

Abstract
Background and Purpose
PDE1, a subfamily of cyclic nucleotide PDEs consisting of three isoforms, PDE1A, PDE1B and PDE1C, has been implicated in the regulation of vascular tone. The PDE1 isoform(s) responsible for tone regulation is unknown. This study used isoform‐preferring PDE1 inhibitors, Lu AF58027, Lu AF64196, Lu AF66896 and Lu AF67897, to investigate the relative contribution of PDE1 isoforms to regulation of vascular tone.
Experimental Approach
In rat mesenteric arteries, expression and localization of Pde1 isoforms were determined by quantitative PCR and in situ hybridization, and physiological impact of PDE1 inhibition was evaluated by isometric tension recordings.
Key Results
In rat mesenteric arteries, Pde1a mRNA expression was higher than Pde1b and Pde1c. In situ hybridization revealed localization of Pde1a to vascular smooth muscle cells (VSMCs) and only minor appearance of Pde1b and Pde1c. The potency of the PDE1 inhibitors at eliciting relaxation showed excellent correlation with their potency at inhibiting PDE1A. Thus, Lu AF58027 was the most potent at inhibiting PDE1A and was also the most potent at eliciting relaxation in mesenteric arteries. Inhibition of NOS with l‐NAME, soluble GC with ODQ or PKG with Rp‐8‐Br‐PET‐cGMP all attenuated the inhibitory effect of PDE1 on relaxation, whereas PKA inhibition with H89 had no effect.
Conclusions and Implications
Pde1a is the dominant PDE1 isoform present in VSMCs, and relaxation mediated by PDE1A inhibition is predominantly driven by enhanced cGMP signalling. These results imply that isoform‐selective PDE1 inhibitors are powerful investigative tools allowing examination of physiological and pathological roles of PDE1 isoforms.
Abbreviations
- CCh
carbachol
- H89
N‐[2‐(p‐bromocinnamylamino)ethyl]‐5‐isoquinolinesulfonamide dihydrochloride
- ODQ
1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxalin‐1‐one
- Rp‐8
Rp‐8‐Br‐PET‐cGMPS
- U46619
9 α‐epoxymethanoPGF2α
- VSMCs
vascular smooth muscle cells
Introduction
cGMP and cAMP are ubiquitous second messengers that regulate numerous cellular functions including vascular smooth muscle cell (VSMC) relaxation (Morgado et al., 2012). The cGMP‐dependent protein kinase PKG and cAMP‐dependent protein kinase PKA are the primary effector molecules of cGMP and cAMP, respectively, and both PKG and PKA activation leads to vasorelaxation by lowering intracellular calcium concentration ([Ca2+]i) (Tsai and Kass, 2009). Intracellular cGMP and cAMP levels depend on the balance between their production, active transport out of the cell and degradation. For example, NOS‐derived NO, a potent endothelial cell‐derived vasodilator, stimulates the production of cGMP from GTP by activating soluble guanylate cyclase (sGC) (Arnold et al., 1977), while cAMP is generated from ATP by the membrane‐bound adenylate cyclase (AC) (Simonds, 1999). The degradation of cGMP and cAMP to inactive 5′GMP and 5′AMP, respectively, is mediated solely by PDEs. Thus, the amplitude, duration and compartmentalization of cyclic nucleotide signalling are profoundly influenced by PDEs (Conti and Beavo, 2007). Twenty‐one PDE genes have been identified in humans and rats. They are categorized into 11 different subfamilies (PDE1–PDE11) depending on their sequence, structure, susceptibility to inhibitors, hydrolysis and regulatory characteristics (Bobin et al., 2016). Some PDEs can hydrolyse both cyclic nucleotides (PDE1–PDE3, PDE10 and PDE11); others exclusively degrade either cGMP (PDE5–PDE6 and PDE9) or cAMP (PDE4, PDE7 and PDE8) (Bobin et al., 2016).
In the vasculature, PDE2–PDE5 expression has been reported in human and bovine endothelial cells (Netherton and Maurice, 2005; Nagel et al., 2006), while PDE1 and PDE3–PDE5 have been identified in VSMCs (Komas et al., 1991; Matsumoto et al., 2003; Rybalkin et al., 2003). Notably, contractile and proliferating VSMCs have different expression profiles of PDE isoforms, emphasizing the potential of different functional roles for distinct variants (Winquist et al., 1984; Maurice et al., 2003). PDE5 is highly abundant in smooth muscle, and by limiting the breakdown of cGMP, PDE5 inhibition potentiates the relaxant effect of the cGMP pathway initiated by NO (Gratzke et al., 2010). Contrary to PDE5, PDE1 activity is Ca2+/calmodulin dependent, and all three PDE1 isoforms (PDE1A, PDE1B and PDE1C) can hydrolyse both cGMP and cAMP. PDE1A and PDE1B preferentially hydrolyse cGMP, while PDE1C hydrolyses cAMP and cGMP equally well (Bender and Beavo, 2006). There is little information on the functional roles of PDE1 isoforms, but PDE1A is hypothesized to be the most important PDE1 isoform with regard to vasodilatation, primarily based on its expression pattern, but not functionally due to the lack of isoform‐selective inhibitors. Most published studies of PDE1 function in the vasculature have used non‐selective PDE1 inhibitors, such as vinpocetine, which elicit concentration‐dependent relaxation of rat mesenteric arteries (Loughney et al., 1996; Kim et al., 2001). However, no studies have tested the effect of PDE1 isoform‐selective inhibitors on vascular tone. Therefore, we investigated the expression profile and localization of Pde1 isoforms, and using four PDE1 inhibitors: Lu AF58027, Lu AF64196, Lu AF66896 and Lu AF67897 with different PDE1 isoform selectivity profiles, we studied the role of PDE1A–C in the regulation of vascular tone in rat mesenteric resistance arteries.
Methods
Recombinant PDE1 assays
PDE1A, PDE1B and PDE1C assays were performed as follows: the assays were performed in 60 μL samples containing a fixed amount of recombinant human PDE1 enzyme (sufficient to convert 20–25% of the cyclic nucleotide substrate), a buffer (50 mM HEPES pH 7.6, 10 mM MgCl2 and 0.02% Tween 20), 0.1 mg·mL−1 BSA, 15 nM tritium‐labelled cAMP and varying amounts of inhibitors. Reactions were initiated by addition of the cyclic nucleotide substrate, and reactions were allowed to proceed for 1 h at room temperature before being terminated through addition of 20 μL (0.2 mg) yttrium silicate SPA beads (PerkinElmer, USA). The beads were allowed to settle for 1 h in the dark before the plates were counted in a Wallac 1450 Microbeta counter (PerkinElmer, Boston, MA, USA). The measured signals were converted to activity relative to an uninhibited control (100%), and IC50 values were calculated using XlFit (model 205, IDBS, Guildford, UK). Inhibition constants (K i) were calculated from the measured IC50 values using the Cheng–Prusoff equation (Yung‐Chi and Prusoff, 1973). GST‐tagged PDE1A1 was purchased from BPS Bioscience (San Diego, CA, USA; catalogue # 60010), HIS‐tagged PDE1B (amino acids 150‐503) was purchased from Biofocus (Chesterford, UK) and HIS‐tagged recombinant PDE1C (amino acids 150‐634) was expressed in Escherichia coli. All other PDE assays were performed in a similar fashion using either tritium‐labelled cAMP or tritium‐labelled cGMP at the following concentrations: hPDE2A: 15 nM [3H]‐cAMP, hPDE3A: 4 nM [3H]‐cAMP, hPDE4D6: 4 nM [3H]‐cAMP, hPDE5A: 15 nM [3H]‐cGMP, hPDE7B: 4 nM [3H]‐cAMP, hPDE8A: 15 nM [3H]‐cAMP, hPDE9A: 15 nM [3H]‐cGMP, hPDE10A: 5 nM [3H]‐cAMP and hPDE11A: 15 nM [3H]‐cGMP.
