

Abstract
Background and Purpose
Atherosclerosis is characterized by a chronic non‐resolving inflammation in the arterial wall. Aspirin‐triggered lipoxin A4 (ATL) is a potent anti‐inflammatory mediator, involved in the resolution of inflammation. However, the therapeutic potential of immune targeting by means of ATL in atherosclerosis has not previously been explored. The aim of the present study was to determine the effects of ATL and its receptor Fpr2 on atherosclerosis development and progression in apolipoprotein E deficient (ApoE−/−) mice.
Experimental Approach
ApoE−/− × Fpr2+/+ and ApoE−/− × Fpr2−/− mice were generated. Four‐week‐old mice fed a high‐fat diet for 4 weeks and 16‐week‐old mice fed chow diet received osmotic pumps containing either vehicle or ATL for 4 weeks. Atherosclerotic lesion size and cellular composition were measured in the aortic root and thoracic aorta. Lipid levels and leukocyte counts were measured in blood and mRNA was isolated from abdominal aorta and spleen.
Key Results
ATL blocked atherosclerosis progression in the aortic root and thoracic aorta of ApoE−/− mice. In addition, ATL reduced macrophage infiltration and apoptotic cells in atherosclerotic lesions. The mRNA levels of several cytokines and chemokines in the spleen and aorta were reduced by ATL, whereas circulating leukocyte levels were unchanged. The ATL‐induced athero‐protection was absent in ApoE−/− mice lacking the Fpr2 receptor.
Conclusion and Implications
ATL blocked atherosclerosis progression by means of an Fpr2‐mediated reduced local and systemic inflammation. These results suggest this anti‐inflammatory and pro‐resolving agent has therapeutic potential for the treatment of atherosclerosis.
Linked Articles
This article is part of a themed section on Targeting Inflammation to Reduce Cardiovascular Disease Risk. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.22/issuetoc and http://onlinelibrary.wiley.com/doi/10.1111/bcp.v82.4/issuetoc
Abbreviations
- ApoE
apolipoprotein E
- ATL
aspirin‐triggered lipoxin A4
- HFD
high‐fat diet
- Ldlr
low‐density lipoprotein receptor
Tables of Links
| TARGETS | |
|---|---|
| FPR2/ALX |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Inflammation plays a well‐established role in the initiation and progression of atherosclerosis and may represent an important therapeutic target in cardiovascular prevention (Libby et al., 2011). Most of the treatment options evaluated, for example, blocking inflammatory mediators and the induction of immunoregulatory responses, aim to inhibit the pro‐inflammatory signalling in atherosclerotic lesions (Bäck and Hansson, 2015). In addition to a continued stimulation of inflammation, chronic inflammation is also favoured by a failure in the resolution of inflammation (Serhan, 2014). As an example, a defective uptake of apoptotic cells (efferocytosis) increases experimental atherosclerosis (Thorp et al., 2008; Van Vre et al., 2012).
The lipoxygenase metabolism of arachidonic acid yields the pro‐inflammatory mediators leukotrienes. Targeting the specific leukotriene receptors reduces atherosclerosis and intimal hyperplasia in different animal models (Bäck et al., 2005; Ketelhuth et al., 2015). In addition, anti‐leukotrienes used in the treatment of asthma have been associated with decreased cardiovascular risk in observational studies (Ingelsson et al., 2012), hence reinforcing the potent pro‐inflammatory and pro‐atherogenic role of this class of lipid mediators. On the other hand, dual lipoxygenation of arachidonic acid yields a group of lipid mediators called lipoxins (Serhan, 1997; Serhan, 2014), which have the opposite effects to leukotrienes. Lipoxins have been shown to induce a resolution of inflammation, by means of, for example, stimulating efferocytosis (Maderna et al., 2010), granulocyte apoptosis (Barnig et al., 2013), leukocyte egress (van Gils et al., 2012), modulate endothelial activation and reduce leukocyte recruitment after ischaemia/reperfusion (Brancaleone et al., 2013; Smith et al., 2015). In addition, the 15‐epimer of lipoxin A4, referred to as aspirin‐triggered lipoxin A4 (ATL) because of the stimulation of its formation by aspirin‐induced cyclooxygenase acetylation (Serhan, 1997), reduces smooth muscle cell responses in vitro and intimal hyperplasia after carotid ligation in mice in vitro (Petri et al., 2015b). However, the therapeutic potential of ATL in atherosclerosis has remained unexplored.
Lipoxin A4 and ATL share their signalling pathway by means of the FPR2/ALX receptor (Bäck et al., 2014) with other both pro‐inflammatory (e.g. amyloid and antibacterial) peptides (Ye et al., 2009) and pro‐resolving mediators, such as annexin A1 (Hayhoe et al., 2006) and resolvin D1 (Krishnamoorthy et al., 2012). Three major cell types within human atherosclerotic lesions express FPR2/ALX, namely, macrophages, vascular smooth muscle cells and endothelial cells (Petri et al., 2015a). In atherosclerosis‐prone low‐density lipoprotein receptor deficient (Ldlr−/−) mice, pro‐inflammatory ligands for the murine homologue of FPR2/ALX (termed Fpr2) appear to dominate to promote leukocyte recruitment and activation in atherosclerotic lesions (Petri et al., 2015a). Increased atherosclerotic lesion size has also been reported in hyperlipidaemic Fpr2−/− mice at early time‐points, associated with defective pro‐resolving signalling of the FPR2/ALX peptide agonist annexin A1 (Drechsler et al., 2015). Although FPR2/ALX signalling apparently is activated in atherosclerosis, the effects of ATL as a pro‐resolving mediator and a possible therapeutic molecule for the treatment of atherosclerosis have not previously been evaluated.
The aim of the present study was to unravel the potential therapeutic value of ATL in atherosclerosis, and to explore the mechanisms involved, in terms of receptor activation and key local and systemic pro‐atherogenic processes during initiation and progression of the disease.
