

Abstract
Background and Purpose
Sustained pulmonary vasoconstriction and excessive pulmonary vascular remodelling are two major causes of elevated pulmonary vascular resistance in patients with pulmonary arterial hypertension. The purpose of this study was to investigate whether chloroquine induced relaxation in the pulmonary artery (PA) and attenuates hypoxia‐induced pulmonary hypertension (HPH).
Experimental Approach
Isometric tension was measured in rat PA rings pre‐constricted with phenylephrine or high K+ solution. PA pressure was measured in mouse isolated, perfused and ventilated lungs. Fura‐2 fluorescence microscopy was used to measure cytosolic free Ca2+ concentration levels in PA smooth muscle cells (PASMCs). Patch‐clamp experiments were performed to assess the activity of voltage‐dependent Ca2+ channels (VDCCs) in PASMC. Rats exposed to hypoxia (10% O2) for 3 weeks were used as the model of HPH or Sugen5416/hypoxia (SuHx) for in vivo experiments.
Key Results
Chloroquine attenuated agonist‐induced and high K+‐induced contraction in isolated rat PA. Pretreatment with l‐NAME or indomethacin and functional removal of endothelium failed to inhibit chloroquine‐induced PA relaxation. In PASMC, extracellular application of chloroquine attenuated store‐operated Ca2+ entry and ATP‐induced Ca2+ entry. Furthermore, chloroquine also inhibited whole‐cell Ba2+ currents through VDCC in PASMC. In vivo experiments demonstrated that chloroquine treatment ameliorated the HPH and SuHx models.
Conclusions and Implications
Chloroquine is a potent pulmonary vasodilator that may directly or indirectly block VDCC, store‐operated Ca2+ channels and receptor‐operated Ca2+ channels in PASMC. The therapeutic potential of chloroquine in pulmonary hypertension is probably due to the combination of its vasodilator, anti‐proliferative and anti‐autophagic effects.
Abbreviations
- [Ca2+]cyt
cytosolic free Ca2+ concentration
- BK
large‐conductance Ca2+‐activated K+
- ChTX
charybdotoxin
- CPA
cyclopiazonic acid
- HPH
hypoxia‐induced pulmonary hypertension
- HPV
hypoxic pulmonary vasoconstriction
- IPAH
idiopathic pulmonary arterial hypertension
- MCT
monocrotaline
- PA
pulmonary artery
- PAH
pulmonary arterial hypertension
- PAP
pulmonary arterial pressure
- PASMCs
pulmonary artery smooth muscle cells
- ROCC
receptor‐operated Ca2+ channel
- ROCE
receptor‐operated Ca2+ entry
- RV/(LV + S)
ratio of the weight of right ventricle to the weight of left ventricle and septum (also referred to as Fulton index)
- RV/BW
ratio of the weight of right ventricle to the body weight
- RVSP
right ventricular systolic pressure
- SNP
sodium nitroprusside
- SOCC
store‐operated Ca2+ channel
- SOCE
store‐operated Ca2+ entry
- SR
sarcoplasmic reticulum
- TAS2R
taste receptor type 2
- TEA
tetraethylammonium
- VDCC
voltage‐dependent Ca2+ channel
Introduction
Idiopathic pulmonary arterial hypertension (IPAH) is a progressive and fatal disease with a 5 year survival rate of only 34% (McLaughlin et al., 2009). Sustained pulmonary vasoconstriction and vascular remodelling characterized by concentric wall thickening and lumen obliteration of small‐sized and medium‐sized pulmonary artery (PA) are the major causes of the elevated pulmonary vascular resistance and pulmonary arterial pressure (PAP) in patients with IPAH (Kuhr et al., 2012; Tuder et al., 2013; Sakao et al., 2015). Increased cytosolic free Ca2+ concentration ([Ca2+]cyt) in PA smooth muscle cells (PASMCs) is an important trigger for pulmonary vasoconstriction and a necessary stimulus for pulmonary vascular remodelling by increasing PASMC proliferation and migration. Thus, enhanced Ca2+ signalling in PASMC is an important therapeutic target for developing novel treatments for pulmonary hypertension (PH).
Chloroquine is mostly known for its use in the treatment for Plasmodium vivax malaria (White, 1996) but it has also been used to treat autoimmune disorders such as rheumatoid arthritis and systemic lupus erythematosus (Solomon and Lee, 2009; Rainsford et al., 2015). Chloroquine is a weak base that can increase the pH in lysosomal lumens, which disrupts lysosomal enzymes, thereby inhibits autophagy (Klionsky et al., 2016). Autophagy is involved in several diseases including pulmonary arterial hypertension (PAH) (Jin and Choi, 2012). Under disease conditions, cells are reliant on autophagy for survival. Therefore, inhibition of autophagy by treatment with chloroquine leads to increased apoptosis. In a recent study, Long et al. (2013) found that in rats treated with monocrotaline (MCT), the induction of PH was associated with increased expression of markers associated with autophagy including LC3B‐II, ATG5 and p62. Chloroquine treatment prevented pulmonary vascular remodelling and inhibited the development of MCT‐induced PH. Furthermore, chloroquine inhibited autophagy and proliferation and increased apoptosis in PASMC in MCT‐PH rats both in vitro and in vivo (Long et al., 2013).
In addition to its anti‐autophagic effects, chloroquine induced vasodilation and decreased BP following treatment for malaria in humans (Looareesuwan et al., 1986; Anigbogu et al., 1993) and animals (Musabayane et al., 1994; McCarthy et al., 2016). In thoracic aortas precontracted by noradrenaline or high K+, treatment with chloroquine resulted in a concentration‐dependent relaxation (Aziba and Okpako, 2003). Furthermore, vasodilatation following chloroquine was associated with blockade of L‐type voltage‐dependent Ca2+ channels (VDCCs) (Manson et al., 2014; Sai et al., 2014). A recent study found that chloroquine induced relaxation in human PAs precontracted by the thromboxane A2 analogue U46619 and phenylephrine (Manson et al., 2014). Chloroquine also induced bronchodilation (Deshpande et al., 2010; Pulkkinen et al., 2012; Zhang et al., 2013; Tan and Sanderson, 2014).
Chloroquine is bitter tasting and an agonist of the G‐protein‐coupled bitter taste receptor type 2 (TAS2R). Bronchodilation is mediated through canonical TAS2R signalling that initially results in a transient increase in [Ca2+]cyt, which subsequently activates the large‐conductance Ca2+‐activated K+ (BK) channels and enhances K+ efflux leading to membrane hyperpolarization and closure of VDCC (Deshpande et al., 2010). Chloroquine also induced membrane hyperpolarization and inhibited L‐type VDCC in airway smooth muscle cells, as observed in thoracic aorta (Sai et al., 2014). This blockage of L‐type VDCC ultimately leads to an overall decrease in Ca2+ influx and bronchodilation or airway smooth muscle relaxation (Zhang et al., 2013). These studies indicated that chloroquine could induce smooth muscle relaxation through activation of BK channels and/or inhibition of VDCC. However, the mechanism by which chloroquine inhibits pulmonary vasoconstriction remains uncertain.
In this study, we have found that short‐term (2–15 min) and long‐term (24–48 h) treatment with chloroquine had different effects. The short‐term treatment rapidly caused relaxation in isolated PA, decreased PAP in isolated perfused and ventilated lung, reduced whole‐cell Ba2+ currents through VDCC in PASMC and inhibited store‐operated Ca2+ entry (SOCE) and ATP‐induced Ca2+ entry in PASMC, but short‐term treatment of PASMC with chloroquine did not affect autophagy. However, long‐term treatment of PASMC with chloroquine, did inhibit autophagy and markedly inhibited PASMC proliferation. In this study, we performed in vitro, ex vivo and in vivo experiments to investigate the potential mechanisms involved in the beneficial effects on PH, mediated by chloroquine.
Methods
Pulmonary hypertension rat model and chloroquine treatment
All animal care and experimental procedures were approved by the Animal Care and Use Committee of Guangzhou Medical University and the University of Arizona. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). All animals were housed in standard plastic cages on sawdust bedding in an air‐conditioned room at 22 ± 1°C under lighting controls with 12 h light and dark cycles. Standard mouse chow and tap water were provided ad libitum.
For the hypoxia‐induced PH (HPH) rat model, adult male Sprague Dawley (SD) rats (175–250 g) were exposed to hypoxia (10% O2) for 21 days as previously described (Wang et al., 2006; Das et al., 2012). For Sugen 5416/hypoxia (SuHx) PH rat model, SD rats were either injected s.c. with 20 mg·kg−1 SU5416 (Sigma‐Aldrich, St. Louis, MO, USA) or vehicle, exposed to hypoxia conditions for 3 weeks (10% O2) and returned to normoxia (21% O2) for 2 weeks. Normoxic controls were kept in room air only (21% O2) for a total of 5 weeks. For the HPH model, rats were housed in sets of three animals per cage and randomly assigned to four different experimental groups (n = 6 each group): (i) normoxic control, (ii) normoxia + chloroquine (CHQ), (iii) hypoxic control and (iv) hypoxia + CHQ. For SuHx model, rats were also randomly assigned to four different experimental groups: (i) normoxia for 5 weeks, (ii) Sugen/hypoxia for 3 weeks, (iii) Sugen/hypoxia for 3 weeks + normoxia for 2 weeks and (iv) Sugen/hypoxia for 3 weeks + CHQ/normoxia for 2 weeks. Chloroquine (C6628, Sigma‐Aldrich) was given to the rats by i.p. injection at 50 mg·kg−1·day−1 for 21 days, while control groups of animals received equivalent amounts of saline.
Haemodynamic evaluation and lung histochemistry experiment
Following hypoxic exposure, rats were anaesthetized with ketamine/xylazine before haemodynamic measurements. Right ventricular systolic pressure (RVSP) and right ventricular hypertrophy were measured using the same method as we described previously (Lu et al., 2010). Blood was collected into EDTA‐treated tubes to quantify haematocrit. The blood was placed in a long capillary tube and centrifuged in a microhaematocrit centrifuge (TG12, Xiangzhi Centrifuge Instrument Co., Changsha, China) at 14 730 × g for 15 min. The HCT level reached by the column of erythrocytes was determined with a scale reader. Paraffin‐embedded lung cross sections at a thickness of 5 μm were stained by haematoxylin and eosin (H&E) and visualized according to common histopathological procedures. PA wall thickness or the thickness of the smooth muscle layer of distal small PA (50–100 μm) was measured in H&E images of lung cross sections using the Image‐Pro Plus software.
Culture of PASMC
Human PASMCs were acquired from Lonza (Walkersville, MD, USA) and cultured in SmGM‐2 medium (Lonza) supplemented with SmGM‐2 SingleQuots (Lonza) according to the manufacturer's instruction. The cells at passages 5–10 were used for the experiments. The human PASMCs were incubated and maintained in a humidified atmosphere of 5% CO2 and 95% air at 37°C.
Measurement of cell proliferation and cytotoxicity
The 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT, from Sigma‐Aldrich) assay was used to quantify the level of cell proliferation in PASMC. For determining the short‐term treatment effect of chloroquine, PASMCs were incubated with different concentrations of chloroquine (0, 10, 20, 50, 100 and 200 μM) for 15 min. Following the treatment, PASMCs were washed with fresh medium to remove chloroquine and further incubated for 48 h after exposure. For determining the long‐term treatment effect of chloroquine, PASMCs were incubated with different concentrations of chloroquine (0, 1, 5, 10, 20, 50 and 100 μM) for 48 h. Cells were incubated with 0.5 mg·mL−1 MTT solution for 4 h at 37°C, followed by replacement with 200 μL DMSO after incubation. The absorbance was measured at 490 nm by an iMark Absorbance Reader (Bio‐Rad, Hercules, CA). For the cytotoxicity assay, the percentage of live cells was counted after treatment with 200 μM chloroquine for 15 min via automated Trypan Blue exclusion with the assistance of Countess II FL Automated Cell Counter (Life Technologies, Grand Island, NY).