Animals
Animal care and experimental procedures were performed in accordance with Guide for the Care and Use of Laboratory Animals, 8th Edition, National Research Council (USA), Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Washington (DC): National Academies Press (USA); 2011. ISBN‐13: 978‐0‐309‐15400‐0ISBN‐10: 0‐309‐15400‐6 and approved by the National Ethics Committee, Denmark (license number: 2014‐15‐2934‐0161). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Adult male Wistar rats (10–12 weeks old, approx 300 g; Janvier, Le Genêt‐St. Isle, France), a recognized animal model used for vascular research, were kept and cared for in standard dual cages under clean conditions in separate quarters in a 12–12 h light–dark cycle with free access to water and food pellets. Rats were killed via cervical dislocation, and the mesenteric vasculature removed and placed in cold physiological saline solution (PSS) of the following composition (mM): NaCl 119, KCl 2.82, KH2PO4 1.18, MgSO4.7H2O 1.17, NaHCO3 25, CaCl2 1.6, EDTA 0.03 and glucose 5.5, saturated with carbogen (O2 95% and CO2 5%) at pH 7.4.
Vessel in situ hybridization
Dual probe in situ hybridization (Quantigene ViewRNA, Affymetrix, CA, USA) was performed to study the expression of Pde1a, Pde1b and Pde1c in rat third‐order mesenteric arteries. The probes sets [Pde1a : VC1‐17446‐01 (covering 149‐1083), Pde1b : VC1‐14401 (covering), Pde1c : VC1‐15788‐01 (covering 362‐1899) and Pecam : VC6‐13457‐01 (covering 671‐1579)] were purchased from Affymetrix, and the in situ hybridization was performed according to the manufacturer's instructions. In brief, a piece of rat mesentery containing both second‐ and third‐order mesenteric arteries with surrounding tissue was dissected and frozen. Third‐order mesenteric arteries were identified, and 20 μm tissue sections were cut and fixed for 30 min in 4% paraformaldehyde. Probes for Pde1a, Pde1b or Pde1c (type 1) were hybridized at 40°C overnight together with a probe for platelet endothelial cell adhesion molecule (PECAM‐1, type 6, vascular endothelial cell marker). Signals were amplified according to the instructions and visualized with fast red and fast blue chromogens. As these chromogens are also fluorescent, the signals can be visualized both in bright field and with a fluorescence microscope. Images were acquired using a Leica DC5500 fluorescence microscope.
Quantitative PCR (qPCR)
Total RNA was purified from isolated mesenteric arteries using the NucleoSpin RNA and Protein Kit according to the manufacturer's instructions (Macherey‐Nagel, Düren, Germany), and the yield was determined using a NanoDrop spectrophotometer (NanoDrop products, Wilmington, DE, USA). RNA, 30–100 ng, was used for first‐strand cDNA synthesis using random hexamer primers and TaqMan reverse transcription reagents according to the manufacturer's protocol (Life Technologies, Paisley, UK).
qPCR was performed on a Bio‐Rad c1000 Touch thermal cycler with a CFX384 optical reaction module using primers at a final concentration of 6 pM and SsofastTM Evagreen Supermix reagent according to the manufacturer's instructions (Bio‐Rad, Hercules, CA, USA) starting with 95°C for 30 s followed by 40 cycles of 95°C for 5 s and 60°C for 10s. Samples were run in technical duplicates, and the mean of the quantification cycle (Cq) values was used for further calculations. The housekeeping genes, ubiquitin C, transferrin receptor and β‐actin, were included in each experiment and the geometric mean of these was used for analysis of gene expression using the ∆∆Ct method. Subsequently, data were normalized to Pde1a expression levels and are presented as mean ± SEM of six biologically independent samples.
Myography
Rat third‐order mesenteric arteries (2 mm length) were dissected and mounted on two 40 μm stainless steel wires in isolated chambers of a multiwire myograph system (model 610, DMT, Aarhus, Denmark). The preparations were allowed to equilibrate in PSS thermoregulated to 37°C for 30 min before they were normalized as previously described (Mulvany and Halpern, 1977). In brief, the internal circumference of each arterial segment when relaxed and under a transmural pressure of 100 mmHg, L 100, was calculated. Subsequently, the internal circumference of the arterial segment was set to L 1, where L 1 = 0.9 × L 100.
To test the viability of the arterial segments, a standard start was performed where the arterial segments were contracted twice with high potassium (K+)‐containing PSS (KPSS), where NaCl had been replaced with KCl giving a final K+ concentration of 124 mM, to test VSMC function. To test endothelial integrity, carbachol (CCh, 1 μM) was added onto arteries pre‐contracted with noradrenaline (NA, 1 μM). Only arterial segments that generated a response greater than 5 mN to the second KPSS stimulus and a CCh‐induced relaxation >50% of the NA‐induced contraction were included. To assess relaxation, cumulative concentration–response curves for the PDE1 inhibitors, Lu AF58027, Lu AF64196, Lu AF66896 and Lu AF67897 (1 nM–10 μM), the AC stimulator, forskolin (1 nM–10 μM), and the sGC stimulator, 3‐(4‐amino‐5‐cyclopropylpyrimidin‐2‐yl)‐1‐(2‐fluorobenzyl)‐1H‐pyrazolo[3,4‐b]pyridine (BAY 41‐2272, 1 nM–10 μM), were generated in arteries pre‐contracted to approx 80% of their maximum contraction with NA or the TxA2‐analogue, 9α‐epoxymethanoPGF2α (U46619). For those experiments investigating the involvement of NOS‐derived NO, sGC, PKG or PKA, the NOS inhibitor, l‐NAME (100 μM) (Graves and Poston, 1993), the sGC inhibitor, 1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxalin‐1‐one (ODQ, 3 μM) (Garthwaite et al., 1995), the PKG inhibitor, Rp‐8‐bromo‐β‐phenyl‐1,N 2‐ethenoguanosine 3′,5′‐cyclic monophosphorothioate sodium salt [Rp‐8‐Br‐PET‐cGMPS (Rp‐8), 10 μM] (Butt et al., 1994), or the PKA inhibitor, [N‐[2‐(p‐bromocinnamylamino)ethyl]‐5‐isoquinolinesulfonamide dihydrochloride (H89), 1 μM] (Chijiwa et al., 1990) were added 15 min prior to contraction with U46619. l‐NAME (100 μM), ODQ (3 μM), Rp‐8 (10 μM) or H89 (1 μM) did not change the concentration of U46619 used to pre‐contract the arteries to approx 80% of their maximum contraction with U46619. For those experiments investigating the involvement of the endothelium, the endothelium was mechanically removed by introducing a hair into the lumen of the mounted segment. Removal of the endothelium was considered successful if CCh (1 μM)‐induced relaxation was less than 10% of the NA (1 μM)‐induced contraction.
Data analysis
Arterial segments were randomly assigned to experimental groups. For the myograph experiments, PowerLab4/25‐Chart7 acquisition systems (ADInstruments Ltd, Oxford, UK) were used for data recording, and the operator and data analyst were blinded to the pK i values for the PDE1 isoforms from the four PDE1 inhibitors, as well as the qPCR and in situ hybridization data. Blinding of the investigators in regard to the treatment of each experimental group was not possible due to the technical layout of the assay used in this study. The mechanical responses of arterial segments were measured as force (mN) and expressed as active wall tension, ΔT, which is the increase in measured force, ΔF, divided by twice the segment length (g) (Mulvany and Halpern, 1977).