Methods
Generation of ApoE and Fpr2 double knockout mice
All experiments were performed according to the animal regulations and guidelines of Karolinska Institutet (ethical permit #N138/12). Fpr2 deficient mice were generated at the William Harvey Institute (London, UK) as previously described through insertion of the gene cassette and a GFP reporter in reverse orientation into intron 1 of Fpr2, which prevented transcriptional read‐through of the Fpr2 as well as Fpr3 genes (Dufton et al., 2010). Therefore, these mice have also been termed Fpr2/Fpr3 knockout mice (Brancaleone et al., 2013), but will be referred to as Fpr2−/− in the present report. The mice were backcrossed for at least seven generations in to a C57BL/6 background before being crossed to apolipoprotein E (ApoE)−/− mice from Taconic (Ry, Denmark) for another two generations to achieve ApoE−/− × Fpr2−/− mice. Female mice were kept in a 12 h light/dark cycle with either chow diet (D12102, Research Diets Inc, New Brunswick, NJ) or high‐fat diet (HFD, 21% fat, D12108C, Research Diets Inc, New Brunswick, NJ) and water ad libitum. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015).
ATL treatment protocol
In an initial set of experiments, four groups of mice were studied. ApoE−/− × Fpr2+/+ and ApoE−/− × Fpr2−/− mice were implanted with osmotic pumps (Alzet Mod #2004) under general anaesthesia (1.5% isoflurane; 1 L·min−1) at 16 weeks of age. Mice were randomized to receive osmotic pumps loaded with either vehicle (15% of ethanol) or ATL (10 μg·kg−1; Millipore) as previously described (Petri et al., 2015b). The skin was sutured with 5–0 Vicryl and 0.1% of buprenorphine injected s.c. at the moment of the pump implantation and every morning for the next 2 days. The mice were kept on chow diet during the whole experiment. After the mice had been put to death by CO2 inhalation, a blood sample was withdrawn by cardiac puncture, and the heart, spleen and aorta were harvested.
In a second set of experiments, three groups of ApoE−/− × Fpr2+/+ mice were studied. The first group was put to death at 16 weeks of age, representing the baseline before the treatment protocols began. The remaining 16‐week‐old mice were implanted with osmotic pumps and randomized to the same treatment protocols as indicated above, to obtain a replication of the findings and to determine if ATL treatment either reduced progression or induced regression of atherosclerotic lesions.
In a third set of experiments, 4‐week‐old ApoE−/− × Fpr2+/+ and ApoE−/− × Fpr2−/− mice were fed a HFD and put to death after 4 weeks to study the genotype‐induced effect on atherosclerosis initiation.
ATL analysis
Osmotic pumps filled with ATL were incubated at 37°C for 4 weeks, and the content was then collected, and two volumes of ice‐cold MeOH was added followed by C18 solid phase extraction for liquid chromatography and tandem mass spectrometry (LC–MS‐MS; Colas et al., 2014; Stanke‐Labesque et al., 2012). The system consisted of Waters Xevo ® TQS triple quadruple equipped with Acquity UPLC System from Waters Corporation and an autosampler cooled to 5°C (Milford, MA, USA). An Acquity UPLC BEH (Ethylene Bridged Hybrid) C18 column (130 Å, 1.7 μm, 2.1 × 150 mm) equipped with a pre‐column (Acquity UPLC C18 VanGuard Pre‐column, 130 Å, 1.7 μm, 2.1 × 5 mm; Milford, US) was used with gradients A (0.1% acetic acid in water) and B [acetonitrile/isopropanol; 90:10 (v.v‐1)] from 80:20 (v.v‐1 ) to 0:100 (v.v‐1) in 17 min at a 0.5 mL·min−1 flow rate, which was then equilibrated to initial conditions for 2.5 min. Data acquisition was performed in negative ionization mode, and identification conducted in accordance with published criteria (Colas et al., 2014) with a minimum of six diagnostic ions.
Blood and plasma analysis
Circulating leukocytes were measured by an automated cell counter (ABC vetpack). Cholesterol and triglycerides were measured in plasma by kits from Randox following the manufacturer's protocol. Protein quantification was performed by elisa following the manufacturer's protocols: IL‐6 (Cat # 431303, BioLegend), MMP13 (cat# LS‐F5519, LSBio) and serum amyloid A (SAA; Cat# KMA0021, Invitrogen).
Evaluation of atherosclerosis
The thoracic aorta was opened longitudinally, pinned and stained with Sudan IV solution. A micrograph of the aortic arch was captured using a Leica Microsystems colour video camera, and the image analysis was performed using ImageJ in a blinded fashion. The area of the aortic arch covered by atherosclerotic lesions divided by the aortic arch area was calculated as previously described (Petri et al., 2015a).
In addition, the upper portion of the heart and aortic root was embedded in optimal cutting temperature compound and frozen at −80°C. Serial 10 μm cryosections were collected beginning at the aortic valve cusp for a distance of 800 μm. Oil red‐O staining was performed on eight formalin‐fixed sections collected every 100 μm from the aortic valve cusps. Lesion area was determined in a blinded fashion by light microscopy using Leica Qwin. The mean value of lipid stained areas (lesion size) of aortic root sections was calculated.
Immunohistochemistry and plaque composition analysis
Antibodies against CD68 (MCA1957, Serotec) and CD4 (553647, BD) were used for the immunodetection of macrophages and T‐cells in sections representing the largest lesion area of each aortic root. One section per animal was analysed, and n indicates the number of animals. In a subset of animals, four sections from each aortic root were stained for CD68, CD4 and apoptosis detection and yielded similar results as those obtained with the largest lesion size (data not shown). Biotinylated secondary antibodies and Nova Red staining were used to identify positive cells/areas, and sections were counterstained with haematoxylin. Apoptotic cells were detected using an in situ apoptosis detection kit (ab206386, Abcam) following the manufacturer's protocol. For collagen quantification, sections were stained with Picrosirius red (Histolab® HL27150) and analysed under polarized light for the amount of red (thick) and green (thin) fibres, as quantified by Leica Qwin. The slides were mounted, and the sections were analysed as described for oil red‐O for quantification of CD68, CD4 and apoptosis detection positive areas.
Real‐time PCR
Total RNA was isolated from the abdominal aorta and spleen using QIAZOL (Qiagen) and concentrations were measured spectrophotometrically using Nanodrop 1000 (Thermo Fisher Scientific). Reverse‐transcription was performed with a high capacity kit (Life Technologies, USA). Real‐time PCR was performed on a 7900HT Fast Real‐Time PCR system (Perkin‐Elmer Applied Biosystems) using TaqMan Assay‐on‐Demand from Applied Biosystems for IL‐6 (Mm00446190_m1), MMP13 (Mm00439491_m1), CCL2 (Mm00441242_m1), CCL5 (Mm01302427_m1), CXCL16 (Mm00469712_m1), IFN γ (Mm01168134_m1), IL‐23A (Mm00518984_m1), SAA (Mm00441203_m1) and TNF (Mm00443258_m1). Results are expressed as 2‐ΔCT obtained by comparing the threshold cycle (CT) for the gene of interest with that obtained using hypoxanthine phosphoribosyl‐transferase (HPRT; Mm01545399_m1) as a housekeeping gene.
