

Abstract
Background and Purpose
Heart failure with preserved ejection fraction (HFpEF) is a systemic syndrome driven by co‐morbidities, and its pathophysiology is poorly understood. Several studies suggesting that dipeptidyl peptidase 4 (DPP4) might be involved in the pathophysiology of heart failure have prompted experimental and clinical investigations of DPP4 inhibitors in the cardiovascular system. Here we have investigated whether the DPP4 inhibitor sitagliptin affected the progression of HFpEF independently of its effects on glycaemia.
Experimental Approach
Seven‐week‐old Dahl salt‐sensitive rats were fed a high‐salt diet for 5 weeks to induce hypertension. Then the rats continued with the high‐salt diet and were treated with either sitagliptin (10 mg·kg−1) or vehicle for the following 8 weeks. Blood pressure and cardiac function were measured in vivo. Histochemical and molecular biology analyses of myocardium were used to assay cytokines, fibrotic markers, DPP4 and glucagon‐like peptide‐1 (GLP‐1)/GLP‐1 receptor.
Key Results
Treatment with sitagliptin attenuated diastolic dysfunction, reduced mortality and reduced cardiac DPP4 activity, along with increased circulating GLP‐1 and myocardial expression of GLP‐1 receptors. Myocardial levels of pro‐inflammatory cytokines (TNF‐α, IL‐6 and CCL2) were reduced. Sitagliptin treatment decreased the levels of endothelial NOS monomer, responsible for generation of ROS, while the amount of NO‐producing dimeric form increased. Markers of oxidative and nitrosative stress were decreased. Moreover, increased collagen deposition and activation of pro‐fibrotic signalling, inducing elevated myocardial stiffness, were attenuated by sitagliptin treatment.
Conclusions and Implications
Sitagliptin positively modulated active relaxation and passive diastolic compliance by decreasing inflammation‐related endothelial dysfunction and fibrosis, associated with HFpEF.
Linked Articles
This article is part of a themed section on Targeting Inflammation to Reduce Cardiovascular Disease Risk. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.22/issuetoc and http://onlinelibrary.wiley.com/doi/10.1111/bcp.v82.4/issuetoc
Abbreviations
- BNP
brain natriuretic peptide
- cGMP
cyclic GMP
- CTGF
connective tissue growth factor
- DHE
dihydroethidium
- DPP4
dipeptidyl peptidase 4
- DPP4i
dipeptidyl peptidase 4 inhibitors
- EDP
end‐diastolic pressure
- EDPVR
end‐diastolic pressure‐volume relationship
- EF
ejection fraction
- eNOS
endothelial NOS
- FS
fractional shortening
- GLP‐1
glucagon‐like peptide‐1
- HF
heart failure
- HFpEF
heart failure with preserved ejection fraction
- HS
high‐salt
- LS
low‐salt
- VASP
vasodilator‐stimulated phosphoprotein
- VCAM‐1
vascular cell adhesion molecule‐1
Tables of Links
| TARGETS |
|---|
| Enzymes a |
| DPP4, dipeptidyl peptidase 4 http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=989 |
| GLP‐1 receptor |
| eNOS, endothelial NOS |
| PKG, protein kinase G |
| Other protein targets b |
| TNF‐α |
| LIGANDS |
|---|
| CCL2 |
| GLP‐1 |
| IL‐6 |
| SDF‐1, CXCL12 |
| Sitagliptin http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=989 |
| VCAM‐1 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Heart failure (HF) with preserved ejection fraction (EF) (HFpEF) is a clinical syndrome that affects about half of all patients with HF. The rising prevalence and significant mortality rate, ranging from 10 to 30% per year, carry a remarkable economic and social burden (Chan and Lam, 2013; Ambrosy et al., 2014). In addition to an overt HFpEF, the growing interest in diastolic performance arises from the observation that, also in non‐HF subjects, diastolic abnormalities are associated with increased mortality (Schwarzl et al., 2016). Diastolic dysfunction in HFpEF is characterized by slower left ventricle (LV) relaxation, elevated stiffness and increased filling pressure. The pathophysiology of HFpEF is incompletely understood. While the vast majority of HFpEF patients do not have a recognized primary cardiac pathology, they are older, more often female and have high prevalence of co‐morbidities, such as hypertension, obesity, diabetes, chronic obstructive pulmonary disease, anaemia and chronic kidney disease (Yancy et al., 2006; Fonorow et al., 2007; Ather et al., 2012).
In a recently proposed new paradigm, HFpEF is regarded as a systemic syndrome mediated in large part by risk factors and co‐morbidities, resulting in a systemic pro‐inflammatory state, which in the heart affects the endothelium and other cellular components of the myocardium. The chain of events that includes inflammation, oxidative stress, coronary endothelial dysfunction and down‐regulation of myocardial NO‐cyclic GMP (cGMP)‐PKG signalling leads to cardiomyocyte stiffness, cellular hypertrophy and enhanced myocardial fibrosis (Paulus and Tschöpe, 2013; Ferrari et al., 2015). Unfortunately, the most recent guidelines confirm that no treatment has been shown to reduce morbidity and mortality in patients with HFpEF, and management is limited to treatment of co‐morbidities and administration of diuretics to relieve symptoms (Ponikowski et al., 2016). While several potential candidates, such as statins, the advanced glycation end‐product breaker alagebrium, the soluble guanylate cyclase pathway stimulator verciguat, the If inhibitor ivabradine and the late I Na inhibitor ranolazine, await proper trials in HFpEF settings, the scientific and clinical community looks forward for the results of the ongoing phase III trial with sacubitril/valsartan (PARAGON‐HF).
The widespread expression of dipeptidyl peptidase 4 (DPP4) in the vasculature, myocardium and immune cells raises the possibility that this protein could play a role in cardiovascular function (Zhong et al., 2013, 2015). In particular, the finding that DPP4 activity is often associated with inflammation and cardiac remodelling points to an involvement of DPP4 in the pathophysiology of HF (Salles et al., 2015). HF patients and animals have increased DPP4 plasma activity that negatively correlates with LV performance (dos Santos et al., 2013), including diastolic function (Shigeta et al., 2012). Moreover, patients with acute HF with high DPP4 levels are at higher risk of death (Lourenco et al., 2013). DPP4 inhibitors (DPP4i) have been increasingly used for their hypoglycaemic effect, due to the prolongation of the action of the incretin hormones, glucagon‐like peptide‐1 (GLP‐1) and glucose insulinotropic peptide. This effect is accompanied by a low risk of hypoglycaemia and weight neutrality. Interestingly, following DPP‐4 inhibition, enhanced levels of GLP‐1 and activation of its receptor provide, independently of glycaemic control, cardiovascular protection probably through their anti‐inflammatory, anti‐atherosclerotic and antioxidant activities (Zerilli and Pyon, 2007; Scheen, 2010; Chang et al., 2015). Unfortunately, DPP4i, by interfering with the hydrolysis of many peptides other than the incretins, such as neuropeptides, cytokines and chemokines, may have pleiotropic effects on cardiovascular system that can be either beneficial (Shah et al., 2011a,b; dos Santos et al., 2013; Takahashi et al., 2013; Miyoshi et al., 2014) or harmful (Zhu et al., 2015; Mulvihill et al., 2016). Most importantly, concerns arise from clinical studies where an increase in the rate of hospitalization for HF has been observed (Scirica et al., 2013; Zannad et al., 2015).
An attractive explanation for these differences emerges from preclinical studies, which suggest that the effects of DPP4i may be context‐dependent, because of different alterations in the levels of substrates and their metabolites following DPP4 inhibition, which may vary depending on patient or animal model characteristics (Jackson et al., 2015; Mulvihill et al., 2016). Taken together, the results available demonstrate that the cardiovascular safety of DPP4i is, at present, uncertain and this uncertainty is far from being resolved.
In light of this complexity, the question of which cardiovascular outcomes can be associated with DPP4i in the presence of diastolic dysfunction or HFpEF remains unanswered. Thus, our aim was to test whether chronic administration of the most widely used DPP4i sitagliptin may affect the course of LV dysfunction, independently of any influence on diabetes, in a hypertensive, non‐diabetic, rat model of HFpEF.
Methods
Animal procedures
All animal care and experimental procedures in the present study have been performed according to the National ethical guidelines (Italian Ministry of Health; D.L.vo 26, March 4, 2014) and were approved by the local ethics committee. All the animal procedures are in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath and Lilley, 2015). Seven‐week‐old male Dahl salt‐sensitive rats (Charles River Laboratories, Wilmington, MA, USA) were maintained on a 12 h–12 h light–dark cycle in temperature‐ and humidity‐controlled room. Animals were fed laboratory chow with a high salt (8% NaCl) content (HS diet; from Safe, Augy, France.) for 5 weeks to induce hypertension. Afterwards, under the same HS diet, they were randomly divided into two groups: sitagliptin‐treated rats (n = 35; HS + SITA group; 10 mg·kg−1 daily by oral gavage) and vehicle‐treated rats (n = 40; HS group). In the control group (n = 10; LS group), rats were fed a low‐salt (LS) diet, containing 0.3% NaCl (standard laboratory chow provided by Envigo, S. Pietro al Natisone, Italy.). All the rats were killed 8 weeks later. The time course of these experimental protocols is set out in Figure 1.
Figure 1.
Experimental design. Temporal scheme of in vivo experiments.
Blood pressure and heart function
Blood pressure was measured weekly in conscious rats with a tail‐cuff (BP‐2000; Visitech Systems, Apex, NC, USA). Echocardiography and haemodynamic analyses were carried out on rats anaesthetized with a mixture of ketamine (100 mg·kg−1) and medetomidine (0.25 mg·kg−1), given i.m. A high‐resolution ultrasound system equipped with a 25 MHz linear transducer (Vevo 770, VisualSonics, Toronto, Ontario, Canada) was used to record serial M‐mode images along the minor axis at the level of the papillary muscles. Diastolic LV diameter was measured, and EF and fractional shortening (FS) were calculated. Prior to killing, a microtip pressure‐volume transducer (SPR‐612, Millar Instruments, Houston, TX, USA) connected to an A/D converter (iWorx 214) was inserted into the right carotid artery and arterial blood pressure was measured. After advancing the catheter into the LV, end‐diastolic pressure (EDP), dP/dt min, Tau and end‐diastolic pressure–volume relationship (EDPVR) were calculated (Cappetta et al., 2016).
