

Abstract
Data from basic science experiments is overwhelmingly supportive of the causal role of immune‐inflammatory response(s) at the core of atherosclerosis, and therefore, the theoretical potential to manipulate the inflammatory response to prevent cardiovascular events. However, extrapolation to humans requires care and we still lack definitive evidence to show that interfering in immune‐inflammatory processes may safely lessen clinical atherosclerosis. In this review, we discuss key therapeutic targets in the treatment of vascular inflammation, placing basic research in a wider clinical perspective, as well as identifying outstanding questions.
Linked Articles
This article is part of a themed section on Targeting Inflammation to Reduce Cardiovascular Disease Risk. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.22/issuetoc and http://onlinelibrary.wiley.com/doi/10.1111/bcp.v82.4/issuetoc
Abbreviations
- ApoB
apolipoprotein B
- ApoE
apolipoprotein E
- CANTOS
The Canakinumab Anti‐inflammatory Thrombosis Outcomes Study
- CHD
coronary heart disease
- CIRT
the Cardiovascular Inflammation Reduction Trial
- CRP
C‐reactive protein
- CVD
cardiovascular diseases
- DCs
dendritic cells
- ECs
endothelial cells
- eNOS
endothelial NOS
- GULO
L‐gulonolactone oxidase
- HLA‐DR
human leukocyte antigen‐DR
- HMG‐CoA
3‐hydroxy 3‐methyl glutaryl CoA
- ICAM‐1
intercellular adhesion molecule 1
- IL‐1RA
IL‐1 receptor antagonist
- LDL‐C
LDL‐cholesterol
- LDLr
LDL receptor
- Lp‐PLA2
lipoprotein‐associated PLA2
- MI
myocardial infarction
- oxLDL
oxidized LDL
- RA
rheumatoid arthritis
- RCTs
randomized controlled trials
- SMCs
smooth muscle cells
- TNFR
TNF receptor
- VCAM‐1
vascular cell adhesion protein 1
- WT
wild type
Introduction
Atherosclerosis‐related cardiovascular diseases (CVD) are the leading cause of mortality worldwide (WHO, 2011). Immune responses play a decisive role in all phases of atherosclerosis (Galkina and Ley, 2009; Libby and Hansson, 2015), and inflammation contributes to plaque vulnerability (Hansson et al., 2015). Atherosclerosis‐prone conditions accelerate immune cell recruitment into the arteries in the early and advanced stages of the pathology (Galkina et al., 2006; Maffia et al., 2007; Swirski et al., 2016), and in experimental models, antigen‐presenting cell/T‐cell interactions have been shown in the arterial wall leading to local T‐cell activation and production of pro‐inflammatory cytokines (Koltsova et al., 2012; Macritchie et al., 2012; Sage et al., 2014). In the advanced stages of the pathology, immune responses are tightly controlled in situ by the formation of artery tertiary lymphoid organs in the adventitial connective tissue adjoining arteries. These lymphocyte aggregates control primary T‐cell responses while bypassing secondary lymphoid organs exerting a protective effect on atherosclerosis in mice (Hu et al., 2015; Srikakulapu et al., 2016). Therefore, there is a range of inflammatory processes underpinning atherogenesis, which might be amenable to interventions.
Data from observational epidemiological studies also give some support to the inflammatory hypothesis of CVD. A host of prospective cohort data show that elevated circulating levels of C‐reactive protein (CRP), or indeed almost any other circulating inflammatory marker, are associated with an increased risk of future CVD events, even after adjusting for established classical CVD risk factors (Woodward et al., 2007; Danesh et al., 2008; Welsh et al., 2011; Kaptoge et al., 2012). These data suggest that low grade systemic inflammation precedes incident cardiovascular events and, as such, also imply that inflammation might cause vascular diseases that lead to major CVD events. Indeed, similar epidemiological associations between elevated cholesterol and blood pressure and risk of CVD have also been established (Lewington et al., 2002; Sniderman et al., 2011), and we know these to be causal risk factors due to supporting data from randomized controlled trials (RCTs) with specific pharmacological interventions [Cholesterol Treatment Trialists' (CTT) Collaborators et al., 2012; Ettehad et al., 2016]. However, although the association of these inflammatory biomarkers with CVD appears to be independent of other risk factors, and the utility of these biomarkers in clinical risk prediction warrants debate, experience tells us to be cautious with interpreting even strong associations as causal, given the potential for residual confounding.
