

Abstract
Background and Purpose
The potential for therapeutic antibody treatment of neurological diseases is limited by poor penetration across the blood–brain barrier. I.c.v. delivery is a promising route to the brain; however, it is unclear how efficiently antibodies delivered i.c.v. penetrate the cerebrospinal spinal fluid (CSF)‐brain barrier and distribute throughout the brain parenchyma.
Experimental Approach
We evaluated the pharmacokinetics and pharmacodynamics of an inhibitory monoclonal antibody against β‐secretase 1 (anti‐BACE1) following continuous infusion into the left lateral ventricle of healthy adult cynomolgus monkeys.
Key Results
Animals infused with anti‐BACE1 i.c.v. showed a robust and sustained reduction (~70%) of CSF amyloid‐β (Aβ) peptides. Antibody distribution was near uniform across the brain parenchyma, ranging from 20 to 40 nM, resulting in a ~50% reduction of Aβ in the cortical parenchyma. In contrast, animals administered anti‐BACE1 i.v. showed no significant change in CSF or cortical Aβ levels and had a low (~0.6 nM) antibody concentration in the brain.
Conclusion and Implications
I.c.v. administration of anti‐BACE1 resulted in enhanced BACE1 target engagement and inhibition, with a corresponding dramatic reduction in CNS Aβ concentrations, due to enhanced brain exposure to antibody.
Abbreviations
- anti‐BACE1
antibody to β‐secretase 1 (also known as β‐site amyloid precursor protein cleaving enzyme 1)
- APP
amyloid precursor protein
- Aβ
amyloid‐β
- BBB
blood–brain barrier
- ECL
electrochemiluminescence
- HE
haematoxylin and eosin
- IHC
immunohistochemistry
- i.t.‐L
i.t. lumbar
- mAbs
monoclonal antibodies
- TBS
Tris‐buffered saline
- TBS‐T
Tris‐buffered saline–Tween 20
Introduction
Monoclonal antibodies (mAbs) are increasingly being explored as a promising therapeutic approach for neurodegenerative diseases, owing to their target specificity and long half‐life. Indeed, mAbs have had a significant impact on treatment of a variety of diseases (Reichert, 2015). However, they have yet to demonstrate marked clinical benefit in neurodegenerative indications, with the notable exception of multiple sclerosis, where the therapeutic target of the mAb is in the periphery. Currently, most therapeutic mAbs for diseases of the CNS are being developed for Alzheimer's disease, and the majority of these are directed against amyloid β (Aβ) peptides. While numerous anti‐Aβ mAbs have shown success in preclinical models, anti‐Aβ immunotherapies have demonstrated limited clinical efficacy (Wisniewski and Goni, 2015). One explanation for the lack of clinical success is the very low level of antibody that is able to penetrate the blood–brain barrier (BBB) after systemic dosing (Bickel et al., 2001; Yu and Watts, 2013; Golde, 2014). Physicochemical properties of antibodies, including high molecular weight and hydrophilicity, make them non‐conducive to passive diffusion across the BBB.
Most mAbs are administered systemically in the clinic, usually by i.v. or s.c. routes. Administration directly to the CNS through the more invasive and technically challenging intracerebral, i.t. or i.c.v. routes is less common, and there is limited characterization of the distribution and pharmacokinetics of mAbs following these routes. Studies in rodents suggest that i.c.v.‐delivered macromolecules can be distributed evenly throughout the brain (Wilcock et al., 2003; Wilcock et al., 2004; Weinmann et al., 2006; Dodge et al., 2009; Thakker et al., 2009). However, delivery in larger animals is predicted to be difficult due to larger brain size and differences in cerebrospinal fluid (CSF) flow (Wolak and Thorne, 2013). Indeed, a study investigating i.c.v. infusion of large molecules in nonhuman primates demonstrated that acute infusion (over approximately 4 h) resulted in limited delivery into the brain parenchyma, with the highest concentrations of the infused molecule in areas in contact with the CSF (ventricular and subarachnoid spaces) (Ziegler et al., 2011). A more systematic assessment of mAb distribution and activity following prolonged delivery to the CSF in nonhuman primates is needed to understand the clinical utility of such techniques.
In this study, we evaluated the distribution, pharmacokinetics (PK) and pharmacodynamic (PD) activity of a monoclonal human IgG1 antibody targeting β‐secretase 1 (BACE1), following sustained i.c.v. administration in nonhuman primates. BACE1 is an aspartyl protease critical for the generation of toxic Aβ peptides from amyloid precursor protein (APP) in the brain (Yan and Vassar, 2014). In Alzheimer's disease (AD), an imbalance in the production and clearance of Aβ peptides is believed to result in amyloid plaque accumulation (Hardy and Selkoe, 2002), and reduced Aβ production has been associated with protection from AD (Jonsson et al., 2012; Maloney et al., 2014), making BACE1 inhibition promising as a disease‐modifying therapy. Indeed, several preclinical studies have demonstrated that BACE1 inhibition can reduce amyloid pathology in transgenic mouse models of AD (Eketjall et al., 2013; Jacobsen et al., 2014; Neumann et al., 2015; Thakker et al., 2015). Anti‐BACE1, an allosteric inhibitor, binds its target with high specificity and affinity (KD = 1.3 nM) and inhibits BACE1 activity both in primary neurons and in vivo (Atwal et al., 2011), providing a robust PD readout of mAb activity. We chose to use anti‐BACE1 in this study, so we could correlate PD responses (reduced Aβ production) with antibody PK and distribution in the brain. We have shown previously that a single i.v. injection of anti‐BACE1 at 30 mg·kg−1 to cynomolgus monkeys reduced plasma Aβ levels but only transiently decreased Aβ concentration in the CSF. This limited PD response is attributed to poor brain antibody penetration (Atwal et al., 2011). In the current study, i.c.v. delivery of anti‐BACE1 by continuous infusion into the left lateral ventricle of healthy adult cynomolgus monkeys for 6 weeks was explored as a means to increase brain uptake of antibody as compared to traditional i.v. administration.
Methods
Nonhuman primate study
The study design is summarized in Supporting Information Figure S1. Cynomolgus monkey (Macaca fascicularis; Cambodian origin) was chosen as the test system because of its established utility and acceptance as a model for toxicological and pharmacological studies in large animal species. The cynomolgus monkey has been used for evaluation of this test article in previous pharmacology studies related to i.v. delivery (Atwal et al., 2011; Yu et al., 2014). The animals were housed in individual stainless steel cages at Northern Biomedical Research (NBR) throughout the study. The housing protocol complied with the Guide for the Care and Use of Laboratory Animals, DHHS (NIH), No. 86‐23 and the Animal Welfare Act (9 CFR 3). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
A prior power analysis was carried out using G*Power (Universitat Dusseldorf). Parameter inputs used for calculation were as follows: α = 0.05, power = 0.8 and effect size = 0.86. The effect size was determined based on estimated 50% reduction in Aβx‐40 in CSF or brain, using means and variance based on historical data. Power analysis indicated a sample size of 16 was appropriate for the study (n = 4 for four groups).
Twenty‐one experimentally‐naïve female cynomolgus monkeys, aged 5–8 years old, were used for this study and randomized based on bodyweight to one of four groups. Group 1 animals (n = 7) received i.c.v. infusions of anti‐BACE1; group 2 animals (n = 3) received i.c.v. infusions of anti‐BACE1 vehicle (20 mM sodium succinate, 6% sucrose, 0.02% Tween‐20, pH 5.0); group 3 animals (n = 6) received i.c.v. infusions of control IgG; and group 4 animals (n = 5) were dosed i.v. with anti‐BACE1. Group numbers were not equal in this study. Twenty‐one animals had originally been designated to this study, and the study design was adapted to meet this number, aiming for a final n = 4–5 for all antibody‐treated groups. The i.c.v.‐infused groups were initially overpowered (n = 6–7) relative to the i.v.‐dosed group (n = 5) to balance any attrition of animals due to complications related to surgery in the i.c.v. groups. Five i.c.v.‐dosed animals (two receiving control IgG and three receiving anti‐BACE1) were excluded based on microscopic analysis of brain sections to evaluate tissue integrity (see below). The vehicle i.c.v. group was underpowered (n = 3) as this group was primarily included as a background control for the immunohistochemical analyses using anti‐human IgG. Animals were assigned individual animal identification numbers and tattoos. These identifiers were associated with samples and raw data, allowing sample handlers and data analysers to be blinded to study grouping.
For animals receiving i.c.v. infusions, a Medtronic's 8910 i.c.v.‐AS catheter was surgically implanted in the left lateral ventricle. The pump reservoir was filled with PBS for infusion at 0.048 mL·day−1 to maintain catheter patency for 10 days while allowing the animals to recover from surgery. After these 10 days, the reservoir was rinsed three times with 3 mL of the formulated test article, filled with 20 mL of the test or control article and used to initiate infusion (Study Day 1). Test or control articles were delivered at a rate of 0.4 mL·day−1, resulting in infusion of 5.2–5.6 mg of antibody day−1. Group 4 animals received an i.v. bolus of anti‐BACE1 via the saphenous vein at 50 mg·kg−1 on study days 15, 22, 29 and 36. All animal protocols were approved by the IACUC of Medtronic and were conducted in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals.