The magnitude of relaxation responses is given as a percentage of the stable pre‐contraction level. Data are expressed as mean ± SEM, n denotes the number of rats and specific n values for each experimental group/condition are provided throughout the manuscript. Sample size was selected based on power analyses of what n would be required to detect a 5% difference with 90% confidence. Data were log transformed to generate a Gaussian‐distributed data set amenable to parametric analysis. Individual cumulative concentration–response curves were fitted to a non‐linear regression curve, from which potency (pEC50) and maximal efficacy (R max) of the PDE1 inhibitors were determined. When the maximum relaxation plateau was undefined, p(50% relax) and the relaxation response to 10 μM of the compounds were compared, where p(50% relax) is the negative log of the concentration of PDE1 inhibitor required to elicit 50% relaxation of the NA‐ or U46619‐induced submaximal contraction. One‐way ANOVA with Bonferroni post hoc test was used to compare the potency and efficacy of the PDE1 inhibitors in the absence and presence of various treatments. A Bonferroni post hoc test was only applied if P < 0.05, and there was homogeneity of variance. Differences in qPCR data were analysed using a one‐way ANOVA with a Bonferroni post hoc test. Differences at the P < 0.05 level were considered significant. All analyses were performed using GraphPad Prism 6.04 (GraphPad Software Inc., San Diego, CA, USA). The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Materials
The following drugs were used: BAY 41‐2272 (Tocris, Bristol, UK), CCh (Sigma‐Aldrich, Copenhagen, Denmark), forskolin (Sigma‐Aldrich), l‐NAME (Sigma‐Aldrich), H89 (Sigma‐Aldrich), NA (Sigma‐Aldrich), ODQ (Tocris), Rp‐8 and U46619 (Sigma‐Aldrich). Lu AF58027 was prepared as described in Li et al. (2016), and Lu AF64196, Lu AF66896 and Lu AF67897 (Figure 1) were synthesized at Lundbeck as described in patent applications WO2015091805, WO2015150254 and WO2016055618 respectively. Lu AF58027, Lu AF64196, Lu AF66896, Lu AF67897, BAY 41‐2272, forskolin, ODQ and U46619 were dissolved in DMSO to a stock concentration of 10 mM. The maximal DMSO concentration applied in vitro did not modulate smooth muscle tone in control experiments. All other drugs were dissolved in distilled water.
Figure 1.
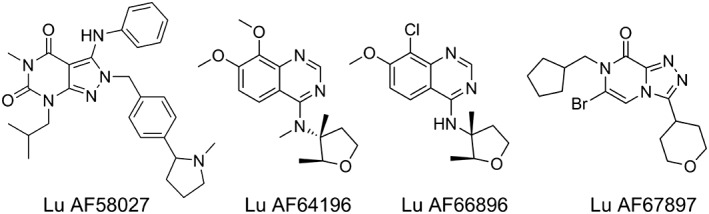
Chemical structure of the four PDE1 inhibitors.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
PDE1 isoform activities of tool compounds
The PDE activity assay showed that Lu AF58027 had a preference for inhibiting PDE1C followed by A and B, with the following K i (nM): PDE1A = 4, PDE1B = 18 and PDE1C = 0.6 (Table 1). Lu AF64196 was slightly PDE1B preferring, K i (nM): PDE1A = 58, PDE1B = 14 and PDE1C = 47 (Table 1). Lu AF66896 had a higher potency for inhibiting PDE1B and C than A, K i (nM): PDE1A = 79, PDE1B = 5 and PDE1C = 3 (Table 1). Lu AF67897 had a higher potency for inhibiting PDE1C than A and B, IC50 (nM): PDE1A = 100, PDE1B = 123 and PDE1C = 6 (Table 1). Lu AF58027, Lu AF64196, Lu AF66896 and Lu AF67897 all had very low affinity towards other PDE enzymes with K i values above 10 μM (Table 1).
Table 1.
pK i values for the four PDE1 inhibitors
| Name | pK i (M) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PDE1A | n | PDE1B | n | PDE1C | n | PDE5 | n | Other PDEs | |
| Lu AF58027 | 8.43 ± 0.12 | 4 | 7.75 ± 0.04 | 8 | 9.25 ± 0.08 | 6 | 6.04 ± 0.02 | 2 | PDE3, PDE4, PDE7 and PDE10, pK i ≈ 5 |
| Lu AF64196 | 7.24 ± 0.04 | 4 | 7.86 ± 0.05 | 9 | 7.33 ± 0.04 | 9 | <5 | 4 | All pK i < 5 |
| Lu AF66896 | 7.10 ± 0.10 | 3 | 8.31 ± 0.04 | 5 | 8.49 ± 0.08 | 5 | <5 | 3 | All pK i < 5 |
| Lu AF67897 | 7.00 ± 0.06 | 3 | 6.91 ± 0.05 | 6 | 8.19 ± 0.02 | 5 | <5 | 3 | Not broadly tested |
n is the number independent experimental determinations of pK i. Values are mean ± SEM.
Localization and expression of Pde1 isoforms in the vascular wall
To investigate the presence and localization of Pde1 isoforms in the vascular wall, rat mesenteric arteries were fixed for in situ hybridization. In situ hybridization analysis revealed that Pde1a was the major Pde1 isoform in rat mesenteric resistance arteries (Figure 2A), with very little Pde1b or Pde1c detected (Figure 2C, E). Dual labelling with the endothelial cell marker, PECAM‐1, showed that Pde1a expression was not localized to endothelial cells, but tended to be confined to VSMCs (Figure 2A). Supporting these results, qPCR data showed low expression of Pde1b and Pde1c and higher expression of Pde1a (Figure 2G).
Figure 2.
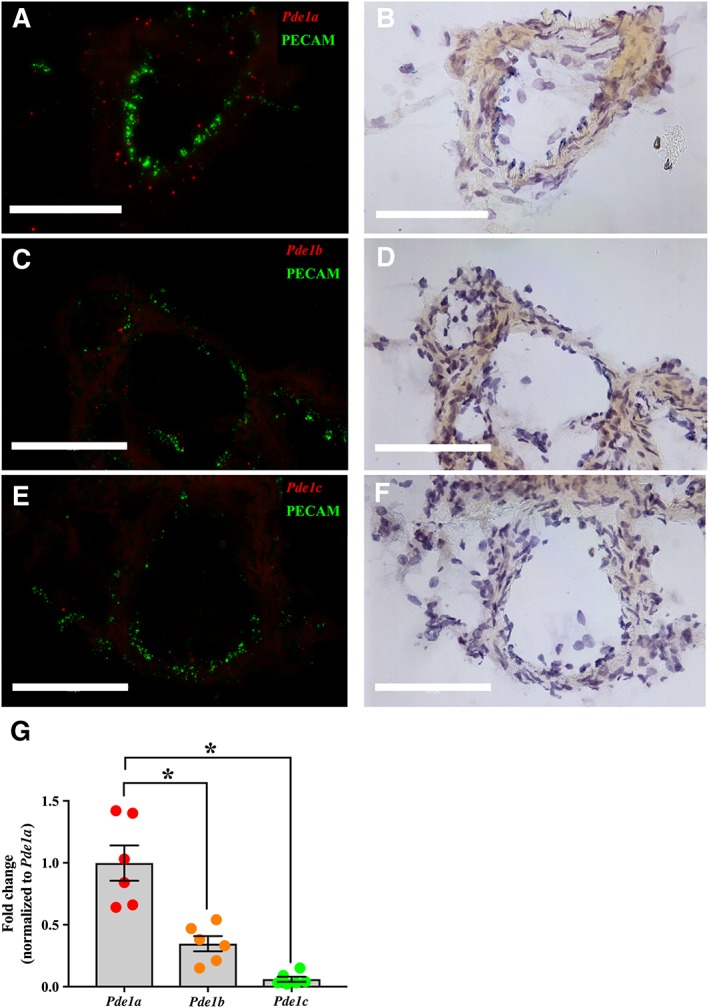
Pde1a was the major isoform expressed in small mesenteric arteries and was predominantly confined to VSMCs. Using in situ hybridization, expression of Pde1a (A), Pde1b (C) and Pde1c (E) was determined in separate cross sections of rat small mesenteric arteries. Red and green indicate the signals for the Pde1 isoforms and the endothelial cell marker, PECAM‐1, respectively. Representative sections from three to five experiments per probe set are shown visualized by both fluorescence microscopy (left panel, A, C, E) and bright‐field microscopy (right panel, B, D, F). Scale bar represents 100 μM. qPCR analysis of relative expression of the Pde1 isoform mRNAs in small mesenteric arteries normalized to the mean of Pde1a (G). Data are mean ± SEM. n = 6. One‐way ANOVA with Bonferroni's multiple comparisons test. *P < 0.05.