In this review, we will discuss the key potential therapeutic targets in the treatment of vascular inflammation (Figure 1), placing basic research in to a wider clinical perspective, as well as identifying questions yet to be addressed.
Figure 1.
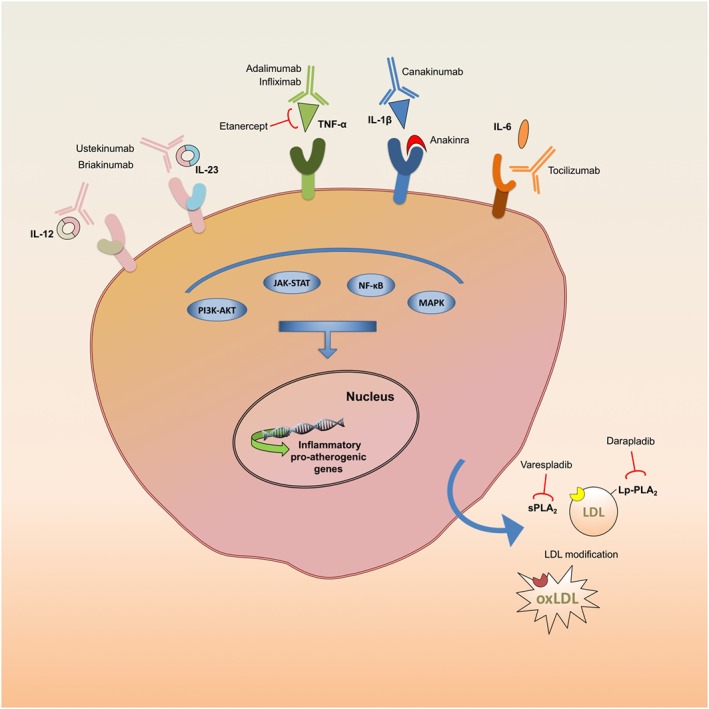
Pro‐atherogenic and inflammatory pathways targeted by prospective anti‐atherosclerotic antibodies and inhibitors.
Lessons from the antioxidant vitamins
Claims about the efficacy of L‐ascorbic acid (vitamin C) in the prevention of the common cold, cancer, and CVD can be traced back to the influence of the double Nobel Laureate, Linus Pauling (Pauling, 1971). Indeed, there is an abundance of literature investigating the important issue of whether localized oxidative stress causes an inflammatory response in the vasculature, and other organs. Under this hypothesis, there may be a vicious positive feedback cycle between inflammation and oxidative stress, causing vascular remodelling and plaque development (Montezano et al., 2015). In support of this idea, basic scientific experiments, and limited trial data suggest that vitamin C prevents free radical‐induced lipid peroxidation (Huang et al., 2002). Vitamin C induces proliferation and concomitantly prevents in vivo apoptosis of endothelial cells (ECs), increasing the recovery of the endothelial layer following vascular damage (Rössig et al., 2001; Saeed et al., 2003). In addition, vitamin C is an essential regulator of collagen synthesis (Murad et al., 1981; Davidson et al., 1997; Qiao et al., 2009). Both humans and guinea pigs are unable to synthetize vitamin C due to an inactivating mutation of the L‐gulonolactone oxidase (GULO), and a supplement of vitamin C in the diet is necessary otherwise the development of scurvy. Interestingly, chronic deprivation of vitamin C in diet, produce intimal lesions in guinea pigs (Willis, 1953). The corresponding KO strain, GULO−/−, fed a diet without vitamin C, showed extensive vascular impairment including disruption of elastin layers and desquamation of ECs (Maeda et al., 2000). Apolipoprotein‐E deficient mice (apoE−/−), also lacking GULO showed a reduction of 40% in plaque collagen content (Nakata and Maeda, 2002), supporting a potential involvement of vitamin C in plaque stability rather than in plaque formation. It was therefore thought that vitamin C might ameliorate some of the downstream effects of an inflammatory response, and therefore prevent atherosclerosis.