Blood glucose determination, brain natriuretic peptide and GLP‐1 analysis
Blood glucose levels were measured from tail‐prick blood samples with a glucometer. Plasma brain natriuretic peptide (BNP) and GLP‐1 levels were determined by using an ELISA kit according to the manufacturer's instructions.
Tissue harvesting
After completion of the functional measurements, the abdominal aorta was cannulated and the heart was arrested in diastole by injection of 100 mM CdCl2. After perfusion with 10% phosphate‐buffered formalin, the heart was dissected and weighed. Finally, tissue specimens were embedded in paraffin, and 5 μm thick histological sections were cut. Alternatively, the heart was fixed in 4% paraformaldehyde for 1 h, immersed in a 30% sucrose solution overnight at 4°C and then embedded in tissue freezing medium (OCT). Tissue sections of 10 μm in thickness were cut (Di Meglio et al., 2012; De Angelis et al., 2015).
In situ DPP4 activity
DPP4 proteolytic activity was detected, in situ, as previously described (Shigeta et al., 2012), with some modifications. Briefly, frozen sections were fixed in a 1:1 mixture of acetone and chloroform for 2 min at 4°C. After several washes, the incubation solution (5 mg Gly‐Pro 4‐methoxy‐β‐naphthylamide hydrochloride in 0.5 mL dimethylformamide; 10 mg Fast Blue Salt; 10 mL PBS; pH 7.4) was applied to each sample. Specimens were incubated overnight at 4°C and mounted in an aqueous medium.
Histochemistry
Tissue fibrosis was detected with Masson's trichrome staining. Interstitial and perivascular fibrosis were measured using the ImageJ software (Media Cybernetics, Rockville, MD, USA). Immunofluorescence labelling and confocal microscopy were used to examine the endothelial localization of vascular cell adhesion molecule‐1 (VCAM‐1) and E‐selectin. Macrophage and neutrophil infiltration was assessed by antibodies against CD68 and myeloperoxidase; GLP‐1 receptor expression was detected in myocardial tissue; myocytes were labelled with α‐sarcomeric actin (α‐SA). Nitrosative stress was assessed by the presence of nitrotyrosine. To assess superoxide generation, frozen sections were incubated with dihydroethidium (DHE). Nuclei were counterstained with DAPI. FITC and tetramethylrhodamine‐5‐(and 6)‐isothiocyanate (TRITC)‐conjugated secondary antibodies were used. Sections were analysed with a Leica DM5000B microscope (Leica Microsystems, Wetzlar, Germany) and a Zeiss LSM700 confocal microscope (Zeiss, Oberkochen, Germany).
Quantitative RT‐PCR
Total RNA was extracted from heart tissue using the TRIzol reagent according to the manufacturer's instructions. Quantitative RT‐PCR was performed by using SYBR Green on a iCycler iQ System (Bio‐Rad Laboratories, Hercules, CA, USA). The transcript levels of connective tissue growth factor (CTGF), collagen I, collagen III and fibronectin were assayed, and the data were normalized to the housekeeping gene hypoxanthine phosphoribosyltransferase. All reactions were carried out in triplicate.
Western blotting
For Western blotting analysis, protein extracts were resolved on gradient (8 to12%) SDS‐PAGE and transferred onto PVDF membranes (Rinaldi et al., 2009). Membranes were probed with primary antibodies against TNF‐α, CCL2, NF‐κB, TGF‐β, IL‐6, myeloperoxidase, NADPH oxidase 2 (NOX‐2), SMAD3, CTGF, VCAM‐1, E‐selectin, DPP4, GLP‐1 receptors, the chemokine SDF‐1, vasodilator‐stimulated phosphoprotein (VASP), phospho‐VASP(Ser239), phospho‐SMAD3(Ser423/425) and collagen I. Loading conditions were determined with GAPDH. Peroxidase‐conjugated secondary antibodies were employed to detect primary antibodies. Antibody binding was visualized by chemiluminescence (ECL), and images were collected and analysed using a Chemidoc‐It Imager (Ultra‐Violet Products, Cambridge, UK).
Western blotting for native eNOS and phospho‐eNOS(Ser1177)
Protein expression of endothelial NOS (eNOS) and phospho‐eNOS(Ser1177) was performed in non‐denaturating conditions as previously described (Yamamoto et al., 2007). Briefly, rat hearts were lysed in ice‐cold protein lysis buffer under native conditions (50 mM Tris‐HCl, pH 7.4; 150 mM NaCl; 5 mM CaCl2; protease and phosphatase inhibitor cocktails). Protein concentration was determined, and 20 μg of native total protein was diluted in 5×non‐denaturating loading buffer (250 mM Tris‐HCl, pH 6.8; 50% glycerol; 0.5% w/v bromophenol blue). Both electrophoresis and blotting procedures were performed at 4°C. Samples were separated by SDS‐PAGE on 10% bis‐acrylamide gels in SDS‐free buffer and transferred onto PVDF membranes. Membranes were probed with anti‐eNOS and anti‐phospho‐eNOS(Ser1177) antibodies.
Data analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The results are presented as mean ± SD and the number of replicates was at least n = 5 per group for each data set. Significant differences between groups was determined by one‐way ANOVA and Bonferroni's post hoc test. Mortality curves were analysed by the standard Kaplan–Meier method with log‐rank test. Data were analysed using GraphPad Prism (GraphPad software, San Diego, CA, USA). To avoid inter‐operator variability, a single investigator, blinded to the treatments of the animal groups, performed all image acquisitions and offline measurements. All P values are two‐sided and P < 0.05 was considered statistically significant.
Materials
Sitagliptin, primary antibodies for TNF‐α, CCL2, NF‐κB and peroxidase‐conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). BNP immunoassay kit, primary antibodies for TGF‐β, IL‐6, NOX‐2, SMAD3, CTGF, VCAM‐1, E‐selectin, DPP4, SDF‐1, CD68, GAPDH and myeloperoxidase were from Abcam (Cambridge, UK). GLP‐1 immunoassay kit was from Tecan (Männedorf, Switzerland). α‐SA antibody, CdCl2, Masson's trichrome kit, DAPI, Tris‐HCl, NaCl, CaCl2, protease and phosphatase inhibitor cocktails, glycerol, bromophenol blue, acetone, chloroform, dimethylformamide; Fast Blue Salt; and DHE were from Sigma‐Aldrich (St. Louis, MO, USA). OCT was from Bio‐Optica (Milan, Italy). Nitrotyrosine antibody and ECL were from Merck Millipore (Milan, Italy). FITC and TRITC‐conjugated secondary antibodies were from Jackson ImmunoResearch (Suffolk, UK). TRIzol and SYBR Green were from Life Technologies Italia (Milan, Italy). PVDF and primary antibodies for eNOS and phospho‐eNOS(Ser1177) were from Thermo Fisher Scientific (Waltham, MA, USA). VASP, phospho‐VASP(Ser239) and phospho‐SMAD3(Ser423/425) antibodies were from Cell Signaling Technology (Danvers, MA, USA). Collagen I and GLP‐1 receptor antibodies were from Novus Biologicals (Littleton, CO, USA).
Results
Dahl salt‐sensitive rats, fed the HS (8% NaCl) diet from 7 weeks of age, progressively developed hypertension (De Angelis et al., 2016). After 5 weeks, before starting the treatment with sitagliptin, mean blood pressure was significantly higher in all animals given the HS diet, compared with those on the LS diet (161 ± 9 mmHg vs. 115 ± 4 mmHg).
Survival, blood pressure and body weight
Kaplan–Meier analysis showed a high mortality of HS animals with a significant difference in the survival rate in favour of the HS + SITA group (68% vs. 32% respectively). The LS group had no deaths during the experimental period (Figure 2A). During 8 weeks of treatment with sitagliptin, blood pressure remained markedly elevated, with a slight but significant reduction observed only at 19 weeks of age (222 ± 8 mmHg vs. 196 ± 14 mmHg) (Figure 2B). A comparable reduction was observed in the SITA‐group when blood pressure was invasively monitored through the carotid artery at 19 weeks of age (Figure 2C). Moreover, sitagliptin treatment had a beneficial effect on the general conditions of the animals and counteracted the loss of body weight (Figure 2D). The levels of blood glucose in all the experimental groups were unchanged, excluding a possible involvement of glycaemia in the pathological state of the animals (Figure 2E).
Figure 2.
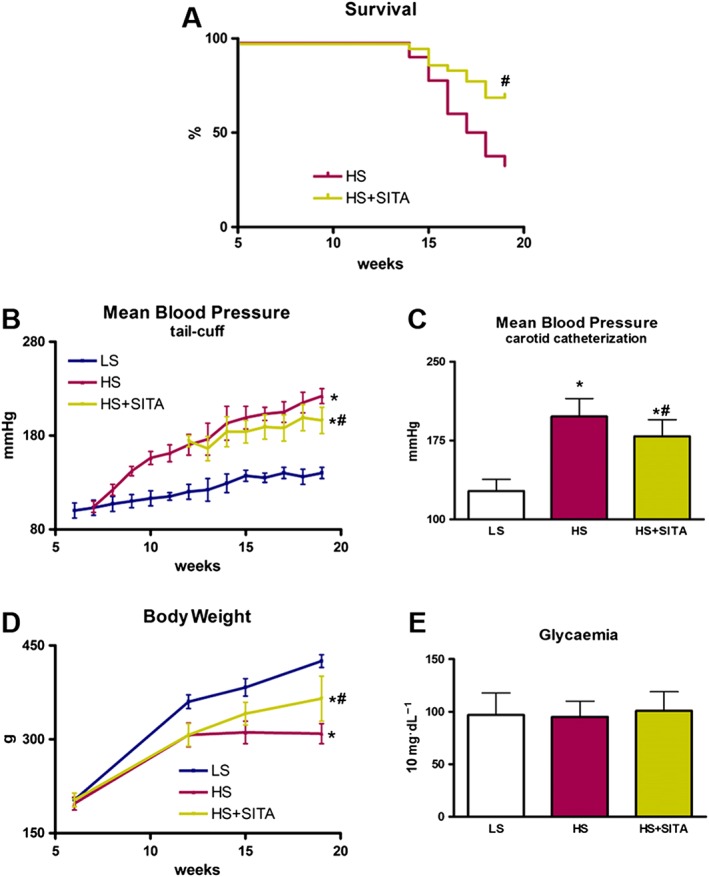
Blood pressure, body weight and animal survival in Dahl SS rats on HS and LS diets; effects of sitagliptin treatment. Changes in Kaplan–Meier survival (A). Mean blood pressure by tail‐cuff method (B) and intra‐arterial analysis (C). Body weight monitoring (D). Levels of glycaemia (E) (n = 10 in each group). Data represent the mean ± SD. *P < 0.05, significantly different from LS; #P < 0.05, significantly different from HS.
