

Abstract
Background and Purpose
The majority of the severe vascular complications in fibrosis are a consequence of a deregulated activity of mediators controlling vasomotor tone. One of the most important of these mediators is endothelin‐1 (ET‐1). Here, we have investigated the role of proteinase‐activated receptor 2 (PAR2) in the vascular dysfunction in a model of fibrosis, using tight‐skin (Tsk) mice.
Experimental Approach
Aortas were collected from Tsk, transgenic over‐expressing PAR2 (TgPAR2), PAR2 deficient (PAR2−/−) or the corresponding WT mice. Histological and immunohistochemistry analysis for α‐smooth muscle actin, PAR2 and ET‐1 receptors were performed on aorta sections. Vascular responses to phenylephrine, ET‐1 and PAR2 activating peptide (PAR2‐AP) were assessed on aortic rings.
Key Results
In aortas from Tsk mice, responses to phenylephrine were reduced, contractions to ET‐1 were increased and vasorelaxation to PAR2‐AP was enhanced. These alterations matched changes observed in whole vessel architecture such as vascular fibre re‐organization, increased collagen deposition and enhanced α‐smooth muscle actin expression. Expression of both ETA receptors and PAR2 was enhanced in Tsk mice. Antagonism of PAR2 potentiated vascular effects of ET‐1, whereas antagonism of ETA receptors increased vasorelaxation induced by PAR2‐AP. In TgPAR2 mice, responses to ET‐1 and ET‐1 plasma levels were reduced. Conversely, PAR2−/− mice showed enhanced ET‐1 induced contraction in aortic rings and higher circulating ET‐1 levels.
Conclusions and Implications
Our data show that PAR2 counterbalanced enhanced contractions to ET‐1 in aortas from Tsk mice. PAR2 could represent a possible target for novel drugs in the treatment of vascular complications in fibrosis.
Linked Articles
This article is part of a themed section on Targeting Inflammation to Reduce Cardiovascular Disease Risk. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.22/issuetoc and http://onlinelibrary.wiley.com/doi/10.1111/bcp.v82.4/issuetoc
Abbreviations
- ET‐1
endothelin‐1
- PAR2‐AP
proteinase activated receptor 2 activating peptide
- α‐SMA
α‐smooth muscle actin
- SLS
scleroderma‐like syndrome
- Tsk
tight skin
Tables of Links
| TARGETS |
|---|
| GPCRs |
| ETA receptor |
| ETB receptor |
| PAR2 |
| LIGANDS |
|---|
| FR139317 |
| Ibuprofen |
| L‐NAME |
| SLIGRL‐NH2 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
The proteinase‐activated receptor 2 (PAR2) belongs to a restricted subgroup of the G‐protein‐coupled receptor superfamily, named PARs. These receptors are activated by certain extracellular proteases derived from the circulation and inflammatory cells. PAR2 is activated by trypsin‐like serine proteases through a proteolytic activation that unmask an N‐terminus sequence that auto‐activates the receptor (Macfarlane et al., 2001). Activation of the receptor is mimicked by small peptides obtained from the active sequence known as PAR2 activating peptides (PAR2‐AP). PAR2 is strongly expressed in the endothelium and in vascular smooth muscle cells (Cottrell et al., 2003; McGuire, 2004). Activation of PAR2 causes acute vasodilatation, lowers blood pressure and protects tissues from ischaemic injury (Cheung et al., 1998; Zhong and Wang, 2009; van den Hengel et al., 2013). Of particular interest is the finding that PAR2‐mediated vasodilatation persists despite endothelial dysfunction (Hamilton and Cocks, 2000). Under normal conditions, PAR2 mediates acute vasodilatation of small calibre resistance arteries via NO and Ca2 +‐activated K+ channels (KCa) (Hennessey et al., 2015). Nevertheless, there are a few changes in the mechanisms underlying PAR2‐AP‐mediated vasodilatation during endothelial dysfunction (McGuire et al., 2002). PAR2 is also up‐regulated in human skin in patients with fibrotic scleroderma‐like syndrome (SLS) (Cevikbas et al., 2011).
SLS is a complex immune‐mediated disease associated with a high mortality rate (Denton et al., 2006). Although SLS exhibits a complex of interlinked vascular, immunological and fibrotic components, one hypothesis suggests that endothelial damage and vascular dysfunction may characterize the earliest pathological alterations (LeRoy, 1996; Kahaleh, 2004). In particular, several functional and structural abnormalities occur within blood vessels, including enhanced vasoconstriction, intimal hyperplasia and vessel media/adventitia fibrosis. These features are clinically relevant and referred to as Raynaud's phenomenon. The majority of severe complications associated to SLS are based on the vasculature, including pulmonary arterial hypertension and scleroderma renal crisis. All these events develop from deregulation of the vasomotor tone, where a key role is played by endothelin‐1 (ET‐1), a mediator inducing potent vasoconstriction through binding to its ETA receptors. ET‐1 also triggers vascular cell proliferation, smooth muscle hypertrophy and irreversible vascular remodelling (Wort et al., 2001; Lambers et al., 2013; Maier et al., 2014; Kim et al., 2015). The relevance of ET‐1 to systemic sclerosis (SSc) is demonstrated by the elevated ET‐1 circulating levels (Morelli et al., 1995) and by the finding that the ET receptor pan‐antagonist, bosentan, is used in patients with arterial pulmonary hypertension, secondary to SSc (Heresi and Minai, 2008; Guiducci et al., 2012; Kawashiri et al., 2014).
A valid preclinical experimental model to study the SLS syndrome is provided by tight‐skin (Tsk) mice. These mice have been successfully used to study the pathological mechanisms underlying the disease and to test potential therapeutic treatments (Iwamoto et al., 2011; Takahashi et al., 2015). This strain of mice expresses an autosomal dominant mutation in the fibrillin‐1 gene, located on chromosome 2, that was first discovered by Bunker in 1976 (Green et al., 1976). Mice homozygous for the mutation (Tsk/Tsk) die in utero by 8–10 days of gestation, whereas heterozygous mice survive to develop a SLS syndrome. Therefore, the Tsk strain has been carried forward and theses mice have been extensively characterized. In particular, these mice typically exhibit marked thickening of subcutaneous dermal tissue, heart fibrotic abnormalities, distended emphysematous lungs with little fibrosis and features of SSc autoimmunity, such as a positive reaction to RNA polymerase 1, anti‐ Scl70 or antinuclear antibodies. This mouse strain provides a well‐known model for sclerodermia, as it shows many features of this pathology associated with abnormalities of connective tissue (Green et al., 1976; Jimenez et al., 1984; Kasturi et al., 1994).
Here, we have defined the role of PAR2 and its interactions with the ET‐1 pathway in the vascular dysfunction associated with SLS in Tsk mice.
