistent with increased hemichannel activity due to the Cx43-G8V mutation [108]. Consistent with these observations, we have found that expression of Cx43-G8V in single Xenopus oocytes (Figure 3, our unpublished data) results in readily detectable single channel currents that have a unitary conductance (215 pS) in accordance with what has been previously reported for wild-type Cx43 hemichannels [171].
Figure 3.
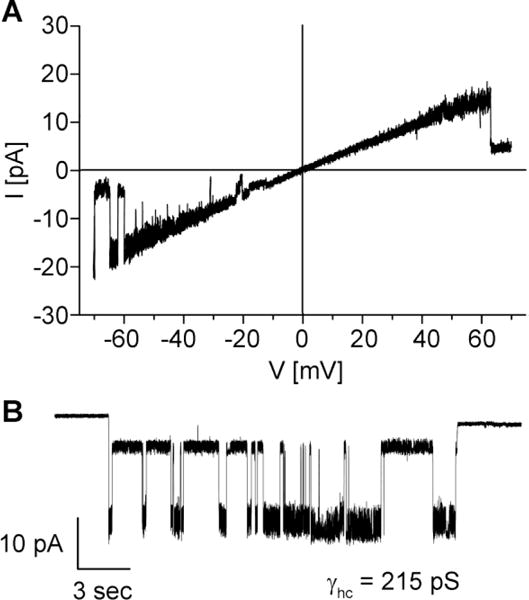
Hemichannel activity of the Cx43-G8V mutation causing PPKCA1 [108]. (A). A recording from a cell attached patch containing a single Cx43-G8V hemichannel obtained in response to an 8s voltage ramp applied between −70 and +70 mV. The leak current was subtracted offline. The hemichannel unitary conductance (γhc) was ~215 pS. (B). Another recording from a cell attached patch containing a single Cx43-G8V hemichannel obtained in response to a constant voltage step of −70 mV. At this voltage a sub conductance state was also evident. All currents were filtered at 1 kHz, data were acquired at 5 kHz and the pipette solution contained 140 mM KCl.
Taken together, these data suggest that there may be a common pathological mechanism across the diverse connexin mediated skin disorders. Thus far, the most consistent gain of function is the formation of dysregulated hemichannels that either increase ATP leakage, or disrupt Ca2+ signaling within keratinocytes. Further study of disease causing mutations in expression systems and transgenic mice will continue to refine our understanding of the epidermal pathophysiology.
4. Summary
Important roles for connexin channels in the generation and maintenance of tissue homeostasis has become increasingly evident through the continuing discovery of human disorders caused by mutations in connexin genes. Of note, the disease phenotypes in many cases are highly restricted, despite the fact that the connexins can be widely expressed. This aspect of connexin-mediated pathology is most evident in disorders like non-syndromic deafness and CMTX1 in which pathology is highly restricted despite the expression of the mutated Cx26 and Cx32 genes across many organ systems. These cases may reflect the possibility that compensatory mechanisms can be at play in a tissue-specific manner. On the other hand, confinement of phenotype to lens cataract for Cx46 and Cx50 mutations is consistent with their expression pattern, both are largely restricted to this organ. Finally, one of the remaining major challenges will be to discern the precise mechanism(s) whereby gene mutation leads to tissue pathology. For connexin channels, this likely comes down to identifying the biological signaling molecules that are transmitted through these channels and how this transmission is altered in mutated channels within an affected tissue.
Highlights.
Twenty-eight human genetic diseases result from connexin mutations
Mutations in two connexin genes encoding Cx26 and Cx43 cause fourteen disorders
Both gap junction and/or hemichannel function can be compromised by mutations
Diseases are usually restricted, despite many connexins being widely expressed
Acknowledgments
Work in our laboratories is supported by NIH grants R01 AR059505 (NIAMS), R01 EY026911, R01 EY013163 (NEI) & R01 GM054179 (NIGMS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- 2.Vinken M. Introduction: connexins, pannexins and their channels as gatekeepers of organ physiology. Cell Mol Life Sci. 2015;72:2775–2778. doi: 10.1007/s00018-015-1958-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris AL. Emerging issues of connexin channels: biophysics fills the gap. Q Rev Biophys. 2001;34:325–472. doi: 10.1017/s0033583501003705. [DOI] [PubMed] [Google Scholar]
- 4.Willecke K, Eiberger J, Degen J, Eckardt D, Romualdi A, Guldenagel M, Deutsch U, Sohl G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem. 2002;383:725–737. doi: 10.1515/BC.2002.076. [DOI] [PubMed] [Google Scholar]
- 5.Das Sarma J, Wang F, Koval M. Targeted gap junction protein constructs reveal connexin-specific differences in oligomerization. J Biol Chem. 2002;277:20911–20918. doi: 10.1074/jbc.M111498200. [DOI] [PubMed] [Google Scholar]
- 6.Laird DW. Life cycle of connexins in health and disease. Biochem J. 2006;394:527–543. doi: 10.1042/BJ20051922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans WH, De Vuyst E, Leybaert L. The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem J. 2006;397:1–14. doi: 10.1042/BJ20060175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Retamal MA, Reyes EP, Garcia IE, Pinto B, Martinez AD, Gonzalez C. Diseases associated with leaky hemichannels. Front Cell Neurosci. 2015;9:267. doi: 10.3389/fncel.2015.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanaporis G, Mese G, Valiuniene L, White TW, Brink PR, Valiunas V. Gap junction channels exhibit connexin-specific permeability to cyclic nucleotides. J Gen Physiol. 2008;131:293–305. doi: 10.1085/jgp.200709934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mese G, Valiunas V, Brink PR, White TW. Connexin26 deafness associated mutations show altered permeability to large cationic molecules. Am J Physiol Cell Physiol. 2008;295:C966–974. doi: 10.1152/ajpcell.00008.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bukauskas FF, Verselis VK. Gap junction channel gating. Biochim Biophys Acta. 2004;1662:42–60. doi: 10.1016/j.bbamem.2004.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris AL. Connexin channel permeability to cytoplasmic molecules. Prog Biophys Mol Biol. 2007;94:120–143. doi: 10.1016/j.pbiomolbio.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slavi N, Rubinos C, Li L, Sellitto C, White TW, Mathias R, Srinivas M. Connexin 46 (cx46) gap junctions provide a pathway for the delivery of glutathione to the lens nucleus. J Biol Chem. 2014;289:32694–32702. doi: 10.1074/jbc.M114.597898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plum A, Hallas G, Magin T, Dombrowski F, Hagendorff A, Schumacher B, Wolpert C, Kim J, Lamers WH, Evert M, Meda P, Traub O, Willecke K. Unique and shared functions of different connexins in mice. Curr Biol. 2000;10:1083–1091. doi: 10.1016/s0960-9822(00)00690-4. [DOI] [PubMed] [Google Scholar]