Systolic and diastolic function
At 19 weeks of age, systolic parameters were unaltered in all the experimental groups, as shown by comparable values of EF, FS and LV diameter (Figure 3A–C). On the other hand, diastolic function was impaired in HS animals and haemodynamic analysis showed an increased EDP, a decreased dP/dt min and a longer Tau, the time constant of LV relaxation. Furthermore, the end‐diastolic stiffness index and the EDPVR slope were increased. The deleterious effects of HS diet‐induced hypertension on diastolic compliance were partly blocked by sitagliptin (Figure 3D–G), supporting the possibility that this DPP4i may positively interfere with the impairment of LV relaxation. Functional improvement in the HS + SITA group was associated with a decrease in plasma BNP concentration, markedly raised in HS rats (Figure 3H). Finally, the lung weight/body weight ratio, an index of pulmonary congestion, was reduced in sitagliptin‐treated animals (Figure 3I).
Figure 3.
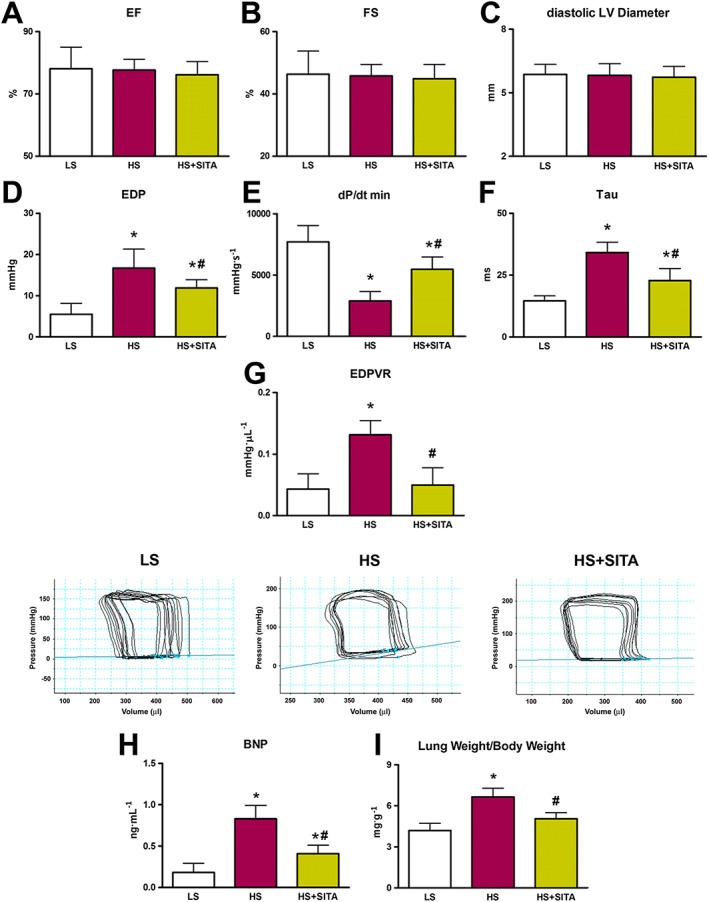
Cardiac function and hypertrophy. Echocardiography showing EF, FS and diastolic LV diameter (A–C). Diastolic indices EDP, dP/dt min and time constant Tau (D–F) together with EDPVR slope values and representative P‐V loops (G) (for cardiac function, n = 5 in LS group; n = 10 in HS and HS + SITA groups). Plasma BNP (H) (n = 5 in each group). Lung weight/body weight ratio (I) (n = 5 in LS group; n = 6 in HS group; n = 8 in HS + SITA group). Data represent the mean ± SD. *P < 0.05, significantly different from LS; #P < 0.05, significantly different from HS.
DPP4 expression and activity, GLP1 and GLP‐1 receptor expression
Myocardial DPP4 expression was elevated in hypertensive rats with respect to normotensive animals and this was not changed after treatment with sitagliptin (Figure 4A). In situ assessment of DPP4 activity showed an enhanced pattern in hearts of HS‐fed animals when compared with HS + SITA group (Figure 4B). The reduced activity of DPP4 was coupled with an increased expression of GLP‐1 receptors in hearts from the SITA group (Figure 4C). Moreover, serum GLP‐1 levels were increased in the HS + SITA group, compared with HS‐treated animals (Figure 4D).
Figure 4.
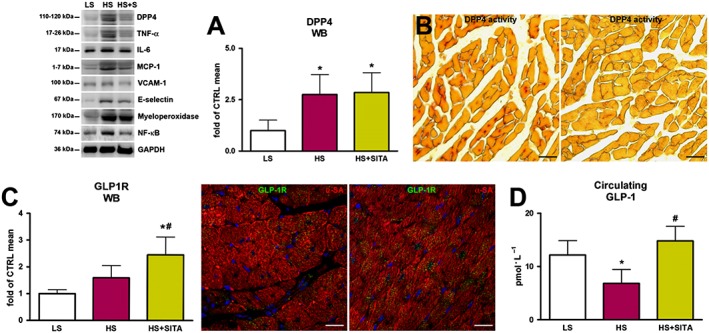
DPP4 and GLP‐1/GLP‐1 receptor axis. DPP4 expression by Western blotting (WB) (A) and activity (red) in HS (left panel) and HS + SITA (right panel) rat hearts (B). Expression of GLP‐1 receptor by WB and immunofluorescence (green) in HS (left panel) and HS + SITA (right panel) hearts (C). Circulating GLP‐1 (D). Cardiomyocytes are labelled with α‐SA (red), nuclei are stained with DAPI (blue). Scale bars (B) 50 μm, (C) 20 μm. Data represent the mean ± SD (n = 5 in each group). *P < 0.05, significantly different from LS; #P < 0.05, significantly different from HS.
Inflammation and activation of coronary endothelium
Myocardial levels of the pro‐inflammatory cytokines TNF‐α, IL‐6 and CCL2 were elevated in HS‐fed animals (Figure 5A–C). Myocardial inflammation was paralleled by vascular activation in HS hearts and was demonstrated by increased expression of adhesion molecules VCAM‐1 and E‐selectin (Figure 5D,E). With respect to the untreated group, sitagliptin treatment lowered the inflammatory status, shown by reduced levels of TNF‐α, IL‐6 and CCL2. Likewise, the expression of VCAM‐1 and E‐selectin was lower in HS + SITA group than in HS rats. Moreover, decreased macrophage infiltration and neutrophil activation, assessed by CD68 and myeloperoxidase expression, as well as macrophage polarization toward an anti‐inflammatory M2‐like phenotype, detected by CD206 expression, were observed in heartf from SITA‐treated rats (Figure 5F–H). These reductions, along with the decreased levels of NF‐κB (Figure 5I), a key target and regulator of inflammatory conditions, show that a comorbidity‐induced inflammatory state was attenuated by sitagliptin in our model.
Figure 5.
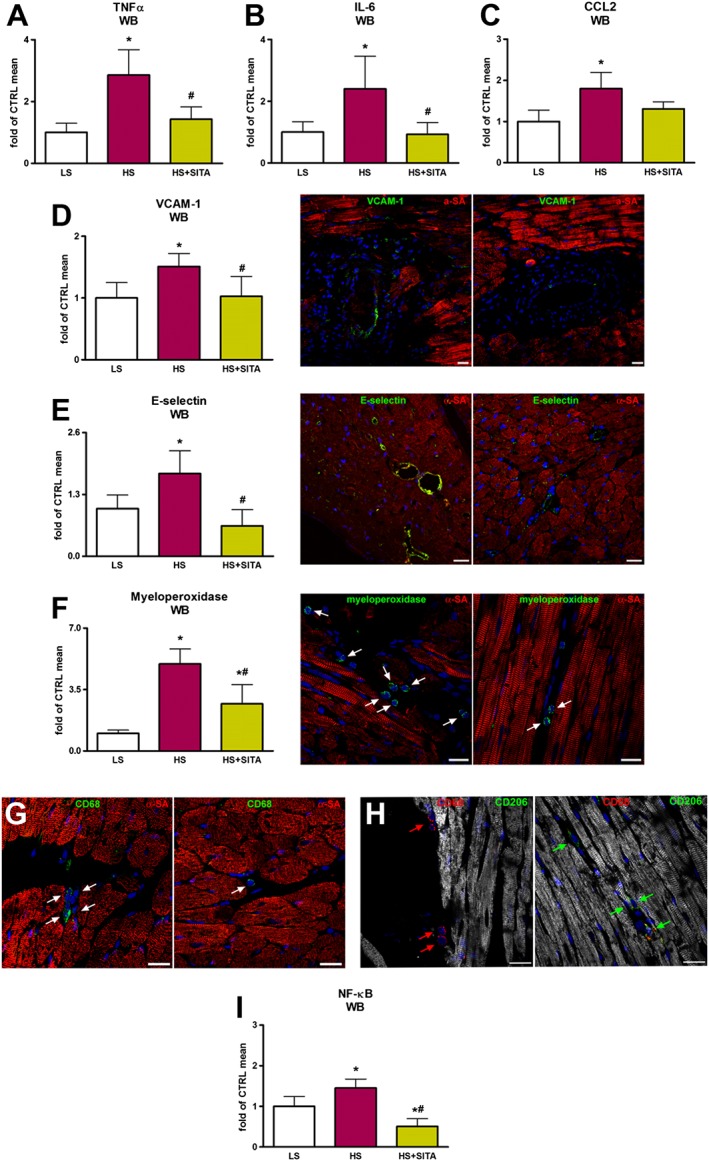
Inflammation and endothelial activation. Myocardial TNF‐α, IL‐6 and CCL2 protein levels (A–C). Expression of adhesion molecules VCAM‐1 and E‐selectin shown by Western blotting (WB) and immunofluorescence. VCAM‐1 and E‐selectin (green) on endothelial layer of the coronary vasculature are shown in HS (left panels) and HS + SITA (right panels) hearts (D,E). Macrophage infiltration and neutrophil activation, as assessed by CD68 and myeloperoxidase expression (green) in HS (left panels) and HS + SITA (right panels) rats (F,G). Macrophage M2‐like phenotype in HS (left panel) and HS + SITA (right panel) hearts (H). Cardiomyocytes are labelled with α‐SA (red); nuclei are stained with DAPI (blue). Myocardial expression of NF‐κB (I). Scale bars (D,H–L) 20 μm, (B) 50 μm. Data represent the mean ± SD (n = 5 in each group). *P < 0.05, significantly different from LS; #P < 0.05, significantly different from HS.