Measurement of [Ca2+]cyt in PASMC
[Ca2+]cyt in human PASMC was measured using a Nikon digital imaging fluorescent microscopy system. Cells grown on 25 mm round coverslips were loaded with Fura‐2/AM for 60 min. Cells were alternately illuminated at 340 and 380 nm wavelengths by a Xenon lamp (Hamamatsu Photonics, Hamamatsu, Japan) connected to an inverted fluorescent microscope (Eclipse Ti‐E; Nikon, Tokyo, Japan). The fluorescence intensity emitted at 520 nm in cells was captured with an EM‐CCD camera (Evolve; Photometrics, Tucson, AZ, USA) and NIS Elements 3.2 software (Nikon). [Ca2+]cyt was measured as the ratio of fluorescence intensity (F 340/F 380) within an area of a region of interest (5 × 5 μm) in a cell recorded every 2 s. All experiments for measurement of [Ca2+]cyt were carried out at room temperature (22–24°C).
Immunocytochemistry
Cells grown on coverslips in chamber slides were fixed in 4% paraformaldehyde for 20 min, permeabilized using 0.2% Triton‐X‐100 for 10 min and incubated overnight with an anti‐LC3B antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA, 1:1000). The specimens were subsequently incubated with Alexa Fluor 488 (1:1000) secondary antibody for 1 h in the dark. DAPI was utilized to stain the nucleus. Images were captured using a fluorescence microscope (Olympus, Tokyo, Japan) and were photographed using a digital camera. The percentage of PASMC containing more than five LC3 dots was used as LC3‐positive cells to quantify autophagy. Selected fields with a minimum of 100 cells were scored.
Western blot analysis
Cells were lysed using ice‐cold RIPA buffer with freshly added protease inhibitor cocktail (Roche, Mannheim, Germany). PASMCs were incubated in lysis buffer for 20 min on ice and centrifuged at 12 000× g for 30 min at 4°C. Protein concentration was determined from the collected supernatant, followed by mixing and boiling samples in Laemmli buffer supplemented with 2‐mercaptoethanol (Basal Medium Eagle, Sigma‐Aldrich). Samples were run on SDS gels and transferred to nitrocellulose membranes according to a standard Western blot protocol. Primary antibodies used in the study include mouse P62 antibody (Santa Cruz Biotechnology, 1:1000), rabbit LC3B antibody (Santa Cruz Biotechnology, 1:1000) and mouse β‐actin antibody (Santa Cruz Biotechnology, 1:5000). Band intensity was quantified with ImageJ and standardized to β‐actin.
Measurement of pulmonary arterial pressure in isolated perfused and ventilated lung
C57BL/6 mice (22–25 g) were anaesthetized with Ketamine (100 mg·kg−1) and Xylazine (10 mg·kg−1) injected intraperitoneally and ventilated (Minivent type 845, Harvard Apparatus, Holliston, MA, USA) after a tracheostomy. Respiratory rate, tidal volume and positive end‐expiratory pressure were maintained at 80 breaths per min, 10 mL·kg−1 and 2 mmH2O respectively. Heparin (20 IU) was injected into the right ventricle to prevent blood coagulation. PAP was measured as previously described (Tang et al., 2015) using a pressure sensor (P75 Type 379, Hugo Sachs Elektronik‐Harvard Apparatus, March, Germany), which was attached to the PA catheter. The other end of the catheter was connected to a tube for PA perfusion with the Krebs–Ringer solution containing (in mM) NaCl 120, KCl 4.3, NaHCO3 19, KH2PO4 1.1, glucose 10, CaCl2 1.8 and MgCl2 1.2 (pH 7.4). The pulmonary circulation was maintained in a closed circuit via a peristaltic pump (ISM 834; Ismatec, Glattbrugg/Switzerland). For isotonic high K+ solutions (40 or 80 mM), NaCl was replaced by an equimolar amount of KCl to maintain normal osmolarity. To elicit hypoxic pulmonary vasoconstriction (HPV), we replaced the normoxic gas mixture (21% O2, 5% CO2 and balanced with N2) with hypoxic gas mixture (1% O2, 5% CO2 and balanced with N2) to ventilate the lung. PowerLab 8/30, Quad Bridge Amp and LabChart (AD Instruments, Inc., Colorado, Springs, CO) were used for data acquisition and storage. Experiments were carried out shortly after basal PAP stabilization, which usually occurred after 40–60 min.
Measurement of isometric tension in isolated rings of rat PA
The intrapulmonary arteries were carefully dissected out from male SD rats under anaesthesia, with a dissecting microscope. Freshly isolated PA rings (2 mm long) were suspended between two tungsten wires to record isometric tension. The two tungsten wires were inserted into the lumen of the PA and fixed respectively to a force transducer and a micrometer and mounted on wire myographs (DMT, Aarhus, Denmark). PA rings were superfused with 5 mL Krebs solution including (in mM): 138 NaCl, 1.8 CaCl2, 4.7 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 5 HEPES and 10 glucose (pH 7.4), maintained at 37°C and aerated with 95% O2/5% CO2.
The passive resting tension was set and maintained at an optimal tension of 300 mg. The rings were allowed to stabilize at the resting tension for approximately 1 h before experimentation. After the 1 h equilibration, the isolated PA rings were stimulated twice with 60 mM K+‐containing solution (60K) to stabilize the vessel and obtain a stable contractile response before further experimentation. The baseline tension of the isolated PA rings usually becomes stable after three challenges with a 60K‐containing solution. The ring was considered non‐viable if the ring could not provide reproducible contractions to 60K or phenylephrine, or because of poor recording quality. The ring was also excluded if it gave poor responses to phenylephrine, due to PA damage. For relaxation studies, rings were contracted with phenylephrine or high K+ solutions containing 40 mM (40K) or 80 mM (80K) of KCl. Selected experiments were performed using vessels with either intact or denuded endothelium. Endothelium was denuded mechanically using forceps before placing the PA rings on tungsten wires. Endothelium‐dependent vasodilation was assessed using ACh (endothelium‐dependent relaxation), while endothelium‐independent vasodilation was assessed using sodium nitroprusside (SNP, endothelium‐independent relaxation) after pre‐constriction of the artery with 1 μM phenylephrine. In some experiments, the rings were treated with l‐NG‐nitroarginine methyl ester (l‐NAME), indomethacin, apamin, paxilline, tetraethylammonium (TEA) and charybdotoxin (ChTX) for 30 min and continuously treated during the experiment to test the effect of chloroquine on phenylephrine‐induced contraction of the PA rings. Data are expressed as % relaxation of phenylephrine‐induced pre‐constriction.
Measurement of whole‐cell inward currents through VDCC in PASMC
Ca2+ currents were recorded with the conventional whole‐cell configuration of the patch‐clamp technique using an Axopatch 200B amplifier and a Digidata 1440A acquisition system (Molecular Devices, Sunnyvale, CA, USA). Low‐resistance patch pipettes (3–4 MΩ) made from borosilicate glass were fabricated on an electrode puller (model P‐97, Sutter Instrument, Novato, CA, USA) and fire polished with a microforge (MF‐63, Narishige Scientific Instrument Laboratories, Tokyo, Japan) for whole‐cell current recordings. Command voltage pulse protocols and data acquisition were performed using pClamp 10.7 (Molecular Devices). Currents were filtered at 1–2 kHz and digitized at 2–5 kHz. The extracellular solution for recording the Ca2+ currents contained the following (in mM): 115 NaCl, 10 BaCl2, 4.7 KCl, 0.5 MgCl2, 10 HEPES and 10 glucose (adjusted to pH 7.4 with NaOH). Cells were continuously superfused in the recording chamber with the extracellular solution at a flow rate of approximately 1.2 mL·min−1. The pipette solution for the Ca2+ current recordings contained the following (in mM): 130 CsCl, 10 EGTA, 5 Mg‐ATP and 10 HEPES (adjusted to 7.2 with CsOH). The P/4 subtraction protocol in the pClamp software was used for the subtraction of leak currents. All electrophysiological recordings were attained at room temperature (22–24°C).
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are expressed as mean ± SEMand analysed for statistical significance between two or among multiple groups using the unpaired Student's t‐test or one‐way ANOVA of the Sigma Plot statistical package (SigmaPlot 11.0; Systat Software, Inc., Chicago, IL, USA). Differences between means were considered to be significant at P < 0.05.
Materials
All drugs were obtained from Sigma Chemical unless otherwise stated. Chloroquine, ACh and SNP were prepared as concentrated stock solutions in saline. TEA was dissolved in distilled water as a stock solution. Paxilline (Cayman Chemical Company, Ann Arbor, MI, USA), ChTX, cyclopiazonic acid (CPA), l‐NAME and indomethacin were prepared as stock solutions in DMSO. Chloroquine, ACh, SNP and TEA were aliquoted at 4°C, and other drug stock solutions were aliquoted and kept frozen at −20°C until use.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
Chloroquine causes endothelium‐independent relaxation in rat PA rings precontracted with phenylephrine or high K+
PA rings were isolated from 7‐ to 9‐week‐old rats. Contraction of the isolated PA ring was induced with 1 μM phenylephrine. After a steady‐state contraction had been achieved, chloroquine was added cumulatively to the organ bath to yield final concentrations of 0.1, 1, 10, 50, 100 and 200 μM. We observed that chloroquine induced a concentration‐dependent relaxation of the PA ring with an EC50 value of 73 ± 2.9 μM (Figure 1A, B). Following treatment with the highest concentration (200 μM) of chloroquine, we observed 97.2% relaxation. After relaxation in the presence of 200 μM chloroquine, the PA rings were washed and then treated again with 1 μM phenylephrine to achieve a steady‐state contraction. The second round of treatment with 200 μM chloroquine resulted again in 97% relaxation (Figure 1C) indicating that chloroquine reversibly induced relaxation in PA rings.
Figure 1.