Data and statistical analysis
Data are expressed as mean ± SEM. When comparing two groups, either a Student's t‐test or Mann–Whitney U‐test was used after a normality test was performed. Either a one or two‐way ANOVA was used for multiple comparisons as indicated, followed by a Dunnett's test versus control. P < 0.05 was considered significant. All analyses were performed using SigmaPlot version 12.5 (Systat Software Inc). The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Results
ATL stability
The stability of ATL was assessed, and the results are shown in Figure 1. ATL remained intact following 4 weeks of incubation in osmotic pump at 37°C, with only small amount (<10%) of potential isomerization as observed by appearance of additional peaks (Figure 1). Its retention time (5.70 min) and MS–MS spectra were identical to that of the 15‐epi‐LXA4 synthetic standard. Furthermore, it retained its tetraene chromophore with λmax at 300 nm and shoulders at 287 and 315 nm, identical to that of the 15‐epi‐LXA4 standard (data not shown).
Figure 1.
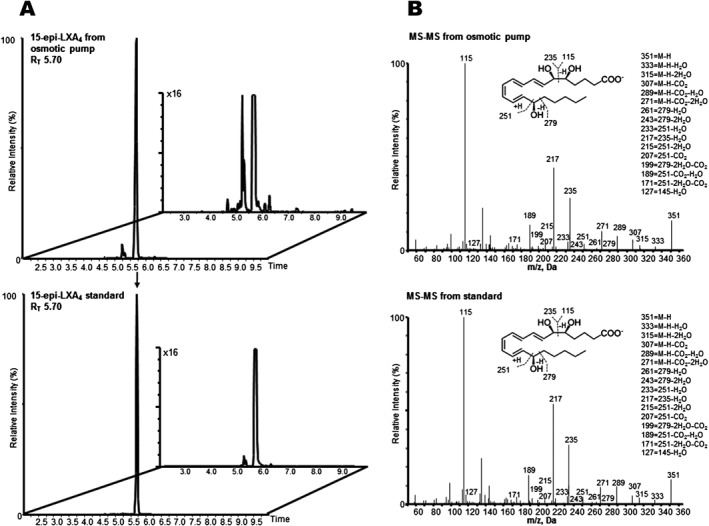
LC–MS‐MS chromatograph of 15‐epi‐LXA4 following incubation in osmotic pump. 15‐epi‐LXA4 in PBS (237.5 ng in 200 μL) was added to an osmotic pump and incubated in PBS at 37°C. After 4 weeks, samples were placed in two volumes of ice‐cold MeOH and taken to solid phase extraction and LC–MS‐MS. (A) Multiple reaction‐monitoring chromatograms of 15‐epi‐LXA4 (m/z 351 > 115) from osmotic pump after 4 weeks (top) and 15‐epi‐LXA4 standard (bottom; Inset: ×16 magnification). (B) Accompanying MS–MS spectra of 15‐epi‐LXA4 from osomotic pump incubation (top) and standard (bottom; inset: diagnostic ions; M, molecular mass).
ATL blocks atherosclerosis progression by means of Fpr2
To study the therapeutic potential of ATL on an established disease, 16‐week‐old ApoE−/− × Fpr2−/− and ApoE−/− × Fpr2+/+ mice were randomized to treatment with either vehicle or ATL for 4 weeks followed by atherosclerosis assessment. At the end of the treatment period, ATL‐treated ApoE−/− × Fpr2+/+ mice exhibited significantly reduced atherosclerosis burden compared with vehicle‐treated mice, both at the aortic root level (Figure 2A) and en face analysis of the aortic arch (Figure 2B). Furthermore, vehicle‐treated ApoE−/− × Fpr2−/− mice exhibited reduced lesion size at both locations compared with vehicle‐treated ApoE−/− × Fpr2+/+ mice (Figure 2A–B). In contrast, atherosclerotic lesion size was not altered by ATL treatment in ApoE−/− × Fpr2−/− mice. There were no significant differences in lipid levels, blood cell counts or body weights between the different groups (Table 1).
Figure 2.
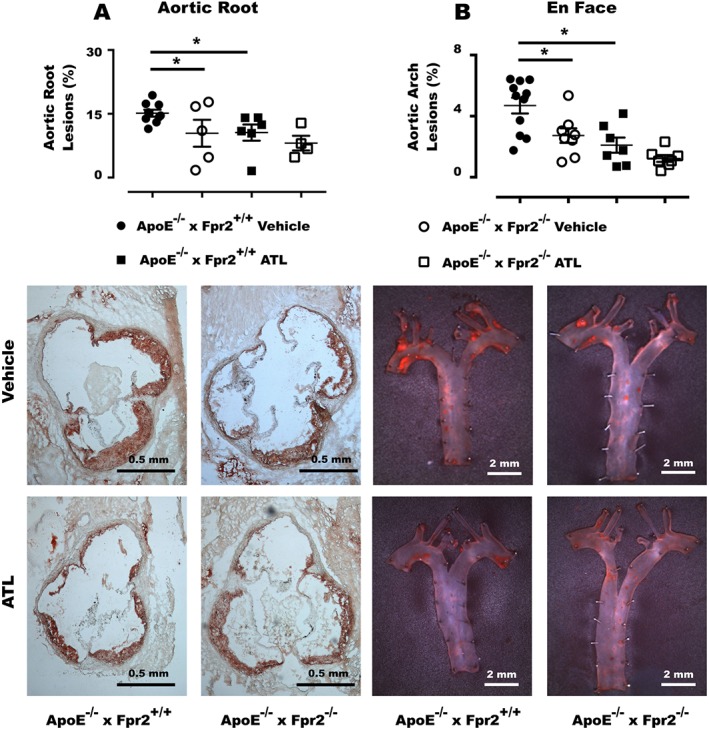
Atherosclerotic lesions in the aortic root (A) and en face analysis of the aortic arch (B) of 20‐week‐old ApoE−/− × Fpr2+/+ and ApoE−/− × Fpr2−/− treated with either vehicle or ATL for 4 weeks. Below the graphs, representative micrographs of aortic root and en face staining of each genotype and treatment are shown. In each group n = at least 6 , and lines represent mean ± SEM. *P < 0.05.