Nonhuman primate surgery
A preoperative MRI was performed to determine the stereotactic coordinates for each animal in the i.c.v. groups. At i.c.v. catheter implantation, the animals were pretreated with atropine sulfate as an s.c. injection at a dose of 0.04 mg·kg−1, followed ~15 min later by an i.m. dose of 8 mg·kg−1of ketamine HCl to induce sedation. The animals were masked to a surgical plane of anaesthesia, intubated and maintained on approximately 1 L·min−1 of oxygen and 2% isoflurane. The anaesthetic gases and mixtures varied as required by each individual animal. Prednisolone sodium succinate, 30 mg·kg−1, i.v., and flunixin meglumine, 2 mg·kg−1, i.m., were administered prior to surgery. A dorsal sagittal incision was made over the calvarium, and a catheter (0.6 mm ID × 0.9 mm OD, polyurethane, 8910 i.c.v.‐AS, Medtronic, Inc.) was inserted through a craniotomy site. The surgical target for the catheter tip was based upon the presurgical MRI mapping coordinates. The proximal end of the catheter was connected to the pump catheter, tunnelled under the skin and connected to a SynchroMed® II pump implanted in the lateral abdominal/flank region between layers of the abdominal oblique muscles through a midline lumbar incision. The pump catheter was primed with PBS before being connected to the i.c.v. catheter. Correct i.c.v. catheter placement was confirmed after implant by a ventriculogram with 0.3 mL of Isovue® injected through the catheter access port of the Synchromed II pump. All animals had a catheter inserted (approximately L4 or L5 vertebra) into the i.t. lumbar (i.t.‐L) space, terminating with an s.c. access port for CSF sampling (catheter 0.038 mm I.D. × 1.0 mm O.D. coextruded polyethylene lined polyurethane catheter, SAI, Inc.). A hemilaminectomy was made at approximately L4–L5, the dura incised and the catheter advanced anterograde approximately 10 cm, terminating near the thoracolumbar junction. The distal end of the catheter terminated in a low volume titanium s.c. access port (MIN LOVOL, Instech Solomon). Correct i.t.‐L catheter placement was verified by the ability to collect CSF from the catheter at surgery. The i.c.v. injected animals had a second polyurethane 8910 i.c.v.‐AS catheter stereotactically inserted with the aid of a fibreoptic scope into the left lateral ventricle of the brain, terminating with a Synchromed II infusion pump. After placement of one or both catheters, depending on group assignment, the skin was closed in layers with sutures and tissue adhesive. Following surgery, i.c.v. animals received PBS at a flow rate of 0.048 mL·day−1 for a minimum of 7 days to allow for surgical recovery and to avoid loss of catheter patency during this period. Upon recovery from anaesthesia, the animals were given butorphanol tartrate i.m., 0.05 mg·kg−1, for analgesia and placed on a post‐surgical antibiotic, and ceftiofur sodium, 5 mg·kg−1 i.m., b.i.d. (one injection during or prior to surgery followed by three injections). Following the ceftiofur regimen, the animals implanted with SynchroMed II pumps (groups 1–3) were placed on a prophylactic regimen of 10 mg·kg−1 linezolid p.o., b.i.d. for approximately 10 days during surgical recovery.
Sample collection
For PK and PD analysis, 0.5 mL CSF was obtained via the i.t.‐L s.c. access port on study days −7, 1 (before pump initiation), 4, 8, 15, 16, 18, 22, 25, 29, 32, 36, 39 and 43. For the i.v. cohort, sampling was performed pre‐dose on dosing days (15, 22, 29 and 36). CSF was collected into polypropylene tubes, centrifuged at 4°C at 4000× g for 4 min, split into five aliquots of 0.1 mL each, quickly frozen in liquid nitrogen and stored at −60°C or colder until analysis.
For PK analysis, 2 mL of blood was collected from each animal into serum separator tubes on study days −7, 1 (pre‐pump initiation), 4, 8, 15, 16, 18, 22, 25, 29, 32, 36, 39 and 43. One extra sample was collected 5 min after the first i.v. dose in group 4 (day 15) to capture Cmax. For PD analysis, 2 mL of blood was collected into tubes containing potassium EDTA at the same time points. All blood was centrifuged at 610 × g at 4°C for 15 min. The plasma or serum was harvested, split into duplicate aliquots, quick frozen in liquid nitrogen and stored at −60°C or colder until shipment.
At the end of the study (42 days after initiation of pump infusion or 7 days after the final i.v. dose), all animals were sedated with 8.0 mg·kg−1of ketamine hydrochloride i.m., maintained on an isoflurane/oxygen mixture and provided with an i.v. bolus of heparin sodium, 200 IU·kg−1. The animals were perfused via the left cardiac ventricle with 0.001% sodium nitrite in PBS (pH ~7.2). Following perfusion, the entire delivery system (pump, pump catheter, connector and i.c.v. catheter) was carefully removed intact. The brains were harvested, placed in a chilled brain matrix to facilitate accurate tissue sectioning and processed. Slices of the brain tissue were taken in the coronal plane, at the following levels: slice I (coronal slice at +45.0 mm), slice II (coronal slice at +17.0 mm), slice III (coronal slice at −6.0 mm), slice IV (coronal slice at −19.0 mm), slice V (coronal slice at −32.0 mm) and slice VI (coronal slice at −45.0 mm). At each slice, two slabs were collected, of 3 and 6 mm thickness. The 3 mm slabs were sampled by tissue punches (4 mm diameter) to collect representative tissue samples from distinct brain structures for antibody PK elisa analysis. In addition, larger tissue samples weighing ~100–200 mg were collected from the cortical region of the 3 mm slabs for Aβ and sAPP elisa analysis. Tissue weights for each sample (slices and punches) were measured and recorded. The tissue samples were stored at approximately −80°C until further processing.
The intermittent 6 mm slabs were processed for immunohistochemistry (IHC). They were saved in individually‐labelled, perforated plastic bags and placed in perfusion fixation solution #2 (phosphate buffered 4% paraformaldehyde, Neuroscience Associates) and stored at room temperature for approximately 48 h with stirring. The volume of fixation solution was approximately 10× the volume of the tissue being fixed. Following 48 h of fixation, the brain slabs were transferred to PBS and stored refrigerated until shipment to Neuroscience Associates at room temperature for processing, sectioning and histochemical and IHC staining.
Brain tissue processing for bioanalytical assays
For the elisa to detect antibody concentrations, brain tissue was weighed and homogenized in 1% NP‐40 in PBS containing Complete Mini EDTA‐free protease inhibitor cocktail tablets (Roche Diagnostics). The homogenized brain samples were then rotated at 4°C for 1 h before spinning at 20 000 × g for 20 min. The supernatant was isolated for brain antibody measurement.
For the assay to detect Aβx‐40, the different brain slices collected allowed us to examine different cortical regions in the brain (Slice II, prefrontal cortex, Slice III, motor cortex, Slice IV, temporal cortex, Slice V, parietal cortex). Brain samples were weighed and homogenized in 10 volumes of 5 M guanidine hydrochloride buffer. The samples were rotated for 3 h at room temperature prior to diluting (1:10) in 0.25% casein buffer with 5 mM EDTA (pH 8.0) in PBS containing freshly added aprotinin (20 mg·mL−1) and leupeptin (10 mg·mL−1). The diluted homogenates were spun at 20 000 × g for 20 min, and supernatants were isolated for Aβx‐40 measurement.
For the assay to detect sAPPα and sAPPβ, cortical tissue was weighed and homogenized in 10 volumes of Tris‐buffered saline (TBS) containing Complete Mini EDTA‐free protease inhibitor cocktail tablets (Roche Diagnostics), as well as freshly added aprotinin (20 mg·mL−1) and leupeptin (10 mg·mL−1). The samples were spun at 20 000 × g for 20 min, and supernatants were isolated for sAPPα and sAPPβ measurement.
PK assays
Total antibody concentrations in cynomolgus monkey serum, CSF and brain homogenates were measured with an elisa using a sheep anti‐human IgG (monkey absorbed) antibody coat, followed by adding serum/brain/CSF samples starting at a dilution of 1:100 and finished by adding a goat anti‐human IgG antibody conjugated to horseradish peroxidase (monkey adsorbed), for detection. The assay has a standard curve range of 0.156−10 ng·mL−1 and a limit of detection of 15.6 ng·mL−1. Results below this concentration were reported as less than reportable.
