

Abstract
T‐cell‐mediated immunity has been linked not only to a variety of heart diseases, including classic inflammatory diseases such as myocarditis and post‐myocardial infarction (Dressler's) syndrome, but also to conditions without an obvious inflammatory component such as idiopathic dilated cardiomyopathy and hypertensive cardiomyopathy. It has been recently proposed that in all these conditions, the heart becomes the focus of T‐cell‐mediated autoimmune inflammation following ischaemic or infectious injury. For example, in acute myocarditis, an inflammatory disease of heart muscle, T‐cell responses are thought to arise as a consequence of a viral infection. In a number of patients, persistent T‐cell‐mediated responses in acute viral myocarditis can lead to autoimmunity and chronic cardiac inflammation resulting in dilated cardiomyopathy. In spite of the major progress made in understanding the mechanisms of pathogenic T‐cell responses, effective and safe therapeutic targeting of the immune system in chronic inflammatory diseases of the heart has not yet been developed due to the lack of specific diagnostic and prognostic biomarkers at an early stage. This has also prevented the identification of targets for patient‐tailored immunomodulatory therapies that are both disease‐ and organ‐selective. In this review, we discuss current knowledge of the development and functional characteristics of pathogenic T‐cell‐mediated immune responses in the heart, and, in particular, in myocarditis, as well as recent advances in experimental models which have the potential to translate into heart‐selective immunomodulation.
Linked Articles
This article is part of a themed section on Targeting Inflammation to Reduce Cardiovascular Disease Risk. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.22/issuetoc and http://onlinelibrary.wiley.com/doi/10.1111/bcp.v82.4/issuetoc
Abbreviations
- Ang
angiotensin
- CS
cardiac sarcoidosis
- DC
dendritic cell
- DCM
dilated cardiomyopathy
- EMB
endomyocardial biopsy
- GCM
giant cell myocarditis
- MI
myocardial infarction
- mTOR
mammalian target of rapamycin
- PRR
pattern recognition receptor
- SCD
sudden cardiac death
- TLR
toll‐like receptor
- Treg
T regulatory cell
- TRIF
TIR‐domain‐containing adapter‐inducing interferon‐β
Tables of Links
| TARGETS | |
|---|---|
| Enzymes a | Catalytic Receptors c |
| mTOR | TLR 2 |
| GPCRs b | TLR 3 |
| AT1 receptor | TLR 4 |
| CCR4 | TLR 7 |
| CXCR3 | TLR 8 |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a,b,c).
Introduction
Inflammatory processes underlie many diseases associated with injury of the heart muscle, which in the long term can cause structural and functional deficits and defects. T‐cell‐mediated immunity has been linked not only to a variety of heart diseases, including classic inflammatory diseases of the heart, such as myocarditis and post‐myocardial infarction (MI) (Dressler's) syndrome, but also to conditions without an obvious inflammatory pathogenic component such as hypertensive and diabetic cardiomyopathy. Each of these pathological states has its own unique features in aetiology, pathogenesis and disease evolution, but they are also characterized by shared inflammatory mechanisms, and, in particular, a pathogenic role of T‐cell responses. T‐cell activation in these conditions results from the interaction of an external environmental trigger (viral infection in myocarditis) or an endogenous stimulus (mechanical or oxidative stress in hypertension and diabetes) with the host's immune system. Persistence of the pro‐inflammatory stimulus or the development of autoimmunity leads to chronic myocardial inflammation, which causes continuous remodelling and matrix turnover leading to pump dysfunction. In this context, the balance of immune response by the host is a major determinant of patient outcome. While the immune response is activated to eliminate as many virus‐infected cells as possible to control the infection, it needs to be modulated and turned off when appropriate. Ineffective immune homeostasis leads to the progression of inflammatory response, resulting in organ dysfunction.
A major clinical challenge in the management of these conditions is the identification of patients in which the immune response has not been turned off. For this reason, therapeutic targeting of the immune response by conventional immunosuppressive agents remains controversial.
In this review, we summarize the immunological checkpoints in the pathogenesis of myocardial inflammation in different conditions, with a focus on myocarditis as a paradigm with a well‐established immunological basis and which remains one of the major clinical challenges in current cardiology. Further, we will discuss potential alternatives to conventional immunosuppression arisen from studies of heart allograft rejection treatment, which, as a consequence of the reduced side effects, might provide a viable option for the management of cardiac inflammation in other conditions. Finally, based on recent findings from our group, we will put forward an alternative strategy to monitor and manipulate heart‐specific T‐cell responses.
Myocarditis: aetiology, pathogenesis and diagnosis
Aetiology
Myocarditis, an inflammatory disease of heart muscle, is a common cause of heart failure in young patients (Andrews et al., 2008). Although cardiotropic viruses are considered to be the leading aetiological agents of myocarditis in North America and Europe (Cooper, 2009), the cause of disease in most patients is unknown (Stewart et al., 2011). Dilated cardiomyopathy (DCM) with chronic congestive heart failure is the major long‐term sequelae of myocarditis in a minority of patients (Cooper, 2009). When the underlying aetiology is discovered, it can be classified into infective or non‐infective causes (Table 1).
Table 1.
Causes of myocarditis
| Idiopathic | ||
|---|---|---|
| Infective | Viral | Coxsackie virus A/B, adenovirus, CMV, echovirus, influenza, polio, hepatitis, HIV, enterovirus, parvovirus B19 |
| Bacterial | Streptococcal, Diphtheria, Spirochetal (Lyme disease, leptospirosis) | |
| Parasitic | Trypanosoma cruzi, Toxoplasma gondii | |
| Fungal | – | |
| Non‐infective | Toxic | Amphetamines, cocaine, ethanol |
| Drugs | Hypersensitivity reactions (methyldopa, penicillin, sulphonamides) | |
| Radiation | – | |
| Autoimmune | Autoactivated T cells and organ specific antibodies | |
(CMV, cytomegalovirus; HIV, human immunodeficiency virus)
Incidence and prognosis
The relative infrequency, heterogeneity and the limitations of current diagnostic strategies make it difficult to diagnose and therefore estimate the incidence of the various presentations of myocarditis. One Scandinavian study looking at autopsy reports from 10 years and applying the Dallas criteria, suggested the incidence is 1.1% in this setting (Gravanis and Sternby, 1991). The most recent Global Burden of Disease Study has suggested a global age‐standardized rate of myocarditis of 21.8 cases per 100 000 patients in 2013 – a reduction of 4% versus 1990 (Global Burden of Disease Study, 2015).
There are few long‐term studies of myocarditis; however, the few studies available suggest that DCM develops in 10 to 20% of patients following an acute illness (Abe et al., 2013; Rose, 2014). Myocarditis is also an important cause of sudden cardiac death (SCD), accounting for between 3 and 15% of SCD cases (Virmani et al., 2001; Fragkouli and Vougiouklakis, 2010; Choi et al., 2013; Suarez‐Mier et al., 2013; Wang et al., 2013). The prevalence of myocarditis as a cause of SCD in children appears to be higher than in adults. A study in Canada reviewed almost 5000 autopsies of 0 to 17 year olds and found myocarditis as a cause of SCD in 35% (Ilina et al., 2011). The prognosis of myocarditis can vary according to underlying aetiology, symptoms and ventricular dysfunction, but it is poorly described.
Pathogenesis: initial innate immune response
Viral infection of the heart is recognized by pattern recognition receptors (PRRs), an example of a PRR is the transmembrane or intracellular toll‐like receptor (TLR), expressed by antigen‐presenting cells such as the dendritic cells (DCs). TLRs recognize simple molecules and patterns of molecular structure known as pathogen‐associated molecular patterns that are part of micro‐organisms but not of self. Both infectious and non‐infectious causes of myocarditis can also trigger TLR signalling through signals of tissue damage – damage‐associated molecular patterns.