PDE1 inhibition induced relaxation of rat mesenteric arteries predominantly through isoform A
To investigate the functional role of the three different PDE1 isoforms in the regulation of vascular tone, rat mesenteric resistance arteries were isolated and contracted with either NA or U46619. All the PDE1 inhibitors, for example, Lu AF58027, Lu AF64196, Lu AF66896 and Lu AF67897 (1 nM–10 μM), induced concentration‐dependent relaxation in pre‐contracted mesenteric arteries (Figure 3). Concentration–response curves generated to Lu AF66896 and Lu AF67897 generally had a poorly defined maximum plateau; in such instances, maximal efficacy and pEC50 could not be properly calculated. However, at the highest concentration tested (10 μM), Lu AF58027 caused complete relaxation of NA‐contracted and U46619‐contracted arteries, while Lu AF66896 elicited almost complete relaxation, although only in NA‐contracted arteries. Comparatively, Lu AF64196 and Lu AF67897 only elicited partial relaxation, irrespective of the contractile agonist (Figure 3 and Table 2).
Figure 3.
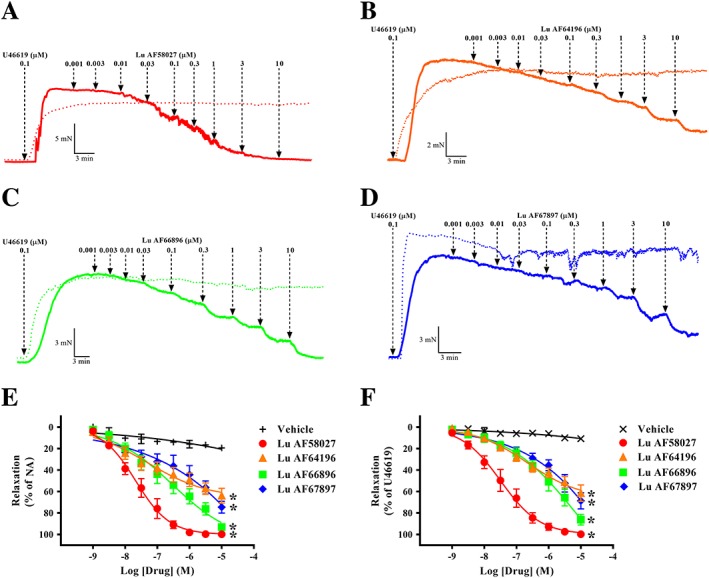
Inhibition of PDE1‐induced potent relaxation in pre‐contracted mesenteric arterial segments. Representative traces of mesenteric arterial segments contracted with U46619 to approximately 80% of maximal contraction following cumulative additions of the PDE1 inhibitors: Lu AF58027 (A), Lu AF64196 (B), Lu AF66896 (C) and Lu AF67897 (D). Summary data of the cumulative concentration–response curves for the PDE1 inhibitors: Lu AF58027, Lu AF66896, Lu AF64196 and Lu AF67897 contracted with NA or U46619 are shown in (E) and (F) respectively. In (A)–(D), solid lines represent experiments, where drug has been added, and dotted lines represent their respective time–control experiments. Data are mean ± SEM. n = 6 arteries from separate rats. One‐way ANOVA with a Bonferroni's multiple comparisons test of the relaxation response to 10 μM of the PDE1 inhibitors. *P < 0.05 from vehicle.
Table 2.
Values for p(50% relax) and efficacy for the four PDE1 inhibitors in mesenteric arteries contracted with NA or U46619
| Name | NA | U46619 | ||||
|---|---|---|---|---|---|---|
| p(50% relax) (M) | Relaxation (%) | n | p(50% relax) (M) | Relaxation (%) | n | |
| Lu AF58027 | 7.6 ± 0.2 | 100 ± 1 | 6 | 7.4 ± 0.2 | 100 ± 1 | 6 |
| Lu AF64196 | 5.7 ± 0.2 | 64 ± 7 | 6 | 6.0 ± 0.2 | 61 ± 4 | 6 |
| Lu AF66896 | 6.6 ± 0.2 | 93 ± 4 | 6 | 5.9 ± 0.2 | 88 ± 3 | 6 |
| Lu AF67897 | 5.8 ± 0.3 | 74 ± 5 | 6 | 5.7 ± 0.1 | 69 ± 4 | 6 |
Data are from Figure 3, and values are mean ± SEM. p(50% relax) is defined as the concentration (−Log) of the PDE1 inhibitors giving a 50% relaxation of the NA‐ or U46619‐mediated pre‐contraction. Efficacy [relaxation (%)] was calculated at the highest concentration (10 μM) of the four PDE1 inhibitors tested.
Furthermore, when comparing the p(50% relax) of all the PDE1 inhibitors, Lu AF58027, the PDE1 inhibitor with the highest affinity for the PDE1A isoform (IC50 = 4 nM, Table 1) was the most potent at inducing relaxation. The order of potency of the PDE1 inhibitors was as follows: in NA‐contracted segments, Lu AF58027 > Lu AF66896 > Lu AF67897 > Lu AF64196 (Table 2), and in U46619‐contracted segments, Lu AF58027 > Lu AF64196 > Lu AF66896 > Lu AF67897 (Table 2). The p(50% relax) values of the PDE1 inhibitors contracted with U46619 correlated well with their inhibitory constants for PDE1A (R 2 = 0.99) as demonstrated in Figure 4A by a linear regression analysis of relaxation p(50% relax) values (Table 2) and PDE1A isoform pK i values obtained from the recombinant PDE1 assay (Table 1). In contrast, linear regression analysis revealed that relaxation p(50% relax) values of the PDE1 inhibitors were poorly correlated with their pK i values for PDE1B (R 2 = 0.03; Figure 4B) or PDE1C (R 2 = 0.50; Figure 4C).
Figure 4.