PD assays
The concentrations of Aβx‐40 in cynomolgus monkey plasma were determined using an electrochemiluminescence (ECL) immunoassay, and the concentration of Aβx‐40 in cynomolgus monkey CSF and brain was determined using an elisa. In both cases, the rabbit polyclonal capture antibody, specific for the C terminus of Aβ 40 (Millipore), was pre‐coated onto the plates, and the anti‐Aβ monoclonal antibody 6E10 (Covance) was used for detection. The assay had limit of quantification values of 60 pg·mL−1 in plasma, 28 pg·mL−1 in CSF and 20 pg·mL−1 in brain lysate.
CSF and brain concentrations of sAPPα and sAPPβ were determined with as sAPPα/sAPPβ multiplex ECL assay. The anti‐Aβ monoclonal antibody 6E10 was used to capture sAPPα, whereas an antibody directed against amino acids 591 to 596 of APP was used to capture sAPPβ. Both analytes were detected with an antibody directed against the N terminus of APP. CSF was thawed on ice, then diluted 1:10 into 1% BSA in TBS–Tween 20 (TBS‐T). The assay had lower limit of quantification values of 0.3 ng·mL−1 for sAPPα and 0.1 ng·mL−1 for sAPPβ.
Because absolute concentrations of Aβx‐40 in plasma and CSF varied greatly between animals, as noted previously (Atwal et al., 2011), Aβx‐40 measurements were normalized as follows: a baseline Aβx‐40 concentration was determined for each animal by averaging the data from two pre‐dose time points. All Aβx‐40 measurements for each animal were then calculated as a percentage of that baseline value. Because absolute sAPPα and sAPPβ values can also vary, we present data as a ratio of sAPPβ/sAPPα, as both analytes are measured within the same sample allowing for internal normalization.
Monkey brain embedding and sectioning
The coronal tissue slabs were treated overnight with 20% glycerol and 2% DMSO to prevent freeze artefacts. Two MultiBrain® blocks were created for each brain. The first block consisted of a single/primary slab designated as the slab adjacent to the infusion site. The second block consisted of the remaining slabs stacked in a serial manner to mimic an intact brain. Each block was then encased in a gelatin matrix using MultiBrain® Technology (NeuroScience Associates, Knoxville, TN, USA). After being cured, the block was rapidly frozen by immersion in isopentane chilled to −70°C with crushed dry ice and mounted on a freezing stage of an AO 860 sliding microtome. Each MultiBrain® block was sectioned coronally at 40 μ. All sections cut (none discarded) were collected sequentially into a series of 24 containers for the single slab and 30 containers for the stacked slabs. The cups were filled with Antigen Preserve solution (50% PBS pH 7.0, 50% ethylene glycol, 1% polyvinyl pyrrolidone) for storage until staining.
For the stains listed below, sections were stained at intervals of 980 μm for the single/primary slab and intervals of 1.96 mm for the stacked slabs.
Histochemical and immunohistochemical staining
For haematoxylin and eosin (HE; Richard‐Allan Scientific®, Thermo Scientific) staining, air‐dried sections on slides were stained using standard histological procedures. For anti‐human IgG and anti‐macaque IgG IHC, the sections were stained free‐floating as follows. All incubation solutions from the blocking serum onward used TBS with Triton X‐100 as the vehicle; all rinses were with TBS. After being treated with hydrogen peroxide and blocking serum, the sections were immunostained with either monkey pre‐adsorbed anti‐human IgG‐HRP (The Binding Site, Cat# AP003CUS01) or anti‐macaque IgG IB3‐HRP (Kerafast. Cat# ET0002) as primary antibody overnight at room temperature. An avidin‐biotin‐HRP complex (details in Vectastain elite ABC kit, Vector, Burlingame, CA, USA) was used to detect antibody binding. After being rinsed, the sections were treated with diaminobenzidine tetrahydrochloride and hydrogen peroxide to create a visible reaction product and mounted on gelatinized (subbed) glass slides, air‐dried, dehydrated in alcohols, cleared in xylene and cover‐slipped.
Animal exclusion based on microscopic evaluation of brain sections
Five animals (three in the anti‐BACE1 i.c.v. group and two in the control IgG i.c.v. group) were eliminated from PK and PD composite data analysis because microscopic examination of the HE and IHC‐stained sections indicated a breakdown in the integrity of the BBB surrounding the left lateral ventricle (site of catheter) or because it was considered likely that the catheter was infusing directly in the periventricular parenchyma rather than into the ventricle. The breakdown of BBB integrity, as determined by parenchymal staining for anti‐macaque IgG, was attributed to complications related to the catheter. In one animal, marked inflammation of and around the lateral ventricle was suggestive of a bacterial infection, and in another animal, mild inflammation was observed around the catheter in the caudate nucleus. The other three animals did not have evidence of BBB breakdown but had intense human IgG staining in the periventricular parenchyma suggesting direct infusion of mAb that was probably due to catheter displacement during the study.
Data analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). PK analysis was carried out as follows: individual anti‐BACE1 and control IgG serum and CSF concentration time profiles were created on a semi‐logarithmic plot using nominal time of sample collection. PK parameters were estimated using WinNonlin® Enterprise, Version 5.2.1 (Pharsight Corporation; Mountain View, CA, USA). Non‐compartmental analysis (WinNonlin® Model 201) was used for PK analysis. The following PK parameters were reported: AUCall [area under the concentration time curve from time 0 (time of drug administration) to time of last measurable concentration was calculated using the linear trapezoidal rule)], Css (steady‐state concentration), CL (clearance) and time to steady state. Antibody concentrations below the limit of detection were interpreted as missing for graphical and summary presentations and not included in any calculations. The nominal dose administered to each group was used for modelling.
Materials
Test articles
Anti‐BACE1 and the control IgG (anti‐gD, targeting the glycoprotein D epitope of Herpes Simplex Virus) have been described previously (Atwal et al., 2011) and were produced at Genentech. The antibodies were formulated at 13–17 mg·mL−1. Prior to administering the antibodies to animals, molecular stability was established by analysing samples that were incubated at 37°C in the Synchromed II infusion system at simulated preclinical use conditions. Analysis included visual inspection, size exclusion chromatography, ion‐exchange chromatography and capillary electrophoresis under reduced and non‐reduced conditions. Both antibodies retained physical and chemical stability over a period of time equal to the infusion time, and anti‐BACE1 retained high activity in cell‐based BACE1 inhibition assays.
Antibodies
elisa: monkey pre‐adsorbed sheep anti‐human IgG (Jackson ImmunResearch, West Grove, PA, USA), monkey pre‐absorbed goat anti‐human IgG‐HRP (Jackson, ImmunoResearch), anti‐Aβ40 (Millipore, Bedford, MA, USA), anti‐Aβ clone 6E10 (Covance, Dedham, MA, USA) and anti‐APP 591–596 (Covance). IHC: monkey pre‐adsorbed anti‐human IgG‐HRP (The Binding Site, Cat# AP003CUS01) and anti‐macaque IgG IB3‐HRP (Kerafast. Cat# ET0002).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
CSF pharmacokinetics and pharmacodynamics
To assess the brain distribution and activity of mAbs after i.c.v. infusion, either anti‐BACE1, a non‐targeting control IgG, or vehicle alone was delivered by continuous infusion into the left lateral ventricle of cynomolgus monkeys over 6 weeks at a rate of 0.4 mL·day−1. This resulted in infusion of approximately 5.2–5.6 mg IgG·day−1, delivering 36.4–39.2 mg IgG over a 7 day period. To compare brain distribution following i.c.v. versus traditional systemic delivery, a separate group of animals was i.v. dosed with 50 mg·kg−1 anti‐BACE1 weekly for 4 weeks, with delivery of 150–220 mg of IgG over 7 days. The i.v. delivery route is commonly used in the clinic, although the doses administered here exceed typical clinical doses in an attempt to maximize passive brain penetration. CSF was sampled throughout the study to measure (i) antibody delivery and (ii) PD response to anti‐BACE1.
Both anti‐BACE1 and control IgG reached steady‐state concentrations of ~1.2 μM in the CSF by the first sample collection 4 days following initiation of i.c.v. drug administration (Figure 1A). Anti‐BACE1‐administered i.v. also reached steady‐state concentrations in the CSF by 4 days after initiation of drug administration. However, this concentration range of 3–5 nM was significantly lower (by 300‐fold) than that achieved with i.c.v. infusion. Antibody exposure, as measured by AUCall, and clearance in CSF were calculated and are reported in Table 1.
Figure 1.