Murine and human studies of myocarditis have shown a role for TLR 2, 3, 4, 7 and 8 in the development of myocarditis in infectious and non‐infectious models of myocarditis (Triantafilou et al., 2005; Negishi et al., 2008). Although TLR expression in mice and humans is not completely overlapping (Bryant and Monie, 2012) and the downstream effects of TLR activation are particular to each TLR, common to all is a pro‐inflammatory response. For example, following TLR2 stimulation by damaged self‐protein (cardiac myosin – used to model experimental autoimmune myocarditis), monocytes have been shown to produce pro‐inflammatory cytokines such as IL‐6, IL‐8 and TNF‐α (Zhang et al., 2009). Engagement of specific TLRs and activation of their downstream mediators can influence the outcome of the myocardial inflammation by activating either protective or deleterious immune responses. Signalling of the adapter protein Myd88 downstream of TLR4 leads to decreased survival in a mouse model of Coxsackie virus B3 (CVB3)‐myocarditis through activation of NF‐κB, while the adapter protein TRIF is important for survival through up‐regulation of the antiviral IFN‐β response (Riad et al., 2011, Fuse et al., 2005). The CVB3 myocarditis model involves intraperitoneal infection with CVB3 – this model can result in two phases of infection, including the late (chronic) stage in some strains of mice – similar to myocarditis infection in humans.
Viral infection of heart muscle also elicits the activation of NK cells – a form of innate lymphoid cell that circulates in the blood responding to viral and intracellular infections. Animal models of viral myocarditis have shown a protective role of NK cells in inhibiting viral replication and eosinophil infiltration into the myocardium – limiting cardiac inflammation and fibrosis (Godeny and Gauntt, 1986; Ong et al., 2015).
Pathogenesis: adaptive immune response
The animal study by Woodruff et al in 1974 provided an insight into the role of the adaptive immune system, and specifically the T‐cell response, in the mouse viral myocarditis model (Woodruff and Woodruff, 1974). Coxsackie viral replication was shown to be identical in mice with or without (thymectomized mice) T‐cell responses during the first week of infection. Administration of anti‐thymocyte serum to those mice with a thymus who had survived the first week of the study then led to reduced inflammation and tissue injury in infected hearts. T‐cell deprivation also protected thymectomized mice from a lethal infection. The authors suggest that the viral replication is halted during the first week of infection by the innate immune system followed by a predominantly β cell response. The subsequent T‐cell response, from 8 to 14 days post‐infection, has then been shown to be associated with myocyte injury.
Whereas both direct injuries due to the infection and host immune responses can cause temporary cardiac dysfunction, strong evidence suggests that T‐cell‐mediated autoimmunity is the mechanism driving events from viral infection to chronic inflammation (Liu et al., 2012; Liu et al., 2013). A large number of studies have characterized the immune response in different murine models of autoimmune myocarditis. For example, cardiac myosin (Myhca)‐induced murine autoimmune myocarditis is considered an accurate model of the pathogenic mechanisms underlying the human condition (Rose, 2014). Myhca is the major autoantigen associated with autoimmune myocarditis both in humans and in mice (Pummerer et al., 1996; Mascaro‐Blanco et al., 2008). All of the mouse strains susceptible to myocarditis following CVB3 infection develop disease approximating to post‐viral myocarditis, following immunization with Myhca (Neu et al., 1987). In contrast, C57/BL10 and C57/BL6 mice, which are resistant to post‐infection chronic myocarditis, fail to develop disease following immunization with purified mouse cardiac myosin. Although the pathogenic events that lead to the development of spontaneous autoimmunity against Myhca in humans are not reproducible in these models, in which active immunization against myosin is required, experimental myocarditis studies have provided important clues on the effector mechanisms of tissue damage.
Studies with knockout mice and adoptive transfer experiments have indicated an instrumental role for CD4+ T cells in the development of myocarditis in this model, similar to human myocarditis (Afanasyeva et al., 2004). In a landmark study of an experimental model of CVB3‐myocarditis, CD4+ deletion led to amelioration of myocarditis whereas CD8+ deletion alone had no effect on the myocardial inflammation; deletion of both CD4+ and CD8+ had the strongest protective effects (Opavsky et al., 1999). These results underline the importance of CD4+ helper T cells in the pathogenesis of myocarditis. Immune responses in myocarditis have been associated with the production of T‐cell cytokines, which, in experimental models, can dictate the course of disease progression and its outcome (Afanasyeva et al., 2001).
There are different ILs involved in the varying forms of myocarditis – for example IL‐12 (an inducer in Th1 responses), IL‐4 (a Th2 inducer) and IL‐23 (this leads to a sustained Th17 response). Although IL‐4 has been linked with more severe myocarditis, another cytokine that is part of the Th2 response (IL‐13) has been associated with diminishing disease (Rose, 2011). Further work from 2010 suggested an important role for IL‐17 (produced by Th17 cells) in the progression to DCM in an experimental autoimmune model of myocarditis in mice, promoting a fibrotic response (Baldeviano et al., 2010).
Another T‐cell subset – the CD4 + CD25 + FoxP3 + T regulatory (Treg) cells (Fontenot et al., 2003) – is increasingly being recognized as crucial to the resolution or progression of myocarditis. Having a higher proportion of Treg cells has been shown to be protective against both viral and autoimmune myocarditis in mouse models. In particular, in autoimmune myocarditis, a lower proportion of Treg cells has been shown to be associated with a greater Th17 response and more severe autoimmune myocarditis (Chen et al., 2012). Prophylactic administration of naive Treg cells in a mouse model of CVB3 myocarditis led to decreased myocardial inflammation and fibrosis mainly through the TGF‐β‐Coxsackie‐adenovirus receptor pathway (Shi et al., 2010).
Patients with idiopathic DCM appear to have a reduced ratio of Treg /Th17 cells in the blood (Li et al., 2010). We also found an imbalance between CD3+ and FoxP3 expression in the endomyocardial biopsy (EMB) from patients with DCM and chronic myocardial inflammation (Savvatis et al., unpublished observations). Furthermore, effector T cells seem to acquire a resistance to the suppressive effects of Treg cells in patients with DCM (Tang et al., 2010). Thus, following acute myocarditis, development of chronic myocardial inflammation may result from an imbalance between pro‐ and anti‐inflammatory T‐cell types (Figure 1).
Figure 1.
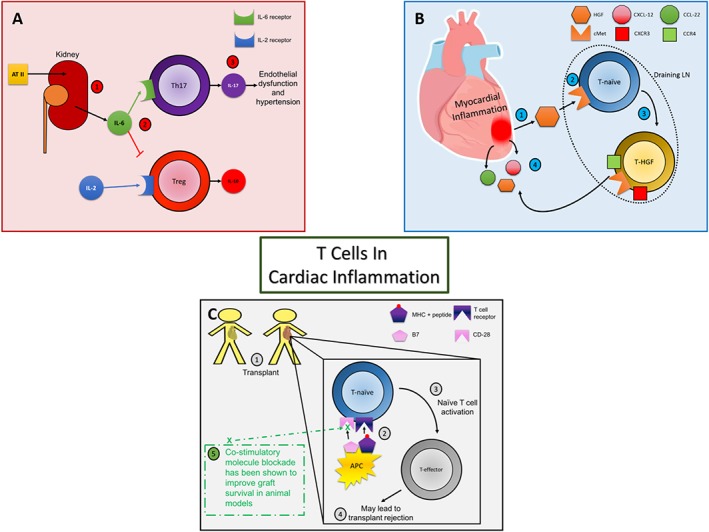
Panel A: Hypertension: (1) Murine models have demonstrated renal production of IL‐6 following exposure to Ang II (AT II). (2) IL‐6 is associated with an enhanced CD4+ Th17 subtype response as well as diminishing the differentiation of T cells towards Treg cells. (3) IL‐17, from Th17 cells, has been shown to contribute to endothelial dysfunction, hypertension as well as deleterious cardiac remodelling in mouse models. Panel B: Myocardial inflammation: (1) HGF has been shown to be present following myocardial inflammation in a mouse model. (2) This can then act on the HGF receptor, c‐Met, on naïve T cells in draining LNs. (3) The HGF–cMet interaction on naïve T cells during priming has then been shown to instruct T cell cardiotropism (4). Panel C: Transplant models: Following transplantation (1), antigen presenting cells (APC) can then activate naïve T cells (2) via both the MHC‐T cell receptor interaction as well as the co‐stimulatory actions of B7 on the APC with CD‐28 on the T cell surface. Naïve T cell activation (3) can then lead to effector T cells that may mediate transplant rejection (4). As described in the text, blockade of the CD‐28 receptor improved graft survival in animal models (5).