Methods
Animals
All animal care and experimental procedures were in compliance with Italian (D.M. 116192) and EEC (O. J. of E. C. L 358/1‐12/18/1986) regulations and followed the ARRIVE guidelines for the handling and use of laboratory animals for scientific purposes (Kilkenny et al., 2010; McGrath and Lilley, 2015). A total of 60 animals have been used in this study and all experimental protocols have been carried out in adherence with the BJP guidelines. Male Tsk (B6.Cg‐Fbn1Tsk+/+ Pldnpa/J), transgenic over‐expressing PAR2 (TgPAR2) and PAR2 deficient mice (PAR2−/−) were supplied by at the Biological Service Unit (Siena, Italy) and originally obtained from Jackson Laboratory (Bar Harbor, USA; Green et al., 1976). C57Bl/6J and FVB/N wild type mice were purchased from Charles River (Calco, Italy). C57Bl/6J were used as controls for Tsk and PAR2−/− mice while the FVB/N mice were used as controls for TgPAR2. Mice were multiply‐housed in macrolon cages in a controlled environment (22–24°C; 40% humidity; 12 h light–dark cycle). All animals were allowed food and water ad libitum.
Randomization and blinding procedures
Isolated aorta was collected from a single mouse for each strain and divided into several rings. Each ring was tested for a different compound in order to have a complete data set for each aorta (mouse) used. The experimenter was given coded vials with the different compounds. The data was analysed without knowledge of the treatments of each group. After analysis, the different treatments were disclosed in order to create the graphs.
Tissue preparation
Male Tsk and wild‐type (WT) control mice were used at different ages (2–10 months old). Animals were anaesthetized by using enflurane (5%) and then euthanized by using CO2 (70%) chamber, thoracic aorta was rapidly dissected and cleaned from fat and connective tissue. Rings of 1.5–2 mm length were cut and mounted on wire myographs (Kent Instruments, Torrington, USA), filled with gassed (O2/CO2 95%/5%) Krebs solution at 37°C. Changes in isometric tension were recorded with PowerLab data acquisition system (Ugo Basile, Varese, Italy). Krebs solution composition was as follows (mol·L−1): NaCl 0.118, KCl 0.0047, MgCl2 0.0012, KH2PO4 0.0012, CaCl2 0.0025, NaHCO3 0.025 and glucose 0.010 (Sigma‐Aldrich, Milano, Italy). Rings were initially stretched to a resting tension of 1.5 g and allowed to equilibrate for 40 min. Bathing solution was periodically changed and tension reset when needed. An optimal resting tension of 1.5 g was determined in preliminary experiments.
In vitro experimental protocol
In each set of experiment, rings from six different animals were first challenged with phenylephrine (1 μM; Sigma‐Aldrich) until the response was reproducible. Cumulative concentration curves to phenylephrine or ET‐1 (Tocris, Bristol, UK) were performed. Conversely, to evaluate tissue vasorelaxation, cumulative concentration–response curves to ACh (10 nM–30 μM; Sigma‐Aldrich) and to the PAR2 tethered ligand peptide (PAR2‐AP, SLIGRL‐NH2, 1 nM–1 μM; synthesized in house) were performed on rings precontracted with phenylephrine. In order to investigate the involvement of NO and COX metabolites, concentration–response curves with ET‐1 or PAR2‐AP were carried out in presence of the NOS inhibitor L‐NG‐nitro‐arginine methyl ester (L‐NAME, 100 μM, 20 min; Sigma‐Aldrich) and the COX inhibitor ibuprofen (10 μM; Sigma‐Aldrich). FR139317 (10 μM; Tocris) was used as antagonist for ETA receptors while ENMD1068 was used to block PAR2 (100 μM; Sigma‐Aldrich).
Western blotting
Samples of the thoracic aorta from six different mouse genotypes were homogenized in lysis buffer containing 0.5 M β‐glycerophosphate, 10 mM sodium orthovanadate, 20 mM MgCl2, 10 mM EGTA, 100 mM DTT and protease inhibitors. Protein concentration was determined by using Bradford assay (Bio‐Rad Laboratories, Milano, Italy) and 30 μg of total proteins were separated by electrophoresis. Proteins were then transferred onto a nitrocellulose membrane (Schleicher&Schuell, Munich, Germany), and immunoblots were incubated as follows: rabbit polyclonal anti‐PAR2 (1:500, Santa Cruz,Biotechnology, Heidelberg, Germany), rabbit anti‐ETA or anti‐ ETB receptor (1:1000, Santa Cruz Biotechnology). Signal detection was performed by using ECL System (Amersham Pharmacia Biotech, Amersham, UK).
Histology
Eight‐month‐old mice (n = 8) were killed, and thoracic aortas were excised and fixed in buffered formalin (5%) for 24 h. All tissues were then dehydrated, cleared in toluene and embedded in paraffin. Transverse sections (6 μm) were cut and stained with Masson's trichrome and Weigert's resorcin‐fucsin method.
ETA, α‐SMA, PAR2 immunostaining
Paraffin‐embedded thoracic aorta sections (6 μm) were stained for ETA receptors, α‐smooth muscle actin (α‐SMA) and PAR2. The sections were pretreated with 3% hydrogen peroxide to block the endogenous peroxidase. For PAR2 detection, no blocking was performed. Antigen retrieval was performed by heating in a microwave oven for 20 min in 0.01 M citrate buffer at pH 6.0 and allowing slow cooling at room temperature. All sections were incubated with 3% bovine serum albumin for 30 min at room temperature to block non‐specific antibody binding. Tissues were incubated overnight at 4°C with primary antibodies: goat‐polyclonal anti‐ETA (1:50, Novus Biologicals, Cambridge, UK); mouse‐monoclonal anti‐α‐SMA (1:200, Sigma‐Aldrich); and rabbit‐polyclonal anti‐PAR2 (1:25, Santa Cruz Biotechnology). For α‐SMA detection, no antigen retrieval was performed. After incubation with primary anti‐ETA, tissue sections were rinsed with TBST and then incubated with peroxidase‐conjugated rabbit anti‐goat IgG (1:200, Sigma‐Aldrich) for 30 min at room temperature. Colour development was performed by using 3,3'‐diaminobenzidine (DAB) as chromogen. The M.O.M. immunodetection kit (Transduction Laboratories, Lexington, USA) was used for α‐SMA determination. The sections incubated with anti‐PAR2 antibody were rinsed with PBS and incubated with biotinylated goat‐polyclonal anti‐rabbit IgG (1:100, Vector Labs, Burlingame, USA) for 40 min at room temperature. The staining was revealed by adding streptavidin‐conjugated AP and NBT/BCIP (BDPharmingen, Buccinasco, Italy). Zeiss confocal microscope with selective multitracking excitation (LSM510, Zeiss, Germany) was used to determine α‐SMA/ETA co‐localization by immunofluorescence. Briefly, antigen retrieval was performed by heating in a microwave oven for 20 min in 0.01 M citrate buffer at pH 6.0 and allowing to cool slowly at room temperature. All sections were blocked with mouse IgG for 60 min at room temperature for non‐specific binding. Sections were incubated overnight at 4°C with both goat‐polyclonal anti‐ETA (1:50, Novus Biologicals) and mouse‐monoclonal anti‐α‐SMA (1:400, Sigma‐Aldrich). The primary antibodies were detected by using a mixture of Alexa546‐labelled donkey anti‐mouse and Alexa488‐labelled donkey anti‐goat antibodies (1:200, 45 min in the dark at room temperature; Molecular Probes, Eugene, USA). Non‐immunized serum was used as negative control for the all immunostaining performed.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are reported as mean ± SEM and the number of independent samples was at least 6 per group, for each data set. Statistical analysis was carried out with GraphPad Prism 5.0 software (San Diego, CA, USA) and performed using Student's t‐test to compare two groups, one‐way ANOVA followed by Dunnett's post test when comparing more than two groups or two‐way ANOVA for multiple comparisons followed by Bonferroni's post test. Post hoc tests were performed when the ANOVA indicated that a significant difference existed between groups. All statistical tests performed showed no significant variance in data set homogeneity. Data were considered statistically significant when a value of at least P < 0.05 was achieved.