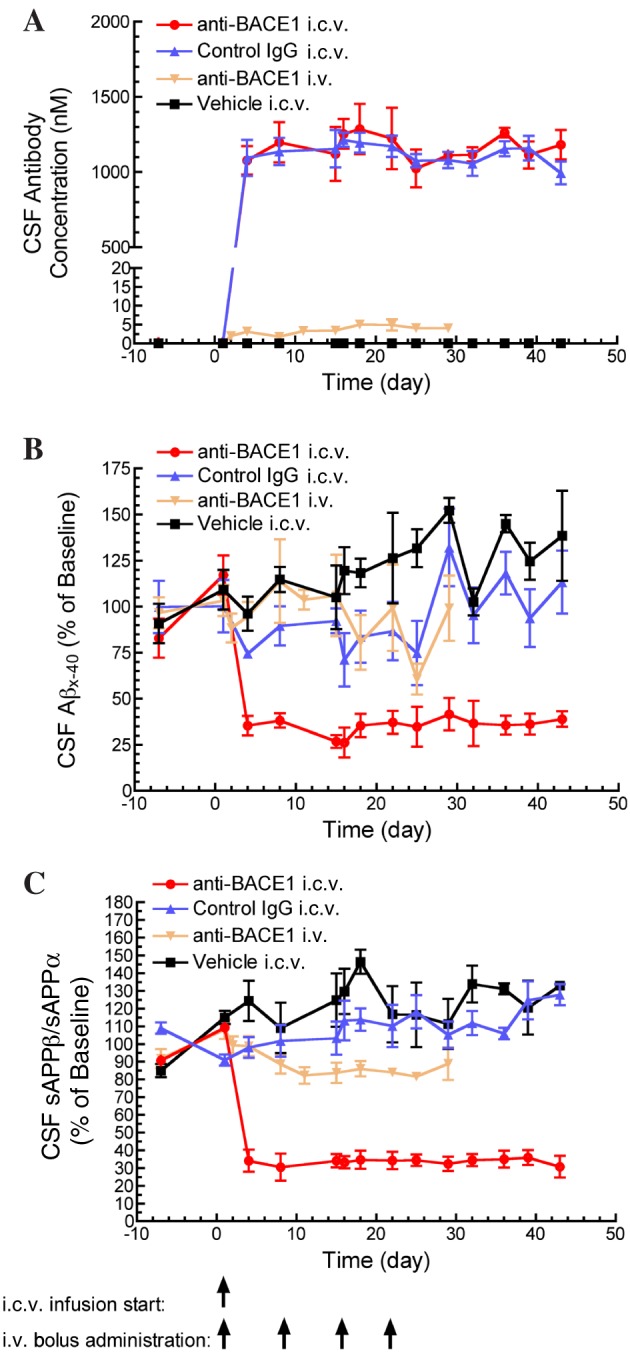
Reduced CSF Aβx‐40 concentration and sAPPβ/sAPPα ratio (amyloid precursor protein cleavage products) following anti‐BACE1 (β‐site amyloid precursor protein cleaving enzyme 1) i.c.v. infusion. (A) Anti‐BACE1 and control IgG reached steady‐state CSF concentrations by the earliest sampling time (4 days) following the start of i.c.v. antibody infusion. Only anti‐BACE1 delivered by i.c.v. infusion significantly achieved and maintained reductions in CSF Aβx‐40 concentration, P < 0.05 (B) and CSF sAPPβ/sAPPα ratio, P < 0.05 (C). Statistical test: one‐way ANOVA with Bonferroni multiple comparisons test compared to control IgG i.c.v. Points represent mean ± SEM. Group (n): anti‐BACE1 i.c.v. (4), control IgG i.c.v. (4), anti‐BACE1 i.v. (5) and vehicle i.c.v. (3).
Table 1.
Pharmacokinetic parameters of antibodies in CSF after a 6 week i.c.v. infusion or four weekly i.v. bolus in cynomolgus monkeys
| anti‐gDa i.c.v. | anti‐BACE1a i.c.v. | anti‐BACE1b i.v. | |
|---|---|---|---|
| Time to steady state (days)c | ≤4 | ≤4 | ≤4 |
| AUC (μM * day)d | 45.6 | 48.2 | 0.0267 |
| CL (mL·day−1·kg−1)e | 6.8 | 7.11 | NAg |
| Css (μM)f | 1.2 | 1.2 | 0.004 |
Each group received their respective dose material as a constant i.c.v. infusion over 6 weeks (n = 4 per group). Twelve samples were collected from each monkey over the 6 week study.
Each monkey received four weekly i.v. injecttions of this dose material (n = 5). Ten samples were collected from each monkey over the 4 week study.
This PK parameter estimates how long it took for the concentration in the CSF to reach steady state.
This PK parameter estimates the area under the concentration–time curve from 0 to the last measured time point.
This PK parameter estimates the clearance of the antibody from the CSF.
This PK parameter estimates the steady‐state concentration in the CSF.
Clearance estimation is not applicable for this dose group.
Animals dosed with anti‐BACE1 by i.c.v. delivery showed a robust reduction in CSF Aβ levels. By 4 days post‐drug administration, CSF Aβ was reduced by about 70% compared to baseline levels, and this level of reduction was maintained for the duration of the study (Figure 1B). This PD response was confirmed by measuring the concentration of the APP cleavage products sAPPβ and sAPPα in CSF. Following i.c.v. anti‐BACE1 dosing, the ratio of sAPPβ/sAPPα was decreased, with a mean reduction of 65% from baseline (Figure 1C). In contrast, i.c.v. infusion of control IgG or vehicle did not alter CSF Aβ levels or the sAPPβ/sAPPα ratio, confirming the specificity of the PD response to anti‐BACE1. Delivery of anti‐BACE1 via systemic (i.v.) administration did not significantly reduce CSF Aβ levels, although there was a trend toward reduction of the sAPPβ/α ratio after multiple doses (Figure 1C).
Systemic pharmacokinetics and pharmacodynamics
We evaluated the concentration of antibody present in circulation following i.c.v. delivery. Both anti‐BACE1 and control IgG redistributed from CSF into the serum at a rate of about 0.02 mL·min−1. Serum steady‐state concentrations were achieved 14 days after the start of i.c.v. drug infusion and were maintained at 2.5‐3 μM for the remainder of the study (Figure 2A). In comparison, direct systemic delivery of anti‐BACE1 by i.v. dosing (50 mg·kg−1) resulted in peak serum antibody concentrations of ~12 μM, which decreased over the dosing period. The peak and trough concentrations of anti‐BACE1 increased with each antibody dose. Some accumulation (accumulation ratio = 2.34) was observed, in line with the weekly administration and the long half‐life of the antibody when administered systemically. The high levels of systemic anti‐BACE1 achieved through both i.c.v. and i.v. delivery led to a ~50% reduction in peripheral Aβ levels at 4 days following antibody administration, which persisted for the duration of the study (Figure 2B).
Figure 2.
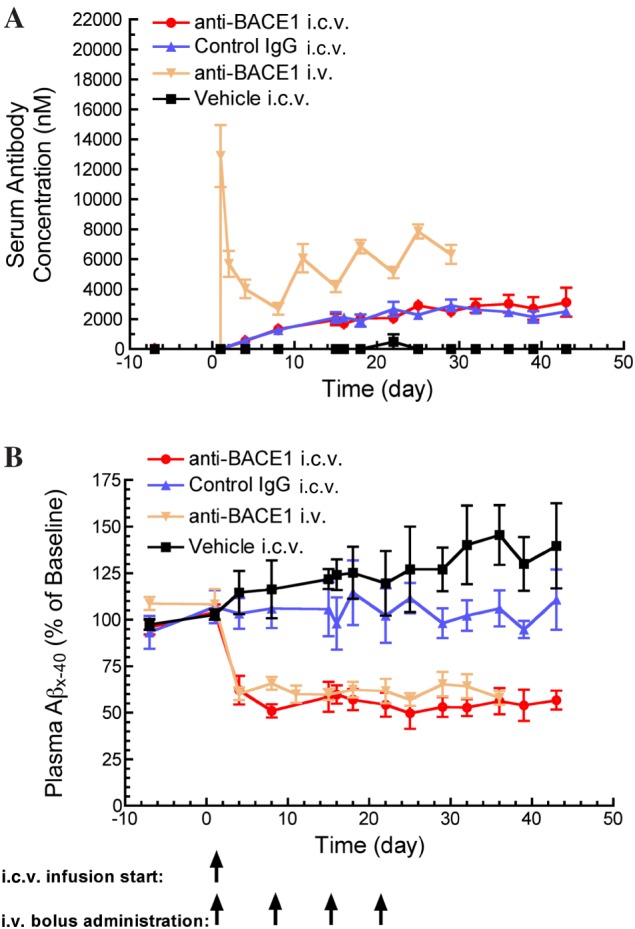
Reduced plasma Aβx‐40 concentration following anti‐BACE1 i.c.v. infusion and i.v. bolus administration. (A) Anti‐BACE1 and control IgG reached steady‐state serum concentrations 2 weeks following the start of i.c.v. antibody infusion. Anti‐BACE1 serum concentrations were maintained at higher levels in animals receiving i.v. dosing compared to those receiving i.c.v. infusion. (B) Anti‐BACE1 delivered by both routes significantly reduced and maintained plasma Aβx‐40 concentrations at ~50% below baseline, P < 0.05 for anti‐BACE1 i.v. and P < 0.05 for anti‐BACE1 i.c.v. Statistical test: one‐way ANOVA with Bonferroni multiple comparisons test compared to control IgG i.c.v. Points represent mean ± SEM. Group (n): anti‐BACE1 i.c.v. (4), control IgG i.c.v. (4), anti‐BACE1 i.v. (5) and vehicle i.c.v. (3).