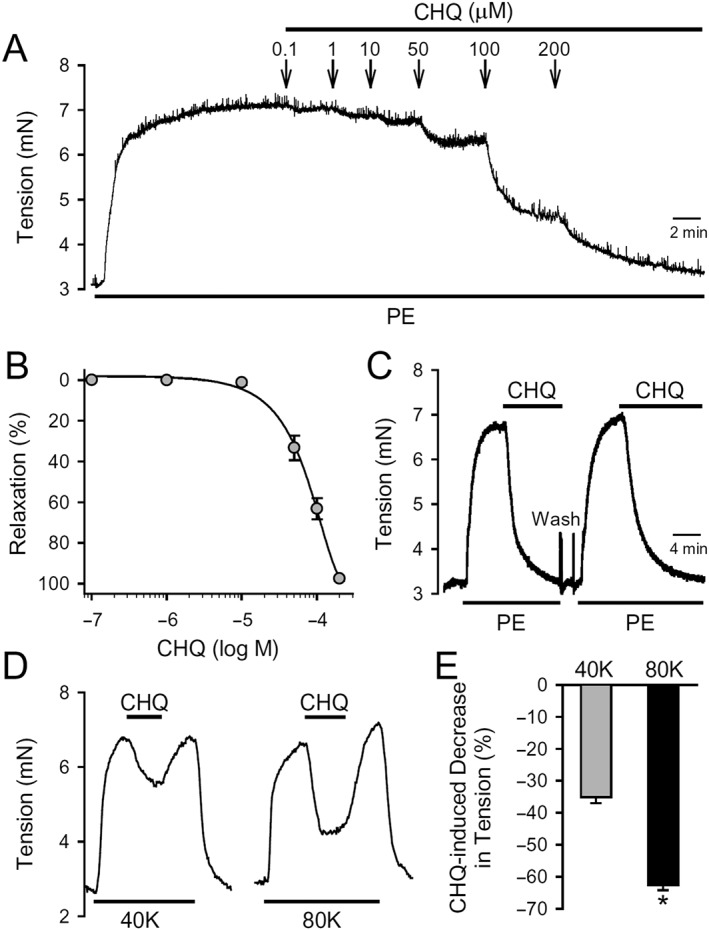
Vasorelaxant effect of chloroquine on rings of rat PA. (A) Representative record of isometric tension in an isolated PA ring constricted with 1 μM phenylephrine (PE) before and during application of 0.1, 1, 10, 50, 100 and 200 μM of chloroquine (CHQ). (B) Dose–response curve of chloroquine‐induced vasorelaxation in rings of rat PA, pre‐constricted with 1 μM phenylephrine. Data are means ± SEM from 12 independent experiments. (C) A representative record of isometric tension in a PA ring consecutively contracted with 1 μM phenylephrine before, during and after application of 200 μM chloroquine. (D) Representative records of isometric tension in a PA ring constricted with 40 mM (40K) or 80 mM (80K) K+‐containing solution before, during and after application of chloroquine (200 μM). (E) Summarized data (mean ± SE) showing decrease of active tension, induced by 200 μM chloroquine, in rings of rat PA, pre‐contracted with 40K or 80K. Data are obtained from six independent experiments (n = 6 for each group). *P < 0.05, significantly different from 40K.
Furthermore, we found that the chloroquine‐mediated relaxation in PA rings precontracted with 80 mM K+ (63 ± 1.6%) was greater than in the rings precontracted with 40 mM K+ (35 ± 1.9%; P < 0.05) (Figure 1D, E).
Next, we evaluated whether chloroquine‐mediated vasodilation of PA rings was dependent on endothelial cells. To test this, we utilized and compared the endothelium‐intact PA rings and the endothelium‐denuded PA rings. Endothelium was considered intact if, following pre‐constriction with 1 μM phenylephrine, 1 μM ACh relaxed the PA ring by greater than 80% of the maximal contraction (Figure 2A) and endothelium was considered functionally removed if the relaxation to 1 μM ACh was less than 20% of the maximal contraction and complete relaxation of pre‐constricted PA rings was obtained with 10 μM SNP (Figure 2B). Treatment with 200 μM chloroquine inhibited phenylephrine‐induced contraction in endothelium‐intact or endothelium‐denuded PA rings to the same extent (Figure 2A, B, C).
Figure 2.
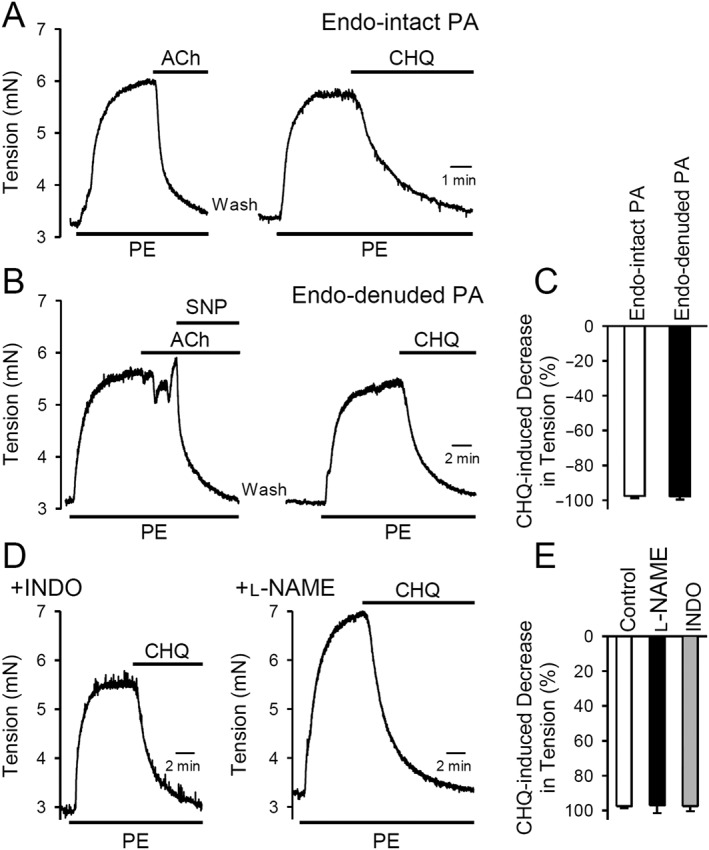
Vasorelaxant effect of chloroquine on isolated rat PA is endothelium‐independent. (A and B) Representative record of the effect of 200 μM chloroquine (CHQ) on isometric tension in endothelium‐intact (A) or endothelium‐denuded (B) PA rings constricted with 1 μM phenylephrine (PE). (C) Summarized data as means ± SEM, showing that chloroquine (200 μM) induced decreases in active tension in endothelium‐intact or endothelium‐denuded rat PA rings pre‐constricted with 1 μM phenylephrine. n = 6 for each group. (D) Representative record showing the effect of 200 μM chloroquine on the phenylephrine‐induced active tension in PA rings pretreated (for 30 min) with the PGI2 synthesis inhibitor, indomethacin (INDO; 10 μM) or the endothelial NOS inhibitor, l‐NAME (100 μM). (E) Summarized data as means ± SEM, showing chloroquine (200 μM)‐induced decrease in phenylephrine‐induced active tension in PA rings pretreated with vehicle (control, n = 6), l‐NAME (n = 8) or indomethacin (n = 6).
To further confirm that chloroquine‐induced PA relaxation was not dependent on endothelial cells, we inhibited the production of NO or PGI2 in the endothelium using the NOS inhibitor, l‐NAME and the PGI2 synthesis inhibitor, indomethacin (Chennupati et al., 2013). As shown in Figure 2D, chloroquine (200 μM) relaxed PA rings pretreated with l‐NAME (100 μM for 30 min) or indomethacin (10 μM for 30 min), to the same extent under all three conditions (control, treated with INDO or treated with l‐NAME (Figure 2E).
Chloroquine inhibits Ca2+ entry through SOCC, ROCC and VDCC in human PASMC
A rise in [Ca2+]cyt in PASMC is a major trigger for pulmonary vasoconstriction. There are at least three pathways for the Ca2+ influx that would cause an increase in [Ca2+]cyt in PASMC: voltage‐dependent Ca2+ entry through VDCC, receptor‐operated Ca2+ entry (ROCE) through the receptor‐operated Ca2+ channels (ROCC) and SOCE through store‐operated Ca2+ channels (SOCC). To examine the effect of chloroquine on SOCE in human PASMC, we used the sarcoplasmic reticulum (SR) Ca2+ ATPase inhibitor, wCPA. Extracellular application of 10 μM CPA in the absence of extracellular Ca2+ (0Ca) resulted in a transient increase in [Ca2+]cyt, likely to be due to Ca2+ release from the SR. Restoration of extracellular Ca2+ (1.8 mM) induced a second increase in [Ca2+]cyt in the presence of CPA, which was mainly due to SOCE (Figure 3A). Treatment with chloroquine decreased the amplitude of SOCE induced by passive store depletion using CPA in human PASMC by about 20 % (Figure 3B).
Figure 3.
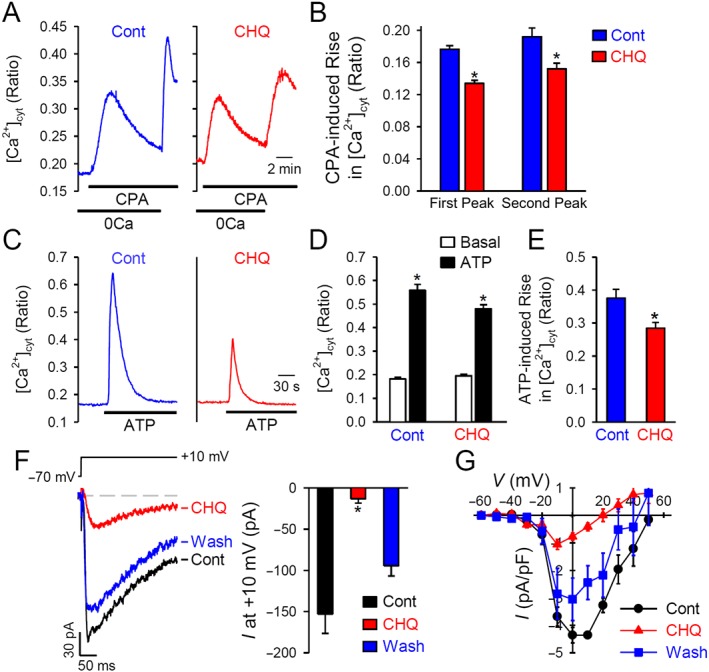
Chloroquine inhibits store‐operated, ROCE and reduces whole‐cell Ba2+ currents in human PASMC. (A) Representative traces showing changes in [Ca2+]cyt in control PASMC and chloroquine (CHQ; 200 μM)‐treated PASMC before, during and after application of 10 μM CPA (an inhibitor of sarcoendoplasmic reticulum Ca2+‐ATPase) in the absence or presence of extracellular Ca2+ (1.8 mM). (B) Summarized data as means ± SEM, showing the amplitude of increase in [Ca2+]cyt induced by application of 10 μM CPA in the absence (first peak) or presence (second peak) of 1.8 mM extracellular Ca2+ (n = 44–54 PASMC). *P < 0.05, significantly different from control (cont). (C) Representative traces showing changes in [Ca2+]cyt before and during application of 100 μM ATP in control PASMC and chloroquine (200 μM)‐treated human PASMC. (D) Summarized data, as means ± SEM, showing [Ca2+]cyt before (basal) and during (ATP) application of ATP in control and chloroquine‐treated PASMC. *P < 0.05, significantly different from basal values. (E) Summarized data, as means ± SEM, showing ATP‐induced increases in [Ca2+]cyt in control and chloroquine‐treated PASMC. *P < 0.05, significantly different from control. (F) Representative record (left panel) of inward currents, elicited by a depolarizing test pulse of +10 mV from a holding potential of −70 mV, in PASMC before (cont), during (CHQ) and after (wash) extracellular application of chloroquine. Right panel: summarized data, as means ± SEM, showing the amplitudes of peak inward currents at +10 mV in PASMC before (cont), during (CHQ) and after (wash) extracellular application of chloroquine (n = 6 PASMC). *P < 0.05, significantly different from control. (G) The current–voltage (I–V) relationship curves constructed from current recordings in PASMC before (cont), during (CHQ) and after (wash) application of chloroquine.
To examine the potential effect of chloroquine on ROCC, we used ATP as the extracellular agonist to induce ROCE. ATP binds to purinergic receptors at the plasma membrane, resulting in the production of IP3 and diacylglycerol (DAG). DAG is a known second messenger that directly opens ROCC to induce ROCE, while IP3 activates the IP3 receptors in the SR producing active Ca2+ release and resulting in SOCE. In control human PASMC, extracellular application of ATP caused a transient increase in [Ca2+]cyt due to both ROCE and SOCE (Figure 3C). Treatment with chloroquine inhibited the amplitude of the ATP‐induced increases in [Ca2+]cyt in human PASMC, by about 25%. Also, we performed the same experiments in HEK293 cells transiently transfected with the gene for the TRPC6 cation channel (Supporting Information Figure S1). Extracellular application of chloroquine significantly inhibited the ATP‐induced increase in TRPC6‐transfected cells.