Results
Vascular reactivity is impaired in Tsk mice
We first addressed the vascular responses in aortas from Tsk mice. Contraction induced by phenylephrine was within physiological range in aorta from 2 and 4‐month‐old mice, while a significant reduction was observed at 6 and 8 months (Figure 1A). Conversely, ET‐1‐induced contraction was significantly enhanced with ageing. Indeed, in 6 to 8‐month‐old mice, the contractile response was twice as much that observed in 2 to 4‐month‐old mice (Figure 1B). Aortas from 10‐month‐old Tsk mice lost their ability to contract to phenylephrine. Conversely, in aortas from control mice there were no changes in either phenylephrine‐ or ET‐1‐induced contraction until 10 months of age (Table 1).
Figure 1.
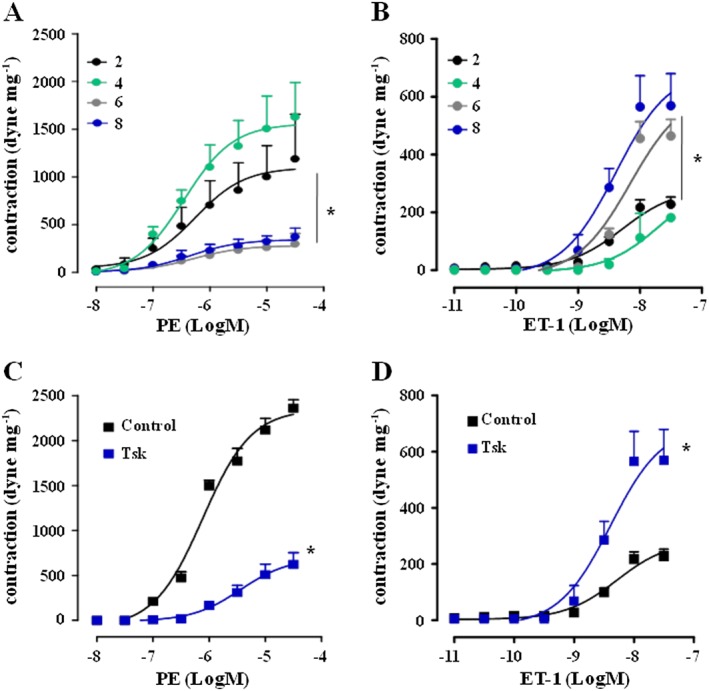
Change in vascular responses to phenylephrine (PE) and ET‐1 in aortas from Tsk mice. (A) Concentration‐response curve for phenylephrine in aortas from 2, 4, 6 and 8‐month‐old Tsk mice; (B) Concentration‐response curve for ET‐1 in aortas from 2, 4, 6 and 8‐month‐old Tsk mice; (C) Concentration‐response curve for phenylephrine in aortas from 8‐month‐old Tsk mice and compared with aortas from control animals; (D) Concentration‐response curve for ET‐1 in aortas from 8‐month‐old Tsk mice and compared with aortas from control animals. *P < 0.05, significantly different from 2‐month‐old mice (A and B) or 8‐month‐old control mice (C and D).
Table 1.
Different relevance of phenylephrine (PE), ET‐1 and PAR2 in vascular function in health or fibrosis
| Health (normal mice) | Fibrosis (Tsk mice) | |
|---|---|---|
| PE‐mediated contraction | Predominant | Heavily impaired |
| ETA receptor‐mediated contraction | Physiological | Heavily increased (predominant) |
| PAR2‐mediated vasodilation | Physiological | Highly enhanced |
In order to further investigate on the molecular mechanisms underlying vascular dysfunction, 8‐month‐old Tsk and control mice were used. Aortic rings from the Tsk mice displayed a significantly reduced phenylephrine‐induced contraction (Figure 1C), compared with age‐matched control vessel (Figure 1C). Conversely, ET‐1 contractile response was significantly augmented, compared with those in control rings (Figure 1D). In addition, no substantial strain‐related difference was found in vasorelaxation induced by ACh (Fig. S1).
Tsk mice show an altered architecture in aorta
Next, we assessed the structural organization of the aorta, using Weigert's resorcin‐fucsin stain to evaluate elastic fibre organization (Figure 2A). Aortas from Tsk mice showed a more pronounced staining (Figure 2A; images iii and iv) compared with control samples (Figure 2A; images i and ii). In particular, Tsk aortas displayed fibre disorganization associated with some rupture points, as indicated by the arrowheads. Masson's assay revealed a more prominent deposition of collagen in aortas from Tsk mice (Figure 2B; images iii and iv), compared with control samples (Figure 2B; images i and ii). In addition, panels C and D show the quantitative results for elastin and hydroxyproline content respectively.
Figure 2.