Indeed, observational studies suggest that low vitamin C levels might predict CVD outcomes. For example, in the EPIC‐Norfolk study, individuals in the highest quartile for plasma vitamin C had 33% lower risk of cardiovascular events (Boekholdt et al., 2006). In a meta‐analysis of 15 studies (375 000 participants), those with the highest third of dietary intake of vitamin C were at 16% lower risk of CVD events (Ye and Song, 2008). Yet, despite these tantalizing data, meta‐analysis of 50 RCTs not only failed to show any benefit of supplementation with vitamin C in the prevention of CVD, but tight confidence intervals also essentially exclude any possibility of clinically meaningful benefits (Myung et al., 2013) (Figure 2). This pattern, whereby basic science and observational data support a protective role of antioxidant vitamins in CVD, but trials of the relevant supplement provide no evidence to support the compelling and coherent theories, has been repeated for other antioxidants (Ye et al., 2013). It could be argued that supplementations trials are more likely to fail when many of the participants are not ‘deficient’ in the vitamin in question. For example, 75% of participants in NHANES took the recommended daily intake of vitamin C (Fulgoni et al., 2011). However, it is also worth noting that, similarly, many of the participants in other observational cohort studies will not be deficient in the studied vitamin. This does not appear to influence the observational association between vitamin status and outcome, and therefore does not offer a satisfactory explanation to resolve the apparent inconsistencies between observations studies and controlled trials. Further arguments about optimal delivery route, and dose of vitamin supplements in trial settings may be pertinent. However, it is worth noting that other pharmacological interventions, such as statins, have a ‘sliding scale’ of biological effects at a wide range of doses (Weng et al., 2010); it is not clear why very specific doses of conventionally dietary antioxidants would be necessary to see any treatment benefit. Therefore, even the most rigorous approaches to data analysis struggle to overcome confounding and the reverse causality inherent in observational studies. While these data do not directly refute the inflammatory hypothesis of CVD, they do illustrate the caution always needed in interpreting observational associations as evidence of causality.
Figure 2.
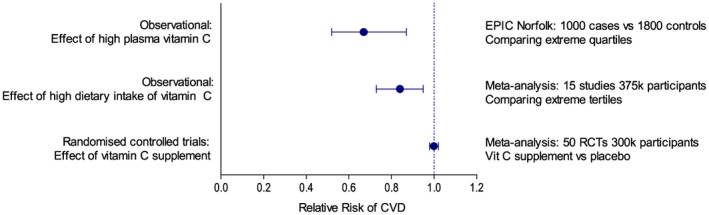
Summary of studies investigating the role of vitamin C in CVD risk. Data from Boekholdt et al. (2006), Ye and Song (2008) and Myung et al. (2013).
Mendelian randomization studies
Mendelian randomization attempts to substantially attenuate or, in some cases, to eliminate the problems of confounding and reverse causality in classical observational epidemiology by exploiting the random allocation of genetic material at conception (Davey Smith and Hemani, 2014). Within a hypothetical population, one group of people has genetic variant(s) that lead to lower average circulating inflammatory markers over their life course, while the other group does not possess these variants. All other traits (such as adiposity, smoking and alcohol intake) should normally be equally distributed in the comparator groups. This situation can then be viewed as analogous to a RCT, and consequently, confers a level of stronger evidence of causality than classical epidemiology. Any differences in health outcomes between these two groups of people can be attributed to the concentration of inflammatory markers.