Pathogenesis: immunological role of gender differences in myocarditis
Another finding from animal studies was that hearts from male mice showed greater inflammation and necrosis following viral infection than those from female mice (Corsten et al., 2012). Along with this difference in animals, male humans are also more likely to be affected by myocarditis. One study reviewed almost 10 years of hospital admissions and found that for every four patients diagnosed with myocarditis, three are male (Kyto et al., 2013). A mechanism underlying this difference was suggested by work from Huber et al in 1990s, suggesting that the reaction of male mice to Coxsackie virus is dominated by CD4+ Th1 cells (leading to increased IFN‐γ and IL‐2), whereas in female mice, it is driven by Th2 cells (IL‐4 and IL‐5 producing cells) (Huber and Pfaeffle, 1994). If the male mice are treated with oestradiol or the female mice with testosterone before infection, this alters the Th cell differentiation. Treatment of female mice with anti‐IL‐4 antibody led to increased susceptibility to myocarditis and greater mortality. In this subgroup, IL‐4 precursors were significantly reduced, as expected; IFN‐γ precursors were increased, suggesting that the Th1 response is suppressed by a Th2 response.
Delving deeper into the causes for these sex differences, more recent studies suggest a role for TLRs. Roberts et al. have demonstrated that both male and female mice show an increased Th1 response following exposure to TLR 2 and four ligands (Roberts et al., 2013). Notably, treatment with the TLR 2 ligand (lipopolysaccharide) resulted in a decreased percentage of Treg cells in male mice; the same treatment did not affect the percentage of Treg cells in females. Whether a similar sex‐related difference can be found in humans is not clear.
Diagnosis
Due to the heterogeneity of symptoms and signs and the absence of reliable and powerful diagnostic tools, myocarditis is a challenging diagnosis (Stewart et al., 2011). It often mimics other common cardiac diseases including acute coronary syndrome, representing the majority of patients with acute cardiac symptoms associated with biochemical evidence of cardiac damage without angiographic evidence of a plaque (Monney et al., 2011). Regardless of the clinical presentation, confirming the absence of angiographically detectable obstructive coronary disease is often an essential step in the diagnostic work‐up. EMB, interpreted according to the Dallas histopathological criteria of 1986 (based on demonstrating inflammatory infiltrate and myocyte necrosis), has been seen as the diagnostic gold standard. The use of more modern techniques such as immunohistochemistry, and PCR can help to improve the sensitivity of EMB and differentiate between causes and types of myocarditis. For example, on EMB in giant cell myocarditis (GCM), there are significantly more eosinophils and necrosis, whereas in cardiac sarcoidosis (CS), there are more signs of fibrosis and granulomas seen. If EMB is able to exclude infective causes of myocarditis (negative viral PCR on EMB), then this can be used as evidence to consider the use of immunosuppressive therapy.
Contribution of immune inflammation to other cardiovascular diseases
Hypertension, cardiac hypertrophy and remodelling
Globally, hypertension is one of the most prevalent chronic diseases, and is associated with atherosclerosis and its complications as well as heart failure. Increasing evidence suggests that immunological mechanisms are involved in mediating the complications of hypertension, including cardiomyopathy (Nataraj et al., 1999; Muller et al., 2002; Crowley et al., 2010; Madhur et al., 2010). For example, macrophage and lymphocyte interstitial infiltration is a key feature of angiotensin (Ang) II‐induced hypertension (Muller et al., 2002), where T cells increase in the adventitia of blood vessels and secrete cytokines. These include TNF‐α and IL‐17, which promote atherosclerosis, as well as NADPH oxidase (Guzik et al., 2007; Madhur et al., 2010), which induces high blood pressure. In line with these observations, immunosuppression reduces hypertension‐induced end‐organ damage while immunodeficiency decreases high blood pressure (Nataraj et al., 1999; Crowley et al., 2010).
A number of studies have investigated the pathogenesis of inflammation in hypertension. Chronic hypertension has been shown to activate DCs (Abbas et al., 2015), the initiators of adaptive immunity. Reactive oxygen species (ROS) produced by DCs lead to the accumulation of proteins that were oxidatively modified by highly reactive γ‐ketoaldehydes (isoketals) (Kirabo et al., 2014). These isoketal‐modified proteins activate DCs and are presented to autoreactive T cells inducing their activation, particularly of CD8+ cells and IFN‐γ and IL‐17A, with the latter known to elevate blood pressure (Nguyen et al., 2013). Plasma F2‐isoprostanes, which are formed in concert with isoketals, were found to be elevated in humans with treated hypertension and were markedly raised in patients with resistant hypertension. Isoketal‐modified proteins were also markedly elevated in circulating monocytes and DCs from humans with hypertension (Kirabo et al., 2014).
Remodelling of the myocardium is a pathological feature of many cardiac diseases including hypertension, cardiomyopathy, valvular dysfunction and MI. Ventricular hypertrophy in these conditions occurs in response to chronic pathological stimuli such as blood pressure and volume overload, sarcomere gene mutations and neurohumoral activation (Frieler and Mortensen, 2015). The major consequence of prolonged hypertrophic remodelling is cardiac dysfunction leading to heart failure or cardiac arrest due to arrhythmia. Although cardiac remodelling is initiated by a variety of pathological stimuli, the hypertrophic response presents shared features regardless of the causative mechanism, including increased cardiomyocyte mass, sarcomere rearrangement and extracellular matrix deposition.
This complex pathological process is also characterized by inflammation and immune cell activation, involving a number of inflammatory cell types, including DCs, macrophages and T lymphocytes (Frieler and Mortensen, 2015). There also appears to be a role of Treg cells in hypertension‐induced cardiac remodelling. In an Ang II‐infusion model of hypertension in mice, adoptive transfer of Treg cells resulted in marked reduction in CD4+ and CD8+ T cells as well as macrophage infiltration (Kvakan et al., 2009). This was associated with a reduction in cardiac hypertrophy and electrical remodelling of the heart despite continuing hypertension.
Although it is known that many of the immune cells involved can be activated by the same pathological stimuli inducing hypertrophy, the molecular interactions involved in the crosstalk between immune cells and cardiomyocytes during homeostasis and in pathology remain largely unknown. In particular, while it is likely that inflammatory cytokines such as TNF‐α, IL‐1β, IL‐6 and TGF‐β, and neurohumoral factors like Ang II and aldosterone are likely mediators of the crosstalk during the pathogenic process and correlate with disease progression (Muller et al., 2002), their effect on target cells including cardiomyocytes has not been investigated.