- 15.White TW. Unique and redundant connexin contributions to lens development. Science. 2002;295:319–320. doi: 10.1126/science.1067582. [DOI] [PubMed] [Google Scholar]
- 16.White TW. Nonredundant gap junction functions. News Physiol Sci. 2003;18:95–99. doi: 10.1152/nips.01430.2002. [DOI] [PubMed] [Google Scholar]
- 17.Kretz M, Euwens C, Hombach S, Eckardt D, Teubner B, Traub O, Willecke K, Ott T. Altered connexin expression and wound healing in the epidermis of connexin-deficient mice. J Cell Sci. 2003;116:3443–3452. doi: 10.1242/jcs.00638. [DOI] [PubMed] [Google Scholar]
- 18.Wiszniewski L, Limat A, Saurat JH, Meda P, Salomon D. Differential expression of connexins during stratification of human keratinocytes. J Invest Dermatol. 2000;115:278–285. doi: 10.1046/j.1523-1747.2000.00043.x. [DOI] [PubMed] [Google Scholar]
- 19.Segretain D, Falk MM. Regulation of connexin biosynthesis, assembly, gap junction formation, and removal. Biochim Biophys Acta. 2004;1662:3–21. doi: 10.1016/j.bbamem.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 20.Bai D, Wang AH. Extracellular domains play different roles in gap junction formation and docking compatibility. Biochem J. 2014;458:1–10. doi: 10.1042/BJ20131162. [DOI] [PubMed] [Google Scholar]
- 21.Koval M, Molina SA, Burt JM. Mix and match: investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 2014;588:1193–1204. doi: 10.1016/j.febslet.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lagree V, Brunschwig K, Lopez P, Gilula NB, Richard G, Falk MM. Specific amino-acid residues in the N-terminus and TM3 implicated in channel function and oligomerization compatibility of connexin43. J Cell Sci. 2003;116:3189–3201. doi: 10.1242/jcs.00604. [DOI] [PubMed] [Google Scholar]
- 23.White TW, Bruzzone R, Wolfram S, Paul DL, Goodenough DA. Selective interactions among the multiple connexin proteins expressed in the vertebrate lens: the second extracellular domain is a determinant of compatibility between connexins. J Cell Biol. 1994;125:879–892. doi: 10.1083/jcb.125.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karademir LB, Aoyama H, Yue B, Chen H, Bai D. Engineered Cx26 variants established functional heterotypic Cx26/Cx43 and Cx26/Cx40 gap junction channels. Biochem J. 2016;473:1391–1403. doi: 10.1042/BCJ20160200. [DOI] [PubMed] [Google Scholar]
- 25.Garcia IE, Prado P, Pupo A, Jara O, Rojas-Gomez D, Mujica P, Flores-Munoz C, Gonzalez-Casanova J, Soto-Riveros C, Pinto BI, Retamal MA, Gonzalez C, Martinez AD. Connexinopathies: a structural and functional glimpse. BMC Cell Biol. 2016;17(Suppl 1):17. doi: 10.1186/s12860-016-0092-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green GE, Scott DA, McDonald JM, Woodworth GG, Sheffield VC, Smith RJ. Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. JAMA. 1999;281:2211–2216. doi: 10.1001/jama.281.23.2211. [DOI] [PubMed] [Google Scholar]
- 27.Han SH, Park HJ, Kang EJ, Ryu JS, Lee A, Yang YH, Lee KR. Carrier frequency of GJB2 (connexin-26) mutations causing inherited deafness in the Korean population. J Hum Genet. 2008;53:1022–1028. doi: 10.1007/s10038-008-0342-7. [DOI] [PubMed] [Google Scholar]
- 28.Lucotte G, Mercier G. Meta-analysis of GJB2 mutation 35delG frequencies in Europe. Genet Test. 2001;5:149–152. doi: 10.1089/109065701753145646. [DOI] [PubMed] [Google Scholar]
- 29.Taniguchi M, Matsuo H, Shimizu S, Nakayama A, Suzuki K, Hamajima N, Shinomiya N, Nishio S, Kosugi S, Usami S, Ito J, Kitajiri S. Carrier frequency of the GJB2 mutations that cause hereditary hearing loss in the Japanese population. J Hum Genet. 2015;60:613–617. doi: 10.1038/jhg.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, Leigh IM. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–83. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- 31.Denoyelle F, Lina-Granade G, Plauchu H, Bruzzone R, Chaib H, Levi-Acobas F, Weil D, Petit C. Connexin 26 gene linked to a dominant deafness. Nature. 1998;393:319–320. doi: 10.1038/30639. [DOI] [PubMed] [Google Scholar]
- 32.Gualandi F, Martini A, Calzolari E. Progress in understanding GJB2-linked deafness. Community Genet. 2003;6:125–132. doi: 10.1159/000078156. [DOI] [PubMed] [Google Scholar]
- 33.Snoeckx RL, Huygen PL, Feldmann D, Marlin S, Denoyelle F, Waligora J, Mueller-Malesinska M, Pollak A, Ploski R, Murgia A, Orzan E, Castorina P, Ambrosetti U, Nowakowska-Szyrwinska E, Bal J, Wiszniewski W, Janecke AR, Nekahm-Heis D, Seeman P, Bendova O, Kenna MA, Frangulov A, Rehm HL, Tekin M, Incesulu A, Dahl HH, du Sart D, Jenkins L, Lucas D, Bitner-Glindzicz M, Avraham KB, Brownstein Z, del Castillo I, Moreno F, Blin N, Pfister M, Sziklai I, Toth T, Kelley PM, Cohn ES, Van Maldergem L, Hilbert P, Roux AF, Mondain M, Hoefsloot LH, Cremers CW, Lopponen T, Lopponen H, Parving A, Gronskov K, Schrijver I, Roberson J, Gualandi F, Martini A, Lina-Granade G, Pallares-Ruiz N, Correia C, Fialho G, Cryns K, Hilgert N, Van de Heyning P, Nishimura CJ, Smith RJ, Van Camp G. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet. 2005;77:945–957. doi: 10.1086/497996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White TW. Functional analysis of human Cx26 mutations associated with deafness. Brain Res Brain Res Rev. 2000;32:181–183. doi: 10.1016/s0165-0173(99)00079-x. [DOI] [PubMed] [Google Scholar]