Oxidative stress and eNOS uncoupling
In our model of HFpEF, the involvement of myocardial oxidative stress was also addressed. Our data showed that the expression of NOX2, an enzyme that produces superoxide anion, was up‐regulated by HS diet and was returned to basal levels after sitagliptin treatment (Figure 6A). Moreover, the levels of DHE oxidation were reduced after the treatment with sitagliptin as well (Figure 6B). Because of inflammation and oxidative stress, a dimeric isoform of eNOS, which produces NO under physiological conditions, is forced to uncouple into monomers, altering the enzymic function and producing more superoxide anion. In our model, the levels of the eNOS monomer in the HS‐fed group increased with respect to control rats, whereas the expression of the dimer decreased (Figure 6C,D). Also, the activation of the eNOS monomeric isoform through phosphorylation of Ser1177 was increased in HS animals (Figure 6E). Conversely, sitagliptin treatment switched the balance of eNOS isoforms toward the dimeric form and significantly reduced the phosphorylation of monomeric eNOS.
Figure 6.
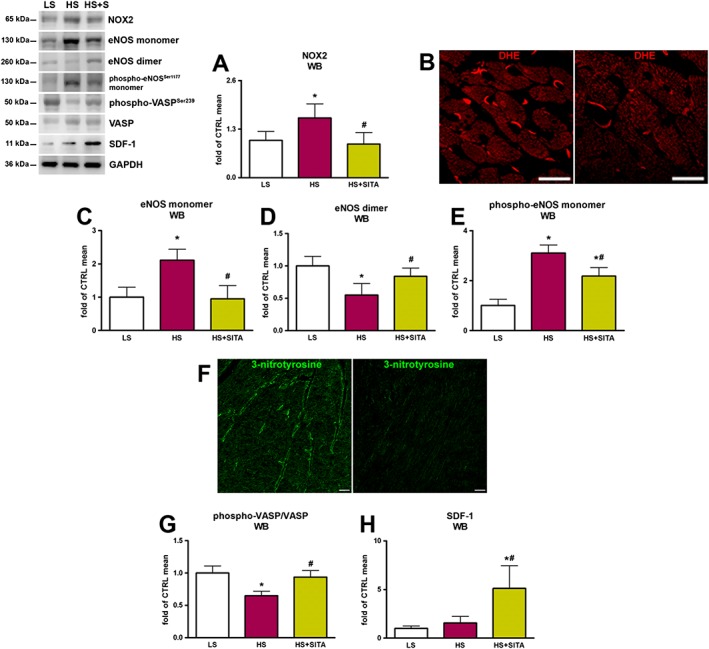
Oxidative stress, eNOS uncoupling and PKG activity. Levels of NOX2 (A) and ROS production as revealed by DHE (red) fluorescence (B) in HS (left panel) and HS + SITA (right panel) myocardium. The expression of eNOS monomer (C), dimer (D) and phosphorylated monomeric isoform (E). 3‐nitrotyrosine (green) in the endothelium in hearts from HS (left panel) and HS + SITA (right panel) mice (F). phospho‐VASP(Ser239)/VASP ratio (G). Myocardial expression of SDF‐1 (H). Scale bars (B) 20 μm, (F) 50 μm. Data represent the mean ± SD (n = 5 in each group). *P < 0.05, significantly different from LS; #P < 0.05, significantly different from HS.
NO/cGMP/PKG signalling
The interaction of superoxide anion with NO to form peroxynitrite and subsequent reaction with proteins to yield 3‐nitrotyrosine constitutes a lowering of NO bioavailability. In our experiments, formation of 3‐nitrotyrosine was increased in myocardial endothelium of HS rats. Treatment with sitagliptin lowered the 3‐nitrotyrosine content of endothelial cells (Figure 6F).
Endothelial dysfunction impairs adjacent cardiomyocytes because the low amount of NO determines reduced cGMP production and activity of its downstream effectors, including PKG. PKG activity can be indirectly detected by measuring VASP phosphorylation rate (Oelze et al., 2000). Our data revealed that the treatment with sitagliptin significantly increased the phospho‐VASP(Ser239)/VASP ratio, reversing the marked lowering observed in the HS group (Figure 6G). Finally, increased myocardial levels of SDF‐1 were only observed in sitagliptin‐treated animals (Figure 6H). Taken together, our results support the hypothesis that sitagliptin has a positive effect on microvascular endothelial dysfunction, the key determinant of a sequence of events leading to LV dysfunction in HFpEF.
Myocardial remodelling
Oxidative stress and pro‐inflammatory status contribute to the activation of pro‐fibrotic pathways in the myocardium (Westermann et al., 2011). Higher levels of perivascular and interstitial fibrosis in HS animals was evident by the accumulation of collagen, shown by Masson's trichrome staining (Figure 7A–C). mRNA and protein expression profiles showed an increase in collagen I mRNA and protein, and collagen type I/III mRNA ratio, along with higher fibronectin mRNA content (Figure 7D–F). Additionally, HS rats exhibited significant increases in the expression of TGF‐β, CTGF and in the phospho‐SMAD3(Ser423/425)/SMAD3 ratio (Figure 7G–I). Sitagliptin treatment markedly attenuated collagen deposition and decreased TGF‐β levels, the phospho‐SMAD3(Ser423/425)/SMAD3 ratio and CTGF expression. Overall, the treatment with sitagliptin, by modulating profibrotic markers, significantly attenuated adverse myocardial remodelling and extracellular matrix accumulation.
Figure 7.
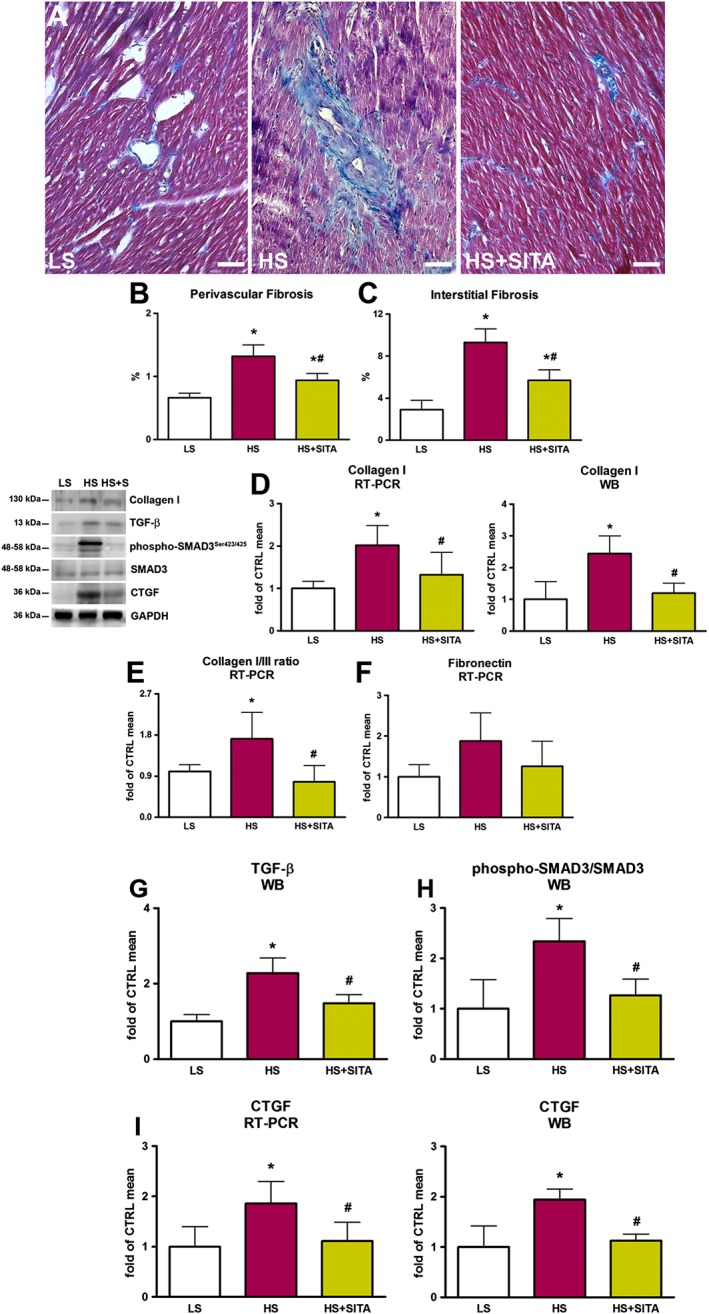
Myocardial fibrosis and extracellular matrix turnover. Masson's trichrome staining showing collagen deposition (blue) in the myocardium of LS (left panel), HS (central panel) and HS + SITA (right panel) animals (A). Perivascular and interstitial collagen fraction (B,C) (n = 5 in LS group; n = 6 in HS group; n = 8 in HS + SITA group). Collagen I mRNA and protein expression (D), collagen I/III mRNA ratio (E) and fibronectin mRNA expression (F). TGF‐β protein expression (G), phospho‐SMAD3(Ser423/425)/SMAD3 ratio (H) and CTGF mRNA and protein myocardial content (I) (for molecular biology, n = 5 in each group). Scale bars 200 μm. Data represent the mean ± SD. *P < 0.05, significantly different from LS; #P < 0.05, significantly different from HS.