To investigate the direct effect of chloroquine on VDCC, we measured the peak inward current that was evoked by a +10 mV depolarizing pulse from a holding potential of −70 mV using whole‐cell configuration of the patch‐clamp technique. In this experiment, we used Ba2+ as the charge carrier to elicit inward currents through L‐type VDCC (Cav1.x channels) and the peak currents through L‐VDCC were normalized to cell capacitance to determine the current density (pA/pF). The whole‐cell inward currents through VDCC were almost abolished when cells were superfused with an external solution containing 200 μM chloroquine (Figure 3F,G) and this inhibition was reversed after washing (Figure 3F).
Activation of the large‐conductance Ca2+‐activated K+ channels is not required for chloroquine‐mediated PA relaxation
Zhang et al., (2012) have shown that the BK channels are activated by chloroquine to exert its relaxing effect on airway smooth muscle. To investigate whether activation of BK channels and voltage‐gated K+ (Kv) channels were involved in chloroquine‐mediated pulmonary vasodilation, we used paxilline and ChTX to block BK channels and TEA to block Kv channels in the PA tension experiments. As shown in Figure 4, blockade of BK and Kv channels using 5 μM paxilline, 10 mM TEA or 100 nM ChTX had a negligible effect on the relaxation induced by 200 μM chloroquine in PA rings precontracted with 1 μM phenylephrine (Figure 4A, B).
Figure 4.
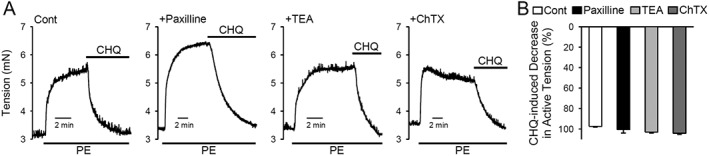
Chloroquine‐induced pulmonary vasodilation is not inhibited by blockade of K+ channels. (A) Representative records of isometric tension before and during application of chloroquine (CHQ; 200 μM) in phenylephrine (PE; 1 μM)‐contracted PA rings that were pretreated (for 30 min) with vehicle (cont), 5 μM paxilline (a BK channel blocker), 10 mM TEA (a non‐selective K+ channel blocker that blocks voltage‐gated K+ channels and BK channels) or 100 nM ChTX (a BK and IK channel blocker). (B) Summarized data, as means ± SEM, showing chloroquine‐induced decrease in active tension in PA rings treated with vehicle (cont, n = 6), 5 μM paxilline (n = 5), 10 mM TEA (n = 5) or 100 nM ChTX (n = 5).
Chloroquine attenuates pulmonary vasoconstriction induced by high K+ or alveolar hypoxia
Using an ex vivo model, we also investigated whether chloroquine directly affects PAP in isolated perfused and ventilated mouse lung. First, we superfused this lung preparation with 40 mM K+ (40K)‐containing solution via a catheter positioned in the right ventricle. The resulting vasoconstriction of the main PA increased PAP by 15–20 mmHg (Figure 5A). When the 40K‐mediated increase in PAP reached a plateau, adding chloroquine (200 μM) to the PA perfusion, for 5 min, attenuated the 40K‐induced increase in PAP (Figure 5A). Pretreatment of the isolated lung by continuous perfusion of chloroquine (200 μM) through the PA almost totally prevented the 40K‐induced increases in PAP. The inhibitory effect of chloroquine was reversed upon washout (Figure 5A, B).
Figure 5.
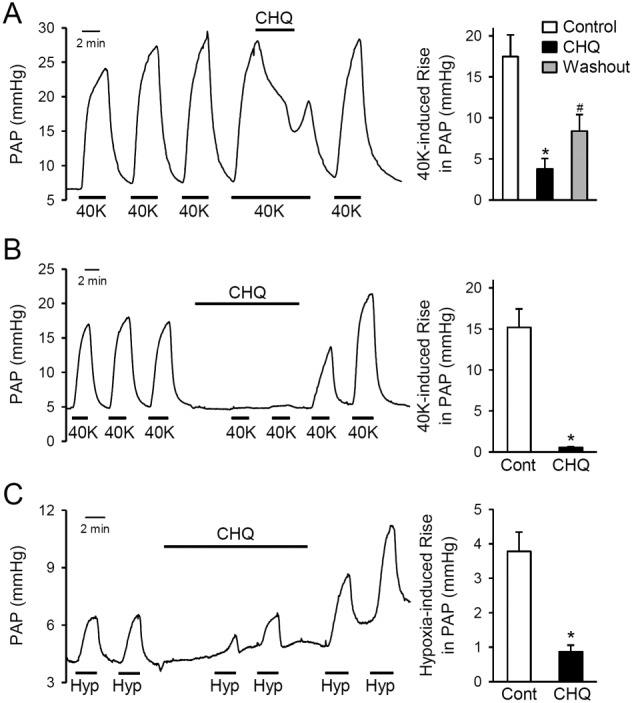
Chloroquine attenuates pulmonary vasoconstriction induced by high K+ concentrations or hypoxia. (A) Representative record (left panel) of PAP in isolated perfused and ventilated lungs before, during and after repeated pulmonary perfusion of 40 mM K+ (40K) solution. Chloroquine (CHQ; 200 μM) was applied to the lung via the intrapulmonary arterial catheter when a 40K‐induced increase in PAP reached its maximal plateau phase. Right panel: summarized data, as means ± SEM (n = 6), showing the 40K‐induced increases in PAP in isolated perfused and ventilated lungs before (control), during (CHQ) and after (washout) perfusion of the pulmonary circulation with chloroquine (200 μM). *P < 0.05 , significantly different from control. # P < 0.05, significantly different from chloroquine. (B) Representative record (left panel) showing 40K‐induced changes of PAP in isolated perfused and ventilated lungs before, during and after intrapulmonary application of chloroquine (200 μM). Right panel: summarized data, as means ± SEM (n = 5), showing the repeated 40K‐induced increases in PAP before (cont) and during (CHQ) application of chloroquine. *P < 0.05, significantly different from control. (C) Representative record (left panel) showing alveolar hypoxia (Hyp)‐induced changes of PAP in isolated perfused and ventilated lungs before, during and after intrapulmonary application of chloroquine (200 μM). Right panel: summarized data, as means ± SE (n = 6), showing the repeated Hyp‐induced increases in PAP before (cont) and during (CHQ) application of chloroquine. *P < 0.05, significantly different from control.
Furthermore, we examined the effect of chloroquine on the alveolar hypoxia‐induced increase in PAP or acute HPV in the isolated perfused and ventilated lung. Alveolar hypoxia was established by ventilating the lung with a hypoxic gas mixture (1% O2 in N2). As shown in Figure 5C, alveolar hypoxia rapidly and reversibly induced pulmonary vasoconstriction and increased PAP. Addition of chloroquine (200 μM) to the pulmonary perfusion attenuated the hypoxia‐induced increases in PAP (Figure 5C) and this effect of chloroquine was reversible.
Expressino of the mRNA for TAS2R in human PASMC, rat PA and mouse PA
In addition to its anti‐malarial, anti‐autophagic and vasodilator effects, chloroquine has been reported to activate TAS2R in various tissues and cells. We conducted a series of RT‐PCR experiments to define the TAS2R transcripts in rat/mouse PA and human PASMC. Based on NCBI.com, we first identified and matched the gene synonym of human TAS2R (hTAS2R), rat TAS2R (rTAS2R) and mouse TAS2R (mTAS2R). We then selected seven matched human, rat and mouse TAS2R subtypes that are suggested to be activated by chloroquine for our RT‐PCR experiments (Supporting Information Table S1). Using the specific primers (Supporting Information Tables S2–S4), we found all seven TAS2R isoforms (hTAS2R3, hTAS2R4, hTAS2R7, hTAS2R10, hTAS2R14, hTAS2R39 and hTAS2R40) are expressed in human PASMC (Supporting Information Figure S2); however, rTAS2R107 (hTAS2R10 and mTAS2R110) in rat PA and mTAS2R108 (hTAS2R4 and rTAS2R108) in mouse PA were not detectable. These data indicate that most of the chloroquine‐sensitive TAS2R are expressed in human, rat and mouse PASMC. However, two TAS2R isoforms, rTAS2R107 and mRAS2R108, are differentially expressed in rat and mouse PA in comparison with human PASMC.
Short‐term treatment with chloroquine has no effect on autophagy or proliferation in human PASMC
Chloroquine inhibits autophagy through lysosome alkalinization, which interferes with the function of the pH‐sensitive lysosomal enzymes. Abundant LC3‐II and increased expression of P62 protein in cells can be used to assess the levels of inhibition of autophagy, after chloroquine treatment. We first assessed the effects of short‐term treatment (2–15 min) with chloroquine (up to 200 μM) on autophagy and cell viability in human PASMC. As shown in Figure 6, short‐term treatment of human PASMC with 200 μM chloroquine did not affect the protein level of LC3‐II and P62 (Figure 6A, B), or the formation of LC3B dots (Figure 6C, D). Also, as shown in Figure 6E, short‐term (5 and 15 min) treatment of PASMC with chloroquine (20–200 μM) did not decrease the number of living cells (% of total cells). Proliferation of human PASMC was similarly not affected by short‐term (15 min) treatment of human PASMC with chloroquine (10‐200 μM), using the MTT assay (Figure 6F).
Figure 6.
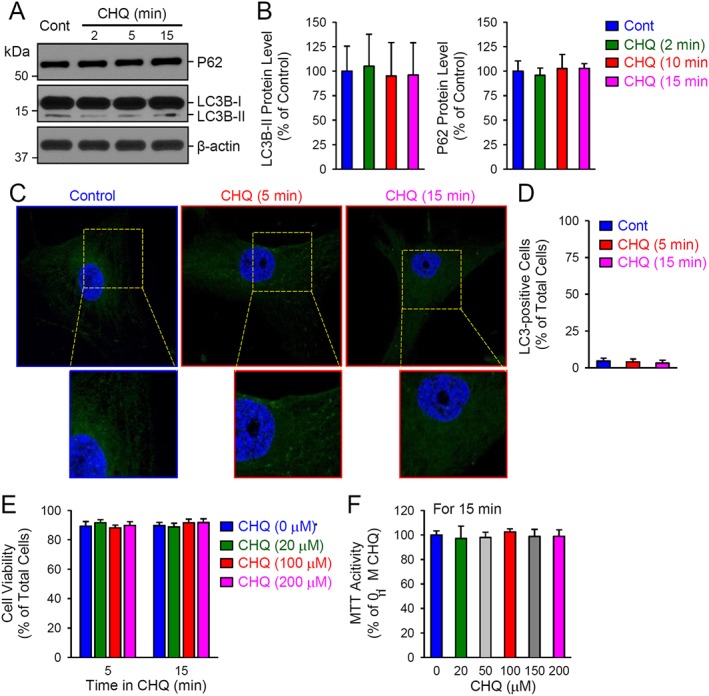
Short‐term treatment of human PASMC with chloroquine does not affect autophagy, cell viability and proliferation. (A and B) Representative image (A) and summarized data (B) as means ± SEM (n = 5), showing Western blot analyses and quantification of P62 and LC3B (LC3B‐I and LC3B‐II) in control PASMC and PASMC treated with 200 μM chloroquine (CHQ) for 2, 5 or 15 min. (C) Representative immunofluorescence images showing control PASMC and PASMC treated with 200 μM chloroquine for 5 or 15 min. (D) Summarized data, as means ± SEM, indicating the percentage of LC3‐positive cells of the total cells in control PASMC and PASMC treated with 200 μM chloroquine for 5 or 15 min. The LC3‐positive cells are defined by the cells with more than five LC3 staining dots. (E) The number of living cells (means ± SEM; n = 6), determined as the percentage of total cells, in control PASMC (CHQ, 0 μM) and PASMC treated (for 5 or 15 min) with 20, 100 or 200 μM of chloroquine. (F) MTT activity (means ± SE, n = 6) in control PASMC (CHQ, 0 μM) and PASMC treated (for 15 min) with 20, 50, 100, 150 or 200 μM of chloroquine.