Strong evidence based on such data indicates that the inflammatory marker CRP does not cause CVD. In a meta‐analysis of nearly 200 000 participants, the relative risk for coronary heart disease (CHD) was 1.00 (0.90 to 1.13) per 1 SD higher genetically raised CRP concentration (Wensley et al., 2011). Whether these polymorphisms might explain subtleties in the biology of atherosclerosis, such as the accumulation of monomeric CRP in plaques (Eisenhardt et al., 2009), is not presently clear. In contrast however, genetic variants which lead to higher circulating concentrations of IL‐6 receptors (IL‐6R) (and consequently less IL‐6 cell signalling and lower circulating CRP) appear protective against CHD (IL6R Mendelian randomisation Consortium, 2012; Sarwar et al., 2012). This has fuelled interest in the hypothesis that upstream key regulator cytokines are players in the development of atherosclerosis, and these two independent studies remain the most relevant findings to date supporting the inflammatory hypothesis.
Lipid lowering therapies, inflammation and CVD
Statins, which act by inhibiting 3‐hydroxy 3‐methyl glutaryl CoA (HMG‐CoA) reductase, are the front‐line drug used for lipid reduction in primary and secondary CVD prevention. Their efficacy in reducing cholesterol and preventing CVD events is widely accepted on the basis of a large body of RCTs (CTT Collaborators et al., 2012). However, basic science has published evidence over many years to suggest that statins might have pleiotropic effects (Bellosta et al., 2000). Specifically of interest for this review, the idea that statins prevent CVD not only through lipid reduction but also through a lipid independent, anti‐inflammatory action has been extensively debated (Kinlay, 2007; Babelova et al., 2013; Sirtori, 2014).
Statins can exert their putative pleiotropic effects through a broad range of mechanisms. They inhibit Rho‐GTPase isoprenylation through reducing geranyl‐geranylpyrophosphate production during cholesterol biosynthesis (Liao and Laufs, 2005; Cai et al., 2015). This is leading to increased expression of endothelial NOS (eNOS) and NO production (Liao and Laufs, 2005). Statins have also been shown to increase NO production via activation of the PI3K‐Akt pathway through phosphorylation of Akt (Kureishi et al., 2000). In vivo, a single dose of simvastatin to either wild type (WT) or apoE−/− mice increased endothelium‐derived NO production (Scalia et al., 2001). Moreover, statins can suppress the activity of pro‐oxidant enzymes (such as NADPH oxidase) in the endothelium (Margaritis et al., 2014).
Statins also affect leukocyte trafficking at the inflammatory site. Atorvastatin, simvastatin and cerivastatin reduced the expression of intercellular adhesion molecule (ICAM)‐1 and lymphocyte function‐associated antigen‐1 on human ECs and circulating peripheral blood mononuclear cells stimulated with TNF‐α (Rezaie‐Majd et al., 2003). Statins have also been shown to reduce, in vitro, in human primary cells, the production of the chemokine CCL2 (Romano et al., 2000), and the secretion of matrix metalloproteinase (MMP)‐9 (Wong et al., 2001; Wang et al., 2016).