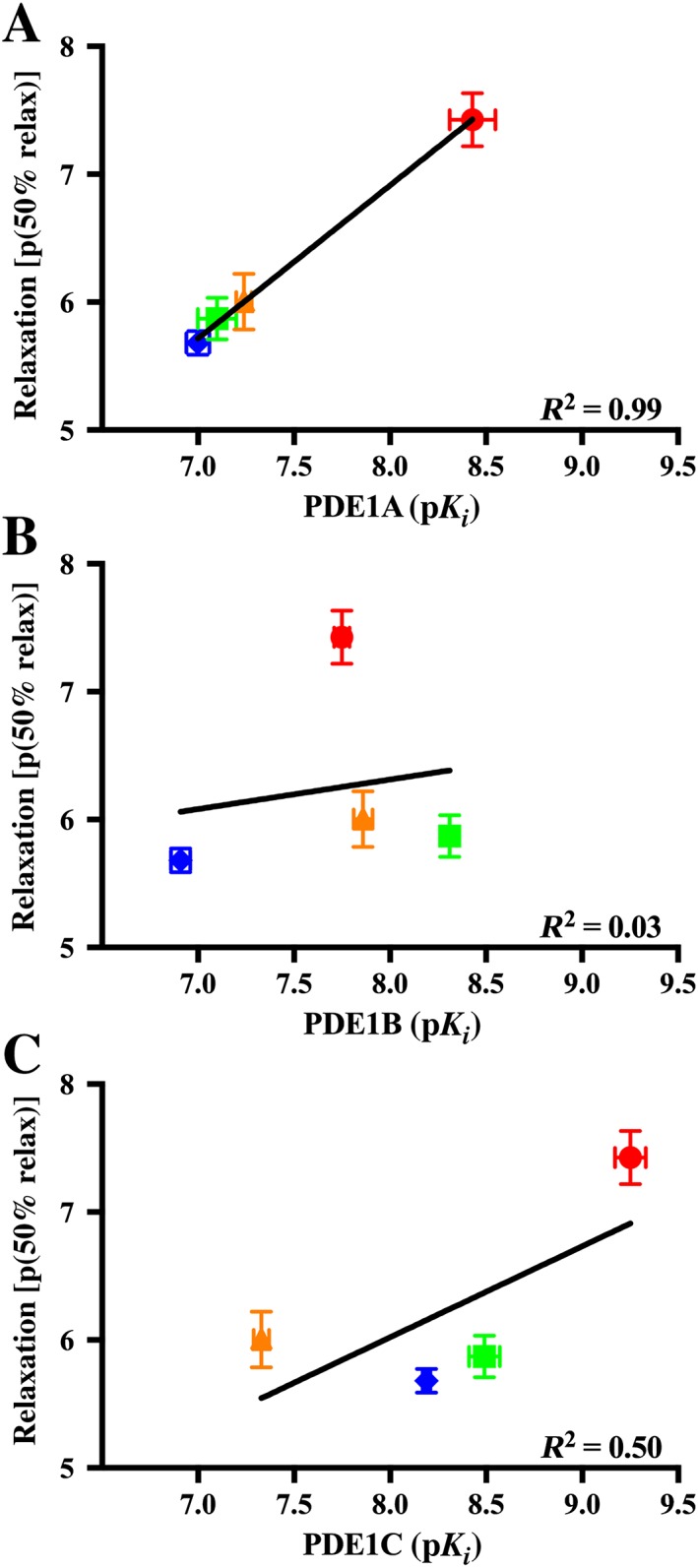
Inhibitory potency for PDE1 isoforms correlated with 50% relaxation potency. Relaxation p(50% relax) values from arteries contracted with U46619 from Table 2 plotted against the pK i values for the PDE1A (A), PDE1B (B) and PDE1C (C) isoforms from Table 1 for the four PDE1 inhibitors. Based on the pK i values for the PDE1A, PDE1B and PDE1C and p(50% relax) values for the four compounds, a linear regression was fitted, and the R 2 value for this linear regression is displayed in the bottom right corner of each graph. Relaxation p(50% relax) values were expressed as the concentration (−Log) of the PDE1 inhibitors giving a 50% relaxation of pre‐contraction with U46619. Data are mean ± SEM. n = 3–9.
PDE1‐inhibition induced relaxation via the the NO‐cGMP‐PKG signalling pathway
To determine whether the relaxation induced by PDE1 inhibition was mediated through a cGMP/PKG‐mediated and/or cAMP/PKA‐mediated pathway, Lu AF58027 cumulative concentration–response curves were obtained in the presence of the PKG inhibitor (Rp‐8, 10 μM), or the PKA inhibitor (H89, 1 μM) in arteries pre‐contracted with U46619. Rp‐8 caused an eightfold rightward shift of the Lu AF58027 concentration–response curve without affecting maximal relaxation. In contrast, H89 did not change the relaxation response to Lu AF58027 (Figure 5A, C), thereby indicating that the PDE1 response was via cGMP.
Figure 5.
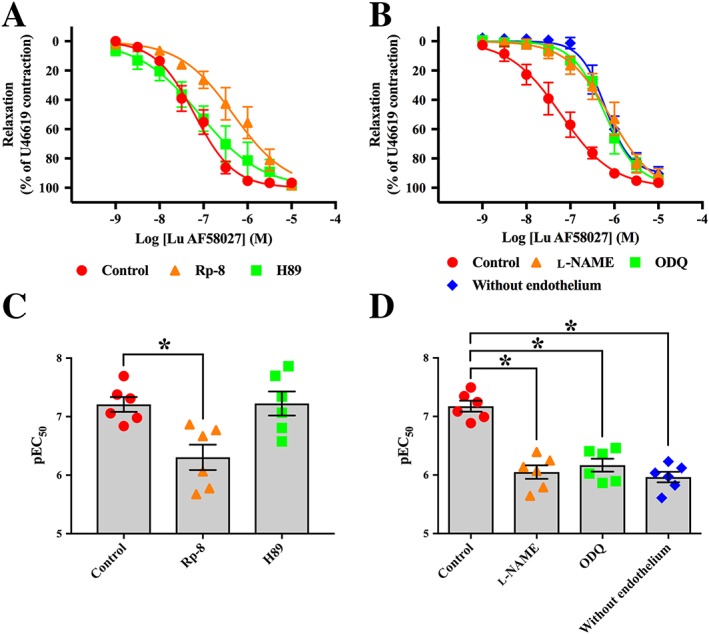
Relaxation induced by PDE1 inhibition is reduced by inhibition of PKG and the NO‐cGMP pathway. Cumulative concentration–response curves for the PDE1 inhibitor, Lu AF58027, in the presence of the PKG inhibitor, Rp‐8 (10 μM), or the PKA inhibitor, H89 (1 μM) (A), or in the presence of the NOS inhibitor, l‐NAME (100 μM), the soluble guanylate cyclase inhibitor, ODQ (3 μM), and in arterial segments without endothelium (B). (C) and (D) illustrate the pEC50 values for Lu AF58027 for the different treatments in (A) and (B). Mesenteric arteries were contracted with U46619 to approximately 80% of maximal contraction. Data are mean ± SEM. n = 6 arteries from separate rats. Student's t‐test with Bonferroni's multiple comparisons test. *P < 0.05 from control.
cGMP levels and hence the activity of PKG may be regulated through the bioavailability of endothelium‐derived NO and activation of sGC. Therefore, to further support that the AF58027‐induced relaxation was mediated through a cGMP/PKG‐mediated pathway, Lu AF58027 concentration–response curves were obtained in the presence of the NOS inhibitor, l‐NAME (100 μM) or the sGC inhibitor, ODQ (3 μM), or in arteries where the endothelium had been removed and pre‐contracted with U46619. In the presence of l‐NAME or ODQ, the Lu AF58027 concentration–response curve was rightward shifted (~16‐fold), and maximal relaxation remained unchanged compared with control (Figure 5B, D). A similar rightward shift of the Lu AF58027 concentration–response curve was observed in endothelium‐denuded arteries (Figure 5B, D). Lu AF58027 (10 nM) potentiated the relaxation response to the sGC activator, BAY 41‐2272 (threefold leftward shift; Figure 6A, C), but not the AC activator, forskolin (Figure 6B, D). The competitive PKG inhibitor, Rp‐8, caused approximately a threefold rightward shift of the BAY 41‐2272 concentration–response curve, an effect that was not observed when Rp‐8 was incubated in combination with Lu AF58027 (Figure 6A, C). The lack of effect of Rp‐8 and H89 on forskolin‐induced and BAY 41‐2272‐induced relaxation, respectively, indicated that at the concentrations used Rp‐8 and H89 did not inhibit PKA and PKG respectively (Figure 6A, C).
Figure 6.