- 35.Zhao HB, Kikuchi T, Ngezahayo A, White TW. Gap junctions and cochlear homeostasis. J Membr Biol. 2006;209:177–186. doi: 10.1007/s00232-005-0832-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richard G, Brown N, Ishida-Yamamoto A, Krol A. Expanding the phenotypic spectrum of Cx26 disorders: Bart-Pumphrey syndrome is caused by a novel missense mutation in GJB2. J Invest Dermatol. 2004;123:856–863. doi: 10.1111/j.0022-202X.2004.23470.x. [DOI] [PubMed] [Google Scholar]
- 37.Maestrini E, Korge BP, Ocana-Sierra J, Calzolari E, Cambiaghi S, Scudder PM, Hovnanian A, Monaco AP, Munro CS. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel’s syndrome) in three unrelated families. Hum Mol Genet. 1999;8:1237–1243. doi: 10.1093/hmg/8.7.1237. [DOI] [PubMed] [Google Scholar]
- 38.Richard G, White TW, Smith LE, Bailey RA, Compton JG, Paul DL, Bale SJ. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet. 1998;103:393–399. doi: 10.1007/s004390050839. [DOI] [PubMed] [Google Scholar]
- 39.Bart RS, Pumphrey RE. Knuckle pads, leukonychia and deafness. A dominantly inherited syndrome. N Engl J Med. 1967;276:202–207. doi: 10.1056/NEJM196701262760403. [DOI] [PubMed] [Google Scholar]
- 40.Vohwinkel KH. Keratoma hereditarium mutilans. Archiv Fur Dermatologie Und Syphilis. 1929;158:354–364. [Google Scholar]
- 41.Sharland M, Bleach NR, Goberdhan PD, Patton MA. Autosomal dominant palmoplantar hyperkeratosis and sensorineural deafness in three generations. J Med Genet. 1992;29:50–52. doi: 10.1136/jmg.29.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lilly E, Sellitto C, Milstone LM, White TW. Connexin channels in congenital skin disorders. Semin Cell Dev Biol. 2016;50:4–12. doi: 10.1016/j.semcdb.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Geel M, van Steensel MA, Kuster W, Hennies HC, Happle R, Steijlen PM, Konig A. HID and KID syndromes are associated with the same connexin 26 mutation. Br J Dermatol. 2002;146:938–942. doi: 10.1046/j.1365-2133.2002.04893.x. [DOI] [PubMed] [Google Scholar]
- 44.van Steensel MA, van Geel M, Nahuys M, Smitt JH, Steijlen PM. A novel connexin 26 mutation in a patient diagnosed with keratitis-ichthyosis-deafness syndrome. J Invest Dermatol. 2002;118:724–727. doi: 10.1046/j.1523-1747.2002.01735.x. [DOI] [PubMed] [Google Scholar]
- 45.Richard G, Rouan F, Willoughby CE, Brown N, Chung P, Ryynanen M, Jabs EW, Bale SJ, DiGiovanna JJ, Uitto J, Russell L. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet. 2002;70:1341–1348. doi: 10.1086/339986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gulzow J, Anton-Lamprecht I. [Ichthyosis hystrix gravior typus Rheydt: an otologic-dermatologic syndrome (author’s transl)] Laryngol Rhinol Otol (Stuttg) 1977;56:949–955. [PubMed] [Google Scholar]
- 47.Traupe H. The Ichthyoses. Springer; Berlin Heidelberg: 1989. Hystrix-like Ichthyosis with Deafness: the HID Syndrome; pp. 193–197. [Google Scholar]
- 48.Skinner BA, Greist MC, Norins AL. The keratitis, ichthyosis, and deafness (KID) syndrome. Arch Dermatol. 1981;117:285–289. [PubMed] [Google Scholar]
- 49.Coggshall K, Farsani T, Ruben B, McCalmont TH, Berger TG, Fox LP, Shinkai K. Keratitis, ichthyosis, and deafness syndrome: a review of infectious and neoplastic complications. J Am Acad Dermatol. 2013;69:127–134. doi: 10.1016/j.jaad.2012.12.965. [DOI] [PubMed] [Google Scholar]
- 50.Mazereeuw-Hautier J, Bitoun E, Chevrant-Breton J, Man SY, Bodemer C, Prins C, Antille C, Saurat JH, Atherton D, Harper JI, Kelsell DP, Hovnanian A. Keratitis-ichthyosis-deafness syndrome: disease expression and spectrum of connexin 26 (GJB2) mutations in 14 patients. Br J Dermatol. 2007;156:1015–1019. doi: 10.1111/j.1365-2133.2007.07806.x. [DOI] [PubMed] [Google Scholar]
- 51.Lee JR, White TW. Connexin-26 mutations in deafness and skin disease. Expert Rev Mol Med. 2009;11:e35. doi: 10.1017/S1462399409001276. [DOI] [PubMed] [Google Scholar]
- 52.Mhaske PV, Levit NA, Li L, Wang HZ, Lee JR, Shuja Z, Brink PR, White TW. The human Cx26-D50A and Cx26-A88V mutations causing keratitis-ichthyosis-deafness syndrome display increased hemichannel activity. Am J Physiol Cell Physiol. 2013;304:C1150–1158. doi: 10.1152/ajpcell.00374.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanchez HA, Verselis VK. Aberrant Cx26 hemichannels and keratitis-ichthyosis-deafness syndrome: insights into syndromic hearing loss. Front Cell Neurosci. 2014;8:354. doi: 10.3389/fncel.2014.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grob JJ, Breton A, Bonafe JL, Sauvan-Ferdani M, Bonerandi JJ. Keratitis, ichthyosis, and deafness (KID) syndrome. Vertical transmission and death from multiple squamous cell carcinomas. Arch Dermatol. 1987;123:777–782. [PubMed] [Google Scholar]
- 55.Haruna K, Suga Y, Oizumi A, Mizuno Y, Endo H, Shimizu T, Hasegawa T, Ikeda S. Severe form of keratitis-ichthyosis-deafness (KID) syndrome associated with septic complications. J Dermatol. 2010;37:680–682. doi: 10.1111/j.1346-8138.2010.00839.x. [DOI] [PubMed] [Google Scholar]
- 56.Jonard L, Feldmann D, Parsy C, Freitag S, Sinico M, Koval C, Grati M, Couderc R, Denoyelle F, Bodemer C, Marlin S, Hadj-Rabia S. A familial case of Keratitis-Ichthyosis-Deafness (KID) syndrome with the GJB2 mutation G45E. Eur J Med Genet. 2008;51:35–43. doi: 10.1016/j.ejmg.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 57.Koppelhus U, Tranebjaerg L, Esberg G, Ramsing M, Lodahl M, Rendtorff ND, Olesen HV, Sommerlund M. A novel mutation in the connexin 26 gene (GJB2) in a child with clinical and histological features of keratitis-ichthyosis-deafness (KID) syndrome. Clin Exp Dermatol. 2011;36:142–148. doi: 10.1111/j.1365-2230.2010.03936.x. [DOI] [PubMed] [Google Scholar]