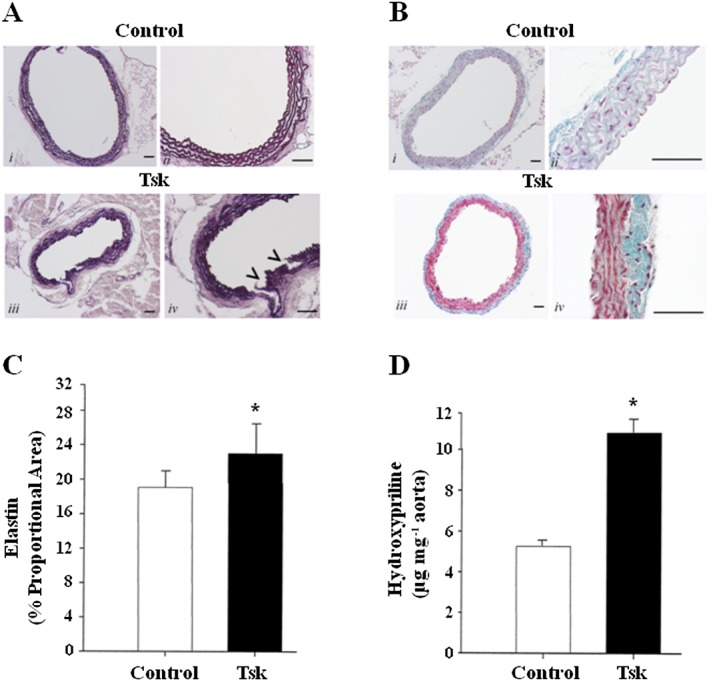
Weigert's and Masson's staining revealed changes in structure of thoracic aortas from 8‐month‐old Tsk mice. (A) Weigert's staining for elastic fibres in control (i–ii) and Tsk (iii–iv) mice; arrowheads indicate rupture points; (B) Collagen distribution (sea green) in control (i–ii) and Tsk mice (iii–iv) after Masson's trichrome staining; (C) Elastin quantification expressed as % of proportional area; (D) Hydroxyproline content shown as μg·mg−1 of aorta tissue. Scale bar indicates 50 μm length.
Responses of aortas from Tsk mice to ET‐1
In order to investigate the role of the ET‐1 pathway, we used the selective ETA receptor antagonist, FR139317, which abolished the contractile response to ET‐1 in aortas from Tsk mice (Figure 3A). We next evaluated the mechanisms involved in ET‐1‐induced contraction, by generating concentration–response curves with Tsk aortas and ET‐1, in presence of L‐NAME or ibuprofen, inhibitors of NOS and COX respectively. ET‐1‐induced vasoconstriction was increased by L‐NAME and inhibited by ibuprofen (Figure 3B). Western blot and immunohistochemistry experiments revealed that expression of ETA receptors in Tsk mice aortas was higher than in control mice (Figure 3C and D) and that this expression was increased throughout the aortic section (Figure 3D and E). No change was found for ETB receptor expression (Fig. S2). As ETA receptors are involved in both α‐SMA production and extracellular matrix contraction, we next evaluated ETA receptor /α‐SMA co‐localization in aorta. Immunofluorescence results showed a marked co‐localization of ETA receptors and α‐SMA in aortas from Tsk mice, compared with the control samples (Figure 4). In particular, in Tsk aorta, positive staining was clearly visible throughout all the vessel layers, in contrast to the staining in control samples, where only a sub‐endothelial staining was evident.
Figure 3.
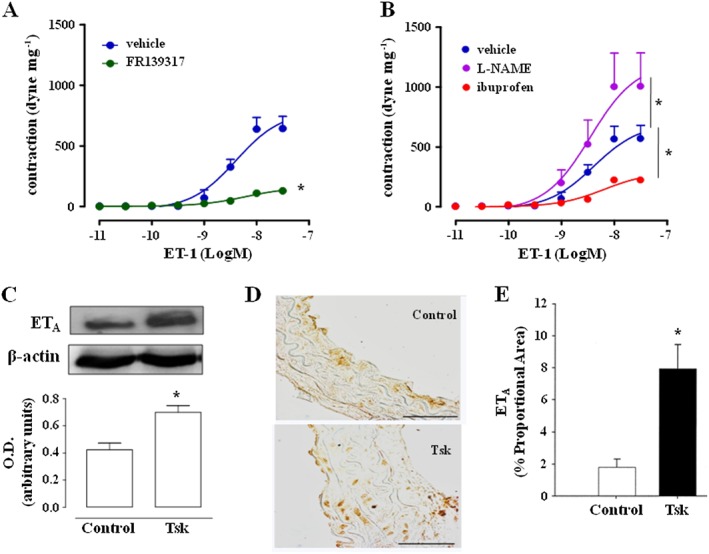
Contraction to ET‐1 in thoracic aorta from 8‐month‐old Tsk mice is mediated by ETA receptors. (A) ET‐1‐induced contraction of Tsk aortas in presence of ETA receptor antagonist FR139317; (B) ET‐1‐induced contraction of Tsk aortas in presence of the COX inhibitor ibuprofen or the NOS inhibitor L‐NAME; (C) Western blot and densitometry analysis for ETA receptor expression in Tsk aortas, compared with control samples. Images are representative of 6 separate experiments; (D) Immunohistochemical analysis for ETA expression in Tsk compared with control mice; (E) ETA receptor quantification reported as % of proportional area. Scale bar indicates 50 μm length. * P < 0.05, significantly different from vehicle (A and B) or control mice (C).
Figure 4.
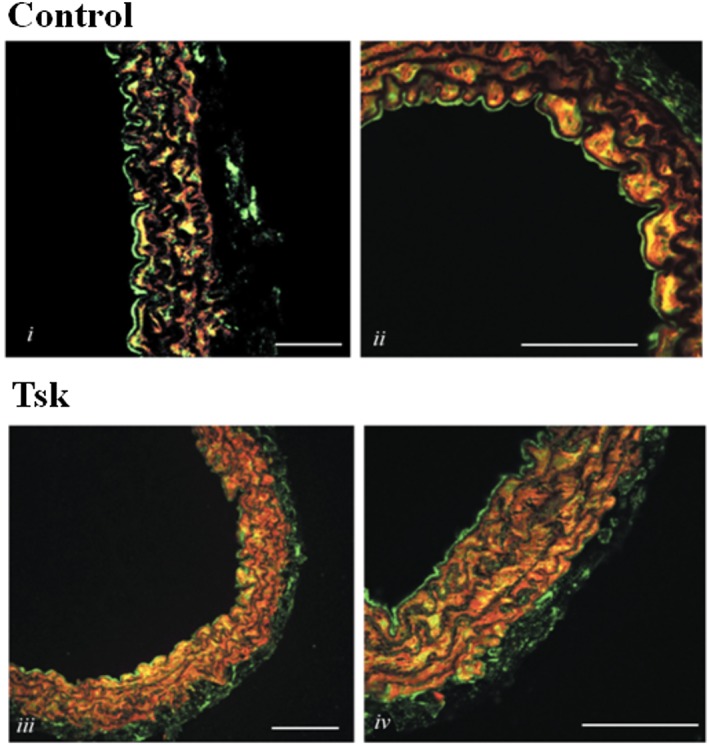
ETA receptors and α‐SMA co‐localize in Tsk mice thoracic aorta. Immunofluorescence images for ETA receptors (green staining) and α‐SMA (red staining) co‐localization (yellow merge) in thoracic aorta sections from control (i–ii) and Tsk (iii–iv) mice. Scale bar indicates 50 μm length.