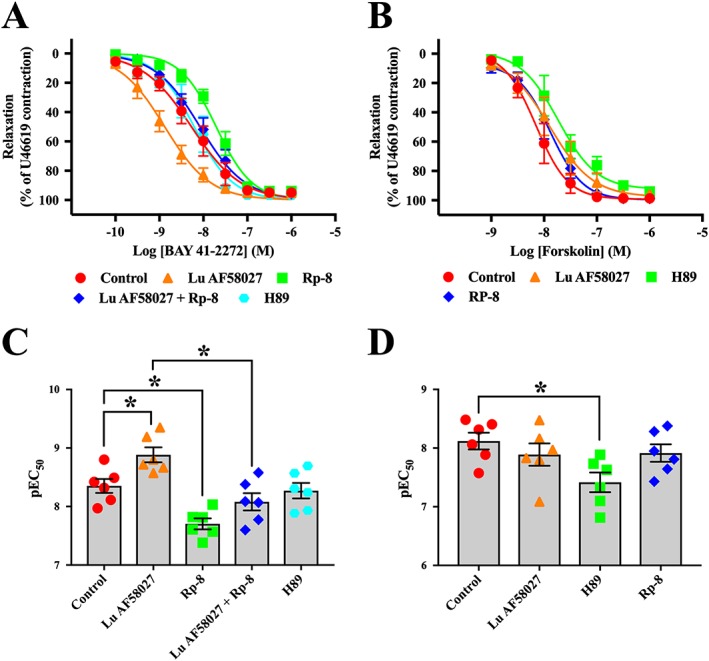
PDE1 inhibition potentiates sGC activator‐induced relaxation, but not AC activator‐induced relaxation. Cumulative concentration–response curves for the sGC activator, BAY 41‐2272, in the presence of the PDE1 inhibitor, Lu AF58027 (10 nM), and/or the PKG inhibitor, Rp‐8 (10 μM) (A). Cumulative concentration–response curves for the AC activator, forskolin, in the presence of the PDE1 inhibitor, Lu AF58027 (10 nM), or the PKA inhibitor, H89 (1 μM) (B). (C) and (D) show pEC50 values for BAY 41‐2272 and forskolin for the different treatments in (A) and (B) respectively. Mesenteric arteries were contracted with U46619 to approximately 80% of maximal contraction. Data are mean ± SEM. n = 6 arteries from separate rats. Student's t‐test with Bonferroni's multiple comparisons test. *P < 0.05, comparisons are indicated by lines.
Discussion
In this study, using in situ hybridization and qPCR for localization and quantification of PDE1 isoforms, and isometric tension recordings for functional investigations, we report the following novel findings: (i) Pde1a was located in VSMCs of the vascular wall; (ii) the relative expression of transcripts of the Pde1 isoforms in rat mesenteric arteries was Pde1a > Pde1b > Pde1c; and (iii) PDE1 inhibition‐induced relaxation of rat mesenteric arteries using PDE1 inhibitors was likely mediated through the PDE1A isoform and was predominantly driven by the NO‐cGMP‐PKG pathway. We propose that these isoform‐selective PDE1 inhibitors are powerful investigative tools that can be exploited to ascertain the physiological and pathological roles of PDE1 isoforms.
The expression of PDE1 isoforms is primarily dependent on species and the state/phenotype of the VSMCs under investigation. PDE1A mRNA and protein are expressed in contractile and proliferative rat and human aortic VSMCs in primary culture (Rybalkin et al., 1997; Nagel et al., 2006) and in mesenteric and femoral arteries of male rats (Giachini et al., 2011; Laursen et al., 2017). Pde1b has also been reported in rat mesenteric arteries (Laursen et al., 2017), as well as in VSMCs from monkeys and baboons (Yan, 2015). In contrast, Pde1c expression is low in medial contractile VSMC and is absent from rat mesenteric arteries (Laursen et al., 2017). It is however induced in proliferative VSMCs in culture and under conditions of vascular injury (Rybalkin et al., 1997; Rybalkin et al., 2002; Cai et al., 2015). In this study, we confirm that Pde1a is the predominant isoform in rat mesenteric resistance arteries (Giachini et al., 2011; Laursen et al., 2017) and furthermore localizes its expression to VSMCs.
The PDE1 subfamily has previously been implicated in the control of vascular tone. The weakly selective PDE1 inhibitors, nimodipine and vinpocetine, have been shown to elicit relaxation in the rat aorta (Noguera et al., 2001) and small mesenteric artery (Giachini et al., 2011). Furthermore, in the presence of vinpocetine, rat small mesenteric arteries are less sensitive to the vasoconstrictor agent, phenylephrine (Giachini et al., 2011). However, the relative contribution of specific PDE1 isoforms to the regulation of vascular tone is unknown, although the prevailing expression of PDE1A in the cytoplasm of contractile VSMCs suggests a role for PDE1A. The inability to assign functional roles to PDE1 isoforms can be principally attributed to a lack of compounds that selectively inhibit specific PDE1 isoforms. Vinpocetine was originally considered to be selective for PDE1 (Hagiwara et al., 1984), but has now been shown to have limited selectivity for PDE1 over PDE2–5 (Saeki and Saito, 1993) and may also activate Ca2+‐activated K+ channels at comparable concentrations (Wu et al., 2001). Although a selective inhibitor of PDE1 exists and studies have successfully reduced PDE1A expression with siRNAs in rat ventricular myocytes and in subcultured VSMCs (Nagel et al., 2006; Miller et al., 2009), the effect of PDE1A inhibition on vascular tone has not been investigated. Using PDE1 isoform‐selective inhibitors, we found that inhibition of PDE1 induced relaxation in pre‐contracted small mesenteric arteries. With the exception of Lu AF66896, the PDE1 inhibitors elicited similar relaxation responses irrespective of the contractile agonist. It is unclear why Lu AF66896 only elicited complete relaxation in arteries pre‐contracted with NA and not U46619. In rat mesenteric arteries, NA and U46619 induce contraction via the α1 adrenoceptor and Tx receptor respectively (Docherty, 2010). Although both receptors are Gq protein coupled and mobilize [Ca2+]i via the PLCβ/inositol 1,4,5‐triphosphate/DAG pathway (Sellers and Stallone, 2008), NA‐mediated contraction additionally entails Ca2+ influx following VSMC membrane depolarization (Plane and Garland, 1996). It is possible that PDE1 activates differently depending on the source of Ca2+, explaining the difference in relaxation by PDE1 inhibition with Lu AF66896 in NA‐contracted and U46619‐contracted arteries. Another explanation stems from the knowledge that NA can activate Gs‐coupled β2‐adrenoceptors on VSMC, thereby activating AC to produce cAMP. By preventing the degradation of cAMP, Lu AF66896 may elicit full relaxation in NA‐contracted but not U46619‐contracted arteries.
Of the PDE1 inhibitors tested, Lu AF58027, which has the greatest affinity for PDE1A out of the PDE1 inhibitors, was the most potent at eliciting relaxation. Although the compound exhibits greater affinity for PDE1C than PDE1A and PDE1B, suggesting a role for PDE1C in the regulation of arterial tone, the lack of PDE1C transcripts in rat mesenteric arteries suggests that the relaxation induced by PDE1 inhibition is unlikely to occur via the PDE1C isoform. Given that the expression of PDE1 in VSMCs of rat mesenteric arteries appears to be limited predominantly to the PDE1A isoform, Lu AF80527 is likely to be mediating relaxation via the PDE1A rather than PDE1C isoform. Furthermore, an excellent linear correlation was observed between each compound's p(50% relax) values for relaxation and their pK i values for PDE1A, whereas a poor linear correlation was observed between p(50% relax) values for relaxation and pK i values for PDE1B and PDE1C. That is, PDE1 inhibitors with a higher potency for inhibiting the PDE1A isoform were more effective at inducing relaxation in rat mesenteric arteries compared with PDE1 inhibitors with a lower affinity for the PDE1A isoform. Further studies are required to confirm whether these PDE1 inhibitors have similar effects in different vascular beds given their different expression profiles of PDE subfamilies (Matsumoto et al., 2003).
To elucidate whether the relaxation induced by PDE1A inhibition was mediated through cAMP/PKA or cGMP/PKG, we used the PKG inhibitor Rp‐8 and found that this drug inhibited Lu AF58027‐induced relaxation, whereas inhibition of PKA with H89 was without effect. ODQ, an inhibitor of sGC, also reduced relaxation induced by PDE1 inhibition. Laursen et al. (2017) similarly demonstrated ODQ‐sensitive relaxation to Lu AF58027 in rat mesenteric arteries pre‐contracted with U46619 and the selective α1‐adrenergic receptor agonist, phenylephrine. Accordingly, PDE1 inhibition potentiated the relaxation induced by the sGC activator BAY 41‐2272, an effect that was abolished with PKG inhibition. In contrast Lu AF58027 did not potentiate cAMP‐mediated relaxation induced by the AC activator, forskolin. Taken together, our results suggest that PDE1 inhibition mediates relaxation predominantly through the cGMP/PKG pathway. However, the contribution of the AC/cAMP pathway cannot be completely dismissed; a recent study by (Laursen et al., 2017) showed that the AC inhibitor, SQ22536, partially inhibited relaxation induced by Lu AF58027 in the rat mesenteric artery.