Statins can also directly affect the adaptive immune response. They have been shown to inhibit the inducible promoter IV of the transactivator CIITA in several cell types, such as the human ECs and monocyte/macrophages, and thereby repress MHC‐II mediated CD4+ T‐cell activation (Kwak et al., 2000). Moreover, statins reduce the expression of CD40 in human vascular ECs, smooth muscle cells (SMCs), macrophages and fibroblasts (Mulhaupt et al., 2003). Simvastatin and atorvastatin have been shown to reduce the expression of other costimulatory molecules such as CD83 and CD86 and human leukocyte antigen‐DR (HLA‐DR) induced by LPS in human monocyte‐derived dendritic cells (DCs) from healthy patients leading to a reduced capability of DCs to induce T‐cell activation, proliferation and Th1 differentiation (Yilmaz et al., 2006). Atorvastatin concomitantly induces activation of STAT‐6 and inhibition of STAT‐4 phosphorylation, leading to secretion of Th2 cytokines (IL‐4, IL‐5 and IL‐10) and TGF‐β and suppression of Th1 cytokines (IL‐2, IL‐12, IFN‐γ and TNF‐α) (Youssef et al., 2002). Lovastatin also increases the recruitment of regulatory T‐cells in inflamed sites. This effect is dependent on the expression of CCL1, a chemokine up‐regulated by statin administration (Mira et al., 2008). More recently, statin‐loaded reconstituted HDL nanoparticles have been shown to inhibit atherosclerotic plaque inflammation in apoE−/− mice, demonstrating that statins can selectively inhibit vascular inflammation in situ, directly in the diseased vessel wall, without any systemic effect such as lipid lowering (Duivenvoorden et al., 2014).
There is therefore a body of evidence showing how statins might exert anti‐inflammatory effects, although one must always bear in mind the potential for publication bias whereby only positive studies fitting a prevailing hypothesis are published whereas negative studies are not easily published or not pushed towards publication in the first place. In epidemiological studies, statin treatment certainly does lower circulating levels of CRP in RCTs (Ridker et al., 1999, 2009; Albert et al., 2001; Sever et al., 2012, 2013; Soedamah‐Muthu et al., 2015), but the underlying mechanism, and whether apparently decreased systemic inflammation translates into a reduction of cardiovascular events is highly controversial. The JUPITER (Justification for the Use of statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) trial suggested that the degree of CRP lowering on statin treatment may offer insight into CVD risk reduction beyond LDL‐cholesterol (LDL‐C) lowering (Ridker et al., 2009). However, this analysis has not been reproduced in other trial data (Sever et al., 2012, 2013; Soedamah‐Muthu et al., 2015); the vast majority of benefit from statins is predictable from the extent of LDL reduction alone. One study level meta‐analysis suggested a strong correlation between change in LDL and change in CRP for a range of lipid lowering agents including statins, ezetimibe, niacin, fibrates and fish oils (r = 0.80) (Kinlay, 2007), perhaps suggesting that CRP lowering is directly or indirectly related to lipid lowering. More recently, however, the new class of lipid lowering drugs, proprotein convertase subtilisin/kexin type 9 inhibitors, have emerged and it appears these have little or no effect on CRP despite reducing circulating LDL by more than 50% (Blom et al., 2014; Cannon et al., 2015). Therefore, CRP reduction is not an inevitable consequence of LDL‐lowering.
The debate about potential pleiotropic effects of statins will continue. However, it is clear that it will be extremely difficult to tease apart lipid‐lowering effects from anti‐inflammatory effects and thus estimate their relative importance for CVD events. Statins studies alone will not prove or disprove the inflammatory hypothesis of CVD. It should be noted that the degree of LDL‐C reduction explains nearly all the CVD benefit seen in clinical trials of statins or indeed other agents so that one does not need to evoke alternative statin effects to explain benefits (Collins et al., 2016).
Autoimmune disease, biological agents and cardiovascular disease
It is well established that patients with a range of chronic systemic autoimmune conditions, including rheumatoid arthritis (RA), ankylosing spondylitis, psoriasis and irritable bowel disease, are at modestly increased risk of cardiovascular events, independent of other traditional risk factors (del Rincón et al., 2001; Andersen and Jess, 2014; Ogdie et al., 2015). The mechanisms by which cardiovascular risk is elevated in RA patients remains unproven, but the primary candidate pathway is that systemic inflammation drives vascular dysfunction and atherosclerosis (Sattar et al., 2003). Indeed, data from the CORRONA database of nearly 25 000 RA patients, followed‐up for median 2.7 years, shows that those with low disease activity or in remission (and therefore with a lower burden of systemic inflammation) have an approximately 60% decrease in risk of CVD events, compared with those classified with high disease activity (Solomon et al., 2015). Use of glucocorticoid treatment has long been a mainstay to reduce the pain and inflammation associated with RA, and it has been hypothesized that these anti‐inflammatory interventions might prevent CVD. Indeed, older studies in animal models suggest dexamethasone reduces atherosclerosis (Makheja et al., 1989). However, the longer‐term side effects of steroid treatment, including potential causes of CVD, like diabetes, central obesity and hypertension, are likely to cause their own cardiovascular risks (Souverein et al., 2004; Walker, 2007). Indeed, evidence from the RA field suggests high‐dose steroids have a net adverse association with CVD risk (Agca et al., 2017).