- 58.Meigh L, Hussain N, Mulkey DK, Dale N. Connexin26 hemichannels with a mutation that causes KID syndrome in humans lack sensitivity to CO2. Elife. 2014;3:e04249. doi: 10.7554/eLife.04249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ogawa Y, Takeichi T, Kono M, Hamajima N, Yamamoto T, Sugiura K, Akiyama M. Revertant mutation releases confined lethal mutation, opening Pandora’s box: a novel genetic pathogenesis. PLoS Genet. 2014;10:e1004276. doi: 10.1371/journal.pgen.1004276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sbidian E, Feldmann D, Bengoa J, Fraitag S, Abadie V, de Prost Y, Bodemer C, Hadj-Rabia S. Germline mosaicism in keratitis-ichthyosis-deafness syndrome: pre-natal diagnosis in a familial lethal form. Clin Genet. 2010;77:587–592. doi: 10.1111/j.1399-0004.2009.01339.x. [DOI] [PubMed] [Google Scholar]
- 61.Esmer C, Salas-Alanis JC, Fajardo-Ramirez OR, Ramirez B, Hua R, Choate K. Lethal Keratitis, Ichthyosis, and Deafness Syndrome Due to the A88V Connexin 26 Mutation. Rev Invest Clin. 2016;68:143–146. [PubMed] [Google Scholar]
- 62.Abell E, Read SI. Porokeratotic eccrine ostial and dermal duct naevus. Br J Dermatol. 1980;103:435–441. doi: 10.1111/j.1365-2133.1980.tb07268.x. [DOI] [PubMed] [Google Scholar]
- 63.Lazic T, Li Q, Frank M, Uitto J, Zhou LH. Extending the phenotypic spectrum of keratitis-ichthyosis-deafness syndrome: report of a patient with GJB2 (G12R) Connexin 26 mutation and unusual clinical findings. Pediatr Dermatol. 2012;29:349–357. doi: 10.1111/j.1525-1470.2011.01425.x. [DOI] [PubMed] [Google Scholar]
- 64.Easton JA, Donnelly S, Kamps MA, Steijlen PM, Martin PE, Tadini G, Janssens R, Happle R, van Geel M, van Steensel MA. Porokeratotic eccrine nevus may be caused by somatic connexin26 mutations. J Invest Dermatol. 2012;132:2184–2191. doi: 10.1038/jid.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Levinsohn JL, McNiff JM, Antaya RJ, Choate KA. A Somatic p.G45E GJB2 Mutation Causing Porokeratotic Eccrine Ostial and Dermal Duct Nevus. JAMA Dermatol. 2015;151:638–641. doi: 10.1001/jamadermatol.2014.5069. [DOI] [PubMed] [Google Scholar]
- 66.Grifa A, Wagner CA, D’Ambrosio L, Melchionda S, Bernardi F, Lopez-Bigas N, Rabionet R, Arbones M, Monica MD, Estivill X, Zelante L, Lang F, Gasparini P. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat Genet. 1999;23:16–18. doi: 10.1038/12612. [DOI] [PubMed] [Google Scholar]
- 67.del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, Menendez I, Moreno F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med. 2002;346:243–249. doi: 10.1056/NEJMoa012052. [DOI] [PubMed] [Google Scholar]
- 68.del Castillo FJ, Rodriguez-Ballesteros M, Alvarez A, Hutchin T, Leonardi E, de Oliveira CA, Azaiez H, Brownstein Z, Avenarius MR, Marlin S, Pandya A, Shahin H, Siemering KR, Weil D, Wuyts W, Aguirre LA, Martin Y, Moreno-Pelayo MA, Villamar M, Avraham KB, Dahl HH, Kanaan M, Nance WE, Petit C, Smith RJ, Van Camp G, Sartorato EL, Murgia A, Moreno F, del Castillo I. A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J Med Genet. 2005;42:588–594. doi: 10.1136/jmg.2004.028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pallares-Ruiz N, Blanchet P, Mondain M, Claustres M, Roux AF. A large deletion including most of GJB6 in recessive non syndromic deafness: a digenic effect? Eur J Hum Genet. 2002;10:72–76. doi: 10.1038/sj.ejhg.5200762. [DOI] [PubMed] [Google Scholar]
- 70.Lamartine J. Towards a new classification of ectodermal dysplasias. Clin Exp Dermatol. 2003;28:351–355. doi: 10.1046/j.1365-2230.2003.01319.x. [DOI] [PubMed] [Google Scholar]
- 71.Lamartine J, Munhoz Essenfelder G, Kibar Z, Lanneluc I, Callouet E, Laoudj D, Lemaitre G, Hand C, Hayflick SJ, Zonana J, Antonarakis S, Radhakrishna U, Kelsell DP, Christianson AL, Pitaval A, Der Kaloustian V, Fraser C, Blanchet-Bardon C, Rouleau GA, Waksman G. Mutations in GJB6 cause hidrotic ectodermal dysplasia. Nat Genet. 2000;26:142–144. doi: 10.1038/79851. [DOI] [PubMed] [Google Scholar]
- 72.Clouston HR. A Hereditary Ectodermal Dystrophy. Can Med Assoc J. 1929;21:18–31. [PMC free article] [PubMed] [Google Scholar]
- 73.Andrade AC, Vieira DC, Harris OM, Pithon MM. Clouston syndrome associated with eccrine syringofibroadenoma. An Bras Dermatol. 2014;89:504–506. doi: 10.1590/abd1806-4841.20142996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Richard G, Brown N, Rouan F, Van der Schroeff JG, Bijlsma E, Eichenfield LF, Sybert VP, Greer KE, Hogan P, Campanelli C, Compton JG, Bale SJ, DiGiovanna JJ, Uitto J. Genetic heterogeneity in erythrokeratodermia variabilis: novel mutations in the connexin gene GJB4 (Cx30.3) and genotype-phenotype correlations. J Invest Dermatol. 2003;120:601–609. doi: 10.1046/j.1523-1747.2003.12080.x. [DOI] [PubMed] [Google Scholar]
- 75.Macari F, Landau M, Cousin P, Mevorah B, Brenner S, Panizzon R, Schorderet DF, Hohl D, Huber M. Mutation in the gene for connexin 30.3 in a family with erythrokeratodermia variabilis. Am J Hum Genet. 2000;67:1296–1301. doi: 10.1016/s0002-9297(07)62957-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ishida-Yamamoto A. Erythrokeratodermia variabilis et progressiva. J Dermatol. 2016;43:280–285. doi: 10.1111/1346-8138.13220. [DOI] [PubMed] [Google Scholar]
- 77.Boyden LM, Craiglow BG, Zhou J, Hu R, Loring EC, Morel KD, Lauren CT, Lifton RP, Bilguvar K, G. Yale Center for Mendelian. Paller AS, Choate KA. Dominant De Novo Mutations in GJA1 Cause Erythrokeratodermia Variabilis et Progressiva, without Features of Oculodentodigital Dysplasia. J Invest Dermatol. 2015;135:1540–1547. doi: 10.1038/jid.2014.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Richard G, Smith LE, Bailey RA, Itin P, Hohl D, Epstein EH, Jr, DiGiovanna JJ, Compton JG, Bale SJ. Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nat Genet. 1998;20:366–369. doi: 10.1038/3840. [DOI] [PubMed] [Google Scholar]
- 79.Duchatelet S, Hovnanian A. Erythrokeratodermia variabilis et progressiva allelic to oculo-dento-digital dysplasia. J Invest Dermatol. 2015;135:1475–1478. doi: 10.1038/jid.2014.535. [DOI] [PubMed] [Google Scholar]
- 80.Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP, Zhang BR, Xie W, Hu DX, Zheng D, Shi XL, Wang DA, Xia K, Yu KP, Liao XD, Feng Y, Yang YF, Xiao JY, Xie DH, Huang JZ. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat Genet. 1998;20:370–373. doi: 10.1038/3845. [DOI] [PubMed] [Google Scholar]