Table 1.
Lipid levels and white blood cell counts for ApoE−/− × Fpr2+/+ and ApoE−/− × Fpr2−/− treated with either Vehicle or ATL
| ApoE−/− × Fpr2+/+ | ApoE−/− × Fpr2−/− | ||||
|---|---|---|---|---|---|
| Vehicle (n = 11) | ATL (n = 8) | Vehicle (n = 7) | ATL (n = 6) | ||
| Lipids | |||||
| Cholesterol (mM) | 9.2±0.5 | 9.4±0.6 | 9.8 ± 0.5 | 7.5 ± 0.7 | NS |
| Triglycerides (mM) | 1.4±0.1 | 0.9±0.1 | 1.2 ± 0.1 | 1.3 ± 0.1 | NS |
| White blood cell count (109 L−1) | 6.2 ± 0.5 | 5.9 ± 0.5 | 7.3 ± 0.7 | 8.6 ± 1.0 | NS |
| Lymphocyte (%) | 70.9 ± 1.0 | 70.8 ± 1.1 | 71.2 ± 1.8 | 72.6 ± 2.4 | NS |
| Monocyte (%) | 4.3 ± 0.2 | 3.5 ± 0.2 | 4.8 ± 0.2 | 4.7 ± 0.1 | NS |
| Granulocyte (%) | 24.5 ± 0.9 | 25.6 ± 1.0 | 23.9 ± 1.8 | 22.6 ± 2.4 | NS |
| Body weight (g) | 25.6 ± 0.4 | 26.7 ± 0.3 | 25.3 ± 0.5 | 26.3 ± 0.7 | NS |
Results are presented as mean ± SEM; NS, non‐significant (P > 0.05; one way ANOVA).
To clarify if ATL either regressed or inhibited the progression of atherosclerosis, the treatment protocol was repeated in another series of ApoE−/− × Fpr2+/+ mice in which the atherosclerotic lesion size after 4 weeks of either ATL or vehicle treatment was compared with that observed in 16‐week‐old littermate mice, which corresponded to the start of the treatments. After 4 weeks of treatment with vehicle, 20‐week‐old mice exhibited significantly increased atherosclerotic lesions in both the aortic root (Figure 3A) and aortic arch (Figure 3B) as compared with the 16‐week‐old mice at baseline. In contrast, no significant differences compared with baseline were observed after 4 weeks of ATL treatment, indicating that ATL prevented plaque progression in both the aortic root and aortic arch (Figure 3). ATL did not significantly alter lipids, blood cell counts or body weight of 20‐week‐old mice (Table 2).
Figure 3.
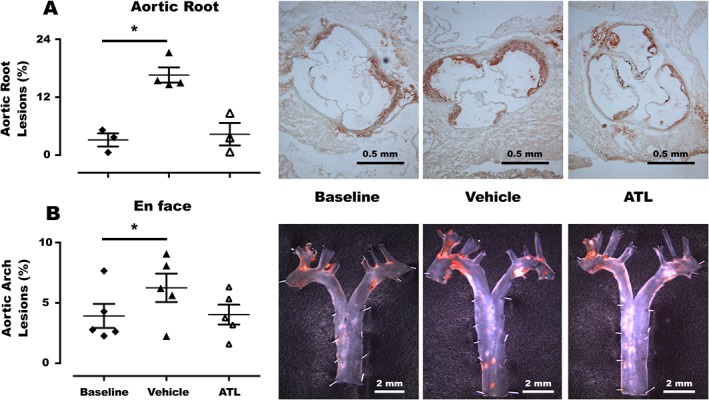
Atherosclerotic lesions in the (A) aortic root and (B) en face analysis of the aortic arch in ApoE−/− × Fpr2+/+ mice at either 16 weeks (baseline, filled diamonds) or 20 weeks (triangles) after 4 weeks of treatment with either vehicle (filled triangles) or ATL (open triangles). Representative micrographs of en face staining of each group are also shown. In each group n = 5, and lines represent mean ± SEM. *P < 0.05
Table 2.
Lipid levels and white blood cell counts for either 16 (baseline) or 20‐week‐old ApoE−/− × Fpr2+/+ mice treated for 4 weeks with either Vehicle or ATL
| Baseline (n = 5) | ATL (n = 5) | Vehicle (n = 5) | ||
|---|---|---|---|---|
| Lipids | ||||
| Cholesterol (mM) | 7.0 ± 0.3 | 7.3 ± 0.3 | 5.6 ± 0.4 | NS |
| Triglycerides (mM) | 1.2 ± 0.1 | 1.2 ± 0.1 | 0.9 ± 0.1 | NS |
| White blood cell count (109 L−1) | 7.1 ± 0.5 | 7.5 ± 1.1 | 8.7 ± 0.9 | NS |
| Lymphocyte (%) | 74.2 ± 1.2 | 66.9 ± 1.5 | 65.7 ± 4.2 | NS |
| Monocyte (%) | 4.5 ± 0.2 | 5.6 ± 0.2 | 5.3 ± 0.3 | NS |
| Granulocyte (%) | 21.2 ± 1.1 | 27.5 ± 1.5 | 28.9 ± 3.9 | NS |
| Body weight (g) | 24.6 ± 0.3 | 28.1 ± 0.5* | 27.8 ± 0.7* |
Results are presented as mean ± SEM; NS, non‐significant (P > 0.05; one way ANOVA),
P < 0.05 versus baseline.
ATL alters plaque composition
The analysis of the atherosclerotic plaque composition in the aortic root of ApoE−/− × Fpr2+/+ mice receiving either ATL or vehicle is shown in Figure 4. Mice receiving ATL exhibited less CD68 positive area compared with vehicle‐treated mice (Figure 4A), whereas there were no significant differences in the amount of CD4 positive cells between ATL and vehicle‐treatment (Figure 4B). In addition, the number of apoptotic cells was reduced in the ATL‐treated mice (Figure 4C). The collagen content, as assessed by Picrosirius red staining, revealed a larger proportion of thick collagen fibres (stained in red) in ATL‐treated mice (Figure 4D).
Figure 4.