Myocardial infarction
Remodelling of myocardial tissue following MI can progressively impair its contractile function, eventually leading to heart failure (Hofmann and Frantz, 2015). Inflammation and adaptive immunity play a major role in the pathogenesis of cardiac remodelling after MI (Anzai et al., 2012). Following MI, autoreactive T cells can be activated by presentation of myocardial peptides by DCs (Hofmann et al., 2012). Hypoxia consequent to ischaemia is a powerful activator of innate immunity and DCs in particular, with the potential to initiate autoimmune responses. Homeostatic mechanisms are in place that can prevent the establishment of autoimmune inflammation, including the presence of Treg cells (Hofmann et al., 2012) and anti‐inflammatory macrophages (Anzai et al., 2012). The failure of these protective mechanisms to control the autoimmune response, with progression to heart failure in some individuals, may be signalled by the presence of autoantibodies against myosin heavy chain, troponins and β1‐adrenoreceptors in patients with DCM or heart failure with a MI history (Kaya et al., 2012). Further information on the role of T cells in MI can be found from a recent excellent review (Hofmann and Frantz, 2016). Examples of immunological and inflammatory processes in myocardial diseases, including those mentioned above, are shown in Table 2.
Table 2.
Myocardial diseases with examples of associated immunological and inflammatory processes
| Inflammatory myocardial disease | Immunopathological factors | References |
|---|---|---|
| Ischaemia‐reperfusion injury | • CD4+ T cells: rapid accumulation of CD4+ T cells soon following reperfusion are associated with build‐up of neutrophils and the release of pro‐inflammatory IFN‐γ | (Yang et al., 2006) |
| • Mast cells: mast cells and their ensuing inflammatory response contribute to IR injury as measured by serum IL‐6 levels | (Bhattacharya et al., 2007) | |
| • TLRs: there is also a role for these receptors in IR injury, for example in mice with non‐functional TLR4 there is a 40% reduction in infarct size | (Nagao et al., 1992) | |
| Myocarditis | • Viral causes: the causative virus has evolved from enteroviral myocarditis to non‐enteroviral myocarditis (for example, increasingly involving Parvovirus, Epstein–Barr Virus and Hepatitis C Virus) | (Schultz et al., 2009) |
| • IL‐17 A: there appears to be a critical role of interleukin‐17 A in the progression to dilated cardiomyopathy | (Baldeviano et al., 2010) | |
| • Th subtypes: higher percentage of CD4+ T cells and their differentiation to a Th17 phenotype contributes to increased susceptibility to murine autoimmune myocarditis | (Chen et al., 2012) | |
| Cardiac allograft rejection | • Blockade of CD40 and CD28 stimulation: Co‐blockade decreases the risk of heart allograft ejection through modifications to T cell regulatory mechanisms | (Guillonneau et al., 2007) |
| • CCR4 receptor: critical role in NK T cell recruitment to cardiac allografts which mediates chronic graft rejection | (Huser et al., 2005) | |
| • CXCL10 receptor: plays a pivotal role in the initiation and development of alloresponses leading to acute transplant rejection | (Hancock et al., 2001) | |
| Sepsis‐induced cardiac dysfunction | • Cell adhesion molecules: Macrophages adhere to cardiomyocytes via ICAM‐1 to cause myocardial dysfunction via reactive oxygen species and nitric oxide | (Simms and Walley, 1999) |
| • TLRs: TLR 2, 4 and 5 are expressed in cardiac muscle and play a pivotal role on cardiac contractility in response to pro‐inflammatory molecules | (Boyd et al., 2006) | |
| • NF‐ĸB: a pleiotropic transcription factor involved in downstream TLR signalling which is implicated with pro‐apoptotic signalling in the heart in relation to sepsis | (Brown and Jones, 2004) | |
| Hypertension | • Lymphocytes: lymphocyte activation contributes to sodium retention through regulation of eNOS and COX‐2 in the kidney leading to hypertensive cardiac and renal injury | (Crowley et al., 2010) |
| • IL‐17: binding of IL‐17 to its endothelial cell receptor with downstream eNOS phosphorylation contributes to endothelial dysfunction and hypertension | (Nguyen et al., 2013) | |
| • Isoketals: Highly reactive lipid‐derived mediators that modify dendritic cell proteins inducing CD8 T cell activation as well as IL‐17 A – known to elevate blood pressure | (Kirabo et al., 2014) | |
| Cardiac hypertrophy and remodelling | • Macrophages: a lack of macrophages as well as abundant inflammatory cell infiltrate within the myocardium is associated with earlier development of myocardial dysfunction in hypertensive rats | (Zandbergen et al., 2009) |
| • IL‐4: exerts pro‐fibrotic effects via increasing the numbers and augmenting the response of myofibroblasts leading to the progression of cardiac fibrosis | (Kanellakis et al., 2012) | |
| • Angiotensin II: induces vascular dysfunction, oxidative stress and hypertension – requiring pro‐inflammatory monocytes for these effects | (Wenzel et al., 2011) |
IR, ischaemia‐reprefusion injury; NK T cell, natural killer T cell; ICAM‐1, intercellular adhesion molecule‐1; eNOS, endothelial nitric oxide synthase
Challenges in the therapeutic modulation of adaptive immunity in the heart: the myocarditis ‘paradigm’
Despite remarkable advances in understanding of the pathophysiological mechanisms of myocarditis, no treatment strategies aimed at eradicating the cause and progression of the inflammatory process are available to date.
The acute management of myocarditis is largely dependent on the severity of the illness at presentation. In severe cases, admission to specialist centres of intensive care for pharmacological or mechanical cardiovascular support or heart transplantation may become necessary. Patients with less severe illness can also be admitted to hospital for management with diuretics, ACE‐inhibitors and β‐blockers. Several animal studies of experimental myocarditis suggest that treatment with ACE‐inhibitors and β‐blockers in the short term reduces left ventricular (LV) mass and myocyte necrosis and improves survival. Antiviral therapy is indicated when evidence of viral infection is available. Treatment with IFN‐α in patients with myocardial enteroviral or adenoviral persistence and LV dysfunction showed an elimination of viral genomes in all patients and an improvement of LV function in clinical trials (Kuhl et al., 2003).
Immunosuppressive treatments should only be instigated after infective causes of acute myocarditis are ruled out by EMB. Treatment with immunosuppressive agents (cyclosporine, prednisolone and azathioprine) in acute myocarditis is controversial, although recent evidence suggests that immunotherapy can be effective for chronic, virus‐negative inflammatory cardiomyopathy. Immunomodulatory treatments, such as intravenous immunoglobulin, prednisone and azathioprine, have led to improvement in left ventricular function in a small number of patients with acute fulminant myocarditis, as well as in clinical trials in chronic heart failure of many causes (Gullestad et al., 2001; Yu et al., 2014). In GCM, immunosuppression by drugs such as cyclosporine and azathioprine has been shown to lead to improved outcomes (Kandolin et al., 2013). In CS, corticosteroid use is often suggested, although a recent systematic review found little evidence for this despite a suggestion that atrioventricular conduction is improved – also reported in many case reports (Nery et al., 2012).
Manipulating the immune response in heart inflammation: lessons from transplantation
The recent advances in heart transplantation are largely based on the understanding of T‐cell‐mediated acute rejection and the development of immunosuppressive drugs targeting crucial signalling pathways of T‐cell activation. These advances might provide viable alternatives for the management of cardiac inflammation.