Long‐term treatment with chloroquine inhibits autophagy and proliferation in human PASMC
We then investigated the effects of long‐term (24h) treatment with chloroquine on autophagy in human PASMC and found an increased expression of P62 and LC3B‐II (Figure 7A, B), with 20 μM chloroquine. This treatment also increased the number of LC3‐positive human PASMC, defined as cells with more than 5 LC3 dots (Figure 7C, D). In terms of proliferation of human PASMC, long‐term treatment (for 48h) with chloroquine (1‐100μM) concentration‐dependently decreased MTT activity.These effects were first significant at 20 μM and reached a maximum at 100 μM (Figure 7E).
Figure 7.
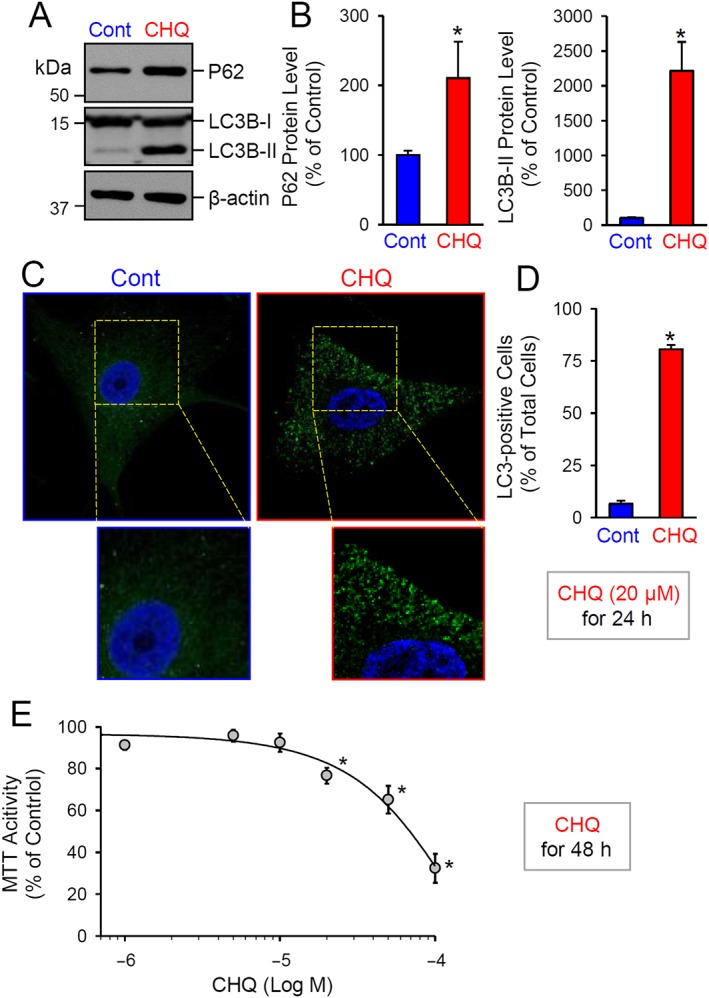
Long‐term treatment of human PASMC with chloroquine inhibits autophagy and decreases cell proliferation. (A and B) Representative image (A) and (B) summarized data, as means ± SEM (n = 5), showing Western blot analyses and quantification of P62 and LC3B (LC3B‐I and LC3B‐II) in control PASMC and PASMC ,treated with 20 μM chloroquine for 24 h. *P < 0.05, significantly different from control. (C) Representative immunofluorescence images showing control PASMC and PASMC treated with 20 μM chloroquine for 24 h. (D) Summarized data, as means ± SEM, showing the percentage of LC3‐positive cells or cells with more than five LC3 staining dots (% of the total cells) in control PASMC and PASMC treated with 20 μM chloroquine for 24 h. (E) Dose–response curve of MTT activity (means ± SEM, n = 6 for each data point) in PASMC treated (for 48 h) with 0 (control), 1, 5, 10, 20, 50 or 100 μM of chloroquine respectively. *P < 0.05, significantly different from control PASMC (or cells not treated with chloroquine).
Chloroquine attenuates experimental pulmonary hypertension models in rats
To determine whether chloroquine can prevent the development of PH or inhibit the progression of established PH in HPH or SuHx reversal rats model. In the HPH model, we examined and compared (a) RVSP; (b) the ratio of the weight of right ventricle (RV) and the weight of left ventricle (LV) and septum (S) [RV/(LV + S)], also referred to as the Fulton Index; and (c) the ratio of the weight of RV to body weight (RV/BW), HCT and the BW change in normoxic control rats treated with vehicle (Nor) and chloroquine (50 mg·kg−1, i.p., daily) (Nor + CHQ) as well as hypoxic rats treated with vehicle (Hyp) and chloroquine (50 mg·kg−1, i.p., daily) (Hyp + CHQ). After 3 week exposure to hypoxia, rats exhibited significantly increased RVSP and RV hypertrophy or increased RV/(LV + S) and RV/BW. Treatment with chloroquine (50 mg·kg−1, i.p., daily, for 3 weeks) prevented the increases in RVSP, RV/(LV + S) and RV/BW in rats exposed to hypoxia (Supporting Information Table S5 and Figure 8A–C). Additionally,the haematocrit was also markedly increased in rats with HPH, compared with that in normoxic control rats (Supporting Information Table S5 and Figure 8D) and this hypoxia‐induced increase in haematocrit was also inhibited by the treatment with chloroquine (Supporting Information Table S5 and Figure 8). In these experiments, chloroquine did not affect any of the parameters measured in the rats exposed to normoxia
Figure 8.
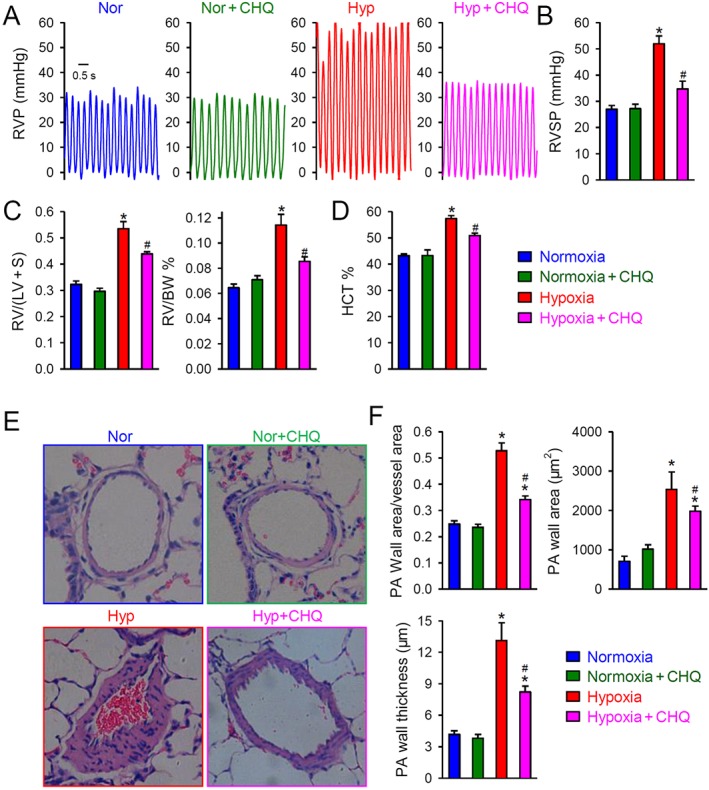
Chloroquine inhibits the development of HPH in rats. (A) Representative record of right ventricular pressure (RVP) in rats exposed to normoxia alone (Nor) or with chloroquine (50 mg·kg−1·day−1; Nor + CHQ) treatment and rats exposed to hypoxia (Hyp; 10% O2 for 21 days) or with chloroquine (Hyp + CHQ) treatment. (B) Summarized data, as means ± SEM, showing the peak value of RVSP in Nor, Nor + CHQ, Hyp and Hyp + CHQ rats (n = 6 in each group). *P < 0.05, significantly different from Nor and Nor + CHQ groups; # P < 0.05, significantly different from Hyp group. (C) Summarized data, as means ± SEM, showing the RV hypertrophy index, determined by the Fulton index [RV/(LV + S)] (left panel) and the ratio of RV/BW (right panel) in Nor, Nor + CHQ, Hyp and Hyp + CHQ rats (n = 6 in each group). (D) Summarized data, as means ± SEM, showing haematocrit (HCT) in Nor, Nor + CHQ, Hyp and Hyp + CHQ rats (n = 6 in each group). *P < 0.05, significantly different from Nor and Nor + CHQ groups; # P < 0.05, significantly different from Hyp group. (E) Representative H&E images of small PAs in Nor, Nor + CHQ, Hyp and Hyp + CHQ rats. (F) Summarized data, as means ± SEM, showing the wall area/vessel area, thickness and area of small PA in Nor, Nor + CHQ, Hyp and Hyp + CHQ rats. *P < 0.05 , significantly different from Nor and Nor + CHQ groups; # P < 0.05, significantly different from Hyp group.
In the model of SuHx, combination of SU5416 with 3 weeks of hypoxia significantly increased RVSP, RV/(LV + S) and RV/BW (Supporting Information Table S6 and Figure 9). After 3 weeks of hypoxia and 2 weeks of follow‐up in normoxia, RVSP, RV/(LV + S) and RV/BW were further increased compared with the corresponding values after 3 weeks of hypoxia exposure, alone. However, after 3 weeks of hypoxia and 2 weeks treatment with chloroquine in normoxia, this further increase in RVSP and RV hypertrophy was blocked (Supporting Information Table S6 and Figure 9).
Figure 9.
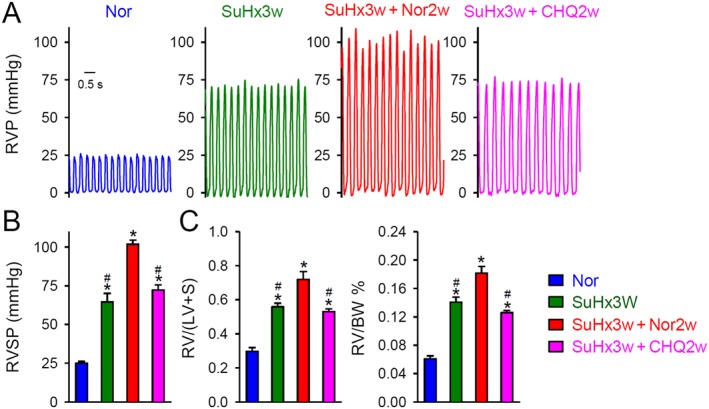
Chloroquine inhibits progression of established PH in the SuHx rat model. (A) Representative record of right ventricular pressure (RVP). Rats were given an s.c. injection of 20 mg·kg−1 SU5416 or vehicle, followed by hypoxia (SuHx) for 3 weeks (10% O2) and normoxia for an additional 2 weeks to elicit severe PH. After 3 weeks of hypoxia, the SuHx rats were divided into three groups: hypoxia with no intervention (SuHx3w), vehicle treatment (SuHx3w + Nor2w) and treatment with chloroquine (CHQ; 50 mg·kg−1·day−1) (SuHx3w + CHQ2w). The normoxic control group was exposed to room air only (21% O2) for a total of 5 weeks (Nor). n = 6 for each group. (B) Summarized data, as means ± SEM, showing the peak value of RVSP in Nor, SuHx3w, SuHx3w + Nor2w and SuHx3w + CHQ2w groups. * P < 0.05, significantly different from Nor; # P < 0.05, significantly different from SuHx3w + Nor2w. (C) Summarized data, as means ± SEM, showing the RV hypertrophy index, determined by the Fulton index [RV/(LV + S)] (left panel) and the ratio of RV/BW (right panel) in Nor, SuHx3w, SuHx3w + Nor2w and SuHx3w + CHQ2w. * P < 0.05, significantly different from Nor; # P < 0.05, significantly different from SuHx3w + Nor2w.