Discussion
There are numerous unresolved issues regarding the underlying pathophysiological mechanisms of HFpEF and a successful clinical approach to this condition is far from being established. In contrast to HF with reduced EF (HFrEF), modern pharmacotherapy has not improved outcome in HFpEF and most of the clinical trials showed negative or inconclusive results (Ponikowski et al., 2016). A possible reason for this failure is the heterogeneity of the cohort of patients with HFpEF and the convergence of multiple abnormalities and different co‐morbidities (Bielecka‐Dabrowa et al., 2016; Shah et al., 2016). Although several pathways have been implicated in the pathogenesis of HFpEF, a low‐grade inflammation, associated with DPP4 up‐regulation and related to co‐morbidities such as obesity, diabetes and hypertension, seems to play an important role (Zhong et al., 2015). Additionally, the higher the activity of DPP4 in the coronary and peripheral circulation, the poorer the diastolic function (Shigeta et al., 2012). These findings prompted us to perform an experimental study exploring the effects of the DPP4i sitagliptin in Dahl SS rats, which, when fed a HS diet, develop hypertension and characteristic metabolic disturbances, such as insulin resistance and dyslipidemia, leading to HFpEF (Zicha et al., 2012; Horgan et al., 2014). The fact that hypertension is a major co‐morbidity in HFpEF, with approximately 70% of HFpEF patients presenting with elevated blood pressure, further validates the model selected for our experiments (Owan et al., 2006; McMurray et al., 2008; Liu et al., 2013; Teo et al., 2016).
Theoretically, inhibition of DPP4 should be beneficial in HF, but concerns about cardiovascular safety of the DPP4i, as a class, have been raised. Several clinical trials and epidemiological studies reported an increased risk of hospitalization for HF, although the findings do not appear fully concordant (Scirica et al., 2013; Wang et al., 2014; Chen et al., 2015; Green et al., 2015; Zannad et al., 2015; Filion et al., 2016). Meta‐analyses evaluating the effects of DPP4i on cardiovascular outcomes in diabetic patients also reported conflicting conclusions (Savarese et al., 2015; Kongwatcharapong et al., 2016). In particular, the risk of hospitalization for HF was higher with saxagliptin (Scirica et al., 2013), while alogliptin showed only a trend toward hospitalization for HF (Zannad et al., 2015). In contrast, no signal for increased risk was detected for sitagliptin (Green et al., 2015). Consequently, unlike sitagliptin, saxagliptin and alogliptin were included in a recent FDA warning about HF for patients with type 2 diabetes with heart or kidney disease (http://www.fda.gov/Drugs/DrugSafety/ucm486096.htm). This difference between DPP4i may suggest a possible drug‐specific effect, rather than a class‐effect.
In our experimental setting, treatment with sitagliptin had beneficial effects on diastolic properties, as shown by the amelioration of haemodynamic parameters EDP, dP/dt min, Tau and EDPVR and, most importantly, reduced mortality. These data are in line with studies supporting a positive role of DPP4 inhibition in diastolic dysfunction (Aroor et al., 2013; Bostick et al., 2014). The functional improvement observed after sitagliptin treatment was associated with decreased plasma levels of BNP that may reflect a better haemodynamic profile and reduced wall stress. Because of the non‐diabetic nature of our model and the unaltered blood glucose levels, these cardioprotective actions of sitagliptin are clearly not related to its effects on glycaemia. In contrast, we observed a slight lowering in blood pressure that started early in the treatment and became significant by the end of the experiment. However, sitagliptin‐treated rats remained severely hypertensive until the end of the study. Thus, these changes cannot exclusively account for the beneficial effects of the drug, although such a reduction in blood pressure may have contributed to the functional and structural modifications. The available data suggest that a large number of biologically active peptides with the capacity of decreasing or increasing blood pressure, are affected by DPP4 inhibition (Aroor et al., 2014), making the effects of DPP4 inhibition strongly context‐dependent (Jackson et al., 2015). The concept of context‐dependence is further supported by the opposite effects of DPP4 inhibition in animal models of HF and diabetic patients. A new DPP4i MK‐0626 ameliorated oxidative stress and fibrosis in diabetic mice (Bostick et al., 2014) but at the same time exerted harmful effects on heart function in a similar animal model (Mulvihill et al., 2016). Similarly, two recent clinical trials conducted on type 2 diabetic patients have reached conflicting conclusions. The PROLOGUE study has shown no additional effect on the progression of carotid artery intima‐media thickness with sitagliptin beyond that achieved with conventional treatment (Oyama et al., 2016), whereas in the SPIKE trial, sitagliptin attenuated the progression of carotid thickening (Mita et al., 2016).
The overall picture emerging from our results is in line with a new paradigm of HFpEF as a systemic inflammatory condition partly mediated by co‐morbidities (Yancy et al., 2006; Ather et al., 2012; Paulus and Tschöpe, 2013). Of note, our hypertensive animals with diastolic dysfunction had an increased myocardial expression of DPP4, which was unchanged by sitagliptin treatment. This can be relevant to the observations that various inflammatory factors can increase DPP4 expression in immune, epithelial or endothelial cells (Stefanovic et al., 1993; Riemann et al., 1995; Cordero et al., 1997; Yamabe et al., 1997). On the contrary, sitagliptin produced a sustained reduction of myocardial DPP4 activity. DPP4 is a principal determinant of the circulating level of GLP‐1, but it also cleaves numerous other biologically active peptide substrates, including chemokines, cytokines, neuropeptides and growth factors (Lambeir et al., 2003). In our model, inhibition of DPP4 was coupled with increased circulating GLP‐1 levels along with the enhancement of GLP‐1 receptors in the myocardium. Apart from the physiological function of GLP‐1 on glycaemic control, there is good evidence for a role in the cardiovascular system. GLP‐1 receptors are expressed in the heart and vasculature, where they modulate heart rate, blood pressure and vascular tone (Grieve et al., 2009). Importantly, GLP‐1 has been found to be cardioprotective in experimental models of dilated cardiomyopathy and hypertensive HF (Nikolaidis et al., 2005; Poornima et al., 2008). Thus, the beneficial effect of DPP4 inhibition is likely to be due, at least partly, to the potentiation of GLP1/GLP‐1 receptor axis.
Experimental and clinical observations have shown that the systemic inflammatory state induced by hypertension and other co‐morbidities is marked by increased levels of the circulating pro‐inflammatory cytokines TNFα and IL‐6, which strongly correlate with the risk of new‐onset HFpEF. Additionally, the levels of inflammatory biomarkers are higher in HFpEF than in hypertension, HFrEF or other conditions (Paulus and Tschöpe, 2013). In our study, sitagliptin treatment reversed TNF‐α and IL‐6 increases in the myocardium. This observation is consistent with previous reports demonstrating an anti‐inflammatory effect of sitagliptin in different experimental models, such as Zucker diabetic fatty rats, ApoE−/− mice, spontaneously hypertensive rats and in a LV radiofrequency ablation model of HF (Ferreira et al., 2010; Ta et al., 2011; Matsubara et al., 2012; Lee et al., 2013; de Almeida Salles et al., 2016).
A pro‐inflammatory state affects the coronary endothelium in HFpEF patients, which show enhanced expression of VCAM‐1 and E‐selectin (Akiyama et al., 2012). Elevated cardiac expression of adhesion molecules and CCL2 favours activation and migration of circulating leukocytes into the subendothelial space. NF‐κB is viewed as the master regulator of the inflammatory process by promoting the transcription of several pro‐inflammatory genes including cytokines, such as TNF‐α, IL‐6, CCL2 and adhesion molecules as VCAM‐1 (Valen et al., 2001; Shi et al., 2010). Our data, showing an increased myocardial expression of NF‐κB coupled with a higher content of inflammatory mediators, the up‐regulation of adhesion molecules and the influx of CD68‐ and myeloperoxidase‐positive cells in the HS group, would be compatible with this proposal. The effects of sitagliptin that include reduced expression of pro‐inflammatory proteins and decreased monocyte migration may be attributed to GLP‐1. In fact, GLP‐1 decreases monocyte migration and can exert anti‐inflammatory effects by reducing NF‐κB activation (Matsubara et al., 2012; Vittone et al., 2012). Additionally, the observed polarization of macrophages toward a M2‐like anti‐inflammatory phenotype, which can also be driven by GLP‐1, could have played a role (Shiraishi et al., 2012; Brenner et al., 2015). Because DPP4 may potentiate the inflammatory process at systemic and local levels by co‐stimulation of T‐cells, macrophage maturation and adhesion of inflammatory cells to extracellular matrix proteins (Zhong et al., 2015), we cannot exclude the potential effects of DPP4i on these non‐catalytic functions of DPP4.
Inflammation‐induced coronary endothelial dysfunction leads to impaired NO bioavailability (Paulus and Tschöpe, 2013), which can also be due to elevated levels of ROS. Pro‐inflammatory cytokines and up‐regulation of adhesion molecules mediate ROS generation (Griendling et al., 2000), which in turn decrease NO production and induce eNOS uncoupling, thus leading to the transition of hypertension to HFpEF (Szelényi et al., 2015). In fact, high nitrotyrosine levels indicate reduced NO bioavailability, as superoxide anion, overproduced in the ‘inflammatory disease’ HFpEF, consumes NO to generate peroxynitrite (Westermann et al., 2011; Ather et al., 2012; Paulus and Tschöpe, 2013). As NO contributes to myocardial relaxation, reduced cardiac NO contributes to diastolic dysfunction (Takimoto et al., 2005). In our study, treatment with sitagliptin counteracted eNOS uncoupling and reduced oxidative/nitrosative stress evident in the myocardium of HS animals. As endothelial cells express both GLP‐1 receptors and DPP4, and DPP4i have been shown to activate eNOS, an improved endothelial function can be attributed to both direct and indirect actions of sitagliptin.