Notably, although Pde1a was found almost exclusively in VSMCs, NOS inhibition and removal of the endothelium reduced the relaxation induced by Lu AF58027. This implies there is a basal production and release of NO from the endothelium in these arteries that is responsible for a cGMP pool that is regulated by PDE1A. This further supports the notion that PDE1 inhibition‐mediated relaxation is predominantly due to the inhibition of the cGMP‐preferring isoform, PDE1A (Loughney et al., 1996), and that PDE1A may play a role in modulating the degree of NO‐mediated vasorelaxation. Further studies are needed to clarify the effect of PDE1A inhibition on intracellular cAMP/cGMP levels and the signalling mechanism/s downstream of PKG. Inhibition of cGMP generation either by NOS inhibition, endothelium removal or sGC inhibition caused a rightward shift of the concentration‐relaxation response curve to Lu AF58027 without suppression of maximal relaxation. This suggests that cGMP is still being generated, albeit at a lower rate, potentially from a small proportion of sGC that has not been inhibited and/or from membrane‐bound GC. We cannot rule out that PDE5 inhibition also contributes to the relaxation response at high concentrations of Lu AF58027 (>1 μM; Table 1).
There is substantial evidence of crosstalk between cyclic nucleotide pathways and such crosstalk can occur at multiple levels (Pelligrino and Wang, 1998). For example, cGMP and cAMP can cross‐activate each other's effector kinases and compete for dual‐specificity PDEs. Furthermore, the activity of several PDEs, such as PDE2 and PDE5, is allosterically activated by cyclic nucleotides (Tsai and Kass, 2009). In addition to Lu AF58027 having no effect on forskolin‐mediated relaxation, H89‐mediated PKA inhibition did not alter cGMP‐dependent relaxation induced by Lu AF58027. The data suggest that there is little reciprocal regulation between PDE1A, the cGMP pool it regulates and the cAMP pool generated by AC activation.
Recently, several studies have implicated PDE1A in the pathogenesis of various cardiovascular diseases. In rats with angiotensin II‐induced hypertension, cGMP levels are attenuated in the aorta and small mesenteric arteries; this was attributed to an up‐regulation of PDE1A (Giachini et al., 2011). These arteries exhibited augmented maximal contraction to phenylephrine compared with arteries from control rats; presumably, this is a functional consequence of increased PDE1 activity and reduced cGMP bioavailability (Giachini et al., 2011). The PDE1 family has also been implicated in the pathogenesis of pulmonary hypertension. PDE1A and PDE5A were found to be up‐regulated in pulmonary arterial smooth muscle cells in both mice and rats of two different experimental models of pulmonary hypertension (Schermuly et al., 2007). Recent single nucleotide polymorphism (SNP) association studies also support that PDE1A could have an impact on BP. A study of 87 736 humans found that the minor allele of the SNP rs16823124 in a PDE1A intron was associated with increased mean arterial pressure and diastolic BP (Tragante et al., 2014). Infusion of Lu AF58027 (3 mg·kg−1·h−1) in anaesthetized rats was recently shown to lower mean arterial pressure by ~4–7 mmHg in anaesthetized rats (Laursen et al., 2017). Furthermore, PDE1 isoforms have been suggested to play a role in atherosclerosis, age‐related vascular disease and VSMC and cardiomyocyte dysfunction (Rybalkin et al., 2002; Miller et al., 2009; Chan and Yan, 2011; Cai et al., 2015; Nino et al., 2015). Taken together, these results identify PDE1 isoforms as a potential treatment target in vascular diseases, where the pathogenesis involves a disturbed NO/cGMP signalling in VSMCs or excessive VSMC proliferation.
In summary, we have shown that Pde1a is the dominant Pde1 isoform present in VSMCs in mesenteric arteries, and using PDE1 subtype selective inhibitors, we show that PDE1A inhibition can induce relaxation of these arteries ex vivo. We further show that this effect is mediated through enhanced intracellular cGMP/PKG signalling. Our study warrants further exploration of PDE1 isoform specific inhibitors as drugs to improve vascular dysfunction. Isoform‐selective PDE1 inhibition is clinically attractive because it provides the possibility for more selective pharmacological interventions. The isoform‐selective inhibitors described here provide the tools to investigate the contributions of different PDE1 isoforms to various biological functions.
Author contributions
M.M.K. and T.D. performed the experiments, analysed the data and drafted the manuscript. P.H.L. performed the experiments, analysed the data and contributed to the manuscript writing. C.T.C. and L.F. analysed the data. D.C. performed the experiments and analysed the data. L.K.R. performed the experiments. M.G. and J.N. analysed the data, provided the project supervision and contributed to the manuscript writing. J.K. and C.A. provided the project supervision and contributed to the manuscript writing.
Conflict of interest
P.H.L., C.T.C., D.C., L.K.R., L.F., M.G., J.K. and J.N. are employed by H. Lundbeck A/S. T.D. has worked as a consultant for the company H. Lundbeck A/S. The PDE1 inhibitors used in this study have been generated by Lundbeck, and some of them are claimed in patent applications from H. Lundbeck A/S.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
We thank Annette Bjørn and Kirsten Jørgensen for their skilful technical assistance.
M.M.K. was supported by the Novo Nordisk Foundation grant (11789) awarded to C.A.