The availability of a range of specific anti‐inflammatory interventions has revolutionized treatment of patients with RA and other autoimmune conditions; TNF‐α blockers and IL‐6 receptor blockers are now common and efficacious (if expensive) second or third line treatment options after use of conventional disease modifying anti‐rheumatic drugs. The availability of these and other biological agents has opened a world of possibilities with regards to testing the anti‐inflammatory hypothesis of CVD using a variety of different pathways.
TNF‐α and IL‐6 in CVD
Experimental data
Several lines of evidence support a pro‐atherogenic role for TNF‐α. TNF‐α binding to the TNF‐α receptors, TNFR1 or TNFR2, activates NF‐κB (Grassia et al., 2010) and p38 MAPK (Sprague and Khalil, 2009) and therefore the transcription of proinflammatory genes including those for IL‐1β, IL‐8, CCL2, ICAM‐1, vascular cell adhesion protein (VCAM)‐1 and MMPs in a variety of cell types including lymphocytes, macrophage, ECs and vascular SMCs (Grassia et al., 2009; Sprague and Khalil, 2009; Grassia et al., 2010; Kalliolias and Ivashkiv, 2016). TNF‐α/apoE double knockout mice showed less atherosclerotic plaque formation compared to apoE−/− mice (Brånén et al., 2004; Ohta et al., 2005), with reduced aortic expression of ICAM‐1, VCAM‐1, CCL2 as well as scavenger receptor class A (Ohta et al., 2005). Treatment with TNF‐α binding protein reduced plaque development in apoE−/− mice (Elhage et al., 1998). Moreover, chimeric LDL receptor knockout (LDLr−/−) mice deficient in p55 TNF receptors (TNFR1) in bone marrow‐derived cells showed a reduction in atherosclerosis and reduced vascular recruitment of immune cells (Xanthoulea et al., 2008). Several immune cells can be a source of TNF‐α in murine atherosclerotic vessels, including macrophages and the pro‐atherogenic B2 cell subset (Tay et al., 2016). Importantly, TNF‐α can strongly influence plaque vulnerability. TNF‐α stimulates MMP production by SMCs as well as SMC activation, proliferation, and migration (Grassia et al., 2009; Grassia et al., 2010; Maddaluno et al., 2012). Intriguingly, TNF‐α can concomitantly induce proliferation of human SMCs and apoptosis of ECs, confirming a scenario in which TNF‐α can alter the fibrous cap composition in the atheroma (Rastogi et al., 2012).
Experimental data show a potential dual effect of IL‐6 on atherogenesis. In vitro, human macrophages stimulated with oxLDL produce IL‐6 (van Tits et al., 2011), while stimulation with IL‐6 enhances the expression of adhesion molecules (ICAM‐1, VCAM‐1 and E‐selectin) in HUVEC (Watson et al., 1996). IL‐6 mRNA is detectable in the aorta of apoE−/− but not in WT mice (Sukovich et al., 1998). The injection of recombinant IL‐6 increased lesion size in the aorta of apoE−/− and C57Bl/6 mice fed a high‐fat diet and increased the expression of tissutal and circulating pro‐inflammatory cytokines (IL‐1β and TNF‐α) (Huber et al., 1999). Moreover, treatment with a fusion protein of the natural IL‐6 trans‐signalling inhibitor soluble glycoprotein 130 reduced atherosclerosis in LDLr−/− mice (Schuett et al., 2012). In contrast, however, serum cholesterol levels and subsequent atherosclerotic lesion formation increased in apoE/IL‐6 double knockout mice compared to control animals, showing a less stable plaque phenotype and reduced circulation levels of IL‐10 (Schieffer et al., 2004).