Another potential mechanism involved in the maintenance of endothelial homeostasis may be related to the increased bioavailability of the chemokine SDF‐1 (CXCL12). DPP4 inhibition prevents the cleavage of SDF‐1 which can activate eNOS in cardiac cells (Zhong and Rajagopalan, 2015). SDF‐1 is also a progenitor cell homing factor. Preclinical studies have shown its therapeutic potential after myocardial injury by recruiting endothelial progenitor cells to the heart (Shigeta et al., 2012). Moreover, sitagliptin increased the number of circulating endothelial progenitor cells in diabetic patients (Fadini et al., 2010), and higher endothelial progenitor cell number and eNOS expression have been found after DPP4 inhibition (Shih et al., 2014).
In HFpEF patients, increased nitrosative/oxidative stress and decreased NO bioavailability translate into lower myocardial cGMP content and reduced PKG activity (Griendling et al., 2000; van Heerebeek et al., 2012). We have measured PKG activity assessing the phosphorylation rate of the VASP protein, one of the PKG specific substrates (Oelze et al., 2000). Treatment with sitagliptin increased the phospho‐VASP(Ser239)/VASP ratio, reversing the negative trend observed in the HS group. This is consistent with the data obtained in a diabetic model, where sitagliptin treatment was associated with the stimulation of the cGMP‐PKG pathway and the decrease in myocardial stiffness (Hamdani et al., 2014). PKG has a number of targets that exert a wide range of downstream effects in cardiovascular physiology. For example, through the phosphorylation of titin, PKG alters the mechanical properties of cardiomyocytes affecting diastolic tone (lowered by PKG), ventricular extensibility (increased by PKG) and relaxation speed (accelerated by PKG) (Linke and Hamdani, 2014). Several PKG targets have also been identified within the intracellular calcium regulatory system (Takimoto, 2012). Additionally, PKG reduces the fibrotic response of the heart by interfering with the activation of the TGF‐β/SMAD3 signalling (Kirk et al., 2016). Increased cardiomyocyte stiffness, in addition to fibrosis, contributes to a reduced LV compliance (Borbely et al., 2005; van Heerebeek et al., 2006; Linke and Hamdani, 2014).
The inflammation‐driven stimulation of TGF‐β/SMAD3 signalling is essential for the activation and transdifferentiation of fibroblasts into myofibroblasts and the extracellular matrix turnover (Kirk et al., 2016). These processes, fuelled by the presence of co‐morbidities such as hypertension and type 2 diabetes, contribute to the increased myocardial stiffness in HFpEF (González et al., 2010; Kasner et al., 2011). In HFpEF patients, in addition to increased circulating biomarkers of inflammation, myocardial expression of TGF‐β and other markers of matrix turnover increased and correlated with diastolic dysfunction (Westermann et al., 2011). In our study, myocardial levels of TGF‐β and its downstream effectors SMAD3 and CTGF decreased following sitagliptin administration. Moreover, we detected a reduction in collagen and fibronectin accumulation. Thus, by interfering with the development of the pro‐fibrotic phenotype, sitagliptin exerted a beneficial modulation of myocardial remodelling.
Limitations of the study
The restricted availability of animal models represents a major limitation in conducting mechanistic studies on the pathophysiology of HFpEF. So far, the Dahl SS rat offers one of the best and most frequently used models with clinically relevant characteristics. These animals, when fed a HS diet from the age of 7 weeks, develop, over time, most of the hallmarks of HFpEF, including diastolic dysfunction, hypertension and inflammation (Zicha et al., 2012; Horgan et al., 2014). However, given the progressive and transitory nature of this model, structural and functional changes significantly vary at different time‐points. A mild myocardial dysfunction, already present at 12‐14 weeks, progresses to a phenotype that resembles HFpEF only at 18‐20 weeks (Doi et al., 2000; Qu et al., 2000). However, at this time, there is also a high mortality in the HS animals, which means that dysfunction can only be assessed in the survivors. This restriction of the experimental sample can be considered as a limitation. Moving the assessment to earlier times when the mortality is lower would have eliminated this bias (selection by survival), but it would preclude the development of the full‐blown HFpEF phenotype. Now that we have shown a clear effect of sitagliptin on the fully developed phenotype, another set of studies with at an earlier time could yield an interesting mechanistic insight into the dynamics of this model.
In conclusion, given the wide range of effects of DPP4i, it is clear that the mechanisms responsible for the beneficial effects of sitagliptin are equally complex and multiple. Although the potentiation of the GLP‐1/GLP‐1 receptor signalling contributes to cardioprotection, our findings show that sitagliptin‐mediated combination of anti‐inflammatory and anti‐fibrotic actions coupled with improved coronary endothelial function and NO bioavailability acts in concert to return the hypertension‐driven HFpEF phenotype towards normality.
Our study reinforces the notion that the effects of DPP4 inhibition may strongly depend on the overall pathophysiological milieu, which, according to a current concept, can be significantly heterogeneous within the HFpEF population. Our findings may therefore be of particular relevance in the upcoming era of personalized medicine.
Author contributions
G.E. conceived and designed the experiments, performed immunofluorescence and molecular biology experiments, and analysed the data. D.C., A.D.A. and K.U. conceived and designed the experiments, performed in vivo experiments, analysed the data and wrote the paper. E.P., R.R., A.R., L.P.C. and F.R. performed histochemistry and molecular biology experiments and analysed the data. L.B. and F.scoR revised the manuscript and gave final approval to the publication. All authors approved the final draft of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by Regione Campania under POR Campania FESR 2007–2013 (Ockey Project, Oncology and Cardiology Key Target).
Esposito, G. , Cappetta, D. , Russo, R. , Rivellino, A. , Ciuffreda, L. P. , Roviezzo, F. , Piegari, E. , Berrino, L. , Rossi, F. , De Angelis, A. , and Urbanek, K. (2017) Sitagliptin reduces inflammation, fibrosis and preserves diastolic function in a rat model of heart failure with preserved ejection fraction. British Journal of Pharmacology, 174: 4070–4086. doi: 10.1111/bph.13686.
References
- Akiyama E, Sugiyama S, Matsuzawa Y, Konishi M, Suzuki H, Nozaki T et al. (2012). Incremental prognostic significance of peripheral endothelial dysfunction in patients with heart failure with normal left ventricular ejection fraction. J Am Coll Cardiol 60: 1778–1786. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosy AP, Fonarow GC, Butler J, Chioncel O, Greene SJ, Vaduganathan M et al. (2014). The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol 63: 1123–1133. [DOI] [PubMed] [Google Scholar]
- Aroor AR, Sowers JR, Jia G, DeMarco VG (2014). Pleiotropic effects of the dipeptidylpeptidase‐4 inhibitors on the cardiovascular system. Am J Physiol Heart Circ Physiol 307: H477–H492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroor AR, Sowers JR, Bender SB, Nistala R, Garro M, Mugerfeld I et al. (2013). Dipeptidylpeptidase inhibition is associated with improvement in blood pressure and diastolic function in insulin‐resistant male Zucker obese rats. Endocrinology 154: 2501–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ather S, Chan W, Bozkurt B, Aguilar D, Ramasubbu K, Zachariah AA et al. (2012). Impact of noncardiac comorbidities on morbidity and mortality in a predominantly male population with heart failure and preserved versus reduced ejection fraction. J Am Coll Cardiol 59: 998–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielecka‐Dabrowa A, Sakowicz A, Misztal M, von Haehling S, Ahmed A, Pietrucha T et al. (2016). Differences in biochemical and genetic biomarkers in patients with heart failure of various etiologies. Int J Cardiol 221: 1073–1080. [DOI] [PubMed] [Google Scholar]
- Borbely A, van der Velden J, Papp Z, Bronzwaer JG, Edes I, Stienen GJ et al. (2005). Cardiomyocyte stiffness in diastolic heart failure. Circulation 111: 774–781. [DOI] [PubMed] [Google Scholar]
- Bostick B, Habibi J, Ma L, Aroor A, Rehmer N, Hayden MR et al. (2014). Dipeptidyl peptidase inhibition prevents diastolic dysfunction and reduces myocardial fibrosis in a mouse model of Western diet induced obesity. Metabolism 63: 1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner C, Franz WM, Kühlenthal S, Kuschnerus K, Remm F, Gross L et al. (2015). DPP‐4 inhibition ameliorates atherosclerosis by priming monocytes into M2 macrophages. Int J Cardiol 199: 163–169. [DOI] [PubMed] [Google Scholar]
- Cappetta D, Esposito G, Piegari E, Russo R, Ciuffreda LP, Rivellino A et al. (2016). SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int J Cardiol 205: 99–110. [DOI] [PubMed] [Google Scholar]
- Chan MM, Lam CS (2013). How do patients with heart failure with preserved ejection fraction die? Eur J Heart Fail 15: 604–613. [DOI] [PubMed] [Google Scholar]