Brain distribution, pharmacokinetics and pharmacodynamics
At the end of the 6 weeks of infusion, the brains were harvested and tissue was isolated for both histological evaluation and PK/PD analysis. Tissue sections were stained with HE to assess tissue morphology and integrity, for macaque IgG to identify disruptions in the BBB or for human IgG to detect presence of infused antibody. In five of the study animals that received i.c.v. infusion (two control IgG i.c.v. dosed and three anti‐BACE1 i.c.v. dosed), there was evidence of either a breakdown in BBB integrity in the periventricular parenchyma around the left ventricle (site of catheter) or of antibody infusion directly into the periventricular parenchyma due to catheter displacement during the study. These animals were excluded from all analysis (see Methods).
To quantify brain penetration of antibody in the different treatment groups, we carried out elisa analysis of human IgG antibody levels in tissue samples taken throughout the brain. elisa results revealed high concentrations of human IgG in brain tissue from animals that received either anti‐BACE1 or control IgG by i.c.v. infusion, with the highest antibody concentration in the left caudate nucleus, near the site of infusion (Figure 3). Notably, both anti‐BACE1 and control IgG antibodies were distributed evenly throughout other regions of the brain and not limited to the periventricular structures. Most brain regions, regardless of position along the rostro‐caudal axis or hemisphere, had approximately 20–40 nM of antibody (Table 2) upon i.c.v. infusion. Exceptions were the deep internal structures of the caudate, putamen and globus pallidus contralateral to catheter placement that had concentrations of 5–10 nM. No notable differences were observed between the control IgG and anti‐BACE1 i.c.v. infusion groups. Animals that received anti‐BACE1 delivered via systemic administration had brain antibody concentrations of one order of magnitude lower, generally ranging from 0.5 to 2 nM (Table 2).
Figure 3.
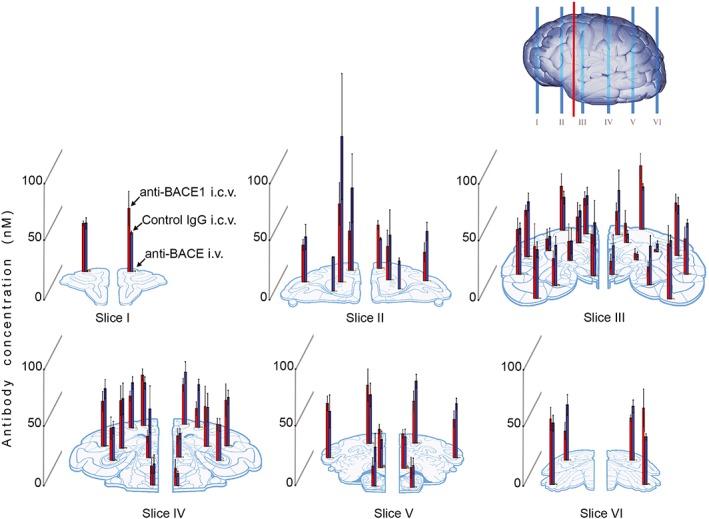
Widespread antibody distribution in brain following anti‐BACE1 i.c.v. infusion. Brain antibody distribution was assessed by elisa measurement of human IgG1 concentration across different brain regions. Bar graphs of antibody concentration are overlaid on the representative brain regions sampled on the schematic slices, taken from rostral (I) to caudal (VI) regions. Red line on brain diagram represents location of i.c.v. cannula placement. Anti‐BACE1 and control IgG delivered by i.c.v. infusion reached similar concentrations (20–40 nM) throughout the brain regions assessed. In comparison, i.v.‐administered anti‐BACE1 achieved brain concentrations approximately one order of magnitude lower. Bars represent mean ± SEM for concentration measured in each brain region. Group (n): anti‐BACE1 i.c.v. (4), control IgG i.c.v. (4), anti‐BACE1 i.v. (5) and vehicle i.c.v. (3).
Table 2.
Antibody brain concentrations (nM) after i.c.v. infusion or four weekly i.v. bolus doses in cynomolgus monkeys
| anti‐BACE1 i.c.v.a | Control IgG i.c.v.a | anti‐BACE1 i.v.b | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Left hemisphere | Right hemisphere | Left hemisphere | Right hemisphere | Left hemisphere | Right hemisphere | ||||||||
| Slicec | Brain regiond | Average | SEM | Average | SEM | Average | SEM | Average | SEM | Average | SEM | Average | SEM |
| Slice I | Prefrontal cortex | 41.0 | 21.0 | 53.7 | 14.3 | 41.0 | 4.55 | 33.0 | 1.24 | 1.96 | 0.505 | 1.41 | 0.242 |
| Cingulate cortex | 33.5 | 7.42 | 36.6 | 3.73 | 69.5 | 20.4 | 25.0 | 0.983 | 0.727 | 0.0492 | 0.969 | 0.168 | |
| Slice II | Prefrontal cortex | 31.1 | 4.79 | 24.3 | 7.93 | 38.1 | 10.5 | 41.7 | 7.75 | 0.627 | 0.135 | 0.543 | 0.0927 |
| Caudate | 65.8 | 17.9 | 28.4 | 13.6 | 123 | 53.1 | 37.6 | 21.6 | 0.562 | 0.0703 | 0.631 | 0.153 | |
| Slice III | Cingulate cortex | 30.0 | 5.69 | 19.8 | 7.31 | 32.1 | 7.1.0 | 37.3 | 15.0 | 0.745 | NA | 0.984 | 0.157 |
| Motor cortex | 37.4 | 10.6 | 53.9 | 10.3 | 27.7 | 5.22 | 35.9 | 2.66 | 0.722 | 0.080 | 0.899 | 0.171 | |
| Somatosensory and insular cortex | 39.5 | 4.19 | 44.6 | 7.42 | 46.3 | 7.27 | 42.2 | 6.8.0 | 0.885 | NA | 0.779 | 0.183 | |
| Temporal cortex | 38.2 | 9.45 | 30.3 | 10.0 | 38.7 | 4.69 | 43.1 | 3.08 | 0.945 | 0.160 | 0.885 | 0.155 | |
| Entorhinal cortex | 44.2 | 19.2 | 46.7 | 15.7 | 41.4 | 5.64 | 49.3 | 4.08 | 0.882 | NA | 0.816 | 0.165 | |
| Caudate | 22.2 | 12.0 | 16.7 | 9.23 | 27.0 | 4.83 | 7.17 | 3.17 | 0.674 | 0.100 | 0.642 | 0.0958 | |
| Putamen | 10.2 | 8.31 | 1.85 | 0.698 | 12.2 | 7.05 | 6.59 | 2.69 | 0.566 | 0.0723 | 0.452 | 0.0451 | |
| Globus pallidus | 16.4 | 14.7 | 5.86 | 4.08 | 16.5 | 10.6 | 4.71 | 2.11 | 0.392 | 0.0839 | 0.371 | 0.0718 | |
| Hypothalamus | 35.0 | 7.14 | 11.5 | 5.99 | 44.8 | 19.0 | 24.1 | 9.56 | 0.709 | 0.133 | 0.731 | 0.247 | |
| Amygdala | 23.2 | 5.78 | 15.5 | 4.2.0 | 34.0 | 7.57 | 32.7 | 8.87 | 1.28 | 0.648 | 1.02 | 0.482 | |
| Slice IV | Cingulate cortex | 42.9 | 5.04 | 33.7 | 5.39 | 36.1 | 4.99 | 44.0 | 8.92 | 0.689 | 0.0182 | 0.936 | 0.185 |
| Parietal cortex | 27.1 | 4.15 | 16.0 | 5.71 | 38.1 | 5.06 | 35.9 | 4.63 | 0.494 | 0.0850 | 0.841 | 0.117 | |
| Temporal cortex | 38.3 | 8.2.0 | 38.7 | 13.8 | 48.5 | 8.08 | 40.7 | 6.06 | 1.03 | 0.396 | 0.974 | 0.307 | |
| Thalamus | 27.7 | 9.35 | 34.1 | 16.9 | 28.0 | 5.10 | 32.9 | 12.0 | 1.37 | 0.788 | 0.879 | 0.200 | |
| Hippocampus | 40.4 | 12.4 | 30.8 | 5.17 | 41.8 | 13.2 | 30.0 | 5.32 | 1.09 | 0.286 | 0.725 | 0.122 | |
| Pontine nuclei | 16.5 | 6.09 | 14.9 | 7.33 | 18.0 | 7.63 | 10.2 | 2.17 | 0.682 | 0.115 | 0.642 | 0.0998 | |
| Dorsal raphe | 18.2 | 4.91 | 18.2 | 6.14 | 41.0 | 17.0 | 19.9 | 4.29 | 0.862 | 0.154 | 0.743 | 0.0925 | |
| Slice V | Parietal cortex | 49.2 | 13.6 | 35.5 | 8.60 | 41.1 | 9.79 | 52.3 | 5.71 | 1.08 | 0.211 | 0.992 | 0.168 |
| Visual cortex | 45.8 | 6.08 | 32.5 | 7.00 | 39.2 | 13.7 | 45.5 | 4.76 | 0.816 | 0.112 | 0.981 | 0.195 | |
| Brainstem | 32.8 | 4.11 | 29.4 | 3.77 | 23.7 | 7.19 | 26.7 | 6.59 | 2.45 | 0.858 | 2.42 | 1.14 | |
| Cerebellum | 17.1 | 7.10 | 16.8 | 8.87 | 32.7 | 11.7 | 18.1 | 6.57 | 0.524 | 0.133 | 0.696 | 0.118 | |
| Slice VI | Visual cortex | 24.6 | 7.31 | 35.7 | 2.76 | 46.7 | 8.62 | 45.5 | 5.36 | 0.829 | 0.0723 | 0.679 | 0.0122 |
| Cerebellum | 55.4 | 8.47 | 64.6 | 13.8 | 52.0 | 5.65 | 40.3 | 2.57 | 0.941 | 0.0759 | 1.24 | 0.379 | |
Results are presented as average and SEM
Each group received their respective dose material as a constant i.c.v. infusion over 6 weeks (n = 4 per group). All brain samples were taken at the end of the 6 week infusion.