One of the characteristics of the adaptive immune system is its ability to recognize a vast number of different antigens. This ability is a consequence of the large lymphocyte repertoire, in which each cell has a different antigen receptor generated by the process of somatic recombination. Since the somatic recombination is a random process, it generates T‐cell clones that can recognize self‐structures or self‐peptides (autoantigens). The mechanism used by the immune system in order to avoid a possible harmful immune response against an individual's own cells and tissues is known as immune tolerance and can be classified into central and peripheral tolerance. Immune tolerance in transplantation is defined as a specific absence of a destructive immune response – mediated by T cells that recognize alloantigens – to a transplanted tissue without immunosuppression. Operative criteria are the complete withdrawal of immunosuppression followed by no evidence for rejection for the transplanted organ for over 1 year. In humans, this is characterized by specific in vitro non‐responsiveness to the donor.
The mammalian target of rapamycin (mTOR) is a regulator protein playing an important role in growth factor‐driven proliferation of T lymphocytes, and in transplantation models, mTOR inhibition has been shown to promote the development of immunological tolerance. Recently, a combination of a reduced cyclosporine dose with mTOR inhibitors (sirolimus or everolimus) has allowed the effective prevention of acute cellular rejection, while reducing the severity of side‐effects including renal dysfunction (Kushwaha et al., 2005) and malignancies (Kauffman et al., 2005).
CD28 is an important co‐stimulatory molecule involved in T‐cell proliferation at the time of antigen recognition. This suggests that inhibition of CD28/B7 T‐cell co‐stimulatory receptors may present a highly specific way of immunosuppression (Adams et al., 2001). In animal models, inhibition of CD28 results in reduced T‐cell proliferation and prolonged graft survival. Belatacept is a humanized immunoglobulin detecting a human homologue of CD28 and has been approved by the US Food and Drug Administration for use in renal transplantation based on its non‐inferiority to standard cyclosporine combination therapy (Archdeacon et al., 2012). More recently, Treg cells were shown to suppress immune response to foreign antigens in the context of transplantation (Wood and Sakaguchi, 2003) and, therefore, may mediate prevention from rejection.
Recent clinical and experimental studies have shown that non‐conventional immunosuppressive therapy might be effective in controlling T‐cell‐mediated chronic myocardial inflammation. For example, the use of cannabidiol has been proposed to reduce T‐cell inflammatory response and injury in myocarditis (Lee et al., 2016). In addition, systemic application of mesenchymal stromal cells in inflammatory cardiomyopathy exerts beneficial effects (Savvatis et al., 2012). Finally, IL‐6 receptor blockade exerts cardiac beneficial effects by antiviral and immunomodulatory actions after induction of an acute murine CVB3 virus myocarditis (Savvatis et al., 2014).
Importantly, experimental models have shown that operational T‐cell tolerance can be induced in myocarditis. Specifically, genetically susceptible mice tolerized to cardiac myosin failed to develop chronic myocarditis following viral infection (Wang et al., 2000; Fousteri et al., 2011). Similarly, it has been reported that suppression of the adaptive immune system can attenuate hypertension in experimental animals and in humans (Tian et al., 2007). Activation of the Ang II receptor in T cells has been associated with the development of hypertension and end‐organ damage, an effect prevented by AT1 receptor antagonists (Kassan et al., 2013). An imbalance in Treg cell activity has been hypothesized to contribute to the immunopathogenesis of hypertension supported by the observation that infusion of Treg cells, which promote endothelium‐dependent relaxation in coronary arterioles, has a therapeutic effect in experimental models of hypertension (Kassan et al., 2013). The latter observations have led to the suggestion that replacement of Treg cells by infusion may represent a new therapeutic strategy for the treatment of cardiovascular diseases, and that these cells could represent an important and highly promising new target for modifying cardiovascular pathophysiology (Kassan et al., 2013).
It is important to carefully consider the feasibility of infusing Treg cells in clinical studies. Use of these cells requires careful separation (usually by FACS or immunomagnetic separation) and then expansion by cell culture. There have already been a number of preliminary safety and dose optimization clinical trials, mainly in the field of graft versus host disease (GvHD) (Trzonkowski et al., 2013). In the context of GvHD, the use of Treg cells is either for prophylaxis or treatment of chronic GvHD. Closer to the field of cardiac transplant, a trial of of Treg cell infusions in non‐human primate heart transplantation has recently been published (Ezzelarab et al., 2016). Interestingly, survival of the graft was longest in the control primates and shortest in those who had received multiple transfusions of Treg cells. The transfusions occurred during the early post‐transplant period (up to 30 days) and was associated with an increase in pro‐inflammatory cytokines, recipient CD8+ T cells and effector T memory cells. These findings are at odds with murine models of transplantation where infusion of Treg cells improved graft survival (Golshayan et al., 2007). The authors of the primate cardiac transplant study suggest that one reason for this discrepancy might be that the Treg cells lose their characteristic FoxP3 expression and then differentiate into Th1 and Th17 subtypes. Although the authors did not demonstrate reduced FoxP3 expression, cytokines such as IFN‐γ and IL‐6, which have been shown to destabilise Treg cells, were elevated (Kimura and Kishimoto, 2010).
Towards cardio‐selective immunosuppression
A key event in the adaptive immune response is the migration of activated T cells to antigen‐rich inflammatory sites. By possessing a unique set of adhesion molecules and chemokine receptors – the ‘homing’ receptors – distinct activated T‐cell populations are able to interact with organ‐specific endothelial cells and are recruited to distinct target tissues (Butcher and Picker, 1996; Mora and von Andrian, 2006). For example, lymphocyte trafficking to the intestinal lamina propria is mediated by the interaction between intestinal mucosal addressin cell adhesion molecule‐1 expressed by gut endothelium and lymphocyte α4β7 integrin (Berlin et al., 1993), and T‐cell migration to the skin is promoted by cutaneous lymphocyte‐associated antigen interaction with vascular E‐selectin with the involvement of chemokine–receptor pairs CCR4–CCL17, CCR10–CCL27 and CCR8–CCL1 (Sigmundsdottir and Butcher, 2008; McCully and Moser, 2011).
Besides homing to the gut and skin, it has been challenging to identify unique tissue‐homing signatures to other solid organs including the heart. The chemokine receptors CCR4 (Huser et al., 2005) and CXCR3 (Hancock et al., 2001) contribute to T‐cell accumulation during heart transplant rejection. We have recently uncovered a molecular mechanism of induction of T‐cell cardiotropism. We found that engagement of the hepatocyte growth factor (HGF) receptor c‐Met by heart‐produced HGF during priming in the lymph nodes (LNs) instructs T‐cell cardiotropism, which was associated with a specialized homing ‘signature’ (c‐Met+CCR4+CXCR3+) (Komarowska et al., 2015). T‐cell priming is the process by which mature naïve T cells are activated by their specific antigen and begin their process of differentiation. HGF is expressed by healthy heart tissue and transported to local draining LNs. Inside heart draining LNs, HGF binds to c‐Met on naive T cells, inducing higher expression of c‐Met itself and of the chemokine receptors CCR4 and CXCR3. Triggering of c‐Met was sufficient to support cardiotropic T‐cell recirculation, while CCR4 and CXCR3 sustained recruitment during heart inflammation. The expression of the chemokine receptors, CCR4 and CXCR3, can mediate T‐cell homing to other sites of inflammation as well, although co‐expression of these receptors is unusual in non‐cardiotropic T cells (Komarowska et al., 2015).
In steady state conditions, engagement of c‐Met induces autocrine release of β chemokines, which favour T‐cell recruitment via their receptor CCR5. Under inflammatory conditions, cardiac tissue releases higher levels of the HGF and chemokines CXCL10 and CCL4, which facilitate HGF‐primed T cells recruitment to the heart (Komarowska et al., 2015). Importantly, transient blockade of c‐Met with a low MW inhibitor during T‐cell priming led to substantially enhanced heart, but not skin, allograft survival associated with impaired localization of alloreactive T‐cells to the heart grafts without tolerance. A humanized in vivo model showed that HGF/cMet‐induced T‐cell cardiotropism is also functional in the human system. In addition to providing an ‘identity’ to T cells mediating immunity in the heart – a potentially valuable diagnostic marker – these findings define a novel heart‐selective T‐cell area code and provide a conceptual framework for the development of cardio‐selective immunomodulation.