- Chang MW, Chen CH, Chen YC, Wu YC, Zhen YY, Leu S et al. (2015). Sitagliptin protects rat kidneys from acute ischemia–reperfusion injury via upregulation of GLP‐1 and GLP‐1 receptors. Acta Pharmacol Sin 36: 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DY, Wang SH, Mao CT, Tsai ML, Lin YS, Chou CC et al. (2015). Sitagliptin and cardiovascular outcomes in diabetic patients with chronic kidney disease and acute myocardial infarction: a nationwide cohort study. Int J Cardiol 181: 200–206. [DOI] [PubMed] [Google Scholar]
- Cordero OJ, Salgado FJ, Vinuela JE, Nogueira M (1997). Interleukin‐12 enhances CD26 expression and dipeptidyl peptidase IV function on human activated lymphocytes. Immunobiology 197: 522–533. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida Salles T, Zogbi C, de Lima TM, de Godoi Carneiro C, Garcez AT, Barbeiro HV et al. (2016). The contributions of dipeptidyl peptidase IV to inflammation in heart failure. Am J Physiol Heart Circ Physiol 310: H1760–H1772. [DOI] [PubMed] [Google Scholar]
- De Angelis A, Cappetta D, Piegari E, Rinaldi B, Ciuffreda LP, Esposito G et al. (2016). Long‐term administration of ranolazine attenuates diastolic dysfunction and adverse myocardial remodeling in a model of heart failure with preserved ejection fraction. Int J Cardiol 217: 69–79. [DOI] [PubMed] [Google Scholar]
- De Angelis A, Piegari E, Cappetta D, Russo R, Esposito G, Ciuffreda LP et al. (2015). SIRT1 activation rescues doxorubicin‐induced loss of functional competence of human cardiac progenitor cells. Int J Cardiol 189: 30–44. [DOI] [PubMed] [Google Scholar]
- Di Meglio F, Nurzynska D, Castaldo C, Miraglia R, Romano V, De Mngelis A et al. (2012). Cardiac shock wave therapy: assessment of safety and new insights into mechanisms of tissue regeneration. J Cell Mol Med 16: 936–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi R, Masuyama T, Yamamoto K, Doi Y, Mano T, Sakata Y et al. (2000). Development of different phenotypes of hypertensive heart failure: systolic versus diastolic failure in Dahl salt‐sensitive rats. J Hypertens 18: 111–120. [DOI] [PubMed] [Google Scholar]
- dos Santos L, Salles TA, Arruda‐Junior DF, Campos LC, Pereira AC, Barreto AL et al. (2013). Circulating dipeptidyl peptidase IV activity correlates with cardiac dysfunction in human and experimental heart failure. Circ Heart Fail 6: 1029–1038. [DOI] [PubMed] [Google Scholar]
- Fadini GP, Boscaro E, Albiero M, Menegazzo L, Frison V, de Kreutzenberg S et al. (2010). The oral dipeptidyl peptidase‐4 inhibitor sitagliptin increases circulating endothelial progenitor cells in patients with type 2 diabetes: possible role of stromal‐derived factor‐1α. Diabetes Care 33: 1607–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari R, Böhm M, Cleland JG, Paulus WJ, Pieske B, Rapezzi C et al. (2015). Heart failure with preserved ejection fraction: uncertainties and dilemmas. Eur J Heart Fail 17: 665–671. [DOI] [PubMed] [Google Scholar]
- Ferreira L, Teixeira‐de‐Lemos E, Pinto F, Parada B, Mega C, Vala H et al. (2010). Effects of sitagliptin treatment on dysmetabolism, inflammation, and oxidative stress in an animal model of type 2 diabetes (ZDF rat). Mediators Inflamm 2010: 592760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion KB, Azoulay L, Platt RW, Dahl M, Dormuth CR, Clemens KK et al. (2016). A multicenter observational study of incretin‐based drugs and heart failure. N Engl J Med 374: 1145–1154. [DOI] [PubMed] [Google Scholar]
- Fonorow GC, Stough WG, Abraham WT, Albert NM, Gheorghiade M, Greenberg BH et al. (2007). OPTIMIZE‐HF investigators and hospitals. Characteristics, treatments and outcomes of patients with preserved systolic function hospitalized for heart failure: a report from the OPTIMIZE‐HF Registry. J Am Coll Cardiol 50: 768–777. [DOI] [PubMed] [Google Scholar]
- González A, López B, Querejeta R, Zubillaga E, Echeverría T, Díez J (2010). Filling pressures and collagen metabolism in hypertensive patients with heart failure and normal ejection fraction. Hypertension 55: 1418–1424. [DOI] [PubMed] [Google Scholar]
- Green JB, Bethel MA, Armstrong PW, for the TECOS Study Group (2015). Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 373: 232–242. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Sorescu D, Ushio‐Fukai M (2000). NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 86: 494–501. [DOI] [PubMed] [Google Scholar]
- Grieve DJ, Cassidy RS, Green BD (2009). Emerging cardiovascular actions of the incretin hormone glucagon‐like peptide‐1: potential therapeutic benefits beyond glycaemic control? Br J Pharmacol 157: 1340–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdani N, Hervent AS, Vandekerckhove L, Matheeussen V, Demolder M, Baerts L et al. (2014). Left ventricular diastolic dysfunction and myocardial stiffness in diabetic mice is attenuated by inhibition of dipeptidyl peptidase 4. Cardiovasc Res 104: 423–431. [DOI] [PubMed] [Google Scholar]
- Horgan S, Watson C, Glezeva N, Baugh J (2014). Murine models of diastolic dysfunction and heart failure with preserved ejection fraction. J Card Fail 20: 984–995. [DOI] [PubMed] [Google Scholar]
- Jackson EK, Mi Z, Tofovic SP, Gillespie DG (2015). Effect of dipeptidyl peptidase 4 inhibition on arterial blood pressure is context dependent. Hypertension 65: 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasner M, Westermann D, Lopez B, Gaub R, Escher F, Kühl U et al. (2011). Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross‐linking in heart failure and normal ejection fraction. J Am Coll Cardiol 57: 977–985. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, NC3Rs Reporting Guidelines Working Group (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk JA, Holewinski RJ, Crowgey EL, Van Eyk JE (2016). Protein kinase G signaling in cardiac pathophysiology: impact of proteomics on clinical trials. Proteomics 16: 894–905. [DOI] [PubMed] [Google Scholar]
- Kongwatcharapong J, Dilokthornsakul P, Nathisuwan S, Phrommintikul A, Chaiyakunapruk N (2016). Effect of dipeptidyl peptidase‐4 inhibitors on heart failure: a meta‐analysis of randomized clinical trials. Int J Cardiol 211: 88–95. [DOI] [PubMed] [Google Scholar]
- Lambeir AM, Durinx C, Scharpe S, de Meester I (2003). Dipeptidyl‐peptidase IV from bench to bedside: an update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit Rev Clin Lab Sci 40: 209–294. [DOI] [PubMed] [Google Scholar]
- Lee TI, Kao YH, Chen YC, Huang JH, Hsu MI, Chen YJ (2013). The dipeptidyl peptidase‐4 inhibitor‐sitagliptin modulates calcium dysregulation, inflammation, and PPARs in hypertensive cardiomyocytes. Int J Cardiol 168: 5390–5395. [DOI] [PubMed] [Google Scholar]
- Linke WA, Hamdani N (2014). Gigantic business: titin properties and function through thick and thin. Circ Res 114: 1052–1068. [DOI] [PubMed] [Google Scholar]
- Liu Y, Haddad T, Dwivedi G (2013). Heart failure with preserved ejection fraction: current understanding and emerging concepts. Curr Opin Cardiol 28: 187–196. [DOI] [PubMed] [Google Scholar]
- Lourenco P, Frioes F, Silva N, Guimaraes JT, Bettencourt P (2013). Dipeptidyl peptidase IV and mortality after an acute heart failure episode. J Cardiovasc Pharmacol 62: 138–142. [DOI] [PubMed] [Google Scholar]
- Matsubara J, Sugiyama S, Sugamura K, Nakamura T, Fujiwara Y, Akiyama E et al. (2012). A dipeptidyl peptidase‐4 inhibitor, des‐fluoro‐sitagliptin, improves endothelial function and reduces atherosclerotic lesion formation in apolipoprotein E‐deficient mice. J Am Coll Cardiol 59: 265–276. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray JJ, Carson PE, Komajda M, McKelvie R, Zile MR, Ptaszynska A et al. (2008). Heart failure with preserved ejection fraction: clinical characteristics of 4133 patients enrolled in the I‐PRESERVE trial. Eur J Heart Fail 10: 149–156. [DOI] [PubMed] [Google Scholar]
- Mita T, Katakami N, Shiraiwa T, Yoshii H, Onuma T, Kuribayashi N et al. (2016). Sitagliptin attenuates the progression of carotid intima‐media thickening in insulin‐treated patients with type 2 diabetes: the sitagliptin preventive study of intima‐media thickness evaluation (SPIKE): a randomized controlled trial. Diabetes Care 39: 455–464. [DOI] [PubMed] [Google Scholar]
- Miyoshi T, Nakamura K, Yoshida M, Miura D, Oe H, Akagi S et al. (2014). Effect of vildagliptin, a dipeptidyl peptidase 4 inhibitor, on cardiac hypertrophy induced by chronic beta‐adrenergic stimulation in rats. Cardiovasc Diabetol 13: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvihill EE, Varin EM, Ussher JR, Campbell JE, Bang KW, Abdullah T et al. (2016). Inhibition of dipeptidyl peptidase‐4 impairs ventricular function and promotes cardiac fibrosis in high fat‐fed diabetic mice. Diabetes 65: 742–754. [DOI] [PubMed] [Google Scholar]
- Nikolaidis LA, Elahi D, Shen YT, Shannon RP (2005). Active metabolite of GLP‐1 mediates myocardial glucose uptake and improves left ventricular performance in conscious dogs with dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 289: H2401–H2408. [DOI] [PubMed] [Google Scholar]
- Oelze M, Mollnau H, Hoffmann N, Warnholtz A, Bodenschatz M, Smolenski A et al. (2000). Vasodilator‐stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circ Res 87: 999–1005. [DOI] [PubMed] [Google Scholar]
- Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM (2006). Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 355: 251–259. [DOI] [PubMed] [Google Scholar]
- Oyama J, Murohara T, Kitakaze M, Ishizu T, Sato Y, Kitagawa K et al. (2016). The effect of sitagliptin on carotid artery atherosclerosis in type 2 diabetes: the PROLOGUE randomized controlled trial. PLoS Med 13: e1002051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus WJ, Tschöpe C (2013). A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62: 263–271. [DOI] [PubMed] [Google Scholar]
- Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ et al. (2016). 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J pii: ehw128. [DOI] [PubMed] [Google Scholar]
- Poornima I, Brown S, Bhashyam S, Parikh P, Bolukoglu H, Shannon RP (2008). Chronic glucagon‐like peptide‐1 (GLP‐1) infusion sustains LV systolic function and prolongs survival in the spontaneously hypertensive‐heart failure prone rat. Circ Heart Fail 1: 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu P, Hamada M, Ikeda S, Hiasa G, Shigematsu Y, Hiwada K (2000). Time‐course changes in left ventricular geometry and function during the development of hypertension in Dahl salt‐sensitive rats. Hypertens Res 23: 613–623. [DOI] [PubMed] [Google Scholar]
- Riemann D, Kehlen A, Langner J (1995). Stimulation of the expression and the enzyme activity of aminopeptidase N/CD13 and dipeptidylpeptidase IV/CD26 on human renal cell carcinoma cells and renal tubular epithelial cells by T cell‐derived cytokines, such as IL‐4 and IL‐13. Clin Exp Immunol 100: 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi B, Pieri L, Donniacuo M, Cappetta D, Capuano A, Domenici L et al. (2009). Rosiglitazone reduces the inflammatory response in a model of vascular injury in rats. Shock 32: 638–644. [DOI] [PubMed] [Google Scholar]
- Salles TA, dos Santos L, Barauna VG, Girardi AC (2015). Potential role of dipeptidyl peptidase IV in the pathophysiology of heart failure. Int J Mol Sci 16: 4226–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarese G, Perrone‐Filardi P, D'Amore C, Vitale C, Trimarco B, Pani L et al. (2015). Cardiovascular effects of dipeptidyl peptidase‐4 inhibitors in diabetic patients: A meta‐analysis. Int J Cardiol 181: 239–244. [DOI] [PubMed] [Google Scholar]
- Scheen AJ (2010). Dipeptidylpeptidase‐4 inhibitors (gliptins): focus on drug–drug interactions. Clin Pharmacokinet 49: 573–588. [DOI] [PubMed] [Google Scholar]
- Schwarzl M, Ojeda F, Zeller T, Seiffert M, Becher PM, Munzel T et al. (2016). Risk factors for heart failure are associated with alterations of the LV end‐diastolic pressure‐volume relationship in non‐heart failure individuals: data from a large‐scale, population‐based cohort. Eur Heart J 37: 1807–1814. [DOI] [PubMed] [Google Scholar]
- Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B et al. (2013). Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 369: 1317–1326. [DOI] [PubMed] [Google Scholar]
- Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA et al. (2016). Phenotype‐Specific treatment of heart failure with preserved ejection fraction: a multiorgan roadmap. Circulation 134: 73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah Z, Pineda C, Kampfrath T, Maiseyeu A, Ying Z, Racoma I et al. (2011a). Acute DPP‐4 inhibition modulates vascular tone through GLP‐1 independent pathways. Vascul Pharmacol 55: 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah Z, Kampfrath T, Deiuliis JA, Zhong J, Pineda C, Ying Z et al. (2011b). Long‐term dipeptidyl‐peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation 124: 2338–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Camici GG, Lüscher TF (2010). Cardiovascular determinants of life span. Pflugers Arch 459: 315–324. [DOI] [PubMed] [Google Scholar]
- Shigeta T, Aoyama M, Bando YK, Monji A, Mitsui T, Takatsu M et al. (2012). Dipeptidyl peptidase‐4 modulates left ventricular dysfunction in chronic heart failure via angiogenesis‐dependent and ‐independent actions. Circulation 126: 1838–1851. [DOI] [PubMed] [Google Scholar]
- Shih CM, Chen YH, Lin YW, Tsao NW, Wu SC, Kao YT et al. (2014). MK‐0626, a dipeptidyl peptidase‐4 inhibitor, improves neovascularization by increasing both the number of circulating endothelial progenitor cells and endothelial nitric oxide synthetase expression. Curr Med Chem 21: 2012–2022. [DOI] [PubMed] [Google Scholar]
- Shiraishi D, Fujiwara Y, Komohara Y, Mizuta H, Takeya M (2012). Glucagon‐like peptide‐1 (GLP‐1) induces M2 polarization of human macrophages via STAT3 activation. Biochem Biophys Res Commun 425: 304–308. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic V, Ardaillou N, Vlahovic P, Placier S, Ronco P, Ardaillou R (1993). Interferon‐γ induces dipeptidylpeptidase IV expression in human glomerular epithelial cells. Immunology 80: 465–470. [PMC free article] [PubMed] [Google Scholar]
- Szelényi Z, Fazakas Á, Szénási G, Kiss M, Tegze N, Fekete BC et al. (2015). Inflammation and oxidative stress caused by nitric oxide synthase uncoupling might lead to left ventricular diastolic and systolic dysfunction in patients with hypertension. J Geriatr Cardiol 12: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ta NN, Schuyler CA, Li Y, Lopes‐Virella MF, Huang Y (2011). DPP‐4 (CD26) inhibitor alogliptin inhibits atherosclerosis in diabetic apolipoprotein E‐deficient mice. J Cardiovasc Pharmacol 58: 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Asakura M, Ito S, Min KD, Shindo K, Yan Y et al. (2013). Dipeptidyl‐peptidase IV inhibition improves pathophysiology of heart failure and increases survival rate in pressure‐overloaded mice. Am J Physiol Heart Circ Physiol 304: H1361–H1369. [DOI] [PubMed] [Google Scholar]
- Takimoto E (2012). Cyclic GMP‐dependent signaling in cardiac myocytes. Circ J 76: 1819–1825. [DOI] [PubMed] [Google Scholar]
- Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B et al. (2005). Oxidant stress from nitric oxide synthase‐3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest 115: 1221–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo LY, Chan LL, Lam CS (2016). Heart failure with preserved ejection fraction in hypertension. Curr Opin Cardiol 31: 410–416. [DOI] [PubMed] [Google Scholar]
- Valen G, Yan ZQ, Hansson GK (2001). Nuclear factor kappa‐B and the heart. J Am Coll Cardiol 38: 307–314. [DOI] [PubMed] [Google Scholar]
- van Heerebeek L, Borbely A, Niessen HWM, Bronzwaer JG, van der Velden J, Stienen GJ et al. (2006). Myocardial structure and function differ in systolic and diastolic heart failure. Circulation 113: 1966–1973. [DOI] [PubMed] [Google Scholar]
- van Heerebeek L, Hamdani N, Falcao‐Pires I, Leite‐Moreira AF, Begieneman MPV, Bronzwaer JGF et al. (2012). Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 126: 830–839. [DOI] [PubMed] [Google Scholar]
- Vittone F, Liberman A, Vasic D, Ostertag R, Esser M, Walcher D et al. (2012). Sitagliptin reduces plaque macrophage content and stabilises arteriosclerotic lesions in Apoe (−/−) mice. Diabetologia 55: 2267–2275. [DOI] [PubMed] [Google Scholar]
- Wang KL, Liu CJ, Chao TF, Huang CM, Wu CH, Chen SJ et al. (2014). Sitagliptin and the risk of hospitalization for heart failure: a population‐based study. Int J Cardiol 177: 86–90. [DOI] [PubMed] [Google Scholar]
- Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F et al. (2011). Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail 4: 44–52. [DOI] [PubMed] [Google Scholar]
- Yamabe T, Takakura K, Sugie K, Kitaoka Y, Takeda S, Okubo Y et al. (1997). Induction of the 2B9 antigen/dipeptidyl peptidase IV/CD26 on human natural killer cells by IL‐2, IL‐12 or IL‐15. Immunology 91: 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto E, Kataoka K, Shintaku H, Yamashita T, Tokutomi Y, Dong YF et al. (2007). Novel mechanism and role of angiotensin II induced vascular endothelial injury in hypertensive diastolic heart failure. Arterioscler Thromb Vasc Biol 27: 2569–2575. [DOI] [PubMed] [Google Scholar]
- Yancy CW, Lopatin M, Stevenson LW, De Marco T, Fonarow GC, ADHERE Scientific Advisory Committee and Investigators (2006). Clinical presentation, management, and in‐hospital outcomes of patients admitted with acute decompensated heart failure with preserved systolic function: a report from the acute decompensated heart failure national registry (ADHERE) database. J Am Coll Cardiol 47: 76–84. [DOI] [PubMed] [Google Scholar]
- Zannad F, Cannon CP, Cushman WC, Bakris GL, Menon V, Perez AT et al. (2015). Heart failure and mortality outcomes on alogliptin versus placebo from the EXAMINE trial. Lancet 385: 2067–2076. [DOI] [PubMed] [Google Scholar]
- Zerilli T, Pyon EY (2007). Sitagliptin phosphate: a DPP‐4 inhibitor for the treatment of type 2 diabetes mellitus. Clin Ther 29: 2614–2634. [DOI] [PubMed] [Google Scholar]
- Zhong J, Maiseyeu A, Davis SN, Rajagopalan S (2015). DPP4 in cardiometabolic disease: recent insights from the laboratory and clinical trials of DPP4 inhibition. Circ Res 116: 1491–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Rajagopalan S (2015). Dipeptidyl peptidase‐4 regulation of SDF‐1/CXCR4 axis: implications for cardiovascular disease. Front Immunol 6: 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Rao X, Rajagopalan S (2013). An emerging role of dipeptidyl peptidase 4 (DPP4) beyond glucose control: potential implications in cardiovascular disease. Atherosclerosis 226: 305–314. [DOI] [PubMed] [Google Scholar]
- Zhu X, Gillespie DG, Jackson EK (2015). NPY1‐36 and PYY1‐36 activate cardiac fibroblasts: an effect enhanced by genetic hypertension and inhibition of dipeptidyl peptidase 4. Am J Physiol Heart Circ Physiol 309: H1528–H1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zicha J, Dobešová Z, Vokurková M, Rauchová H, Hojná S, Kadlecová M et al. (2012). Age‐dependent salt hypertension in Dahl rats: fifty years of research. Physiol Res 61: S35–S87. [DOI] [PubMed] [Google Scholar]