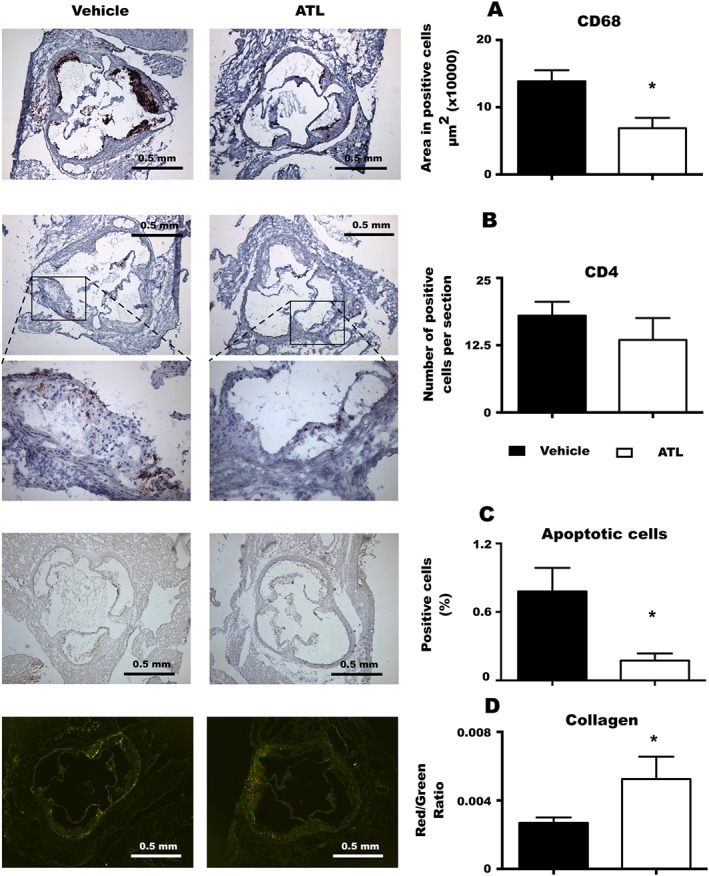
Plaque composition. Quantification of macrophages, CD68 positive (A); T‐lymphocytes, CD4 positive (B); apoptotic cells (in situ apoptosis detection) (C); and collagen fibres (Picrosirium red staining (D), in aortic roots of ApoE−/− × Fpr2+/+ treated with either vehicle or ATL. In all the experiments n = at least 6, and bars represent mean ± SEM. *P < 0.05.
Atherosclerosis initiation is not dependent on Fpr2
In order to clarify whether Fpr2 signalling was important for the initiation of atherosclerosis, 4‐week‐old ApoE−/− × Fpr2−/− and ApoE−/− × Fpr2+/+ mice were fed a HFD for 4 weeks. No differences were observed between the two genotypes in terms of atherosclerotic lesion size (Figure 5), lipid levels or blood count (Table 3).
Figure 5.
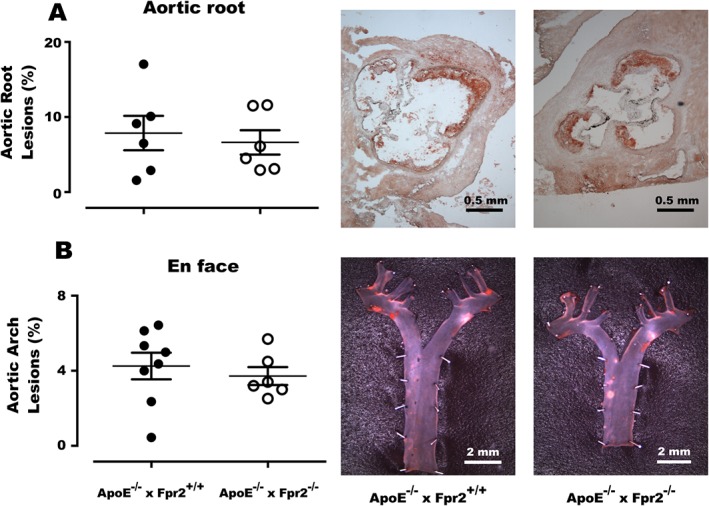
Atherosclerotic lesions in the aortic root (A) and en face analysis of the aortic arch (B) of 8‐week‐old ApoE−/− × Fpr2+/+ (filled circles) and ApoE−/− × Fpr2−/− (open circles) after 4 weeks of HFD. Next to the graphs, representative micrographs of aortic root and en face staining of each genotype are shown. In each group n = at least 6, and lines represent mean ± SEM.
Table 3.
Lipid levels and white blood cell counts for ApoE−/− × Fpr2+/+ and ApoE−/− × Fpr2−/− on HFD for 4 weeks
| ApoE−/− × Fpr2+/+ (n = 8) | ApoE−/− × Fpr2−/− (n = 6) | ||
|---|---|---|---|
| Lipids | |||
| Cholesterol (mM) | 18.6 ± 1.6 | 15.9 ± 0.8 | NS |
| Triglycerides (mM) | 1.7 ± 0.1 | 1.6 ± 0.2 | NS |
| White blood cell count (109 L−1) | 10.8 ± 0.9 | 12.4 ± 0.7 | NS |
| Lymphocyte (%) | 52.9 ± 2.9 | 55.5 ± 1.6 | NS |
| Monocyte (%) | 3.9 ± 0.7 | 3.4 ± 0.2 | NS |
| Granulocyte (%) | 43.1 ± 3.0 | 41.1 ± 1.6 | NS |
Results are presented as mean ± SEM; NS, non‐significant (P > 0.05; Student's t‐test).
The reduction of systemic inflammation induced by ATL is dependent on Fpr2
In order to address whether the observed ATL‐induced effects were limited to the vasculature, or if they were observed systemically, gene expression profiles were compared between the aorta and the spleen. ATL‐treated mice exhibited significantly lower levels of a number of cytokines and chemokines in both the aorta and the spleen, as indicated in Table 4. Further examples of the anti‐inflammatory effects of ATL are shown in Figure 6, namely, the decreased levels of IL‐6 and MMP13 mRNA in the spleen and the aorta and their concomitant decreased circulating protein levels in plasma (Figure 6). In contrast, ATL did not significantly reduce the IL‐6 and MMP13 mRNA levels in organs derived from ApoE−/− × Fpr2−/− mice (data not shown), indicating that the anti‐inflammatory effects of ATL require a functional Fpr2 signalling pathway. Finally, mice treated with SAA, another pro‐inflammatory mediator, which is also an Fpr2 agonist, exhibited significantly reduced mRNA levels in the aorta (Figure 6G) and the spleen (Figure 6H) in response to ATL‐treatment. Also the circulating protein levels in SAA‐treated mice tended to be lower (Figure 6I)
Table 4.