Tsk mice show enhanced vascular responses to PAR2‐AP
In order to assess the role of PAR2 in aorta from Tsk mice, we tested the effect of vasodilating peptide PAR2‐AP. We found that the PAR2‐AP‐induced vasodilation was increased, compared with age‐matched control animals (Figure 5A) and that this effect was inhibited by the PAR2 antagonist ENMD1068 (Figure 5B). Similarly, L‐NAME significantly inhibited PAR2‐AP‐induced vasorelaxation (Figure 5B). Expression of PAR2 receptors was increased in aortas from Tsk mice, compared with those from control mice (Figure 5C). Immunohistochemical analysis showed that PAR2 receptor expression was enhanced throughout the whole vessel thickness in Tsk mice aorta (Figure 5D and E; images iii and iv), while, in aorta from control mice, these receptors were expressed only within the endothelial area (Figure 5D and E; images i and ii).
Figure 5.
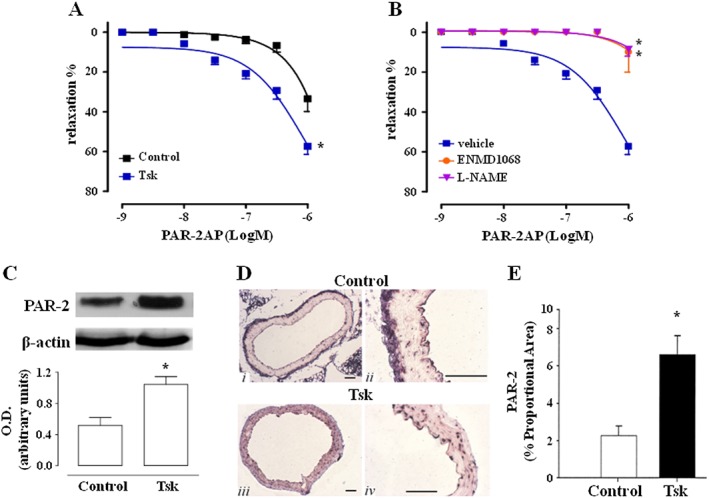
Aortas from 8‐month‐old Tsk mice display enhanced PAR2‐AP‐induced vasorelaxation, as well as up‐regulation of PAR2 receptor expression. (A) PAR2‐AP‐induced vasorelaxation of Tsk aortas, compared with control mice; (B) PAR2‐AP‐induced vasorelaxation in presence of PAR2 antagonist ENMD1068 or NOS inhibitor L‐NAME; (C) Western blot and densitometry analysis of PAR2 expression in oartas from Tsk or control mice. Images are representative of 6 separate experiments; (D) Immunohistochemical analysis of PAR2 in control (i–ii) and Tsk (iii–iv) mice; (E) ETA receptor quantification reported as % of proportional area. Scale bar indicates 50 μm length. *P < 0.05, significantly different from control mice (A and C) or vehicle group (B).
PAR2 counterbalances the vascular effects of ET‐1
Next, we wanted to examine the crosslinkage between the PAR2 and ET‐1 pathways. We found that PAR2‐AP‐induced vasorelaxation was significantly increased in presence of the ETA receptor antagonist FR139317 (Figure 6A). Conversely, blockade of PAR2, by ENMD1068, significantly increased ET‐1 induced contraction in aortas from Tsk mice (Figure 6B). In order to further support the hypothesis of interactions between the PAR2 and ET‐1 pathways, we evaluated ET‐1‐induced contraction in aortas from mice over‐expressing (TgPAR2) or lacking PAR2 receptors (PAR2−/−). ET‐1‐induced contractions were significantly enhanced in aortas from PAR2−/− mice, while a significant reduction was observed in vessels from TgPAR2 (Figure 6C). In parallel, we also measured the circulating levels of ET‐1 in these mice and found that the plasma concentration of ET‐1 was higher in PAR2−/− mice and lower in TgPAR2 mice, compared with their respective matched WT controls (Figure 6D). In addition, there were no significant differences, in terms of ETA receptor expression, between aortas from PAR2−/− and TgPAR2 mice.
Figure 6.
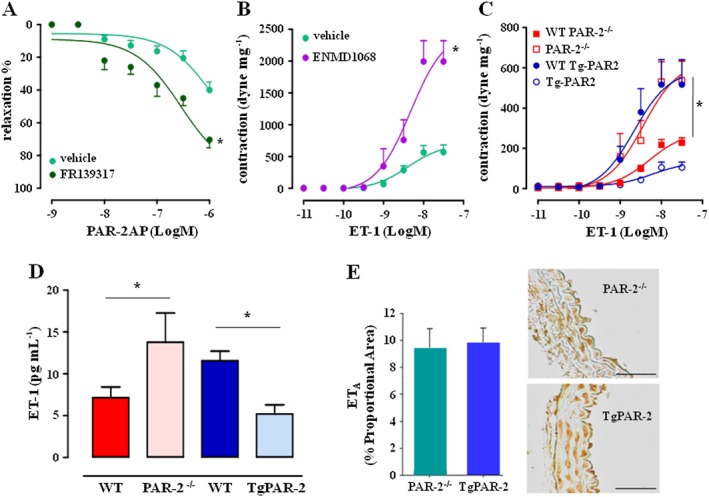
PAR2 activation counterbalances ET‐1/ETA signalling in aortas from Tsk mice. (A) PAR2‐AP‐induced vasorelaxation of Tsk aortas in presence of the ETA receptor antagonist FR139317; (B) ET‐1‐induced contraction of Tsk aortas in presence of PAR2 antagonist ENMD1068; (C) ET‐1 induced contraction in aortas from PAR2−/− and TgPAR2 mice compared with their matched control (WT); (D) ET‐1 plasma level in PAR2−/− and TgPAR2 mice compared with their matched control (WT); (E) ETA receptor quantification reported as % of proportional area in PAR2−/− and TgPAR2 mice. *P < 0.05, significantly different from vehicle group (A and B) or corresponding WT mice (C and D).