Each monkey received four weekly i.v. injections of this dose material (n = 5). All brain samples were taken a week after the fourth dose was administered.
Indicates the depth within the brain from which the samples were harvested. The slice number refers to the depth as indicated in Figure 3.
Indicates which brain region was in the samples within a given brain slice.
To corroborate increased brain exposure and distribution by i.c.v. antibody delivery over i.v., as demonstrated by elisa analysis, we examined brain tissue sections from the animals to see if we could visually detect higher levels of antibody in brain following i.c.v. infusion. Representative brain sections stained for anti‐macaque IgG, anti‐human IgG or with HE from one animal from each group are shown in Figure 4. In general, animals dosed i.c.v. with either anti‐BACE1 or control IgG showed a slight increase in anti‐human IgG staining above background levels (as determined by group administered vehicle i.c.v.). Staining was fairly comparable between these two groups and widespread throughout the grey matter. The highest intensity of staining was observed in the periventricular parenchyma of the left ventricle. Human IgG immunostaining was not detectable above background in animals dosed i.v. with anti‐BACE1.
Figure 4.
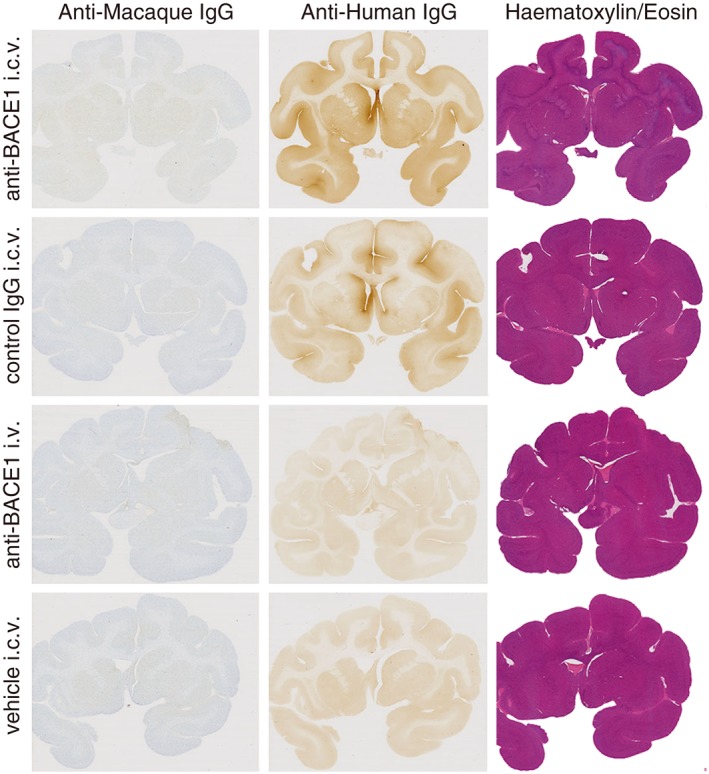
Brain uptake of IgG following i.c.v. infusion. Representative brain sections of one animal in each group show higher and comparable grey matter anti‐human IgG IHC staining over nonspecific background staining in i.c.v.‐dosed groups. Anti‐human IgG IHC was not sensitive enough to detect low levels of anti‐BACE1 following i.v. administration over background staining. HE staining was used to assess tissue morphology and integrity, and anti‐macaque IgG IHC was used to identify disruptions in the BBB. Animals with abnormalities identified with these stains were eliminated from all PK/PD analyses. Group (n): anti‐BACE1 i.c.v. (4), control IgG i.c.v. (4), anti‐BACE1 i.v. (5) and vehicle i.c.v. (3).
Brain Aβ levels were also assessed from several cortical regions by elisa. Animals dosed with anti‐BACE1 by i.c.v. infusion had ~50% reduction in cortical Aβ concentrations compared to control IgG or vehicle‐infused animals throughout multiple cortical regions assessed (prefrontal, motor, temporal and parietal) (Figure 5A). Consistent with lower brain antibody concentrations, animals that received i.v. anti‐BACE1 had no reduction in cortical Aβ levels compared to control animals that received either vehicle or control IgG. We again confirmed these results by looking at another measure of BACE1 target engagement, the sAPPβ/sAPPα ratio, in TBS‐soluble brain lysates from prefrontal cortex. Following i.c.v. anti‐BACE1 dosing, the ratio of sAPPβ/sAPPα was dramatically reduced, reflecting robust inhibition of BACE1 (Figure 5B). In contrast, i.v. delivery of anti‐BACE1 did not alter the sAPPβ/sAPPα ratio in cortical homogenates, consistent with the lack of reduction in Aβ concentrations.
Figure 5.
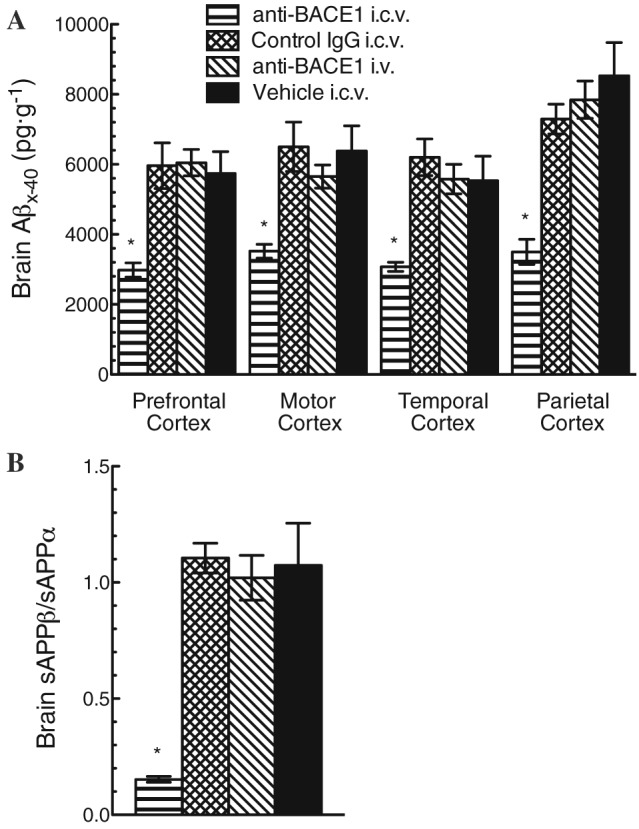
Reduced cortical Aβx‐40 and sAPPβ/sAPPα ratio following anti‐BACE1 i.c.v. infusion. (A) Aβx‐40 concentrations in the brain were measured by elisa of tissue homogenates prepared from different regions of cortex (prefrontal, motor, temporal and parietal). Only anti‐BACE1 delivered by i.c.v. infusion was able to significantly reduce cortical Aβx‐40 concentrations; Aβx‐40 was reduced by ~50% compared to all other group cortical regions assayed. (B) sAPPβ/sAPPα ratio measured in TBS‐soluble lysates from temporal cortex. A significant reduction was seen only in the anti‐BACE1 i.c.v. infusion group. *P < 0.05. Statistical test: two‐way ANOVA with Bonferroni multiple comparisons test compared to control IgG i.c.v. Bars represent mean ± SEM. Group (n): anti‐BACE1 i.c.v. (4), control IgG i.c.v. (4), anti‐BACE1 i.v. (5) and vehicle i.c.v. (3).