- 81.Liu XZ, Yuan Y, Yan D, Ding EH, Ouyang XM, Fei Y, Tang W, Yuan H, Chang Q, Du LL, Zhang X, Wang G, Ahmad S, Kang DY, Lin X, Dai P. Digenic inheritance of non-syndromic deafness caused by mutations at the gap junction proteins Cx26 and Cx31. Hum Genet. 2009;125:53–62. doi: 10.1007/s00439-008-0602-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim SY, Kim AR, Kim NK, Lee C, Kim MY, Jeon EH, Park WY, Choi BY. Unraveling of Enigmatic Hearing-Impaired GJB2 Single Heterozygotes by Massive Parallel Sequencing: DFNB1 or Not? Medicine (Baltimore) 2016;95:e3029. doi: 10.1097/MD.0000000000003029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fischbeck KH. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- 84.White TW, Paul DL. Genetic diseases and gene knockouts reveal diverse connexin functions. Annu Rev Physiol. 1999;61:283–310. doi: 10.1146/annurev.physiol.61.1.283. [DOI] [PubMed] [Google Scholar]
- 85.Wang Y, Yin F. A Review of X-linked Charcot-Marie-Tooth Disease. J Child Neurol. 2016;31:761–772. doi: 10.1177/0883073815604227. [DOI] [PubMed] [Google Scholar]
- 86.Kleopa KA, Sargiannidou I. Connexins, gap junctions and peripheral neuropathy. Neurosci Lett. 2015;596:27–32. doi: 10.1016/j.neulet.2014.10.033. [DOI] [PubMed] [Google Scholar]
- 87.Shy ME, Siskind C, Swan ER, Krajewski KM, Doherty T, Fuerst DR, Ainsworth PJ, Lewis RA, Scherer SS, Hahn AF. CMT1X phenotypes represent loss of GJB1 gene function. Neurology. 2007;68:849–855. doi: 10.1212/01.wnl.0000256709.08271.4d. [DOI] [PubMed] [Google Scholar]
- 88.Sun B, Chen ZH, Ling L, Li YF, Liu LZ, Yang F, Huang XS. Mutation Analysis of Gap Junction Protein Beta 1 and Genotype-Phenotype Correlation in X-linked Charcot-Marie-Tooth Disease in Chinese Patients. Chin Med J (Engl) 2016;129:1011–1016. doi: 10.4103/0366-6999.180511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gollob MH, Jones DL, Krahn AD, Danis L, Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, Tesson F, Klein GJ, Yee R, Skanes AC, Guiraudon GM, Ebihara L, Bai D. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med. 2006;354:2677–2688. doi: 10.1056/NEJMoa052800. [DOI] [PubMed] [Google Scholar]
- 90.Yang YQ, Liu X, Zhang XL, Wang XH, Tan HW, Shi HF, Jiang WF, Fang WY. Novel connexin40 missense mutations in patients with familial atrial fibrillation. Europace. 2010;12:1421–1427. doi: 10.1093/europace/euq274. [DOI] [PubMed] [Google Scholar]
- 91.Yang YQ, Zhang XL, Wang XH, Tan HW, Shi HF, Jiang WF, Fang WY, Liu X. Connexin40 nonsense mutation in familial atrial fibrillation. Int J Mol Med. 2010;26:605–610. doi: 10.3892/ijmm_00000505. [DOI] [PubMed] [Google Scholar]
- 92.Sun Y, Yang YQ, Gong XQ, Wang XH, Li RG, Tan HW, Liu X, Fang WY, Bai D. Novel germline GJA5/connexin40 mutations associated with lone atrial fibrillation impair gap junctional intercellular communication. Hum Mutat. 2013;34:603–609. doi: 10.1002/humu.22278. [DOI] [PubMed] [Google Scholar]
- 93.Shi HF, Yang JF, Wang Q, Li RG, Xu YJ, Qu XK, Fang WY, Liu X, Yang YQ. Prevalence and spectrum of GJA5 mutations associated with lone atrial fibrillation. Mol Med Rep. 2013;7:767–774. doi: 10.3892/mmr.2012.1252. [DOI] [PubMed] [Google Scholar]
- 94.Molica F, Meens MJ, Morel S, Kwak BR. Mutations in cardiovascular connexin genes. Biol Cell. 2014;106:269–293. doi: 10.1111/boc.201400038. [DOI] [PubMed] [Google Scholar]
- 95.Gros D, Jarry-Guichard T, Ten Velde I, de Maziere A, van Kempen MJ, Davoust J, Briand JP, Moorman AF, Jongsma HJ. Restricted distribution of connexin40, a gap junctional protein, in mammalian heart. Circ Res. 1994;74:839–851. doi: 10.1161/01.res.74.5.839. [DOI] [PubMed] [Google Scholar]
- 96.Vozzi C, Dupont E, Coppen SR, Yeh HI, Severs NJ. Chamber-related differences in connexin expression in the human heart. J Mol Cell Cardiol. 1999;31:991–1003. doi: 10.1006/jmcc.1999.0937. [DOI] [PubMed] [Google Scholar]
- 97.Groenewegen WA, Firouzi M, Bezzina CR, Vliex S, van Langen IM, Sandkuijl L, Smits JP, Hulsbeek M, Rook MB, Jongsma HJ, Wilde AA. A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ Res. 2003;92:14–22. doi: 10.1161/01.res.0000050585.07097.d7. [DOI] [PubMed] [Google Scholar]
- 98.Fazelifar AF, Arya A, Haghjoo M, Sadr-Ameli MA. Familial atrial standstill in association with dilated cardiomyopathy. Pacing Clin Electrophysiol. 2005;28:1005–1008. doi: 10.1111/j.1540-8159.2005.00198.x. [DOI] [PubMed] [Google Scholar]
- 99.Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, Innis JW, Dinulos MB, Christian C, Hannibal MC, Jabs EW. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72:408–418. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Laird DW. Syndromic and non-syndromic disease-linked Cx43 mutations. FEBS Lett. 2014;588:1339–1348. doi: 10.1016/j.febslet.2013.12.022. [DOI] [PubMed] [Google Scholar]
- 101.Richardson RJ, Joss S, Tomkin S, Ahmed M, Sheridan E, Dixon MJ. A nonsense mutation in the first transmembrane domain of connexin 43 underlies autosomal recessive oculodentodigital syndrome. J Med Genet. 2006;43:e37. doi: 10.1136/jmg.2005.037655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Paznekas WA, Karczeski B, Vermeer S, Lowry RB, Delatycki M, Laurence F, Koivisto PA, Van Maldergem L, Boyadjiev SA, Bodurtha JN, Jabs EW. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum Mutat. 2009;30:724–733. doi: 10.1002/humu.20958. [DOI] [PubMed] [Google Scholar]
- 103.Kogame T, Dainichi T, Shimomura Y, Tanioka M, Kabashima K, Miyachi Y. Palmoplantar keratosis in oculodentodigital dysplasia with a GJA1 point mutation out of the C-terminal region of connexin 43. J Dermatol. 2014;41:1095–1097. doi: 10.1111/1346-8138.12682. [DOI] [PubMed] [Google Scholar]