Discussion
SLS is a complex fibrotic autoimmune disease, in which early vascular and inflammatory changes lead to endothelial damage and vascular dysfunction (Richard et al., 2008). In particular, several functional and structural abnormalities occur within blood vessels, including an enhanced vasoconstriction (LeRoy, 1996; Kahaleh, 2004). Here, we investigated on the role of PAR2 in vascular homeostasis in the Tsk mouse model of SLS. These mice have been widely reported as the experimental model that, more than others, displays analogies with the clinical features observed in the human pathology (Iwamoto et al., 2011; Takahashi et al., 2015). These features originate from the mutation for the fibrillin‐1 gene. This mutation causes abnormalities in connective tissue and collagen deposition in different tissues, including the vascular system (Green et al., 1976). In particular, three main phases in the evolution of systemic illness in Tsk mice have been reported (Kielty et al., 1998; O'Donnell et al., 1999). During the first phase, until the second month after the birth, there is a rapid disease progression, followed by a second phase of stabilization, or slower progression, occurring between 2 and 8 months. A third phase, between 8 and 16 months, is characterized by a further exacerbation and an irreversible progression.
To date, the studies performed with this model have been mainly focused on the endothelial function, showing that the vascular NO pathway plays a major role (Marie and Beny, 2002; Dooley et al., 2008). Furthermore, these studies have been concentrated on the early phase of the disease ‐ 2–4 months. Here, we have evaluated the vascular changes in Tsk mice during the disease progression over a greater age range ‐‐ 2–10 months. Vascular reactivity in response to adrenergic challenge (phenylephrine) decreases in an age‐dependent manner. Indeed, 10‐month‐old mice were barely responsive to phenylephrine. This progressive loss of response to the adrenergic stimulus was paralleled by an increased vascular susceptibility to the contraction induced by ET‐1. Thus, there is a link between the reduced adrenergic tone and the increased response to ET‐1. This functional interaction between ET‐1 and the adrenergic system has been already described in other pathological conditions characterized by vascular dysfunction (Bender and Klabunde, 2007). In order to further investigate the molecular and cellular mechanisms involved, we selected 8‐month‐old Tsk mice, as they still have a residual ability to contract to phenylephrine and show an enhanced contractile response to ET‐1, compared with age‐matched WT control animals. Changes in vascular reactivity were coupled to structural modifications that were also observed in the aortas from Tsk mice. Indeed, the immunohistochemical analysis performed on aortic sections highlighted a consistent fibre disorganization, associated with several rupture points, and a prominent collagen deposition in Tsk‐derived aorta, but not in age‐matched control mice. This type of re‐organization occurring in smooth muscle layer was paralleled by an increased expression of the pro‐contractile protein α‐SMA, mainly localized within the adventitial layers. Similarly, we found an increased expression of ETA receptors and the immunohistochemistry studies clearly showed that ETA receptors co‐localized with α‐SMA. In addition, this staining was clearly visible throughout the structural layers of aortas from Tsk mice. Therefore, vascular alterations occurring in Tsk mice are mainly associated with activation of the ET‐1/ETA receptor axis.
Interestingly, the changes in vasculature that we observed were also found to be relevant in many clinical investigations and gene association studies published in the recent literature that defines a key role for endothelin in fibrosis and SLS. Thus, in the human pathology there is an increase of the circulating and tissue ET‐1 levels coupled to a parallel over‐expression of ETA receptors (Vancheeswaran et al., 1994; Silver, 2008). These clinical findings have been substantiated by the therapeutic application of ET receptor antagonists to treat the vascular complications associated with SSc (Heresi and Minai, 2008; Cutolo et al., 2013). Indeed, bosentan, a non‐selective endothelin receptor antagonist, shows beneficial therapeutic effects in the treatment of vasculopathy associated with fibrosis by improving peripheral circulation and restoring the natural blood flow (Guiducci et al., 2012). However, there are two major concerns related to the use of bosentan, namely potential liver injury and teratogenicity.
Activation of ETA receptors has been associated with COX‐2 driven prostanoid release, which might be responsible for contraction of vascular tissues(Zhou et al., 2006), though other mechanisms involved cannot be ruled out (Plante et al., 2002). Conversely, activation of ETB receptors has an opposite beneficial effect, triggering NO release. Therefore, a selective blockade of ETA receptors should remove the unwanted effect associated with its activation but, at the same time, should spare the NO component triggered by ET‐1 through ETB receptors. The existence of such a mechanism within the vasculature was confirmed by the finding that L‐NAME, an inhibitor of the NO biosynthesis, further increased ET‐1‐induced contraction, while COX inhibition significantly suppressed ET‐1 contractile response. Therefore, a selective inhibition of ETA receptors blocks the deleterious contractile effect sparing, at the same time, the beneficial effect operated by ET‐1 through ETB receptors. Such a key role for ETA receptors in our experimental model finds a match in the human therapeutic approach. Indeed, clinical studies have shown that the selective ETA receptor antagonist, sitaxsentan, improved the clinical status in more than one third of patients with pulmonary hypertension, secondary to SSc, where the non‐selective antagonist bosentan was ineffective (Barst et al., 2004, 2006).
In several pathological conditions, the existence of some type of backup system is quite intuitive. Indeed, it is reasonable that changes, occurring during development of a disease, can, in turn, activate alternative pathways to mitigate or counterbalance the pathological effect(s). In this context, the PAR2 receptor represents a feasible candidate to be taken in consideration. Indeed, its expression is up‐regulated in vascular tissue in different diseases (Roviezzo et al., 2005; Cevikbas et al., 2011; Kagota et al., 2011), and recently, it has been shown that PAR2 levels are markedly enhanced in the skin of patients affected by SSc (Soumyakrishnan et al., 2014). Taking advantage of Tsk mice as a feasible predictive model for vascular changes in SSc, we evaluated the involvement of PAR2 in Tsk aortas. The enhanced vasorelaxation to PAR2‐AP, coupled to an increased expression of the receptor observed in Tsk mice confirmed that PAR2 plays a role in the control of vascular homeostasis. In particular, the finding that PAR2‐AP mediated vasorelaxation in Tsk aorta started at concentrations (30–100 nM) which were ineffective in aortas from WT control mice implies that the increased expression of PAR2, during disease progression, may represent an endogenous functional response triggered by the disease itself. Therefore, we hypothesized that the over‐expression of PAR2 could counterbalance the excessive activation of the ET‐1/ETA receptor axis. This hypothesis is sustained by pharmacological modulation studies, where we found that the ETA receptor antagonist FR139317 significantly potentiated PAR2‐AP‐mediated relaxation and the PAR2 antagonist ENMD1068 exacerbates ET‐1‐induced contraction. In other words, the removal of the PAR2 endogenous “tone” translates into an enhancement of the ET‐1 contractile effect, highlighting a functional antagonism between ETA receptors and PAR2 in regulation of vascular reactivity. In order to further confirm the presence of this interaction, we evaluated the response to ET‐1 in aortas from mice over‐expressing (TgPAR2) or lacking PAR2 (PAR2−/−). Our hypothesis of a compensatory role for PAR2 when the ET‐1 system was over‐active was confirmed by the finding that aortaa from mice with PAR2 deletion showed an increased contraction to ET‐1. Conversely, aortas from mice over‐expressing PAR2 receptors displayed a significant reduction of ET‐1‐induced contractile response. The alterations of ET‐1 effects observed at vascular level also matched the changes in circulating ET‐1 levels, which were reduced in TgPAR2 mice whereas they were increased in PAR2−/− mice.