Histological examination revealed that chronic exposure to hypoxia significantly increased the wall thickness of the small PA (Supporting Information Table S5 and Figure 8E, F). Treatment of hypoxic rats with chloroquine (Hyp + CHQ, 50 mg·kg−1, i.p., daily for 3 weeks) significantly reduced the small PA wall thickness, in comparison with the hypoxia only (Hyp) group of rats (Supporting Information Table S5 and Figure 8). Chloroquine treatment under normoxic conditions (Nor + CHQ) did change any of the haematological (Figure 8D) and histological (Figure 8E/F) parameters (Supporting Information Table S5).
In all in vivo experiments, the BW of all rats was measured every other day during the continuous 21 day exposure to normoxia or hypoxia. The i.p. injection of chloroquine (50 mg·kg−1, daily) resulted in a slight decrease in BW in Nor + CHQ and Hyp + CHQ groups. however, the ratio of the liver weight/BW, the kidney weight/BW and the spleen weight/BW was not significant between Nor and Nor + CHQ groups and between Hyp and Hyp + CHQ groups (data not shown).
Discussion
The results from the present study demonstrated that (i) chloroquine induced endothelium‐independent pulmonary vasodilation in a concentration‐dependent manner, and blockade of the BK channels and voltage‐gated K+ channels negligibly affected the chloroquine ‐induced relaxation of the PA; (ii) chloroquine rapidly and reversibly induced vasorelaxation in PA rings pre‐contracted by 80 mM K+‐containing solution and reduced whole‐cell inward currents through L‐type VDCC in PASMC; (iii) chloroquine significantly inhibited SOCE and ROCE in PASMC; (iv) perfusion of chloroquine through the pulmonary circulation significantly inhibited alveolar hypoxia‐induced pulmonary vasoconstriction in isolated perfused and ventilated mouse lungs; (v) short‐term (2–15 min) treatment of human PASMC with chloroquine (up to 200 μM) did not affect cell autophagy and cell viability in PASMC, while long‐term (24–48 h) treatment with chloroquine (≥20 μM) inhibited autophagy and attenuated serum‐associated PASMC proliferation; and (vi) i.p. injection of chloroquine (50 mg·kg−1, daily, for 3 weeks) significantly inhibited the development and progression of HPH in rats. These data provide strong evidence that chloroquine is a potent, endothelium‐independent, pulmonary vasodilator that relaxes PA by directly and/or indirectly by inhibiting various Ca2+ channels (e.g. VDCC, SOCC and ROCC) in PASMC. The therapeutic potential of chloroquine in the treatment of PAH and other forms of PH is based on its vasodilator effect on PA smooth muscle as well as its anti‐autophagic and anti‐proliferative effects in PASMC.
Chloroquine (Aralen™, C18H26CIN3), 4‐N‐(7‐chloroquinolin‐4‐yl)‐1‐N,1‐N‐diethylpentane‐1,4‐diamine, is a synthetic compound, best known for treatment of malaria (Pascolo, 2016). Chloroquine also inhibits autophagy, by increasing lysosomal pH and disrupting lysosomal enzymes so cells under stress cannot activate the autophagy pathway. A recent study showed that prolonged treatment (≥24 h) of animals with chloroquine decreased markers of autophagy, inhibited lysosomal degradation of BMPR‐II, inhibited PASMC proliferation and ameliorated MCT‐induced PH in rats (Long et al., 2013). Consistent with the anti‐autophagic and anti‐proliferative effects of chloroquine, we also found that short‐term (≤15 min) treatment with chloroquine had no effect on PASMC autophagy and proliferation, but exerted significant and rapid vasorelaxant effects on PA by inhibiting Ca2+ influx in PASMC. The acute pulmonary vasodilator effect of chloroquine seemed to be independent of its anti‐autophagic effect.
Chloroquine also has a bitter taste and it can activate the bitter taste receptor (TAS2R), a seven transmembrane domain GPCR. Several bitter taste receptors are expressed in airway smooth muscle cells (Deshpande et al., 2010). TAS2R3, TAS2R10, TAS2R39 and TAS2R7 are reported to respond to chloroquine (Sainz et al., 2007; Meyerhof et al., 2010). We found that mRNA of TAS2R3, TAS2R10, TAS2R39 and TAS2R7 are expressed in human PASMC, rat PA and mouse PA. Treatment of airway smooth muscle cells in vitro with chloroquine results in an increase in [Ca2+]cyt (Deshpande et al., 2010; Zhang et al., 2012) that would be expected to induce contraction. However, the authors actually observed smooth muscle relaxation and dilation of the airways. To better understand the mechanism behind this paradox, two separate groups tested the hypothesis that increased [Ca2+]cyt triggers the opening of BK channels resulting in membrane hyperpolarization. Deshpande et al. (2010) reported that chloroquine treatment of isolated human airway smooth muscle cells led to membrane hyperpolarization via a Gβγ‐dependent, PLCβ‐dependent and IP3 receptor‐dependent manner. In contrast, Zhang et al. (2012) demonstrated that bitter‐tasting compounds, including chloroquine inhibited BK channel activity. However, it is not clear why and how BK channel inhibition led to the observed bronchodilation. These discrepant effects of chloroquine on BK channels are most likely due to differences in species (human vs. mouse), tissue (airway vs. vessel) and cell phenotype (freshly dispersed cells vs. primary cultured cells). Activation of BK channels is known to play a major role in PA relaxation (Dong et al., 2016). However, our study showed that BK and non‐selective K+ channel blockers did not significantly inhibit chloroquine‐mediated relaxation of the PA.
Intracellular Ca2+ is one of the most important regulators of cell function in PASMC. The increase in [Ca2+]cyt is known to stimulate cell contraction, migration and proliferation (Somlyo and Somlyo, 1994; Mandegar et al., 2004; Hill et al., 2006). Given that chloroquine does not affect Ca2+ extrusion (Tan and Sanderson, 2014), we hypothesized that this compound might induce PA relaxation by inhibiting Ca2+‐permeable channels in PASMC. There are at least three families of Ca2+‐permeable channels in PASMC: (i) SOCCs, (ii) ROCCs and (iii) VDCCs. The opening of any of these three families of Ca2+ channels would increase [Ca2+]cyt and cause PA contraction. It is known that increasing extracellular K+ (e.g. from 4.7 to 40 or 80 mM) results in membrane depolarization due to a shift of the membrane potential (E m) to the equilibrium potential for K+ (E K), which subsequently opens VDCC, augments Ca2+ influx, increases [Ca2+]cyt and ultimately causes PASMC contraction and pulmonary vasoconstriction. The high K+‐mediated PA contraction is thus mainly due to Ca2+ influx through VDCC in PASMC (Mandegar and Yuan, 2002; Hall et al., 2009). Sai et al. (2014) observed that chloroquine resulted in vasorelaxation in aortas precontracted with a high K+‐concentrations, while pretreatment with chloroquine inhibited the high K+‐induced increases in [Ca2+]cyt in rat thoracic aortic smooth muscle cells. In the current study, we found that chloroquine rapidly and reversibly caused vasorelaxation in PA rings precontracted by 40K and 80K and inhibited 40K‐induced increases in PAP in isolated perfused and ventilated lungs. These data suggest that blockade of VDCC is an important mechanism by which chloroquine induces pulmonary vasodilation.
In PA rings constricted by 40K, the equilibrium potential for K+ (E K) is predicted to be approximately −31 mV based on the Nernst equation. Opening of K+ channels would shift E m toward the E K (−31 mV) and away from the activation threshold of VDCC (the window current of L‐type VDCC is approximately −25 to +15 mV) (Nelson et al., 1990; Fleischmann et al., 1994; Ko et al., 2013), which results in closure of VDCC. In PA rings constricted by 80K, the E K is predicted to be approximately −14 mV. Activation of K+ channels would shift E m toward the E K (−14 mV), which is above the activation threshold of VDCC; this prevents VDCC closure and results in vasodilation. In the current study, we demonstrated that chloroquine rapidly relaxed PA rings precontracted by 40K or 80K and significantly inhibited the 40K‐induced increase in PAP in isolated perfused and ventilated lungs. These data strongly indicate that chloroquine blocks VDCC in PASMC, inhibits Ca2+ influx and elicits pulmonary vasodilation. Our patch‐clamp experiments suggested that chloroquine may directly block L‐type VDCC in human PASMC, as it significantly and reversibly attenuated the maximal inward current. Consistent with our findings, chloroquine inhibited VDCC via Gβγ and Gα1 signalling pathways in airway smooth muscle cells (Zhang et al., 2013). Thus, the chloroquine‐mediated blockade of VDCC could be a potential mechanism involved in inhibiting agonist‐induced PA contraction.
Alveolar hypoxia‐induced pulmonary vasoconstriction (HPV) is an important physiological mechanism that directs blood flow from poorly ventilated regions to well‐ventilated areas in the lungs to maximize oxygenation of the venous blood in the pulmonary circulation. Persistent HPV in patients with obstructive lung diseases and chronic mountain sickness may lead to sustained pulmonary vasoconstriction, vascular remodelling and, eventually, to chronic PH. Alveolar HPV requires an influx of extracellular Ca2+ through various Ca2+ channels in PASMC (Yoo et al., 2013), specifically, Ca2+ influx through VDCC opened by hypoxia‐mediated membrane depolarization (Yuan et al., 1993; Wan et al., 2013) and Ca2+ influx through SOCC and ROCC (Wang et al., 2005; Wang et al., 2006). It has been demonstrated that up‐regulated CaV1.2 or CaV3.2 channels (Wan et al., 2013) and TRPC1and TRPC6 channels (Wang et al., 2006) in PASMC contribute to increased HPV in the development of HPH. Overexpression of TRPC6 also increased [Ca2+]cyt in pulmonary venous and arterial SMC exposed to chronic hypoxia (Lin et al., 2004; Peng et al., 2013; Peng et al., 2015). Using the isolated perfused and ventilated lungs, we found that adding chloroquine to the perfusing solution rapidly and reversibly inhibited the alveolar hypoxia‐induced increase in PAP (Figure 5). The inhibitory effect of chloroquine on acute HPV is due most likely to its blockade of VDCC, ROCC and SOCC in PASMC. Further studies are needed to identify the particular channels targeted by chloroquine to exert its inhibitory effect on acute HPV and define the cellular mechanisms involved in chloroquine‐mediated inhibition of HPV.