- 104.Schrander-Stumpel CT, De Groot-Wijnands JB, De Die-Smulders C, Fryns JP. Type III syndactyly and oculodentodigital dysplasia: a clinical spectrum. Genet Couns. 1993;4:271–276. [PubMed] [Google Scholar]
- 105.Hu Y, Chen IP, de Almeida S, Tiziani V, Do Amaral CM, Gowrishankar K, Passos-Bueno MR, Reichenberger EJ. A novel autosomal recessive GJA1 missense mutation linked to Craniometaphyseal dysplasia. PLoS One. 2013;8:e73576. doi: 10.1371/journal.pone.0073576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Elcioglu N, Hall CM. Temporal aspects in craniometaphyseal dysplasia: autosomal recessive type. Am J Med Genet. 1998;76:245–251. doi: 10.1002/(sici)1096-8628(19980319)76:3<245::aid-ajmg8>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 107.Faruqi T, Dhawan N, Bahl J, Gupta V, Vohra S, Tu K, Abdelmagid SM. Molecular, phenotypic aspects and therapeutic horizons of rare genetic bone disorders. Biomed Res Int. 2014;2014:670842. doi: 10.1155/2014/670842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang H, Cao X, Lin Z, Lee M, Jia X, Ren Y, Dai L, Guan L, Zhang J, Lin X, Zhang J, Chen Q, Feng C, Zhou EY, Yin J, Xu G, Yang Y. Exome sequencing reveals mutation in GJA1 as a cause of keratoderma-hypotrichosis-leukonychia totalis syndrome. Hum Mol Genet. 2015;24:243–250. doi: 10.1093/hmg/ddu442. [DOI] [PubMed] [Google Scholar]
- 109.Basaran E, Yilmaz E, Alpsoy E, Yilmaz GG. Keratoderma, hypotrichosis and leukonychia totalis: a new syndrome? Br J Dermatol. 1995;133:636–638. doi: 10.1111/j.1365-2133.1995.tb02720.x. [DOI] [PubMed] [Google Scholar]
- 110.Mackay D, Ionides A, Kibar Z, Rouleau G, Berry V, Moore A, Shiels A, Bhattacharya S. Connexin46 mutations in autosomal dominant congenital cataract. Am J Hum Genet. 1999;64:1357–1364. doi: 10.1086/302383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li Y, Wang J, Dong B, Man H. A novel connexin46 (GJA3) mutation in autosomal dominant congenital nuclear pulverulent cataract. Mol Vis. 2004;10:668–671. [PubMed] [Google Scholar]
- 112.Zhang L, Qu X, Su S, Guan L, Liu P. A novel mutation in GJA3 associated with congenital Coppock-like cataract in a large Chinese family. Mol Vis. 2012;18:2114–2118. [PMC free article] [PubMed] [Google Scholar]
- 113.Shiels A, Hejtmancik JF. Genetics of human cataract. Clin Genet. 2013;84:120–127. doi: 10.1111/cge.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shiels A, Hejtmancik JF. Mutations and mechanisms in congenital and age-related cataracts. Exp Eye Res. 2016 doi: 10.1016/j.exer.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Uhlenberg B, Schuelke M, Ruschendorf F, Ruf N, Kaindl AM, Henneke M, Thiele H, Stoltenburg-Didinger G, Aksu F, Topaloglu H, Nurnberg P, Hubner C, Weschke B, Gartner J. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet. 2004;75:251–260. doi: 10.1086/422763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Henneke M, Combes P, Diekmann S, Bertini E, Brockmann K, Burlina AP, Kaiser J, Ohlenbusch A, Plecko B, Rodriguez D, Boespflug-Tanguy O, Gartner J. GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease. Neurology. 2008;70:748–754. doi: 10.1212/01.wnl.0000284828.84464.35. [DOI] [PubMed] [Google Scholar]
- 117.Bugiani M, Al Shahwan S, Lamantea E, Bizzi A, Bakhsh E, Moroni I, Balestrini MR, Uziel G, Zeviani M. GJA12 mutations in children with recessive hypomyelinating leukoencephalopathy. Neurology. 2006;67:273–279. doi: 10.1212/01.wnl.0000223832.66286.e4. [DOI] [PubMed] [Google Scholar]
- 118.Hobson GM, Garbern JY. Pelizaeus-Merzbacher disease, Pelizaeus-Merzbacher-like disease 1, and related hypomyelinating disorders. Semin Neurol. 2012;32:62–67. doi: 10.1055/s-0032-1306388. [DOI] [PubMed] [Google Scholar]
- 119.Orthmann-Murphy JL, Salsano E, Abrams CK, Bizzi A, Uziel G, Freidin MM, Lamantea E, Zeviani M, Scherer SS, Pareyson D. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain. 2009;132:426–438. doi: 10.1093/brain/awn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ferrell RE, Baty CJ, Kimak MA, Karlsson JM, Lawrence EC, Franke-Snyder M, Meriney SD, Feingold E, Finegold DN. GJC2 missense mutations cause human lymphedema. Am J Hum Genet. 2010;86:943–948. doi: 10.1016/j.ajhg.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ostergaard P, Simpson MA, Brice G, Mansour S, Connell FC, Onoufriadis A, Child AH, Hwang J, Kalidas K, Mortimer PS, Trembath R, Jeffery S. Rapid identification of mutations in GJC2 in primary lymphoedema using whole exome sequencing combined with linkage analysis with delineation of the phenotype. J Med Genet. 2011;48:251–255. doi: 10.1136/jmg.2010.085563. [DOI] [PubMed] [Google Scholar]
- 122.Connell FC, Gordon K, Brice G, Keeley V, Jeffery S, Mortimer PS, Mansour S, Ostergaard P. The classification and diagnostic algorithm for primary lymphatic dysplasia: an update from 2010 to include molecular findings. Clin Genet. 2013;84:303–314. doi: 10.1111/cge.12173. [DOI] [PubMed] [Google Scholar]
- 123.Shiels A, Mackay D, Ionides A, Berry V, Moore A, Bhattacharya S. A missense mutation in the human connexin50 gene (GJA8) underlies autosomal dominant “zonular pulverulent” cataract, on chromosome 1q. Am J Hum Genet. 1998;62:526–532. doi: 10.1086/301762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shiels A, Bennett TM, Hejtmancik JF. Cat-Map: putting cataract on the map. Mol Vis. 2010;16:2007–2015. [PMC free article] [PubMed] [Google Scholar]
- 125.Devi RR, Vijayalakshmi P. Novel mutations in GJA8 associated with autosomal dominant congenital cataract and microcornea. Mol Vis. 2006;12:190–195. [PubMed] [Google Scholar]
- 126.Donahue RP, Bias WB, Renwick JH, McKusick VA. Probable assignment of the Duffy blood group locus to chromosome 1 in man. Proc Natl Acad Sci U S A. 1968;61:949–955. doi: 10.1073/pnas.61.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Renwick JH, Lawler SD. Probable Linkage between a Congenital Cataract Locus and the Duffy Blood Group Locus. Ann Hum Genet. 1963;27:67–84. doi: 10.1111/j.1469-1809.1963.tb00782.x. [DOI] [PubMed] [Google Scholar]