In conclusion, the vascular dysfunction occurring in Tsk mice is an age dependent process that mimics the alterations present in fibrotic scleroderma syndrome in humans. The finding that the altered vascular response is endogenously counterbalanced by PAR2 defines a novel pharmacological target to develop as an alternative and/or additive therapeutic approach to endothelin antagonists in order to control vascular function.
Author contributions
F.R. conceived, designed experiments and wrote the manuscript; V.B. performed experiments, analysed the data and contributed to write the manuscript; V.M.I. performed vascular functional experiments; A.B. performed vascular functional experiments; G.D. performed immunohistochemistry; V.V. analysed the data; G.L. contributed to data analysis; M.L. performed vascular staining experiments; G.C. supervised all experiments, revised critically for intellectual contribution to the manuscript and gave final approval to publication.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Western blot and densitometry analysis for ETB expression in Tsk compared with control mice. Images are representative of 6 separate experiments.
Figure S2 Concentration‐response curve for ACh in aortas from 8‐month old Tsk mice and compared with aortas from control animals. Data are shown as mean ± SEM of % of relaxation against ACh concentration.
Supporting info item
Roviezzo, F. , Brancaleone, V. , Mattera Iacono, V. , Bertolino, A. , De Cunto, G. , Vellecco, V. , Lungarella, G. , Lucattelli, M. , and Cirino, G. (2017) Proteinase activated receptor‐2 counterbalances the vascular effects of endothelin‐1 in fibrotic tight‐skin mice. British Journal of Pharmacology, 174: 4032–4042. doi: 10.1111/bph.13618.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barst RJ, Langleben D, Badesch D, Frost A, Lawrence EC, Shapiro S et al. (2006). Treatment of pulmonary arterial hypertension with the selective endothelin‐A receptor antagonist sitaxsentan. J Am Coll Cardiol 47: 2049–2056. [DOI] [PubMed] [Google Scholar]
- Barst RJ, Langleben D, Frost A, Horn EM, Oudiz R, Shapiro S et al. (2004). Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med 169: 441–447. [DOI] [PubMed] [Google Scholar]
- Bender SB, Klabunde RE (2007). Altered role of smooth muscle endothelin receptors in coronary endothelin‐1 and alpha1‐adrenoceptor‐mediated vasoconstriction in Type 2 diabetes. Am J Physiol Heart Circ Physiol 293: H2281–H2288. [DOI] [PubMed] [Google Scholar]
- Cevikbas F, Seeliger S, Fastrich M, Hinte H, Metze D, Kempkes C et al. (2011). Role of protease‐activated receptors in human skin fibrosis and scleroderma. Exp Dermatol 20: 69–71. [DOI] [PubMed] [Google Scholar]
- Cheung WM, Andrade‐Gordon P, Derian CK, Damiano BP (1998). Receptor‐activating peptides distinguish thrombin receptor (PAR‐1) and protease activated receptor 2 (PAR‐2) mediated hemodynamic responses in vivo. Can J Physiol Pharmacol 76: 16–25. [DOI] [PubMed] [Google Scholar]
- Cottrell GS, Amadesi S, Schmidlin F, Bunnett N (2003). Protease‐activated receptor 2: activation, signalling and function. Biochem Soc Trans 31 (Pt 6): 1191–1197. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutolo M, Zampogna G, Vremis L, Smith V, Pizzorni C, Sulli A (2013). Longterm effects of endothelin receptor antagonism on microvascular damage evaluated by nailfold capillaroscopic analysis in systemic sclerosis. J Rheumatol 40: 40–45. [DOI] [PubMed] [Google Scholar]
- Denton CP, Black CM, Abraham DJ (2006). Mechanisms and consequences of fibrosis in systemic sclerosis. Nat Clin Pract Rheumatol 2: 134–144. [DOI] [PubMed] [Google Scholar]
- Dooley A, Low SY, Holmes A, Kidane AG, Abraham DJ, Black CM et al. (2008). Nitric oxide synthase expression and activity in the tight‐skin mouse model of fibrosis. Rheumatology (Oxford) 47: 272–280. [DOI] [PubMed] [Google Scholar]
- Green MC, Sweet HO, Bunker LE (1976). Tight‐skin, a new mutation of the mouse causing excessive growth of connective tissue and skeleton. Am J Pathol 82: 493–512. [PMC free article] [PubMed] [Google Scholar]
- Guiducci S, Bellando Randone S, Bruni C, Carnesecchi G, Maresta A, Iannone F et al. (2012). Bosentan fosters microvascular de‐remodelling in systemic sclerosis. Clin Rheumatol 31: 1723–1725. [DOI] [PubMed] [Google Scholar]
- Hamilton JR, Cocks TM (2000). Heterogeneous mechanisms of endothelium‐dependent relaxation for thrombin and peptide activators of protease‐activated receptor‐1 in porcine isolated coronary artery. Br J Pharmacol 130: 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessey JC, Stuyvers BD, McGuire JJ (2015). Small caliber arterial endothelial cells calcium signals elicited by PAR2 are preserved from endothelial dysfunction. Pharmacol Res Perspect 3: e00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heresi GA, Minai OA (2008). Bosentan in systemic sclerosis. Drugs Today (Barc) 44: 415–428. [DOI] [PubMed] [Google Scholar]
- Iwamoto N, Distler JH, Distler O (2011). Tyrosine kinase inhibitors in the treatment of systemic sclerosis: from animal models to clinical trials. Curr Rheumatol Rep 13: 21–27. [DOI] [PubMed] [Google Scholar]
- Jimenez SA, Millan A, Bashey RI (1984). Scleroderma‐like alterations in collagen metabolism occurring in the TSK (tight skin) mouse. Arthritis Rheum 27: 180–185. [DOI] [PubMed] [Google Scholar]
- Kagota S, Maruyama K, Wakuda H, McGuire JJ, Yoshikawa N, Nakamura K et al. (2011). Disturbance of vasodilation via protease‐activated receptor 2 in SHRSP.Z‐Lepr fa/IzmDmcr rats with metabolic syndrome. Vascul Pharmacol 63: 46–54. [DOI] [PubMed] [Google Scholar]
- Kahaleh MB (2004). Vascular involvement in systemic sclerosis (SSc. Clin Exp Rheumatol 22 (3 Suppl. 33): S19–S23. [PubMed] [Google Scholar]
- Kasturi KN, Shibata S, Muryoi T, Bona CA (1994). Tight‐skin mouse an experimental model for scleroderma. Int Rev Immunol 11: 253–271. [DOI] [PubMed] [Google Scholar]