Our results showed that chloroquine inhibited not only high K+‐mediated PA contraction but also phenylephrine‐induced pulmonary vasoconstriction. In PASMC, we also demonstrated that chloroquine significantly inhibited CPA‐mediated SOCE and ATP‐mediated ROCE (Figure 3A–E), suggesting that chloroquine may directly or indirectly block SOCC and ROCC in PASMC. Chloroquine inhibits IP3 receptors (Tan and Sanderson, 2014) and decreases Ca2+ influx through TRPC3 and STIM/Orai channels (Zhang et al., 2014; Xu et al., 2015). These results imply that chloroquine may inhibit many different channel proteins to reduce Ca2+ influx in PASMC and induce pulmonary vasodilation. In our study, ATP‐mediated activation of P2Y receptors, a family of GPCRs, increased [Ca2+]cyt by causing SOCE (due to IP3‐mediated active store depletion and the opening of SOCC) and ROCE (due to the DAG‐mediated opening of ROCC). ATP‐mediated activation of P2X receptors, a family of cation‐permeable ligand‐gated ion channels, increases [Ca2+]cyt by directly inducing Ca2+ influx through the channels and/or indirectly inducing Ca2+ influx through VDCC via membrane depolarization (Gilbert et al., 2016). In HEK293 cells traniently transfected with the gene encoding TRPC6 channels, chloroquine attenuated the ATP‐mediated increase in [Ca2+]cyt (Supporting Information Figure S1), indicating that chloroquine may partly block TRPC6 channels, which contribute to forming ROCC and SOCC (Thebault et al., 2005; Albarran et al., 2014), and thus induce PA relaxation. Further studies are needed to specify the types of cation channels that can be blocked by chloroquine and to define the precise mechanisms by which chloroquine exerts its inhibitory effect on Ca2+ channels in PASMC.
We noted that the concentration of chloroquine (50 to 200 μM) required for acutely inducing PA relaxation (see Figure 1B) was much higher than the therapeutic plasma concentration observed in patients (Phillips et al., 1986). Similar concentrations of chloroquine have been used by other investigators to test its pharmacological effect in different tissues and cells. For example, 3 mM chloroquine was used for experiments in rat thoracic aorta (Sai et al., 2014), 1 mM in human airway smooth muscle cells and mouse tracheas (Deshpande et al., 2010), 500 μM in mouse airway smooth muscle cells (Tan and Sanderson, 2014), 100–300 μM in rat ileum (Jing et al., 2013) and 100 μM in guinea pig aorta (Manson et al., 2014). We found that chloroquine‐mediated PA relaxation started at approximately 20–50 μM and maximized at about 200 μM when this compound was applied to PA rings or to isolated perfused and ventilated lungs) for 2–15 min. The vasodilator effect of chloroquine was rapid in onset (effective within 1–2 min) and reversible (see Figures 1C/D and 5). In our in vitro experiments, we found that short‐term (2, 5 and 15 min) treatment with chloroquine (20, 100 and 200 μM) had neither toxic effect nor anti‐autophagic and anti‐proliferative effects on PASMC (Figure 6), whereas long‐term (24 and 48 h) treatment with chloroquine inhibited autophagy (20 μM for 24 h) and cell proliferation (20 μM for 48 h) in PASMC (Figure 7). These observations imply that chloroquine‐mediated pulmonary vasodilation is a separate process from its anti‐autophagic effect on PASMC. The cellular mechanisms involved in the therapeutic action of chloroquine for PH include its vasodilator effect (due to blockade of Ca2+ channels) on the PA, anti‐autophagic effect and anti‐proliferative effect (due to inhibition of Ca2+ entry and autophagy) on PASMC.
Indeed, in our in vivo experiments, we showed that i.p. injection of chloroquine (50 mg·kg−1, daily for 3 weeks) significantly attenuated the development of PH in rats exposed to hypoxia (10% O2 for 3 weeks). The dose (50 mg·kg−1) we chose in the in vivo experiments was similar to the dose used for anti‐tumour and anti‐angiogenesis studies in tumour‐bearing mice (Zheng et al., 2009; Jiang et al., 2010; Maes et al., 2014). This dose of chloroquine, as reported previously, is known to inhibit autophagy and lead to increased expression of p53 resulting in increased cell apoptosis (Amaravadi et al., 2007). We observed that chloroquine (50 mg·kg−1 daily for 3 weeks) led to an overall loss of BW in both normoxic and hypoxic rats. However, the ratios of liver weight to BW (liver/BW), kidney weight to BW (kidney/BW) and spleen weight to BW (spleen/BW) were not significantly changed by chloroquine treatment at this dose. Measured PA wall area/vessel area alone is not enough to evaluate PA remodelling due to the heterogeneity of the vessel area and lumen area during constriction or vasodilation. However, the PA wall thickness due to vascular remodelling would not change during acute vascular constriction and vasodilation. We found that PA wall thickness, PA wall area and the ratio of PA wall area to vessel external area were significantly decreased after chloroquine treatment, compared with vehicle under hypoxic conditions in rats, suggestingthat chloroquine attenuated PA remodelling in HPH. In consistent with earlier data (Long et al., 2013), we also found that chloroquine can inhibit the progression of established PH in the SuHx rat model. Chloroquine is an FDA approved drug that is easy to use and has minimal side effects. As chloroquine has inhibitory effects on the development of PH and was able to reverses established PH, it could be an effective treatment for PAH, in particular for the patients who do not respond to the conventional therapies such as prostacyclin receptor activators, NO‐cyclic GMP enhancement or endothelin receptor antagonism. These data indicate that chronic treatment with chloroquine may be an effective and safe therapeutic approach for PH.
In summary, our in vitro, ex vivo and in vivo studies all indicate that chloroquine is a potent pulmonary vasodilator that, in combination with its anti‐autophagic and anti‐proliferative effect on PASMC, can significantly attenuate the development and progression of experimental PH in animal models. The cellular mechanisms by which chloroquine induces PA relaxation (and inhibition of PASMC proliferation) may involve blockade of various Ca2+ channels in PASMC.
Author contributions
K.W. conceived the study, designed the experiments, carried out the work, analysed the data and drafted the manuscript. Q.Z. conducted the tension experiments and conceived and designed the in vitro experiments. X.W., Z.L., Y.G., A.B., C.W. and Z.W. carried out part of the in vitro work. S.S., R.J.A. and K.M.M. carried out part of the in vivo work. H.T. kindly provided the human PASMCs. J.X.J.Y., A.M., S.M.B., W.L., H.T. and J.G.N.G. drafted and revised critically for important intellectual content. J.W. conceived the study, designed the experiments and supervised K.W. All authors critically revised the manuscript for important intellectual content and approved its final version.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 RT‐PCR analysis of TAS2R in human PASMC (A), rat pulmonary arteries (B) and mouse pulmonary arteries. Transcripts of seven subtypes of hTAS2Rs (3, 4, 7, 10, 14, 39, 40) (A), rTAS2Rs (137, 108, 121, 107, 123, 139, 144) (B) and mTAS2Rs (137, 108, 130, 110, 114, 139 140) (C) are identified in human PASMC, rat pulmonary arteries and mouse pulmonary arteries, respectively.
{kind=link}
Figure S2 Chloroquine inhibits ATP‐induced increase in [Ca2+]cyt in HEK293 cells transiently transfected with the TRPC6 gene. A: Representative traces showing changes of [Ca2+]cyt before and during extracellular application of ATP (100 μM) in HEK293 cells transfected with an empty vector (GFP) and the TRPC6 gene (Trpc6) in the absence (GFP, Trpc6) or presence (Trpc6 + CHQ) of 200 μM chloroquine (CHQ). B: Summarized data (mean ± SE) showing [Ca2+]cyt before (Basal) and after (ATP) extracellular application of ATP in GFP‐transfected and Trpc6‐transfected cells in the absence (GFP, Trpc6) or presence (Trpc6 + CHQ) of chloroquine. *P < 0.05 vs. Basal. C: Summarized data (mean ± SE) showing ATP‐induced increases in [Ca2+]cyt in GFP‐transfected cells and Trpc6‐transfected cells treated with (Trpc6 + CHQ) or without (Trpc6) chloroquine. *P < 0.05 vs. Control; #P < 0.05 vs. Trpc6.
{kind=link}
Table S1 The gene synonym between human, rat and mouse TAS2Rs.
Table S2 DNA primer sequences for the hTAS2Rs.
Table S3 DNA primer sequences for the mTAS2Rs.
Table S4 DNA primer sequences for the rTAS2Rs.
Table S5 Chloroquine inhibits the development of hypoxia‐induced pulmonary hypertension in rats.
Table S6 Chloroquine inhibits the progression of established pulmonary hypertension in Sugen5416/hypoxia rat model.
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (81630004, 81470246, 81220108001 and 81520108001), the Guangzhou Department of Education Yangcheng Scholarship (12A001S), the Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme of China, China Scholarship Council (201608440324) and the Actelion ENTELLIGENCE Young Investigator Award. The authors also thank Shane G. Carr and Keeley S. Ravellette for critically reviewing the manuscript.
Wu, K. , Zhang, Q. , Wu, X. , Lu, W. , Tang, H. , Liang, Z. , Gu, Y. , Song, S. , Ayon, R. J. , Wang, Z. , McDermott, K. M. , Balistrieri, A. , Wang, C. , Black, S. M. , Garcia, J. G. N. , Makino, A. , Yuan, J. X.‐J. , and Wang, J. (2017) Chloroquine is a potent pulmonary vasodilator that attenuates hypoxia‐induced pulmonary hypertension. British Journal of Pharmacology, 174: 4155–4172. doi: 10.1111/bph.13990.