Khammy, M. M. , Dalsgaard, T. , Larsen, P. H. , Christoffersen, C. T. , Clausen, D. , Rasmussen, L. K. , Folkersen, L. , Grunnet, M. , Kehler, J. , Aalkjær, C. , and Nielsen, J. (2017) PDE1A inhibition elicits cGMP‐dependent relaxation of rat mesenteric arteries. British Journal of Pharmacology, 174: 4186–4198. doi: 10.1111/bph.14034.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold WP, Mittal CK, Katsuki S, Murad F (1977). Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′‐cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci U S A 74: 3203–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AT, Beavo JA (2006). Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58: 488–520. [DOI] [PubMed] [Google Scholar]
- Bobin P, Belacel‐Ouari M, Bedioune I, Zhang L, Leroy J, Leblais V et al (2016). Cyclic nucleotide phosphodiesterases in heart and vessels: a therapeutic perspective. Arch Cardiovasc Dis 109: 431–443. [DOI] [PubMed] [Google Scholar]
- Butt E, Eigenthaler M, Genieser H‐G (1994). (Rp)‐8‐pCPT‐cGMPS, a novel cGMP‐dependent protein kinase inhibitor. Eur J Pharmacol Mol Pharmacol 269: 265–268. [DOI] [PubMed] [Google Scholar]
- Cai Y, Nagel DJ, Zhou Q, Cygnar KD, Zhao H, Li F et al (2015). Role of cAMP‐phosphodiesterase 1C signaling in regulating growth factor receptor stability, vascular smooth muscle cell growth, migration, and neointimal hyperplasia. Circ Res 116: 1120–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Yan C (2011). PDE1 isozymes, key regulators of pathological vascular remodeling. Curr Opin Pharmacol 11: 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T et al (1990). Inhibition of forskolin‐induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP‐dependent protein kinase, N‐[2‐(p‐bromocinnamylamino) ethyl]‐5‐isoquinolinesulfonamide (H‐89), of PC12D pheochromocytoma cells. J Biol Chem 265: 5267–5272. [PubMed] [Google Scholar]
- Conti M, Beavo J (2007). Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 76: 481–511. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander S, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docherty JR (2010). Subtypes of functional α1‐adrenoceptor. Cell Mol Life Sci 67: 405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B (1995). Potent and selective inhibition of nitric oxide‐sensitive guanylyl cyclase by 1H‐[1, 2, 4] oxadiazolo [4, 3‐a] quinoxalin‐1‐one. Mol Pharmacol 48: 184–188. [PubMed] [Google Scholar]
- Giachini FR, Lima VV, Carneiro FS, Tostes RC, Webb RC (2011). Decreased cGMP level contributes to increased contraction in arteries from hypertensive rats: role of phosphodiesterase 1. Hypertension 57: 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratzke C, Angulo J, Chitaley K, Dai Y, Kim NN, Paick JS et al (2010). Anatomy, physiology, and pathophysiology of erectile dysfunction. J Sex Med 7 (1pt2): 445–475. [DOI] [PubMed] [Google Scholar]
- Graves J, Poston L (1993). β‐Adrenoceptor agonist mediated relaxation of rat isolated resistance arteries: a role for the endothelium and nitric oxide. Br J Pharmacol 108: 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara M, Endo T, Hidaka H (1984). Effects of vinpocetine on cyclic nucleotide metabolism in vascular smooth muscle. Biochem Pharmacol 33: 453–457. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Rybalkin SD, Pi X, Wang Y, Zhang C, Munzel T et al (2001). Upregulation of phosphodiesterase 1A1 expression is associated with the development of nitrate tolerance. Circulation 104: 2338–2343. [DOI] [PubMed] [Google Scholar]
- Komas N, Lugnier C, Andriantsitohaina R, Stoclet J‐C (1991). Characterisation of cyclic nucleotide phosphodiesterases from rat mesenteric artery. Eur J Pharmacol 208: 85–87. [DOI] [PubMed] [Google Scholar]
- Laursen M, Beck L, Kehler J, Christoffersen CT, Bundgaard C, Mogensen S et al (2017). Novel selective phosphodiesterase type 1 inhibitors cause vasodilatation and lower blood pressure in rats. Br J Pharmacol 174: 2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Zheng H, Zhao J, Zhang L, Yao W, Zhu H et al (2016). Discovery of potent and selective inhibitors of phosphodiesterase 1 for the treatment of cognitive impairment associated with neurodegenerative and neuropsychiatric diseases. J Med Chem 59: 1149–1164. [DOI] [PubMed] [Google Scholar]
- Loughney K, Martins TJ, Harris EAS, Sadhu K, Hicks JB, Sonnenburg WK et al (1996). Isolation and characterization of cDNAs corresponding to two human calcium, calmodulin‐regulated, 3′,5′‐cyclic nucleotide phosphodiesterases. J Biol Chem 271: 796–806. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Kobayashi T, Kamata K (2003). Phosphodiesterases in the vascular system. J Smooth Muscle Res 39: 67–86. [DOI] [PubMed] [Google Scholar]
- Maurice DH, Palmer D, Tilley DG, Dunkerley HA, Netherton SJ, Raymond DR et al (2003). Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol Pharmacol 64: 533–546. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CL, Oikawa M, Cai Y, Wojtovich AP, Nagel DJ, Xu X et al (2009). Role of Ca2+/calmodulin‐stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ Res 105: 956–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgado M, Cairrão E, Santos‐Silva AJ, Verde I (2012). Cyclic nucleotide‐dependent relaxation pathways in vascular smooth muscle. Cell Mol Life Sci 69: 247–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W (1977). Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res 41: 19–26. [DOI] [PubMed] [Google Scholar]
- Nagel DJ, Aizawa T, Jeon K‐I, Liu W, Mohan A, Wei H et al (2006). Role of nuclear Ca2+/calmodulin‐stimulated phosphodiesterase 1A in vascular smooth muscle cell growth and survival. Circ Res 98: 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netherton SJ, Maurice DH (2005). Vascular endothelial cell cyclic nucleotide phosphodiesterases and regulated cell migration: implications in angiogenesis. Mol Pharmacol 67: 263–272. [DOI] [PubMed] [Google Scholar]
- Nino PKB, Durik M, Danser AHJ, De Vries R, Musterd‐Bhaggoe UM, Meima ME et al (2015). Phosphodiesterase 1 regulation is a key mechanism in vascular aging. Clin Sci 129: 1061–1075. [DOI] [PubMed] [Google Scholar]
- Noguera M, Ivorra M, Lugnier C, D'Ocon P (2001). Role of cyclic nucleotide phosphodiesterase isoenzymes in contractile responses of denuded rat aorta related to various Ca2+ sources. Naunyn Schmiedebergs Arch Pharmacol 363: 612–619. [DOI] [PubMed] [Google Scholar]
- Pelligrino DA, Wang Q (1998). Cyclic nucleotide crosstalk and the regulation of cerebral vasodilation. Prog Neurobiol 56: 1–18. [DOI] [PubMed] [Google Scholar]
- Plane F, Garland CJ (1996). Influence of contractile agonists on the mechanism of endothelium‐dependent relaxation in rat isolated mesenteric artery. Br J Pharmacol 119: 191–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalkin SD, Bornfeldt KE, Sonnenburg WK, Rybalkina IG, Kwak KS, Hanson K et al (1997). Calmodulin‐stimulated cyclic nucleotide phosphodiesterase (PDE1C) is induced in human arterial smooth muscle cells of the synthetic, proliferative phenotype. J Clin Invest 100: 2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalkin SD, Rybalkina I, Beavo JA, Bornfeldt KE (2002). Cyclic nucleotide phosphodiesterase 1C promotes human arterial smooth muscle cell proliferation. Circ Res 90: 151–157. [DOI] [PubMed] [Google Scholar]
- Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA (2003). Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res 93: 280–291. [DOI] [PubMed] [Google Scholar]
- Saeki T, Saito I (1993). Isolation of cyclic nucleotide phosphodiesterase isozymes from pig aorta. Biochem Pharmacol 46: 833–839. [DOI] [PubMed] [Google Scholar]
- Schermuly RT, Pullamsetti SS, Kwapiszewska G, Dumitrascu R, Tian X, Weissmann N et al (2007). Phosphodiesterase 1 upregulation in pulmonary arterial hypertension. Circulation 115: 2331–2339. [DOI] [PubMed] [Google Scholar]
- Sellers MM, Stallone JN (2008). Sympathy for the devil: the role of thromboxane in the regulation of vascular tone and blood pressure. Am J Physiol Heart Circ Physiol 294: H1978. [DOI] [PubMed] [Google Scholar]
- Simonds WF (1999). G protein regulation of adenylate cyclase. Trends Pharmacol Sci 20: 66–73. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tragante V, Barnes MR, Ganesh SK, Lanktree MB, Guo W, Franceschini N et al (2014). Gene‐centric meta‐analysis in 87,736 individuals of European ancestry identifies multiple blood‐pressure‐related loci. Am J Hum Genet 94: 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai EJ, Kass DA (2009). Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther 122: 216–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winquist RJ, Faison EP, Waldman SA, Schwartz K, Murad F, Rapoport RM (1984). Atrial natriuretic factor elicits an endothelium‐independent relaxation and activates particulate guanylate cyclase in vascular smooth muscle. Proc Natl Acad Sci 81: 7661–7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S‐N, Li H‐F, Chiang H‐T (2001). Vinpocetine‐induced stimulation of calcium‐activated potassium currents in rat pituitary GH3 cells. Biochem Pharmacol 61: 877–892. [DOI] [PubMed] [Google Scholar]
- Yan C (2015). Cyclic nucleotide phosphodiesterase 1 and vascular aging. Clin Sci 129: 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung‐Chi C, Prusoff WH (1973). Relationship between the inhibition constant (K i) and the concentration of inhibitor which causes 50 per cent inhibition (I 50) of an enzymatic reaction. Biochem Pharmacol 22: 3099–3108. [DOI] [PubMed] [Google Scholar]