Discussion
In this study, the extent of brain antibody distribution was determined following 6 weeks of continuous i.c.v. infusion of anti‐BACE1 or a control IgG antibody into the left lateral ventricle of cynomolgus monkeys. Steady‐state CSF antibody concentrations of 1.2 μM were reached as early as 4 days after infusion was initiated, consistent with the rapid turnover rate of the CSF in nonhuman primates. Widespread distribution of antibody was observed throughout the brain after 6 weeks, as determined quantitatively by elisa and qualitatively by IHC. Antibody concentrations in the brain after i.c.v. infusion were approximately uniform, with most regions reaching 20–40 nM. Notable exceptions to this were the deep internal structures of the caudate, putamen and the globus pallidus contralateral to catheter placement, which had concentrations of 5–10 nM. These deeper lying brain regions may either require a longer infusion time to achieve antibody concentrations comparable to external structures or may simply be less accessible to large molecules.
The i.v. administration of antibodies has been reported to achieve antibody concentrations in human CSF and rodent brain of about 0.1% that of serum (Pestalozzi and Brignoli, 2000; Banks et al., 2007; Petereit and Rubbert‐Roth, 2009). In our study, i.v.‐administered anti‐BACE1 resulted in steady‐state CSF antibody concentrations of ~0.1% of steady‐state serum concentrations; however, terminal brain antibody concentrations only reached ~0.01–0.02% of steady‐state serum concentrations. This lower serum : brain ratio in primates is consistent with our previous report (Yu et al., 2014). In contrast, the brain concentrations of anti‐BACE1 and control IgG following continuous i.c.v. infusion were much higher, reaching ~1.7–3.3% of CSF steady‐state concentrations and ~0.8–1.6% of corresponding serum concentrations (due to efflux from the CSF). These results emphasize how dramatically the BBB limits the amount of antibody that reaches brain tissue.
The bilateral and widespread brain distribution of antibody we observed after unilateral i.c.v. infusion was remarkable. Antibody distribution by diffusion alone, from the CSF across the pial and ependymal surfaces into the brain, would not predict such broad and uniform brain antibody concentrations (Wolak and Thorne, 2013). These data suggest that additional transport pathways exist that help distribute antibody from CSF into the brain parenchyma. Recently, the perivascular spaces that surround the blood vessels of the brain have been identified as serving a critical function in the interaction of CSF and brain interstitial fluid and thereby drug distribution (Ziegler et al., 2011; Iliff et al., 2012). Subarachnoid CSF transport into periarterial and periarteriolar spaces is enhanced by cardiac‐driven pulsations of these vessels (Hadaczek et al., 2006; Schley et al., 2006). This perivascular path allows bulk CSF to be transported deeper into the brain tissue. The outer boundary of the perivascular space is lined by astrocytic foot processes with approximately 20 nm diameter channels (aquaporin‐4) that allow water and even large solutes to move from the periarterial space into the interstitial spaces of the brain parenchyma (Mathiisen et al., 2010).
Antibody infused into CSF within the left lateral ventricle is transported by bulk flow through the ventricles and around the subarachnoid space overlying the surface of the brain where the major arteries penetrate. These periarterial pathways are likely the basis for the broad and bilateral distribution of antibodies that we observe in the brain parenchyma. Although solute size has been shown to affect movement into the interstitial space (Iliff et al., 2012), our results demonstrate that IgG antibodies infused by i.c.v. were able to traverse and exit the perivascular spaces and thus enter the tissue of the brain where the target enzyme is located. It is unknown whether the perivascular transport is affected in Alzheimer's disease, although at least one report suggests it may be reduced (Weller et al., 2008). Further investigations into alterations of this transport pathway in different disease states will influence clinical use of i.c.v. delivery.
The i.c.v. infusion of anti‐BACE1 reduced CSF Aβ concentrations by ~70%, whereas cortical brain Aβ concentrations only decreased by ~50%. Similar discrepancies in CSF versus brain Aβ reductions have been observed previously following systemic delivery of BACE1 inhibitors (Yu et al., 2014). Since CSF turns over rapidly, CSF Aβ concentrations are likely to represent a labile pool of Aβ in the brain that is cleared to the venous blood via the CSF. In contrast, total Aβ concentrations in brain tissue may include soluble labile pools as well as a less dynamic pool, perhaps including intracellular or membrane‐bound Aβ. These differences warrant further investigation and should be incorporated into PK/PD models for translation to clinical dosing.
Interestingly, we observed equivalent reductions in peripheral (plasma) Aβ concentrations following both i.v. and i.c.v. dosing, but CSF and brain Aβ concentrations were only reduced with i.c.v. dosing. These observations dispute the peripheral sink hypothesis, which proposes that reducing peripheral levels of Aβ will lead to reductions in brain Aβ by drawing Aβ out of the brain. Our data clearly demonstrate the independence of the peripheral and CNS pools of Aβ and reveal the necessity of CNS drug action in altering brain Aβ concentrations. The 0.5–2 nM concentrations of anti‐BACE1 in the cortex following i.v. administration are simply not sufficient to result in significant BACE1 inhibition or lead to cortical Aβ reductions.
I.c.v. infusion pumps have been used in the clinic to deliver small molecule cancer drugs such as methotrexate and cytosine arabinoside for brain tumours (Chandler et al., 1988; Bakhshi and North, 1995) and to deliver opioids to treat refractory head and neck pain (Weigl et al., 1987; Dennis and DeWitty, 1990). Risks associated with i.c.v. brain infusion include tissue damage and haemorrhage due to catheter placement and infection at the implantation site. More recently, the Medtronic SynchroMed® II pump was implanted in 12 patients with Parkinson's disease in order to deliver recombinant human PDGF directly to the brain (Paul et al., 2015), and no unresolved adverse events due to the infusion system or implant procedure were noted. Taken together, these reports support the clinical feasibility of using i.c.v. infusion pumps to deliver therapeutics directly into the brain. Because of the perivascular transport pathway, it is possible that i.t. delivery, potentially with the catheter tip placed in the cervical area (Albright et al., 2006), may also produce effective levels of antibody in the brain (Calias et al., 2014). Advantages of the i.t. route of administration would include (i) the use of the commercially available SynchroMed system that has been used for years clinically to treat chronic pain and spasticity and (ii) the opportunity to avoid brain surgery and its inherent risks.
In summary, these results demonstrate that i.c.v. infusion can produce high concentrations and widespread distribution of antibodies in the brain of nonhuman primates relative to i.v. administration. Moreover, anti‐BACE1 antibodies infused i.c.v. resulted in a significant decrease in Aβ levels in the cerebral cortices and CSF of these animals. Because i.v. administration of antibodies designed to bind Aβ in humans has demonstrated limited success in multiple large‐scale clinical trials to date, a direct route of administration that bypasses the BBB such as i.c.v. or i.t. infusion should be considered. However, additional preclinical development is needed to further assess the effects of delivery methods and antibody doses on brain distribution and Aβ levels and to evaluate safety before translation to the clinic. This i.c.v. delivery approach could help enable the clinical translation for multiple mAbs that have shown preclinical efficacy against various neurodegenerative disease targets, including τ and α‐synuclein (Yu and Watts, 2013). The widespread distribution of therapeutically active drugs could potentially lead to the broad application of the i.c.v. infusion method for treating a variety of neurodegenerative diseases as well as brain tumours.
Author contributions
D.B.Y., K.R.W., R.N.F., A.D., J.A.C., K.S.‐L., S.P., T.K., K.H., L.J.N., D.L., L.S., D.R.T. and J.K.A. designed the experiment. A.K., M.G. and J.A.M. performed stability testing on test articles. E.A. performed and oversaw the in vivo surgery and experiments. R.S. performed and oversaw the brain processing and histochemical and immunohistochemical staining. J.A.M., W.J.M. and H.S. processed tissue samples for analysis. J.A.M., K.R.W., Y.L., K.P., B.W. and P.C. performed the PK and PD assays. D.B.Y., K.R.W., K.G. and J.K.A. analysed the PK and PD samples. J.K.A., D.B.Y. and S.P. wrote the manuscript with extensive comments from K.R.W., R.N.F., K.S.L., K.H. and D.R.T.
Conflict of interest
B.Y., K.R.W., R.N.F., J.A.M., W.J.M., H.S., Y.L., K.P., B.W., P.C., K.P., A.K., M.G., A.D., J.A.C., R.J.W., K.S.‐L., S.P. and J.K.A. are full‐time paid employees of Genentech. T.K., K.H., L.J.N., D.L., K.H., L.S. and D.R.T. are full‐time paid employees of Medtronic.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 i.c.v. infusion study design. (A) The implantable drug pump (SynchroMed® II) used for this study. This is the identical pump used in patients to treat chronic pain and severe spasticity; pump is shown with a commercial intrathecal catheter attached. (B) A schematic of the implanted pump and a unilateral i.c.v. catheter implanted in the cynomolgus model. (C) Diagram of the study design showing the timing of major study events. (D) Study groups.