Biological agents and hard CVD endpoints
Given the strong data on the role of these cytokines in atherosclerosis, the effect of blockade of these pathways on CVD risk in people with autoimmune disease is thus clearly of high interest. Despite this, RCTs of anti‐inflammatory therapies in RA patients and other inflammatory conditions have been powered to demonstrate improvements in disease activity rather than CVD endpoints, which requires far smaller sample sizes. Indeed, even meta‐analysis of randomized placebo controlled trials yields nowhere near sufficient power to investigate the effects of these biological agents on CVD events (Ryan et al., 2011). Given that these drugs are now a cornerstone of treatment in RA patients, practicalities aside, a placebo controlled trial large enough to investigate the impact of these agents on CVD events is unfeasible due to the ethical implications of restricting some patients to placebo. The forthcoming ENTRACTE trial results (https://clinicaltrials.gov/ct2/show/NCT01331837), are highly anticipated and will directly compare the IL‐6 receptor blocker tocilizumab with the TNF‐α blocker etanercept in the prevention of CVD events in RA patients for the first time, but will not be able to test the inflammatory hypothesis directly. It is notable this study is powered only to rule out an upper hazard ratio of risk of 1.8 so it may not be powered sufficiently to provide a robust answer of CVD risk with tocilizumab, compared with that with etanercept.
Thus, given the lack of hard outcomes in trial data, the literature has turned to pharmaco‐epidemiological studies to investigate the effect of biological agents on CVD risk in patients with chronic autoimmune diseases. Meta‐analysis of observational studies and registries in RA, psoriasis and psoriatic arthritis patients suggest that those receiving TNF‐α blockers are at 30% lower risk (95% CI 0.54–0.90) of CVD than patients taking non‐biological therapies (Roubille et al., 2015), perhaps offering some support to the notion that TNF‐α blockade is efficacious in reducing CVD events in people with systemic inflammatory conditions. However, pharmaco‐epidemiological studies are prone not only to confounding by established risk factors but also to confounding by indication. In the CORRONA database, for instance, patients taking TNF‐α blockers (compared to patients on non‐biological and non‐methotrexate based therapies) were at substantially lower risk of CVD events (HR 0.39, 95% CI 0.19 to 0.82) (Greenberg et al., 2011). However, they were also slightly more likely to be female, were less likely to be tertiary educated, had higher scores on general health questionnaires, and were less likely to have had a previous myocardial infarction (MI). The authors adjust for these differences in many of the constituent studies of the meta‐analysis, but the fundamental problem remains that somewhere in the past an informed clinical judgement has been made; those prescribed biologics have fundamental differences in their characteristics from those not prescribed biologics, and these are impossible to fully measure, much less adjust for.