References
- Albarran L, Dionisio N, Lopez E, Salido GM, Redondo PC, Rosado JA (2014). STIM1 regulates TRPC6 heteromultimerization and subcellular location. Biochem J 463: 373–381. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI et al (2007). Autophagy inhibition enhances therapy‐induced apoptosis in a Myc‐induced model of lymphoma. J Clin Invest 117: 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anigbogu CN, Adigun SA, Inyang I, Adegunloye BJ (1993). Chloroquine reduces blood pressure and forearm vascular resistance and increases forearm blood flow in healthy young adults. Clin Physiol 13: 209–216. [DOI] [PubMed] [Google Scholar]
- Aziba PI, Okpako DT (2003). Effects of chloroquine on smooth muscle contracted with noradrenaline or high‐potassium solutions in the rat thoracic aorta. J Smooth Muscle Res 39 (3): 31–37. [DOI] [PubMed] [Google Scholar]
- Chennupati R, Lamers WH, Koehler SE, De Mey JG (2013). Endothelium‐dependent hyperpolarization‐related relaxations diminish with age in murine saphenous arteries of both sexes. Br J Pharmacol 169: 1486–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M, Fessel J, Tang H, West J (2012). A process‐based review of mouse models of pulmonary hypertension. Pulm Circ 2: 415–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande DA, Wang WC, McIlmoyle EL, Robinett KS, Schillinger RM, An SS et al (2010). Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nat Med 16: 1299–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong DL, Bai YL, Cai BZ (2016). Calcium‐activated potassium channels: potential target for cardiovascular diseases. Adv Protein Chem Struct Biol 104: 233–261. [DOI] [PubMed] [Google Scholar]
- Fleischmann BK, Murray RK, Kotlikoff MI (1994). Voltage window for sustained elevation of cytosolic calcium in smooth muscle cells. Proc Natl Acad Sci 91: 11914–11918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert DF, Stebbing MJ, Kuenzel K, Murphy RM, Zacharewicz E, Buttgereit A et al (2016). Store‐operated Ca2+ entry (SOCE) and purinergic receptor‐mediated Ca2+ homeostasis in murine bv2 microglia cells: early cellular responses to ATP‐mediated microglia activation. Front Mol Neurosci 9: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Jones TH, Channer KS, Jones RD (2009). Mechanisms of agonist‐induced constriction in isolated human pulmonary arteries. Vascul Pharmacol 51: 8–12. [DOI] [PubMed] [Google Scholar]
- Hill MA, Davis MJ, Meininger GA, Potocnik SJ, Murphy TV (2006). Arteriolar myogenic signalling mechanisms: implications for local vascular function. Clin Hemorheol Microcirc 34: 67–79. [PubMed] [Google Scholar]
- Jiang PD, Zhao YL, Deng XQ, Mao YQ, Shi W, Tang QQ et al (2010). Antitumor and antimetastatic activities of chloroquine diphosphate in a murine model of breast cancer. Biomed Pharmacother 64: 609–614. [DOI] [PubMed] [Google Scholar]
- Jin Y, Choi AM (2012). Cross talk between autophagy and apoptosis in pulmonary hypertension. Pulm Circ 2: 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing F, Liu M, Yang N, Liu Y, Li X, Li J (2013). Relaxant effect of chloroquine in rat ileum: possible involvement of nitric oxide and BKCa . J Pharm Pharmacol 65: 847–854. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Arozena AA et al (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12: 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko EA, Wan J, Yamamura A, Zimnicka AM, Yamamura H, Yoo HY et al (2013). Functional characterization of voltage‐dependent Ca2+ channels in mouse pulmonary arterial smooth muscle cells: divergent effect of ROS. Am J Physiol Cell Physiol 304: C1042–C1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhr FK, Smith KA, Song MY, Levitan I, Yuan JX (2012). New mechanisms of pulmonary arterial hypertension: role of Ca(2)(+) signaling. Am J Physiol Heart Circ Physiol 302: H1546–H1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM et al (2004). Chronic hypoxia‐induced upregulation of store‐operated and receptor‐operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 95: 496–505. [DOI] [PubMed] [Google Scholar]
- Long L, Yang X, Southwood M, Lu J, Marciniak SJ, Dunmore BJ et al (2013). Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ Res 112: 1159–1170. [DOI] [PubMed] [Google Scholar]
- Looareesuwan S, White NJ, Chanthavanich P, Edwards G, Nicholl DD, Bunch C et al (1986). Cardiovascular toxicity and distribution kinetics of intravenous chloroquine. Br J Clin Pharmacol 22: 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Ran P, Zhang D, Peng G, Li B, Zhong N et al (2010). Sildenafil inhibits chronically hypoxic upregulation of canonical transient receptor potential expression in rat pulmonary arterial smooth muscle. Am J Physiol Cell Physiol 298: C114–C123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K et al (2014). Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 26: 190–206. [DOI] [PubMed] [Google Scholar]
- Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX (2004). Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res 68: 75–103. [DOI] [PubMed] [Google Scholar]
- Mandegar M, Yuan JX (2002). Role of K+ channels in pulmonary hypertension. Vascul Pharmacol 38: 25–33. [DOI] [PubMed] [Google Scholar]
- Manson ML, Safholm J, Al‐Ameri M, Bergman P, Orre AC, Sward K et al (2014). Bitter taste receptor agonists mediate relaxation of human and rodent vascular smooth muscle. Eur J Pharmacol 740: 302–311. [DOI] [PubMed] [Google Scholar]
- McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Matsumoto T, Webb RC (2016). Autoimmune therapeutic chloroquine lowers blood pressure and improves endothelial function in spontaneously hypertensive rats. Pharmacol Res 113 (Pt A): 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR et al (2009). ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association. Circulation 119: 2250–2294. [DOI] [PubMed] [Google Scholar]
- Meyerhof W, Batram C, Kuhn C, Brockhoff A, Chudoba E, Bufe B et al (2010). The molecular receptive ranges of human TAS2R bitter taste receptors. Chem Senses 35: 157–170. [DOI] [PubMed] [Google Scholar]
- Musabayane CT, Ndhlovu CE, Balment RJ (1994). The effects of oral chloroquine administration on kidney function. Ren Fail 16: 221–228. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF, Standen NB (1990). Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol 259 (1 Pt 1): C3–18. [DOI] [PubMed] [Google Scholar]
- Pascolo S (2016). Time to use a dose of chloroquine as an adjuvant to anti‐cancer chemotherapies. Eur J Pharmacol 771: 139–144. [DOI] [PubMed] [Google Scholar]
- Peng G, Li S, Hong W, Hu J, Jiang Y, Hu G et al (2015). Chronic hypoxia increases intracellular Ca2+ concentration via enhanced Ca2+ entry through receptor‐operated Ca2+ channels in pulmonary venous smooth muscle cells. Circ J 79: 2058–2068. [DOI] [PubMed] [Google Scholar]
- Peng G, Ran P, Lu W, Zhong N, Wang J (2013). Acute hypoxia activates store‐operated Ca2+ entry and increases intracellular Ca2+ concentration in rat distal pulmonary venous smooth muscle cells. J Thorac Dis 5: 605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RE, Warrell DA, Edwards G, Galagedera Y, Theakston RD, Abeysekera DT et al (1986). Divided dose intramuscular regimen and single dose subcutaneous regimen for chloroquine: plasma concentrations and toxicity in patients with malaria. Br Med J (Clin Res Ed) 293: 13–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulkkinen V, Manson ML, Safholm J, Adner M, Dahlen SE (2012). The bitter taste receptor (TAS2R) agonists denatonium and chloroquine display distinct patterns of relaxation of the guinea pig trachea. Am J Physiol Lung Cell Mol Physiol 303: L956–L966. [DOI] [PubMed] [Google Scholar]
- Rainsford KD, Parke AL, Clifford‐Rashotte M, Kean WF (2015). Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology 23: 231–269. [DOI] [PubMed] [Google Scholar]
- Sai WB, Yu MF, Wei MY, Lu Z, Zheng YM, Wang YX et al (2014). Bitter tastants induce relaxation of rat thoracic aorta precontracted with high K+ . Clin Exp Pharmacol Physiol 41: 301–308. [DOI] [PubMed] [Google Scholar]
- Sainz E, Cavenagh MM, Gutierrez J, Battey JF, Northup JK, Sullivan SL (2007). Functional characterization of human bitter taste receptors. Biochem J 403: 537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakao S, Voelkel NF, Tanabe N, Tatsumi K (2015). Determinants of an elevated pulmonary arterial pressure in patients with pulmonary arterial hypertension. Respir Res 16: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon VR, Lee H (2009). Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol 625: 220–233. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV (1994). Signal transduction and regulation in smooth muscle. Nature 372: 231–236. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Sanderson MJ (2014). Bitter tasting compounds dilate airways by inhibiting airway smooth muscle calcium oscillations and calcium sensitivity. Br J Pharmacol 171: 646–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Chen J, Fraidenburg DR, Song S, Sysol JR, Drennan AR et al (2015). Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L208–L220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thebault S, Zholos A, Enfissi A, Slomianny C, Dewailly E, Roudbaraki M et al (2005). Receptor‐operated Ca2+ entry mediated by TRPC3/TRPC6 proteins in rat prostate smooth muscle (PS1) cell line. J Cell Physiol 204: 320–328. [DOI] [PubMed] [Google Scholar]
- Tuder RM, Archer SL, Dorfmuller P, Erzurum SC, Guignabert C, Michelakis E et al (2013). Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 62 (25 Suppl): D4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Yamamura A, Zimnicka AM, Voiriot G, Smith KA, Tang H et al (2013). Chronic hypoxia selectively enhances L‐ and T‐type voltage‐dependent Ca2+ channel activity in pulmonary artery by upregulating Cav1.2 and Cav3.2. Am J Physiol Lung Cell Mol Physiol 305: L154–L164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Shimoda LA, Weigand L, Wang W, Sun D, Sylvester JT (2005). Acute hypoxia increases intracellular [Ca2+] in pulmonary arterial smooth muscle by enhancing capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol 288: L1059–L1069. [DOI] [PubMed] [Google Scholar]
- Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA (2006). Hypoxia inducible factor 1 mediates hypoxia‐induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98: 1528–1537. [DOI] [PubMed] [Google Scholar]
- White NJ (1996). The treatment of malaria. N Engl J Med 335: 800–806. [DOI] [PubMed] [Google Scholar]
- Xu JC, Peng YB, Wei MY, Wu YF, Guo D, Qin G et al (2015). Chloroquine inhibits Ca2+ signaling in murine CD4+ thymocytes. Cell Physiol Biochem 36: 133–140. [DOI] [PubMed] [Google Scholar]
- Yoo HY, Zeifman A, Ko EA, Smith KA, Chen J, Machado RF et al (2013). Optimization of isolated perfused/ventilated mouse lung to study hypoxic pulmonary vasoconstriction. Pulm Circ 3: 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan XJ, Goldman WF, Tod ML, Rubin LJ, Blaustein MP (1993). Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am J Physiol 264 (2 Pt 1): L116–L123. [DOI] [PubMed] [Google Scholar]
- Zhang CH, Chen C, Lifshitz LM, Fogarty KE, Zhu MS, ZhuGe R (2012). Activation of BK channels may not be required for bitter tastant‐induced bronchodilation. Nat Med 18: 648–650 author reply 650‐641. [DOI] [PubMed] [Google Scholar]
- Zhang CH, Lifshitz LM, Uy KF, Ikebe M, Fogarty KE, ZhuGe R (2013). The cellular and molecular basis of bitter tastant‐induced bronchodilation. PLoS Biol 11: e1001501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Luo XJ, Sai WB, Yu MF, Li WE, Ma YF et al (2014). Non‐selective cation channels mediate chloroquine‐induced relaxation in precontracted mouse airway smooth muscle. PLoS One 9: e101578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Zhao YL, Deng X, Yang S, Mao Y, Li Z et al (2009). Chloroquine inhibits colon cancer cell growth in vitro and tumor growth in vivo via induction of apoptosis. Cancer Invest 27: 286–292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 RT‐PCR analysis of TAS2R in human PASMC (A), rat pulmonary arteries (B) and mouse pulmonary arteries. Transcripts of seven subtypes of hTAS2Rs (3, 4, 7, 10, 14, 39, 40) (A), rTAS2Rs (137, 108, 121, 107, 123, 139, 144) (B) and mTAS2Rs (137, 108, 130, 110, 114, 139 140) (C) are identified in human PASMC, rat pulmonary arteries and mouse pulmonary arteries, respectively.
Figure S2 Chloroquine inhibits ATP‐induced increase in [Ca2+]cyt in HEK293 cells transiently transfected with the TRPC6 gene. A: Representative traces showing changes of [Ca2+]cyt before and during extracellular application of ATP (100 μM) in HEK293 cells transfected with an empty vector (GFP) and the TRPC6 gene (Trpc6) in the absence (GFP, Trpc6) or presence (Trpc6 + CHQ) of 200 μM chloroquine (CHQ). B: Summarized data (mean ± SE) showing [Ca2+]cyt before (Basal) and after (ATP) extracellular application of ATP in GFP‐transfected and Trpc6‐transfected cells in the absence (GFP, Trpc6) or presence (Trpc6 + CHQ) of chloroquine. *P < 0.05 vs. Basal. C: Summarized data (mean ± SE) showing ATP‐induced increases in [Ca2+]cyt in GFP‐transfected cells and Trpc6‐transfected cells treated with (Trpc6 + CHQ) or without (Trpc6) chloroquine. *P < 0.05 vs. Control; #P < 0.05 vs. Trpc6.
Table S1 The gene synonym between human, rat and mouse TAS2Rs.
Table S2 DNA primer sequences for the hTAS2Rs.
Table S3 DNA primer sequences for the mTAS2Rs.
Table S4 DNA primer sequences for the rTAS2Rs.
Table S5 Chloroquine inhibits the development of hypoxia‐induced pulmonary hypertension in rats.
Table S6 Chloroquine inhibits the progression of established pulmonary hypertension in Sugen5416/hypoxia rat model.