Cytokine and chemokine mRNA levels in the aorta derived from mice treated with either vehicle or ATL
| Aorta | Spleen | |||||
|---|---|---|---|---|---|---|
| Vehicle (n = 11) | ATL (n = 8) | P | Vehicle (n = 11) | ATL (n = 8) | P | |
| IFN‐γ | 0.07 ± 0.01 | 0.001 ± 0.00 | <0.001 | 0.03 ± 0.00 | 0.01 ± 0.00 | 0.02 |
| TNF | 0.28 ± 0.10 | 0.05 ± 0.01 | 0.028 | 2.12 ± 0.12 | 1.19 ± 0.08 | <0.001 |
| CCL2 | 0.77 ± 0.17 | 0.29 ± 0.04 | 0.007 | 0.43 ± 0.02 | 0.29 ± 0.04 | 0.009 |
| CCL5 | 0.84 ± 0.56 | 0.12 ± 0.01 | NS | 6.75 ± 0.27 | 4.62 ± 0.29 | <0.001 |
| CXCL16 | 0.27 ± 0.09 | 0.13 ± 0.04 | 0.021 | 2.41 ± 0.10 | 1.44 ± 0.08 | <0.001 |
| IL‐23a | 0.78 ± 0.76 | 0.01 ± 0.01 | NS | 0.01 ± 0.00 | 0.00 ± 0.00 | 0.016 |
Data (mean ± SEM) are expressed as 2‐ΔCt. NS, non‐significant (P > 0.05; Student's t‐test).
Figure 6.
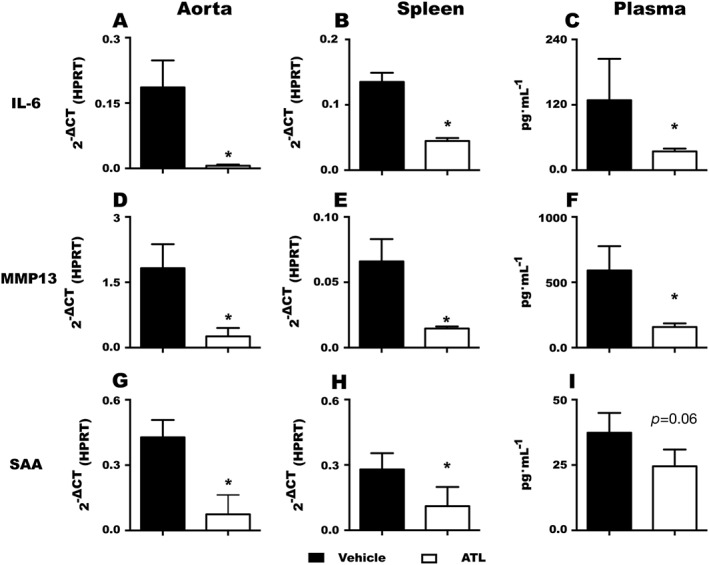
Effects of ATL treatment on IL‐6 (A–C) and MMP13 (D–F) and serum amyloid A (SAA; G‐I) mRNA levels in the aorta (A, D and G), and spleen (B, E and H) and protein levels in plasma (C, F and I). Data (mean ± SEM) are expressed as either 2‐ΔCT or pg·mL−1 for n = at least 6 in each group. *P < 0.05.
Discussion
The present study raises the notion of a therapeutic potential for ATL in stopping the progression of established atherosclerosis. Furthermore, we show that the effects of ATL were transduced by means of Fpr2, reducing vascular inflammation in terms of for example lowering the number of plaque macrophages and inhibiting pro‐inflammatory gene expression. We also show that ATL reduced the systemic inflammation. Taken together, these results suggest that ATL treatment could represent a novel therapeutic strategy to prevent atherosclerosis progression.
Whereas the inhibitory effects of ATL on acute inflammation, such as inflammatory cell recruitment in inflamed dorsal air pouches (Dufton et al., 2010) and microvascular dysfunction after ischaemia–reperfusion (Brancaleone et al., 2013; Vital et al., 2016), are well established, its role for resolving chronic inflammation has been less extensively explored. Long‐term ATL treatment to mice by means of osmotic pumps has however been established to reveal that this lipid mediator reduces intimal hyperplasia after carotid artery ligation (Petri et al., 2015b). Adopting the latter therapeutic strategy, the present study demonstrates the stability of ATL administered under those conditions and for the first time reveals that 4 weeks of ATL treatment to ApoE‐deficient mice with established atherosclerosis significantly reduces lesion size in the aortic root and arch compared with vehicle‐treated mice.
The ATL‐induced effects on atherosclerosis were observed in the absence of significant alterations of the plasma lipid profile, suggesting that pro‐inflammatory pathways rather than lipid metabolism were targeted by this treatment. In line with this, a reduced number of macrophages was observed in atherosclerotic lesions derived from ATL‐treated mice compared with vehicle‐treated mice, whereas lesion T‐lymphocyte infiltration was not altered by ATL. Macrophages in human atherosclerotic lesions indeed express the ATL receptor FPR2/ALX (Petri et al., 2015a), and lipoxin stimulation of human and murine myeloid cells reduces inflammation in vivo and in vitro (Devchand et al., 2003; Maderna et al., 2010; Wu et al., 2011; Petri et al., 2015a). Efferocytosis by macrophages is enhanced by lipoxins (Godson et al., 2000) and inhibited in Fpr2−/− mice (Petri et al., 2015a), suggesting that ATL‐induced efferocytosis by means of Fpr2 signalling may contribute to its athero‐protective effects. In support of the latter, a reduced number of apoptotic cells was observed in atherosclerotic lesions derived from ATL‐treated mice in the present study. An increased proportion of thick collagen fibres was also observed in atherosclerotic lesions after ATL‐treatment, which is line with previous findings in Fpr2−/− mice (Petri et al., 2015a) and supports the notion that ATL may also induce a more stable lesion phenotype, which is further supported by the decrease in collagenase MMP13 after ATL treatment observed in the present study.