- 128.Kelly JJ, Simek J, Laird DW. Mechanisms linking connexin mutations to human diseases. Cell Tissue Res. 2015;360:701–721. doi: 10.1007/s00441-014-2024-4. [DOI] [PubMed] [Google Scholar]
- 129.Abrams CK, Freidin MM, Verselis VK, Bennett MV, Bargiello TA. Functional alterations in gap junction channels formed by mutant forms of connexin 32: evidence for loss of function as a pathogenic mechanism in the X-linked form of Charcot-Marie-Tooth disease. Brain Res. 2001;900:9–25. doi: 10.1016/s0006-8993(00)03327-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bruzzone R, White TW, Scherer SS, Fischbeck KH, Paul DL. Null mutations of connexin32 in patients with X-linked Charcot-Marie-Tooth disease. Neuron. 1994;13:1253–1260. doi: 10.1016/0896-6273(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 131.Lautermann J, ten Cate WJ, Altenhoff P, Grummer R, Traub O, Frank H, Jahnke K, Winterhager E. Expression of the gap-junction connexins 26 and 30 in the rat cochlea. Cell Tissue Res. 1998;294:415–420. doi: 10.1007/s004410051192. [DOI] [PubMed] [Google Scholar]
- 132.Ahmad S, Chen S, Sun J, Lin X. Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mice. Biochem Biophys Res Commun. 2003;307:362–368. doi: 10.1016/s0006-291x(03)01166-5. [DOI] [PubMed] [Google Scholar]
- 133.Forge A, Becker D, Casalotti S, Edwards J, Marziano N, Nevill G. Gap junctions in the inner ear: comparison of distribution patterns in different vertebrates and assessement of connexin composition in mammals. J Comp Neurol. 2003;467:207–231. doi: 10.1002/cne.10916. [DOI] [PubMed] [Google Scholar]
- 134.Zhao HB, Yu N. Distinct and gradient distributions of connexin26 and connexin30 in the cochlear sensory epithelium of guinea pigs. J Comp Neurol. 2006;499:506–518. doi: 10.1002/cne.21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu YP, Zhao HB. Cellular characterization of Connexin26 and Connnexin30 expression in the cochlear lateral wall. Cell Tissue Res. 2008;333:395–403. doi: 10.1007/s00441-008-0641-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cohen-Salmon M, Ott T, Michel V, Hardelin JP, Perfettini I, Eybalin M, Wu T, Marcus DC, Wangemann P, Willecke K, Petit C. Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr Biol. 2002;12:1106–1111. doi: 10.1016/s0960-9822(02)00904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kudo T, Kure S, Ikeda K, Xia AP, Katori Y, Suzuki M, Kojima K, Ichinohe A, Suzuki Y, Aoki Y, Kobayashi T, Matsubara Y. Transgenic expression of a dominant-negative connexin26 causes degeneration of the organ of Corti and non-syndromic deafness. Hum Mol Genet. 2003;12:995–1004. doi: 10.1093/hmg/ddg116. [DOI] [PubMed] [Google Scholar]
- 138.Wangemann P. Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol. 2006;576:11–21. doi: 10.1113/jphysiol.2006.112888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zdebik AA, Wangemann P, Jentsch TJ. Potassium ion movement in the inner ear: insights from genetic disease and mouse models. Physiology (Bethesda) 2009;24:307–316. doi: 10.1152/physiol.00018.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Patuzzi R. Ion flow in stria vascularis and the production and regulation of cochlear endolymph and the endolymphatic potential. Hear Res. 2011;277:4–19. doi: 10.1016/j.heares.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 141.Zhang Y, Tang W, Ahmad S, Sipp JA, Chen P, Lin X. Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc Natl Acad Sci U S A. 2005;102:15201–15206. doi: 10.1073/pnas.0501859102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Beltramello M, Piazza V, Bukauskas FF, Pozzan T, Mammano F. Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness. Nat Cell Biol. 2005;7:63–69. doi: 10.1038/ncb1205. [DOI] [PubMed] [Google Scholar]
- 143.Chang Q, Tang W, Ahmad S, Zhou B, Lin X. Gap junction mediated intercellular metabolite transfer in the cochlea is compromised in connexin30 null mice. PLoS One. 2008;3:e4088. doi: 10.1371/journal.pone.0004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chang Q, Tang W, Ahmad S, Stong B, Leu G, Lin X. Functional studies reveal new mechanisms for deafness caused by connexin mutations. Otol Neurotol. 2009;30:237–240. doi: 10.1097/MAO.0b013e318194f774. [DOI] [PubMed] [Google Scholar]
- 145.Majumder P, Crispino G, Rodriguez L, Ciubotaru CD, Anselmi F, Piazza V, Bortolozzi M, Mammano F. ATP-mediated cell-cell signaling in the organ of Corti: the role of connexin channels. Purinergic Signal. 2010;6:167–187. doi: 10.1007/s11302-010-9192-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Xu J, Nicholson BJ. The role of connexins in ear and skin physiology - functional insights from disease-associated mutations. Biochim Biophys Acta. 2013;1828:167–178. doi: 10.1016/j.bbamem.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yum SW, Zhang J, Scherer SS. Dominant connexin26 mutants associated with human hearing loss have trans-dominant effects on connexin30. Neurobiol Dis. 2010;38:226–236. doi: 10.1016/j.nbd.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhang J, Scherer SS, Yum SW. Dominant Cx26 mutants associated with hearing loss have dominant-negative effects on wild type Cx26. Mol Cell Neurosci. 2011;47:71–78. doi: 10.1016/j.mcn.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Thomas T, Telford D, Laird DW. Functional domain mapping and selective trans-dominant effects exhibited by Cx26 disease-causing mutations. J Biol Chem. 2004;279:19157–19168. doi: 10.1074/jbc.M314117200. [DOI] [PubMed] [Google Scholar]