Concluding remarks
Numerous studies over the years have defined the immunological pathways that determine the progression of environmental and endogenous stimuli to chronic autoimmune inflammation of the heart and will allow predicting the eventual outcome of cardiac inflammation. The main challenge remains to determine, at the earliest stages, when an immune response has exceeded the boundaries of physiological homeostatic immunoregulation and is progressing towards chronic inflammation. Recognition of patients at risk of this process before irreversible damage has occurred will be crucial to interrupt the pathological process. Novel biomarkers are needed that warn of a pathogenic deviation from immunological homeostasis. The identification of immunological parameters including the imbalance between Th17 and Treg cells, the presence of certain pro‐inflammatory cytokines and the definition of cardiotropic T cells in experimental models have the potential to become invaluable diagnostic and prognostic markers as well as therapeutic targets, once they have been validated in human pathology.
Acknowledgements
F.M.B. is recipient of the British Heart Foundation Personal Chair in Cardiovascular Immunology (CH/15/32064). E.S. is supported by a British Heart Foundation clinical research training fellowship (FS/16/18/31973).
Conflict of interest
The authors declare no conflicts of interest.
Stephenson, E. , Savvatis, K. , Mohiddin, S. A. , and Marelli‐Berg, F. M. (2017) T‐cell immunity in myocardial inflammation: pathogenic role and therapeutic manipulation. British Journal of Pharmacology, 174: 3914–3925. doi: 10.1111/bph.13613.
References
- Abbas A, Gregersen I, Holm S, Daissormont I, Bjerkeli V, Krohg‐Sorensen K et al. (2015). Interleukin 23 levels are increased in carotid atherosclerosis: possible role for the interleukin 23/interleukin 17 axis. Stroke 46: 793–799. [DOI] [PubMed] [Google Scholar]
- Abe T, Tsuda E, Miyazaki A, Ishibashi‐Ueda H, Yamada O (2013). Clinical characteristics and long‐term outcome of acute myocarditis in children. Heart Vessels 28: 632–638. [DOI] [PubMed] [Google Scholar]
- Adams AB, Pearson TC, Larsen CP (2001). Conventional immunosuppression and co‐stimulation blockade. Philos Trans R Soc Lond B Biol Sci 356: 703–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afanasyeva M, Wang Y, Kaya Z, Park S, Zilliox MJ, Schofield BH et al. (2001). Experimental autoimmune myocarditis in A/J mice is an interleukin‐4‐dependent disease with a Th2 phenotype. Am J Pathol 159: 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afanasyeva M, Georgakopoulos D, Fairweather D, Caturegli P, Kass DA, Rose NR (2004). Novel model of constrictive pericarditis associated with autoimmune heart disease in interferon‐gamma‐knockout mice. Circulation 110: 2910–2917. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews RE, Fenton MJ, Ridout DA, Burch M, British Congenital Cardiac A (2008). New‐onset heart failure due to heart muscle disease in childhood: a prospective study in the United kingdom and Ireland. Circulation 117: 79–84. [DOI] [PubMed] [Google Scholar]
- Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H et al. (2012). Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 125: 1234–1245. [DOI] [PubMed] [Google Scholar]
- Archdeacon P, Dixon C, Belen O, Albrecht R, Meyer J (2012). Summary of the US FDA approval of belatacept. Am J Transplant 12: 554–562. [DOI] [PubMed] [Google Scholar]
- Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D et al. (2010). Interleukin‐17 A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res 106: 1646–1655. [DOI] [PubMed] [Google Scholar]
- Berlin C, Berg EL, Briskin MJ, Andrew DP, Kilshaw PJ, Holzmann B et al. (1993). Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM‐1. Cell 74: 185–195. [DOI] [PubMed] [Google Scholar]
- Bhattacharya K, Farwell K, Huang M, Kempuraj D, Donelan J, Papaliodis D et al. (2007). Mast cell deficient W/Wv mice have lower serum IL‐6 and less cardiac tissue necrosis than their normal littermates following myocardial ischemia–reperfusion. Int J Immunopathol Pharmacol 20: 69–74. [DOI] [PubMed] [Google Scholar]
- Boyd JH, Mathur S, Wang Y, Bateman RM, Walley KR (2006). Toll‐like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF‐kappaB dependent inflammatory response. Cardiovasc Res 72: 384–393. [DOI] [PubMed] [Google Scholar]
- Brown MA, Jones WK (2004). NF‐kappaB action in sepsis: the innate immune system and the heart. Front Biosci 9: 1201–1217. [DOI] [PubMed] [Google Scholar]
- Bryant CE, Monie TP (2012). Mice, men and the relatives: cross‐species studies underpin innate immunity. Open biol 2: 120015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher EC, Picker LJ (1996). Lymphocyte homing and homeostasis. Science 272: 60–66. [DOI] [PubMed] [Google Scholar]
- Chen P, Baldeviano GC, Ligons DL, Talor MV, Barin JG, Rose NR et al. (2012). Susceptibility to autoimmune myocarditis is associated with intrinsic differences in CD4(+) T cells. Clin Exp Immunol 169: 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K, Pan YP, Pock M, Chang RK (2013). Active surveillance of sudden cardiac death in young athletes by periodic Internet searches. Pediatr Cardiol 34: 1816–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper LT, Jr. (2009). Myocarditis N Engl J Med 360: 1526–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsten MF, Schroen B, Heymans S (2012). Inflammation in viral myocarditis: friend or foe? Trends Mol Med 18: 426–437. [DOI] [PubMed] [Google Scholar]
- Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P (2010). Lymphocyte responses exacerbate angiotensin II‐dependent hypertension. Am J Physiol Regul Integr Comp Physiol 298: R1089–R1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzelarab MB, Zhang H, Guo H, Lu L, Zahorchak AF, Wiseman RW et al. (2016). Regulatory T Cell Infusion Can Enhance Memory T Cell and Alloantibody Responses in Lymphodepleted Nonhuman Primate Heart Allograft Recipients. Am J Transplant 16: 1999–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY (2003). Foxp3 programs the development and function of CD4 + CD25+ regulatory T cells. Nat Immunol 4: 330–336. [DOI] [PubMed] [Google Scholar]
- Fousteri G, Dave A, Morin B, Omid S, Croft M, von Herrath MG (2011). Nasal cardiac myosin peptide treatment and OX40 blockade protect mice from acute and chronic virally‐induced myocarditis. J Autoimmun 36: 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkouli K, Vougiouklakis T (2010). Sudden cardiac death: an 11‐year postmortem analysis in the region of Epirus, Greece. Pathol Res Pract 206: 690–694. [DOI] [PubMed] [Google Scholar]
- Frieler RA, Mortensen RM (2015). Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation 131: 1019–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuse K, Chan G, Liu Y, Gudgeon P, Husain M, Chen M et al (2005). Myeloid differentiation factor‐88 plays a crucial role in the pathogenesis of Coxsackievirus B3‐induced myocarditis and influences type I interferon production. Circulation 112: 2276–2285. [DOI] [PubMed] [Google Scholar]
- Global Burden of Disease Study C (2015). Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990‐2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 386: 743–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godeny EK, Gauntt CJ (1986). Involvement of natural killer cells in coxsackievirus B3‐induced murine myocarditis. J Immunol 137: 1695–1702. [PubMed] [Google Scholar]