These beneficial effects of ATL were reproduced in a second series of mice, in which the atherosclerotic lesion size at 20 weeks after 4 weeks of osmotic pump ATL treatment was unchanged compared with that observed in 16‐week‐old mice, corresponding to the start of the treatment period. Based on the latter experiments, we can conclude that ATL treatment blocked the significant progression of atherosclerosis that was observed in vehicle‐treated mice. It is also important to stress that the mice had atherosclerotic plaques when treatment was initiated, hence extending previous observations that the Fpr2 agonist annexin A1 prevented atherosclerosis development in younger ApoE knockout mice fed a HFD (Drechsler et al., 2015).
Studies in other animal models have shown that lipoxin‐induced effects are abolished in mice lacking the murine FPR2/ALX orthologue Fpr2 (Dufton et al., 2010; Brancaleone et al., 2013; Petri et al., 2015b; Vital et al., 2016). To address the signalling pathways involved in the observed reduction of atherosclerosis induced by ATL, we generated ApoE and Fpr2 double knockout mice for the present study. Importantly, ATL‐treatment was ineffective in reducing atherosclerosis in ApoE−/− mice lacking Fpr2. This latter finding indicates that Fpr2 is the receptor exclusively mediating the beneficial effects of ATL in terms of atherosclerosis development.
Another important finding that emerged from the latter analysis was that Fpr2‐deficient ApoE−/− mice exhibited reduced atherosclerosis. These results confirm our previous results in Ldlr and Fpr2 double knockout mice at similar time points and lesion sizes (Petri et al., 2015a). The similar results obtained with Fpr2 deletion in two different murine atherosclerosis models strongly corroborate the notion that pro‐inflammatory Fpr2 agonists prevail over the anti‐inflammatory receptor ligands in atherosclerosis. Indeed, our previous study showed low levels of lipoxins in Ldlr−/− mice fed a HFD compared with the pro‐inflammatory Fpr2 ligand SAA (Petri et al., 2015a). Taken together, these observations support the hypothesis that murine atherosclerosis models are associated with low pro‐resolving Fpr2 agonists, which may contribute to a failure in the resolution of inflammation. Interestingly, ATL‐treatment decreased the levels of SAA in the present study, hence potentially further facilitating its pro‐resolving Fpr2 signalling.
The lack of effects at early stages of atherosclerosis development in the present study are in contrast to a study using a different Fpr2 gene targeting strategy, which reported exacerbated atherosclerosis in ApoE−/− mice after 4 weeks of HFD (Drechsler et al., 2015). While it remains to be established whether the different Fpr2 genetic targeting techniques or other experimental conditions accounted for these differential results, it should be pointed out that the results of the present study are in line with previous findings that did not reveal any significant effects of Fpr2 deletion at early stages of atherosclerosis in either Ldlr−/− mice (Petri et al., 2015a) or after bone marrow transplantation of Fpr2−/− bone marrow to Ldlr−/− mice (Fredman et al., 2015).
Taken together with the above‐mentioned findings, the results of the present study support the postulate that ATL treatment enhances the resolution of inflammation and inhibits the development of atherosclerosis. In support of the latter, a failure in the resolution of inflammation in terms of reduced lipoxin levels has indeed been established in both experimental atherosclerosis (Petri et al., 2015a) and clinical biomarker studies (Ho et al., 2010). Although not specifically addressed in the present study, it should be noted that aspirin enhances ATL production in atherosclerosis (Brezinski et al., 1992; Ho et al., 2010), and it can hence be anticipated that ATL production may represent an additional beneficial effect of aspirin in cardiovascular prevention, which goes beyond its anti‐aggregatory effects on platelets.
As a final approach in the present study, we sought to determine whether the observed Fpr2‐mediated reduction in atherosclerosis and lesion macrophage content induced by ATL solely represented a local vascular response or if systemic inflammation was also altered in ATL‐treated mice. Based on the lack of significant alterations of the peripheral white blood cell count, we conclude that no apparent myeloid dysfunction was induced by ATL. Nevertheless, ATL reduced the expression levels of a number of pro‐inflammatory cytokines, chemokines and enzymes in both the spleen and the aorta, indicating that ATL decreased systemic inflammation. This observation is supported by the decreased circulating proteins levels of Il‐6 and MMP13.
In summary, ATL treatment initiated after the establishment of atherosclerosis stopped the progression of the disease over 4 weeks. These effects of ATL were absent in mice lacking Fpr2, the murine orthologue of the human FPR2/ALX receptor, indicating that the beneficial effects of ATL on atherosclerosis are mediated through this signalling pathway. However, in the absence of ATL, a deficiency in Fpr2 protected against atherosclerosis progression, supporting the notion that stimulation of this receptor is involved in the failure in the resolution of inflammation, which can be reversed by the treatment of atherosclerotic mice with the pro‐resolving mediator ATL. This is further supported by the reduced macrophage infiltration and inflammatory mRNA levels in ATL‐treated mice. In conclusion, stopping atherosclerosis progression by means of the ATL and FPR2/ALX pathway may represent a novel therapeutic option for immune targeting in cardiovascular prevention.
Author contributions
M.H.P., A.L.F. and M.B. planned and conceived the study. M.H.P., A.L.F. and H.A. performed the experiments. M.H.P., A.L.F., H.A., C.E.W. and M.B. performed the data analysis. M.H.P. and M.B. wrote the manuscript, and A.L.F., H.H., M.P., C.E.W. and G.K.H. revised it critically for important intellectual content.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
The authors would like to acknowledge Jessica Lundgren, Selameyhune Assefa, Sandra Olsson and Ann‐Christine Eklöf for the valuable support in the animal facility. This work was supported by the Swedish Research Council (Grant number 2014–2312); the Swedish Heart and Lung Foundation (grant numbers 20150600 and 20150683); and the Stockholm County Council (grant number 20140222). Mauro Perretti was supported by the Wellcome Trust Programme (grant number 086867/Z/08/Z). Marcelo Heron Petri was supported by a KID PhD‐fellowship from Karolinska Institutet.