- 150.Shuja Z, Li L, Gupta S, Mese G, White TW. Connexin26 Mutations Causing Palmoplantar Keratoderma and Deafness Interact with Connexin43, Modifying Gap Junction and Hemichannel Properties. J Invest Dermatol. 2016;136:225–235. doi: 10.1038/JID.2015.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rouan F, White TW, Brown N, Taylor AM, Lucke TW, Paul DL, Munro CS, Uitto J, Hodgins MB, Richard G. trans-dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation. J Cell Sci. 2001;114:2105–2113. doi: 10.1242/jcs.114.11.2105. [DOI] [PubMed] [Google Scholar]
- 152.Donnelly S, English G, de Zwart-Storm EA, Lang S, van Steensel MA, Martin PE. Differential susceptibility of Cx26 mutations associated with epidermal dysplasias to peptidoglycan derived from Staphylococcus aureus and Staphylococcus epidermidis. Exp Dermatol. 2012;21:592–598. doi: 10.1111/j.1600-0625.2012.01521.x. [DOI] [PubMed] [Google Scholar]
- 153.Gerido DA, DeRosa AM, Richard G, White TW. Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am J Physiol Cell Physiol. 2007;293:C337–345. doi: 10.1152/ajpcell.00626.2006. [DOI] [PubMed] [Google Scholar]
- 154.Lee JR, Derosa AM, White TW. Connexin mutations causing skin disease and deafness increase hemichannel activity and cell death when expressed in Xenopus oocytes. J Invest Dermatol. 2009;129:870–878. doi: 10.1038/jid.2008.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sanchez HA, Mese G, Srinivas M, White TW, Verselis VK. Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J Gen Physiol. 2010;136:47–62. doi: 10.1085/jgp.201010433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sanchez HA, Villone K, Srinivas M, Verselis VK. The D50N mutation and syndromic deafness: altered Cx26 hemichannel properties caused by effects on the pore and intersubunit interactions. J Gen Physiol. 2013;142:3–22. doi: 10.1085/jgp.201310962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Stong BC, Chang Q, Ahmad S, Lin X. A novel mechanism for connexin 26 mutation linked deafness: cell death caused by leaky gap junction hemichannels. Laryngoscope. 2006;116:2205–2210. doi: 10.1097/01.mlg.0000241944.77192.d2. [DOI] [PubMed] [Google Scholar]
- 158.Montgomery JR, White TW, Martin BL, Turner ML, Holland SM. A novel connexin 26 gene mutation associated with features of the keratitis-ichthyosis-deafness syndrome and the follicular occlusion triad. J Am Acad Dermatol. 2004;51:377–382. doi: 10.1016/j.jaad.2003.12.042. [DOI] [PubMed] [Google Scholar]
- 159.Aypek H, Bay V, Mese G. Altered cellular localization and hemichannel activities of KID syndrome associated connexin26 I30N and D50Y mutations. BMC Cell Biol. 2016;17:5. doi: 10.1186/s12860-016-0081-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Garcia IE, Maripillan J, Jara O, Ceriani R, Palacios-Munoz A, Ramachandran J, Olivero P, Perez-Acle T, Gonzalez C, Saez JC, Contreras JE, Martinez AD. Keratitis-ichthyosis-deafness syndrome-associated cx26 mutants produce nonfunctional gap junctions but hyperactive hemichannels when co-expressed with wild type cx43. J Invest Dermatol. 2015;135:1338–1347. doi: 10.1038/jid.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sanchez HA, Slavi N, Srinivas M, Verselis VK. Syndromic deafness mutations at Asn 14 differentially alter the open stability of Cx26 hemichannels. J Gen Physiol. 2016;148:25–42. doi: 10.1085/jgp.201611585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Housley G, Gale J. Purinergic signalling in the inner ear-perspectives and progress. Purinergic Signal. 2010;6:151–153. doi: 10.1007/s11302-010-9196-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tritsch NX, Yi E, Gale JE, Glowatzki E, Bergles DE. The origin of spontaneous activity in the developing auditory system. Nature. 2007;450:50–55. doi: 10.1038/nature06233. [DOI] [PubMed] [Google Scholar]
- 164.Anselmi F, Hernandez VH, Crispino G, Seydel A, Ortolano S, Roper SD, Kessaris N, Richardson W, Rickheit G, Filippov MA, Monyer H, Mammano F. ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc Natl Acad Sci U S A. 2008;105:18770–18775. doi: 10.1073/pnas.0800793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Elsholz F, Harteneck C, Muller W, Friedland K. Calcium–a central regulator of keratinocyte differentiation in health and disease. Eur J Dermatol. 2014;24:650–661. doi: 10.1684/ejd.2014.2452. [DOI] [PubMed] [Google Scholar]
- 166.Mese G, Sellitto C, Li L, Wang HZ, Valiunas V, Richard G, Brink PR, White TW. The Cx26-G45E mutation displays increased hemichannel activity in a mouse model of the lethal form of keratitis-ichthyosis-deafness syndrome. Mol Biol Cell. 2011;22:4776–4786. doi: 10.1091/mbc.E11-09-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Schutz M, Auth T, Gehrt A, Bosen F, Korber I, Strenzke N, Moser T, Willecke K. The connexin26 S17F mouse mutant represents a model for the human hereditary keratitis-ichthyosis-deafness syndrome. Hum Mol Genet. 2011;20:28–39. doi: 10.1093/hmg/ddq429. [DOI] [PubMed] [Google Scholar]
- 168.Bosen F, Celli A, Crumrine D, Vom Dorp K, Ebel P, Jastrow H, Dormann P, Winterhager E, Mauro T, Willecke K. Altered epidermal lipid processing and calcium distribution in the KID syndrome mouse model Cx26S17F. FEBS Lett. 2015 doi: 10.1016/j.febslet.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Essenfelder GM, Bruzzone R, Lamartine J, Charollais A, Blanchet-Bardon C, Barbe MT, Meda P, Waksman G. Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activity. Hum Mol Genet. 2004;13:1703–1714. doi: 10.1093/hmg/ddh191. [DOI] [PubMed] [Google Scholar]
- 170.Chi J, Li L, Liu M, Tan J, Tang C, Pan Q, Wang D, Zhang Z. Pathogenic connexin-31 forms constitutively active hemichannels to promote necrotic cell death. PLoS One. 2012;7:e32531. doi: 10.1371/journal.pone.0032531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Contreras JE, Saez JC, Bukauskas FF, Bennett MV. Gating and regulation of connexin 43 (Cx43) hemichannels. Proc Natl Acad Sci U S A. 2003;100:11388–11393. doi: 10.1073/pnas.1434298100. [DOI] [PMC free article] [PubMed] [Google Scholar]