- Golshayan D, Jiang S, Tsang J, Garin MI, Mottet C, Lechler RI (2007). In vitro‐expanded donor alloantigen‐specific CD4 + CD25+ regulatory T cells promote experimental transplantation tolerance. Blood 109: 827–835. [DOI] [PubMed] [Google Scholar]
- Gravanis MB, Sternby NH (1991). Incidence of myocarditis. A 10‐year autopsy study from Malmo, Sweden. Arch Pathol Lab Med 115: 390–392. [PubMed] [Google Scholar]
- Guillonneau C, Seveno C, Dugast AS, Li XL, Renaudin K, Haspot F et al. (2007). Anti‐CD28 antibodies modify regulatory mechanisms and reinforce tolerance in CD40Ig‐treated heart allograft recipients. J Immunol 179: 8164–8171. [DOI] [PubMed] [Google Scholar]
- Gullestad L, Aass H, Fjeld JG, Wikeby L, Andreassen AK, Ihlen H et al. (2001). Immunomodulating therapy with intravenous immunoglobulin in patients with chronic heart failure. Circulation 103: 220–225. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S et al. (2007). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock WW, Gao W, Csizmadia V, Faia KL, Shemmeri N, Luster AD (2001). Donor‐derived IP‐10 initiates development of acute allograft rejection. J Exp Med 193: 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann U, Frantz S (2015). Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ Res 116: 354–367. [DOI] [PubMed] [Google Scholar]
- Hofmann U, Frantz S (2016). Role of T‐cells in myocardial infarction. Eur Heart J 37: 873–879. [DOI] [PubMed] [Google Scholar]
- Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G et al. (2012). Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 125: 1652–1663. [DOI] [PubMed] [Google Scholar]
- Huber SA, Pfaeffle B (1994). Differential Th1 and Th2 cell responses in male and female BALB/c mice infected with coxsackievirus group B type 3. J Virol 68: 5126–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huser N, Tertilt C, Gerauer K, Maier S, Traeger T, Assfalg V et al. (2005). CCR4‐deficient mice show prolonged graft survival in a chronic cardiac transplant rejection model. Eur J Immunol 35: 128–138. [DOI] [PubMed] [Google Scholar]
- Ilina MV, Kepron CA, Taylor GP, Perrin DG, Kantor PF, Somers GR (2011). Undiagnosed heart disease leading to sudden unexpected death in childhood: a retrospective study. Pediatrics 128: e513–e520. [DOI] [PubMed] [Google Scholar]
- Kandolin R, Lehtonen J, Salmenkivi K, Raisanen‐Sokolowski A, Lommi J, Kupari M (2013). Diagnosis, treatment, and outcome of giant‐cell myocarditis in the era of combined immunosuppression. Circ Heart Fail 6: 15–22. [DOI] [PubMed] [Google Scholar]
- Kanellakis P, Ditiatkovski M, Kostolias G, Bobik A (2012). A pro‐fibrotic role for interleukin‐4 in cardiac pressure overload. Cardiovasc Res 95: 77–85. [DOI] [PubMed] [Google Scholar]
- Kassan M, Wecker A, Kadowitz P, Trebak M, Matrougui K (2013). CD4 + CD25 + Foxp3 regulatory T cells and vascular dysfunction in hypertension. J Hypertens 31: 1939–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman HM, Cherikh WS, Cheng Y, Hanto DW, Kahan BD (2005). Maintenance immunosuppression with target‐of‐rapamycin inhibitors is associated with a reduced incidence of de novo malignancies. Transplantation 80: 883–889. [DOI] [PubMed] [Google Scholar]
- Kaya Z, Leib C, Katus HA (2012). Autoantibodies in heart failure and cardiac dysfunction. Circ Res 110: 145–158. [DOI] [PubMed] [Google Scholar]
- Kimura A, Kishimoto T (2010). IL‐6: regulator of Treg/Th17 balance. Eur J Immunol 40: 1830–1835. [DOI] [PubMed] [Google Scholar]
- Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J et al. (2014). DC isoketal‐modified proteins activate T cells and promote hypertension. J Clin Invest 124: 4642–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarowska I, Coe D, Wang G, Haas R, Mauro C, Kishore M et al. (2015). Hepatocyte Growth Factor Receptor c‐Met Instructs T Cell Cardiotropism and Promotes T Cell Migration to the Heart via Autocrine Chemokine Release. Immunity 42: 1087–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl U, Pauschinger M, Schwimmbeck PL, Seeberg B, Lober C, Noutsias M et al. (2003). Interferon‐beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation 107: 2793–2798. [DOI] [PubMed] [Google Scholar]
- Kushwaha SS, Khalpey Z, Frantz RP, Rodeheffer RJ, Clavell AL, Daly RC et al. (2005). Sirolimus in cardiac transplantation: use as a primary immunosuppressant in calcineurin inhibitor‐induced nephrotoxicity. J Heart Lung Transplant 24: 2129–2136. [DOI] [PubMed] [Google Scholar]
- Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I et al. (2009). Regulatory T cells ameliorate angiotensin II‐induced cardiac damage. Circulation 119: 2904–2912. [DOI] [PubMed] [Google Scholar]
- Kyto V, Sipila J, Rautava P (2013). The effects of gender and age on occurrence of clinically suspected myocarditis in adulthood. Heart 99: 1681–1684. [DOI] [PubMed] [Google Scholar]
- Lee WS, Erdelyi K, Matyas C, Mukhopadhyay P, Varga ZV, Liaudet L et al. (2016). Cannabidiol limits Tcell‐mediated chronic autoimmune myocarditis: implications to autoimmune disorders and organ transplantation. Mol Med 22: 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang L, Wang S, Zhu H, Ye P, Xie A et al. (2010). The Treg/Th17 imbalance in patients with idiopathic dilated cardiomyopathy. Scand J Immunol 71: 298–303. [DOI] [PubMed] [Google Scholar]
- Liu W, Dienz O, Roberts B, Moussawi M, Rincon M, Huber SA (2012). IL‐21R expression on CD8+ T cells promotes CD8+ T cell activation in coxsackievirus B3 induced myocarditis. Exp Mol Pathol 92: 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Moussawi M, Roberts B, Boyson JE, Huber SA (2013). Cross‐regulation of T regulatory‐cell response after coxsackievirus B3 infection by NKT and gammadelta T cells in the mouse. Am J Pathol 183: 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ et al. (2010). Interleukin 17 promotes angiotensin II‐induced hypertension and vascular dysfunction. Hypertension 55: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascaro‐Blanco A, Alvarez K, Yu X, Lindenfeld J, Olansky L, Lyons T et al. (2008). Consequences of unlocking the cardiac myosin molecule in human myocarditis and cardiomyopathies. Autoimmunity 41: 442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCully ML, Moser B (2011). The human cutaneous chemokine system. Front Immunol 2: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monney PA, Sekhri N, Burchell T, Knight C, Davies C, Deaner A et al (2011). Acute myocarditis presenting as acute coronary syndrome: role of early cardiac magnetic resonance in its diagnosis. Heart 97: 1312–1318. [DOI] [PubMed] [Google Scholar]
- Mora JR, von Andrian UH (2006). T‐cell homing specificity and plasticity: new concepts and future challenges. Trends Immunol 27: 235–243. [DOI] [PubMed] [Google Scholar]
- Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F et al. (2002). Immunosuppressive treatment protects against angiotensin II‐induced renal damage. Am J Pathol 161: 1679–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao M, Kita Y, Kamo H (1992). Haemorrhage in the inferior colliculus. Neuroradiology 34: 347. [DOI] [PubMed] [Google Scholar]