Petri, M. H. , Laguna‐Fernandez, A. , Arnardottir, H. , Wheelock, C. E. , Perretti, M. , Hansson, G. K. , and Bäck, M. (2017) Aspirin‐triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E−/− mice. British Journal of Pharmacology, 174: 4043–4054. doi: 10.1111/bph.13707.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäck M, Hansson GK (2015). Anti‐inflammatory therapies for atherosclerosis. Nat Rev Cardiol 12: 199–211. [DOI] [PubMed] [Google Scholar]
- Bäck M, Bu DX, Branstrom R, Sheikine Y, Yan ZQ, Hansson GK (2005). Leukotriene B4 signaling through NF‐kappaB‐dependent BLT1 receptors on vascular smooth muscle cells in atherosclerosis and intimal hyperplasia. Proc Natl Acad Sci U S A 102: 17501–17506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäck M, Powell WS, Dahlen SE, Drazen JM, Evans JF, Serhan CN et al. (2014). Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. Br J Pharmacol 171: 3551–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S et al. (2013). Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med 5 .174ra126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancaleone V, Gobbetti T, Cenac N, le Faouder P, Colom B, Flower RJ et al. (2013). A vasculo‐protective circuit centered on lipoxin A4 and aspirin‐triggered 15‐epi‐lipoxin A4 operative in murine microcirculation. Blood 122: 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brezinski DA, Nesto RW, Serhan CN (1992). Angioplasty triggers intracoronary leukotrienes and lipoxin A4. Impact of aspirin therapy. Circulation 86: 56–63. [DOI] [PubMed] [Google Scholar]
- Colas RA, Shinohara M, Dalli J, Chiang N, Serhan CN (2014). Identification and signature profiles for pro‐resolving and inflammatory lipid mediators in human tissue. Am J Physiol Cell Physiol 307: C39–C54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devchand PR, Arita M, Hong S, Bannenberg G, Moussignac RL, Gronert K et al. (2003). Human ALX receptor regulates neutrophil recruitment in transgenic mice: roles in inflammation and host defense. FASEB J 17: 652–659. [DOI] [PubMed] [Google Scholar]
- Drechsler M, de Jong R, Rossaint J, Viola JR, Leoni G, Wang JM et al. (2015). Annexin A1 counteracts chemokine‐induced arterial myeloid cell recruitment. Circ Res 116: 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M et al. (2010). Anti‐inflammatory role of the murine formyl‐peptide receptor 2: ligand‐specific effects on leukocyte responses and experimental inflammation. J Immunol 184: 2611–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredman G, Kamaly N, Spolitu S, Milton J, Ghorpade D, Chiasson R et al. (2015). Targeted nanoparticles containing the proresolving peptide Ac2‐26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci Transl Med 7 .275ra220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR (2000). Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte‐derived macrophages. J Immunol 164: 1663–1667. [DOI] [PubMed] [Google Scholar]
- Hayhoe RP, Kamal AM, Solito E, Flower RJ, Cooper D, Perretti M (2006). Annexin 1 and its bioactive peptide inhibit neutrophil‐endothelium interactions under flow: indication of distinct receptor involvement. Blood 107: 2123–2130. [DOI] [PubMed] [Google Scholar]
- Ho KJ, Spite M, Owens CD, Lancero H, Kroemer AH, Pande R et al. (2010). Aspirin‐triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am J Pathol 177: 2116–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelsson E, Yin L, Back M (2012). Nationwide cohort study of the leukotriene receptor antagonist montelukast and incident or recurrent cardiovascular disease. J Allergy Clin Immunol 129: 702–707 .e702 [DOI] [PubMed] [Google Scholar]
- Ketelhuth DF, Hermansson A, Hlawaty H, Letourneur D, Yan ZQ, & Back M (2015). The leukotriene B receptor (BLT) antagonist BIIL284 decreases atherosclerosis in ApoE mice. Prostaglandins & other lipid mediators. [DOI] [PubMed]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy S, Recchiuti A, Chiang N, Fredman G, Serhan CN (2012). Resolvin D1 receptor stereoselectivity and regulation of inflammation and proresolving microRNAs. Am J Pathol 180: 2018–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Hansson GK (2011). Progress and challenges in translating the biology of atherosclerosis. Nature 473: 317–325. [DOI] [PubMed] [Google Scholar]
- Maderna P, Cottell DC, Toivonen T, Dufton N, Dalli J, Perretti M et al. (2010). FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin‐derived peptide‐stimulated phagocytosis. FASEB J 24: 4240–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petri MH, Laguna‐Fernandez A, Gonzalez‐Diez M, Paulsson‐Berne G, Hansson GK, Bäck M (2015a). The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc Res 105: 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petri MH, Laguna‐Fernandez A, Tseng CN, Hedin U, Perretti M, Bäck M (2015b). Aspirin‐triggered 15‐epi‐lipoxin A(4) signals through FPR2/ALX in vascular smooth muscle cells and protects against intimal hyperplasia after carotid ligation. Int J Cardiol 179: 370–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN (1997). Lipoxins and novel aspirin‐triggered 15‐epi‐lipoxins (ATL): a jungle of cell–cell interactions or a therapeutic opportunity? Prostaglandins 53: 107–137. [DOI] [PubMed] [Google Scholar]
- Serhan CN (2014). Pro‐resolving lipid mediators are leads for resolution physiology. Nature 510: 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HK, Gil CD, Oliani SM, Gavins FN (2015). Targeting formyl peptide receptor 2 reduces leukocyte‐endothelial interactions in a murine model of stroke. FASEB J 29: 2161–2171. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke‐Labesque F, Pepin JL, de Jouvencel T, Arnaud C, Baguet JP, Petri MH et al. (2012). Leukotriene B4 pathway activation and atherosclerosis in obstructive sleep apnea. J Lipid Res 53: 1944–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I (2008). Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of ApoE−/− mice. Arterioscler Thromb Vasc Biol 28: 1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gils JM, Derby MC, Fernandes LR, Ramkhelawon B, Ray TD, Rayner KJ et al. (2012). The neuroimmune guidance cue netrin‐1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat Immunol 13: 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vre EA, Ait‐Oufella H, Tedgui A, Mallat Z (2012). Apoptotic cell death and efferocytosis in atherosclerosis. Arterioscler Thromb Vasc Biol 32: 887–893. [DOI] [PubMed] [Google Scholar]
- Vital SA, Becker F, Holloway PM, Russell J, Perretti M, Granger DN et al. (2016). Formyl‐peptide receptor 2/3/lipoxin A4 receptor regulates neutrophil–platelet aggregation and attenuates cerebral inflammation: impact for therapy in cardiovascular disease. Circulation 133: 2169–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Wang A, Min Z, Xiong Y, Yan Q, Zhang J et al. (2011). Lipoxin A4 inhibits the production of proinflammatory cytokines induced by beta‐amyloid in vitro and in vivo. Biochem Biophys Res Commun 408: 382–387. [DOI] [PubMed] [Google Scholar]
- Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M et al. (2009). International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev 61: 119–161. [DOI] [PMC free article] [PubMed] [Google Scholar]