- Kawashiri SY, Ueki Y, Terada K, Yamasaki S, Aoyagi K, Kawakami A (2014). Improvement of plasma endothelin‐1 and nitric oxide in patients with systemic sclerosis by bosentan therapy. Rheumatol Int 34: 221–225. [DOI] [PubMed] [Google Scholar]
- Kielty CM, Raghunath M, Siracusa LD, Sherratt MJ, Peters R, Shuttleworth CA et al. (1998). The Tight skin mouse: demonstration of mutant fibrillin‐1 production and assembly into abnormal microfibrils. J Cell Biol 140: 1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim FY, Barnes EA, Ying L, Chen C, Lee L, Alvira CM et al. (2015). Pulmonary artery smooth muscle cell endothelin‐1 expression modulates the pulmonary vascular response to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 308: L368–L377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambers C, Roth M, Zhong J, Campregher C, Binder P, Burian B et al. (2013). The interaction of endothelin‐1 and TGF‐beta1 mediates vascular cell remodeling. PLoS One 8: e73399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy EC (1996). Systemic sclerosis. a vascular perspective. Rheum Dis Clin North Am 22: 675–694. [DOI] [PubMed] [Google Scholar]
- Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R (2001). Proteinase‐activated receptors. Pharmacol Rev 53: 245–282. [PubMed] [Google Scholar]
- Maier C, Distler JH, Beyer C (2014). Deciphering the pro‐fibrotic phenotype of fibroblasts in systemic sclerosis. Exp Dermatol 23: 99–100. [DOI] [PubMed] [Google Scholar]
- Marie I, Beny JL (2002). Endothelial dysfunction in murine model of systemic sclerosis: tight‐skin mice 1. J Invest Dermatol 119: 1379–1387. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JJ (2004). Proteinase‐activated Receptor 2 (PAR2): a challenging new target for treatment of vascular diseases. Curr Pharm Des 10: 2769–2778. [DOI] [PubMed] [Google Scholar]
- McGuire JJ, Dai J, Andrade‐Gordon P, Triggle CR, Hollenberg MD (2002). Proteinase‐activated receptor‐2 (PAR2): vascular effects of a PAR2‐derived activating peptide via a receptor different than PAR2. J Pharmacol Exp Ther 303: 985–992. [DOI] [PubMed] [Google Scholar]
- Morelli S, Ferri C, Polettini E, Bellini C, Gualdi GF, Pittoni V et al. (1995). Plasma endothelin‐1 levels, pulmonary hypertension, and lung fibrosis in patients with systemic sclerosis. Am J Med 99: 255–260. [DOI] [PubMed] [Google Scholar]
- O'Donnell MD, O'Connor CM, FitzGerald MX, Lungarella G, Cavarra E, Martorana PA (1999). Ultrastructure of lung elastin and collagen in mouse models of spontaneous emphysema. Matrix Biol 18: 357–360. [DOI] [PubMed] [Google Scholar]
- Plante M, Honore JC, Neugebauer W, D'Orleans‐Juste P (2002). Endothelin‐1 (1–31) induces a thiorphan‐sensitive release of eicosanoids via ET(B) receptors in the guinea pig perfused lung. Clin Sci (Lond) 103 (Suppl. 48): 128S–131S. [DOI] [PubMed] [Google Scholar]
- Richard V, Solans V, Favre J, Henry JP, Lallemand F, Thuillez C et al. (2008). Role of endogenous endothelin in endothelial dysfunction in murine model of systemic sclerosis: tight skin mice 1. Fundam Clin Pharmacol 22: 649–655. [DOI] [PubMed] [Google Scholar]
- Roviezzo F, Bucci M, Brancaleone V, Di Lorenzo A, Geppetti P, Farneti S et al. (2005). Proteinase‐activated receptor‐2 mediates arterial vasodilation in diabetes. Arterioscler Thromb Vasc Biol 25: 2349–2354. [DOI] [PubMed] [Google Scholar]
- Silver RM (2008). Endothelin and scleroderma lung disease. Rheumatology (Oxford) 47 (Suppl. 5): v25–v26. [DOI] [PubMed] [Google Scholar]
- Soumyakrishnan S, Divya T, Kalayarasan S, Sriram N, Sudhandiran G (2014). Daidzein exhibits anti‐fibrotic effect by reducing the expressions of Proteinase activated receptor 2 and TGFbeta1/smad mediated inflammation and apoptosis in Bleomycin‐induced experimental pulmonary fibrosis. Biochimie 103: 23–36. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Asano Y, Ichimura Y, Toyama T, Taniguchi T, Noda S et al. (2015). Amelioration of tissue fibrosis by toll‐like receptor 4 knockout in murine models of systemic sclerosis. Arthritis Rheumatol 67: 254–265. [DOI] [PubMed] [Google Scholar]
- van den Hengel LG, Hellingman AA, Nossent AY, van Oeveren‐Rietdijk AM, de Vries MR, Spek CA et al. (2013). Protease‐activated receptor (PAR)2, but not PAR1, is involved in collateral formation and anti‐inflammatory monocyte polarization in a mouse hind limb ischemia model. PLoS One 8: e61923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vancheeswaran R, Magoulas T, Efrat G, Wheeler‐Jones C, Olsen I, Penny R et al. (1994). Circulating endothelin‐1 levels in systemic sclerosis subsets – a marker of fibrosis or vascular dysfunction? J Rheumatol 21: 1838–1844. [PubMed] [Google Scholar]
- Wort SJ, Woods M, Warner TD, Evans TW, Mitchell JA (2001). Endogenously released endothelin‐1 from human pulmonary artery smooth muscle promotes cellular proliferation: relevance to pathogenesis of pulmonary hypertension and vascular remodeling. Am J Respir Cell Mol Biol 25: 104–110. [DOI] [PubMed] [Google Scholar]
- Zhong B, Wang DH (2009). Protease‐activated receptor 2‐mediated protection of myocardial ischemia‐reperfusion injury: role of transient receptor potential vanilloid receptors. Am J Physiol Regul Integr Comp Physiol 297: R1681–R1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Mitra S, Varadharaj S, Parinandi N, Zweier JL, Flavahan NA (2006). Increased expression of cyclooxygenase‐2 mediates enhanced contraction to endothelin ETA receptor stimulation in endothelial nitric oxide synthase knockout mice. Circ Res 98: 1439–1445. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Western blot and densitometry analysis for ETB expression in Tsk compared with control mice. Images are representative of 6 separate experiments.
Figure S2 Concentration‐response curve for ACh in aortas from 8‐month old Tsk mice and compared with aortas from control animals. Data are shown as mean ± SEM of % of relaxation against ACh concentration.
Supporting info item