- Nataraj C, Oliverio MI, Mannon RB, Mannon PJ, Audoly LP, Amuchastegui CS et al. (1999). Angiotensin II regulates cellular immune responses through a calcineurin‐dependent pathway. J Clin Invest 104: 1693–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi H, Osawa T, Ogami K, Ouyang X, Sakaguchi S, Koshiba R et al. (2008). A critical link between Toll‐like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc Natl Acad Sci U S A 105: 20446–20451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nery PB, Leung E, Birnie DH (2012). Arrhythmias in cardiac sarcoidosis: diagnosis and treatment. Curr Opin Cardiol 27: 181–189. [DOI] [PubMed] [Google Scholar]
- Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri‐Glass G, Craig SW (1987). Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol 139: 3630–3636. [PubMed] [Google Scholar]
- Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM (2013). Interleukin‐17 causes Rho‐kinase‐mediated endothelial dysfunction and hypertension. Cardiovasc Res 97: 696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S, Ligons DL, Barin JG, Wu L, Talor MV, Diny N et al. (2015). Natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration. Am J Pathol 185: 847–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opavsky MA, Penninger J, Aitken K, Wen WH, Dawood F, Mak T et al. (1999). Susceptibility to myocarditis is dependent on the response of alphabeta T lymphocytes to coxsackieviral infection. Circ Res 85: 551–558. [DOI] [PubMed] [Google Scholar]
- Pummerer CL, Luze K, Grassl G, Bachmaier K, Offner F, Burrell SK et al. (1996). Identification of cardiac myosin peptides capable of inducing autoimmune myocarditis in BALB/c mice. J Clin Invest 97: 2057–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riad A, Westermann D, Zietsch C, Savvatis K, Becher PM, Bereswill S et al. (2011). TRIF is a critical survival factor in viral cardiomyopathy. J Immunol 186: 2561–2570. [DOI] [PubMed] [Google Scholar]
- Roberts BJ, Moussawi M, Huber SA (2013). Sex differences in TLR2 and TLR4 expression and their effect on coxsackievirus‐induced autoimmune myocarditis. Exp Mol Pathol 94: 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose NR (2011). Critical cytokine pathways to cardiac inflammation. J Interferon Cytokine Res 31: 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose NR (2014). Learning from myocarditis: mimicry, chaos and black holes. F1000Prime Rep 6: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savvatis K, van Linthout S, Miteva K, Pappritz K, Westermann D, Schefold JC et al. (2012). Mesenchymal stromal cells but not cardiac fibroblasts exert beneficial systemic immunomodulatory effects in experimental myocarditis. PLoS One 7: e41047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savvatis K, Muller I, Frohlich M, Pappritz K, Zietsch C, Hamdani N et al. (2014). Interleukin‐6 receptor inhibition modulates the immune reaction and restores titin phosphorylation in experimental myocarditis. Basic Res Cardiol 109: 449. [DOI] [PubMed] [Google Scholar]
- Schultz JC, Hilliard AA, Cooper LT Jr, Rihal CS (2009). Diagnosis and treatment of viral myocarditis. Mayo Clin Proc 84: 1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Fukuoka M, Li G, Liu Y, Chen M, Konviser M et al. (2010). Regulatory T cells protect mice against coxsackievirus‐induced myocarditis through the transforming growth factor beta‐coxsackie‐adenovirus receptor pathway. Circulation 121: 2624–2634. [DOI] [PubMed] [Google Scholar]
- Sigmundsdottir H, Butcher EC (2008). Environmental cues, dendritic cells and the programming of tissue‐selective lymphocyte trafficking. Nat Immunol 9: 981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms MG, Walley KR (1999). Activated macrophages decrease rat cardiac myocyte contractility: importance of ICAM‐1‐dependent adhesion. Am J Physiol 277: H253–H260. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GC, Lopez‐Molina J, Gottumukkala RV, Rosner GF, Anello MS, Hecht JL et al. (2011). Myocardial parvovirus B19 persistence: lack of association with clinicopathologic phenotype in adults with heart failure. Circ Heart Fail 4: 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez‐Mier MP, Aguilera B, Mosquera RM, Sanchez‐de‐Leon MS (2013). Pathology of sudden death during recreational sports in Spain. Forensic Sci Int 226: 188–196. [DOI] [PubMed] [Google Scholar]
- Tang H, Zhong Y, Zhu Y, Zhao F, Cui X, Wang Z (2010). Low responder T cell susceptibility to the suppressive function of regulatory T cells in patients with dilated cardiomyopathy. Heart 96: 765–771. [DOI] [PubMed] [Google Scholar]
- Tian N, Gu JW, Jordan S, Rose RA, Hughson MD, Manning RD Jr (2007). Immune suppression prevents renal damage and dysfunction and reduces arterial pressure in salt‐sensitive hypertension. Am J Physiol Heart Circ Physiol 292: H1018–H1025. [DOI] [PubMed] [Google Scholar]
- Triantafilou K, Orthopoulos G, Vakakis E, Ahmed MA, Golenbock DT, Lepper PM et al. (2005). Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll‐like receptor (TLR) 8‐dependent. Cell Microbiol 7: 1117–1126. [DOI] [PubMed] [Google Scholar]
- Trzonkowski P, Dukat‐Mazurek A, Bieniaszewska M, Marek‐Trzonkowska N, Dobyszuk A, Juscinska J et al. (2013). Treatment of graft‐versus‐host disease with naturally occurring T regulatory cells. BioDrugs 27: 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virmani R, Burke AP, Farb A (2001). Sudden cardiac death. Cardiovasc Pathol 10: 211–218. [DOI] [PubMed] [Google Scholar]
- Wang Y, Afanasyeva M, Hill SL, Kaya Z, Rose NR (2000). Nasal administration of cardiac myosin suppresses autoimmune myocarditis in mice. J Am Coll Cardiol 36: 1992–1999. [DOI] [PubMed] [Google Scholar]
- Wang H, Yao Q, Zhu S, Zhang G, Wang Z, Li Z et al. (2013). The autopsy study of 553 cases of sudden cardiac death in Chinese adults. Heart Vessels 29: 486–495. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S et al. (2011). Lysozyme M‐positive monocytes mediate angiotensin II‐induced arterial hypertension and vascular dysfunction. Circulation 124: 1370–1381. [DOI] [PubMed] [Google Scholar]
- Wood KJ, Sakaguchi S (2003). Regulatory T cells in transplantation tolerance. Nat Rev Immunol 3: 199–210. [DOI] [PubMed] [Google Scholar]
- Woodruff JF, Woodruff JJ (1974). Involvement of T lymphocytes in the pathogenesis of coxsackie virus B3 heart disease. J Immunol 113: 1726–1734. [PubMed] [Google Scholar]
- Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA et al. (2006). Myocardial infarct‐sparing effect of adenosine A2 A receptor activation is due to its action on CD4+ T lymphocytes. Circulation 114: 2056–2064. [DOI] [PubMed] [Google Scholar]
- Yu DQ, Wang Y, Ma GZ, Xu RH, Cai ZX, Ni CM et al. (2014). Intravenous immunoglobulin in the therapy of adult acute fulminant myocarditis: A retrospective study. Exp Ther Med 7: 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandbergen HR, Sharma UC, Gupta S, Verjans JW, van den Borne S, Pokharel S et al. (2009). Macrophage depletion in hypertensive rats accelerates development of cardiomyopathy. J Cardiovasc Pharmacol Ther 14: 68–75. [DOI] [PubMed] [Google Scholar]
- Zhang P, Cox CJ, Alvarez KM, Cunningham MW (2009). Cutting edge: cardiac myosin activates innate immune responses through TLRs. J Immunol 183: 27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]