Acknowledgements
This work was fully funded by Medtronic, Inc., and Genentech, Inc., through joint research collaboration. Support for third‐party writing assistance for this article, furnished by Anshin BioSolutions, was provided by Genentech, a member of the Roche Group.
Yadav, D. B. , Maloney, J. A. , Wildsmith, K. R. , Fuji, R. N. , Meilandt, W. J. , Solanoy, H. , Lu, Y. , Peng, K. , Wilson, B. , Chan, P. , Gadkar, K. , Kosky, A. , Goo, M. , Daugherty, A. , Couch, J. A. , Keene, T. , Hayes, K. , Nikolas, L. J. , Lane, D. , Switzer, R. , Adams, E. , Watts, R. J. , Scearce‐Levie, K. , Prabhu, S. , Shafer, L. , Thakker, D. R. , Hildebrand, K. , and Atwal, J. K. (2017) Widespread brain distribution and activity following i.c.v. infusion of anti‐β‐secretase (BACE1) in nonhuman primates. British Journal of Pharmacology, 174: 4173–4185. doi: 10.1111/bph.14021.
Contributor Information
Keith Hildebrand, Email: keith.hildebrand@medtronic.com.
Jasvinder K Atwal, Email: jatwal@gene.com.
References
- Albright AL, Turner M, Pattisapu JV (2006). Best‐practice surgical techniques for intrathecal baclofen therapy. J Neurosurg 104: 233–239. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwal JK, Chen Y, Chiu C, Mortensen DL, Meilandt WJ, Liu Y et al (2011). A therapeutic antibody targeting BACE1 inhibits amyloid‐beta production in vivo. Sci Transl Med 3: 84ra43. [DOI] [PubMed] [Google Scholar]
- Bakhshi S, North RB (1995). Implantable pumps for drug delivery to the brain. J Neurooncol 26: 133–139. [DOI] [PubMed] [Google Scholar]
- Banks WA, Farr SA, Morley JE, Wolf KM, Geylis V, Steinitz M (2007). Anti‐amyloid beta protein antibody passage across the blood‐brain barrier in the SAMP8 mouse model of Alzheimer's disease: an age‐related selective uptake with reversal of learning impairment. Exp Neurol 206: 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickel U, Yoshikawa T, Pardridge WM (2001). Delivery of peptides and proteins through the blood–brain barrier. Adv Drug Deliv Rev 46: 247–279. [DOI] [PubMed] [Google Scholar]
- Calias P, Banks WA, Begley D, Scarpa M, Dickson P (2014). Intrathecal delivery of protein therapeutics to the brain: a critical reassessment. Pharmacol Ther 144: 114–122. [DOI] [PubMed] [Google Scholar]
- Chandler WF, Greenberg HS, Ensminger WD, Diaz RF, Junck LR, Hood TW et al (1988). Use of implantable pump systems for intraarterial, intraventricular and intratumoral treatment of malignant brain tumors. Ann N Y Acad Sci 531: 206–212. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis GC, DeWitty RL (1990). Long‐term intraventricular infusion of morphine for intractable pain in cancer of the head and neck. Neurosurgery 26: 404–407 discussion 407‐408. [PubMed] [Google Scholar]
- Dodge JC, Clarke J, Treleaven CM, Taksir TV, Griffiths DA, Yang W et al (2009). Intracerebroventricular infusion of acid sphingomyelinase corrects CNS manifestations in a mouse model of Niemann‐Pick A disease. Exp Neurol 215: 349–357. [DOI] [PubMed] [Google Scholar]
- Eketjall S, Janson J, Jeppsson F, Svanhagen A, Kolmodin K, Gustavsson S et al (2013). AZ‐4217: a high potency BACE inhibitor displaying acute central efficacy in different in vivo models and reduced amyloid deposition in Tg2576 mice. J Neurosci 33: 10075–10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde TE (2014). Open questions for Alzheimer's disease immunotherapy. Alzheimer's Res Ther 6: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadaczek P, Yamashita Y, Mirek H, Tamas L, Bohn MC, Noble C et al (2006). The “perivascular pump” driven by arterial pulsation is a powerful mechanism for the distribution of therapeutic molecules within the brain. Mol Ther 14: 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353–356. [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA et al (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4: 147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen H, Ozmen L, Caruso A, Narquizian R, Hilpert H, Jacobsen B et al (2014). Combined treatment with a BACE inhibitor and anti‐Abeta antibody gantenerumab enhances amyloid reduction in APPLondon mice. J Neurosci 34: 11621–11630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S et al (2012). A mutation in APP protects against Alzheimer's disease and age‐related cognitive decline. Nature 488: 96–99. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney JA, Bainbridge T, Gustafson A, Zhang S, Kyauk R, Steiner P et al (2014). Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J Biol Chem 289: 30990–31000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP (2010). The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia 58: 1094–1103. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann U, Rueeger H, Machauer R, Veenstra SJ, Lueoend RM, Tintelnot‐Blomley M et al (2015). A novel BACE inhibitor NB‐360 shows a superior pharmacological profile and robust reduction of amyloid‐beta and neuroinflammation in APP transgenic mice. Mol Neurodegener 10: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul G, Zachrisson O, Varrone A, Almqvist P, Jerling M, Lind G et al (2015). Safety and tolerability of intracerebroventricular PDGF‐BB in Parkinson's disease patients. J Clin Invest 125: 1339–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestalozzi BC, Brignoli S (2000). Trastuzumab in CSF. J Clin Oncol 18: 2349–2351. [DOI] [PubMed] [Google Scholar]
- Petereit HF, Rubbert‐Roth A (2009). Rituximab levels in cerebrospinal fluid of patients with neurological autoimmune disorders. Mult Scler 15: 189–192. [DOI] [PubMed] [Google Scholar]
- Reichert JM (2015). Antibodies to watch in 2015. MAbs 7: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schley D, Carare‐Nnadi R, Please CP, Perry VH, Weller RO (2006). Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J Theor Biol 238: 962–974. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakker DR, Sankaranarayanan S, Weatherspoon MR, Harrison J, Pierdomenico M, Heisel JM et al (2015). Centrally delivered BACE1 inhibitor activates microglia, and reverses amyloid pathology and cognitive deficit in aged Tg2576 mice. J Neurosci 35: 6931–6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakker DR, Weatherspoon MR, Harrison J, Keene TE, Lane DS, Kaemmerer WF et al (2009). Intracerebroventricular amyloid‐beta antibodies reduce cerebral amyloid angiopathy and associated micro‐hemorrhages in aged Tg2576 mice. Proc Natl Acad Sci U S A 106: 4501–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigl K, Mundinger F, Chrubasik J (1987). Continuous intraventricular morphine‐ or peptide‐infusion for intractable cancer pain. Acta Neurochir Suppl 39: 163–165. [DOI] [PubMed] [Google Scholar]
- Weinmann O, Schnell L, Ghosh A, Montani L, Wiessner C, Wannier T et al (2006). Intrathecally infused antibodies against Nogo‐A penetrate the CNS and downregulate the endogenous neurite growth inhibitor Nogo‐A. Mol Cell Neurosci 32: 161–173. [DOI] [PubMed] [Google Scholar]
- Weller RO, Subash M, Preston SD, Mazanti I, Carare RO (2008). Perivascular drainage of amyloid‐beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol 18: 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE et al (2003). Intracranially administered anti‐Abeta antibodies reduce beta‐amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci 23: 3745–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Munireddy SK, Rosenthal A, Ugen KE, Gordon MN, Morgan D (2004). Microglial activation facilitates Abeta plaque removal following intracranial anti‐Abeta antibody administration. Neurobiol Dis 15: 11–20. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Goni F (2015). Immunotherapeutic approaches for Alzheimer's disease. Neuron 85: 1162–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolak DJ, Thorne RG (2013). Diffusion of macromolecules in the brain: implications for drug delivery. Mol Pharm 10: 1492–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R, Vassar R (2014). Targeting the beta secretase BACE1 for Alzheimer's disease therapy. Lancet Neurol 13: 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YJ, Atwal JK, Zhang Y, Tong RK, Wildsmith KR, Tan C et al (2014). Therapeutic bispecific antibodies cross the blood‐brain barrier in nonhuman primates. Sci Transl Med 6: 261ra154. [DOI] [PubMed] [Google Scholar]
- Yu YJ, Watts RJ (2013). Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 10: 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler RJ, Salegio EA, Dodge JC, Bringas J, Treleaven CM, Bercury SD et al (2011). Distribution of acid sphingomyelinase in rodent and non‐human primate brain after intracerebroventricular infusion. Exp Neurol 231: 261–271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 i.c.v. infusion study design. (A) The implantable drug pump (SynchroMed® II) used for this study. This is the identical pump used in patients to treat chronic pain and severe spasticity; pump is shown with a commercial intrathecal catheter attached. (B) A schematic of the implanted pump and a unilateral i.c.v. catheter implanted in the cynomolgus model. (C) Diagram of the study design showing the timing of major study events. (D) Study groups.