Biological agents and surrogates of CVD
Further data on surrogate biomarkers of CVD in RA patients might be useful to infer the effects of administration of biological agents on CVD risk. The chronic inflammatory burden of RA patients depresses circulating total cholesterol and other lipid concentrations (Myasoedova et al., 2010), a feature commonly seen in many chronic and acute inflammatory illnesses. Treatment with biological agents may be considered to ‘normalize’ total cholesterol, although some could nevertheless argue that an increase in cholesterol however achieved could still be potentially harmful. These drugs also have effects on several other pathways, as we recently demonstrated (Robertson et al., 2013). In a post hoc study of the MEASURE trial of tocilizumab or placebo in 132 RA patients, total‐cholesterol, LDL‐C and triglyceride levels all increased in tocilizumab treated patients by week 12 (12.6, 28.1 and 10.6%, respectively), although there was no increase in small dense LDL or oxidized (ox)LDL (McInnes et al., 2015). In addition, tocilizumab decreased lipoprotein (a) and decreased D‐dimer (McInnes et al., 2015), a marker of thrombosis and fibrinolysis as well as CVD risk (Willeit et al., 2013). Examination of downstream biomarkers of CVD may also be informative; for instance natriuretic peptides (such as N‐terminal pro B‐type natriuretic peptide, NT‐proBNP) are released during cardiac overload and are strong predictors of CVD risk (Welsh et al., 2013, 2016a,b). Prospective data from an adalimumab (a TNF‐α blocker) treated cohort suggested that therapy reduced the cardiac biomarker and strong predictor of CVD risk, NT‐proBNP, but that study lacked a control arm (Peters et al., 2010). However, this effect was not supported in a post hoc analysis of a RCT, where cardiac biomarkers were lowered by both tocilizumab and the standard care comparator (Welsh et al., 2016a,b). Thus, the net effect of anti‐inflammatories on CVD risk is difficult to interpret from biomarkers alone.
Safety profile of biological agents
Despite the efficacy of biological agents in chronic inflammatory conditions, their immunosuppressive properties have raised safety concerns, and consequently, they have been carefully evaluated in RCTs and by using registry data.
In a meta‐analysis of RCTs and open label studies, the TNFα blocker infliximab was associated with slightly more adverse events compared to placebo (OR 1.55, 95% CI 1.01–2.35), although this did not reach statistical significance for other biologics (Singh et al., 2011). Non‐significant trends towards increases in pulmonary infections and tuberculosis reactivation were also noted. However, small numbers of incident malignancies preclude useful analyses and TNF‐α blockers are contraindicated in patients with heart failure. Attempts to use registry data for long‐term follow‐up of patients treated with biologics are likely to be subject to the same limitations described above.
Despite a predominantly encouraging safety profile in people with chronic diseases, there is a considerable ethical difference in giving immunosuppressive drugs to patients with chronic illness that may limit their quality of life (such as RA), and giving such drugs for the prevention of CVD, in which case the subclinical phase has little effect on quality of life, and a hard clinical event may never occur. For this reason, it is unlikely that the present generation of systemic anti‐inflammatory drugs will ever be prescribed in a primary prevention setting.
PLA2
Experimental data
The PLA2 superfamily are enzymes able to specifically hydrolyse fatty acids at the sn‐2 position of glycophospholipids releasing bioactive lipids, most importantly arachidonic acid and lysophospholipids. There are 15 different groups of PLA2 enzymes, each containing subgroups. Between them, the most studied in atherosclerosis include group II secretory PLA2 (sPLA2), and PAF acetylhydrolase, also known as lipoprotein‐associated (Lp)‐PLA2 (Burke and Dennis, 2009; Rosenson, 2010). Hydrolysis of membrane phospholipids by PLA2 is a key step in the production of precursors for eicosanoids and the potent inflammatory agent PAF. The development of sPLA2 inhibitors as possible anti‐inflammatory agents represents an interesting and active research field.
The proatherogenic activities of sPLA2 enzymes include the modification to circulating LDL, with conformational changes in apolipoprotein B (apoB)‐100 that impairs clearance by LDLr (Kleinman et al., 1988). Thus, in mice overexpressing sPLA2, the time spent by LDL in the circulation increased, along with its susceptibility to oxidation and the loading of arterial macrophages with cholesterol (Ivandic et al., 1999). Interestingly, phospholipid hydrolysis by sPLA2 causes conformational changes in apoB‐100 resulting in increased proteoglycan‐binding activity which facilitates LDL diffusion into the vessel wall (Flood et al., 2004). In addition to these effects on LDL, the hydrolysis of phospholipids from cell membranes and lipoproteins increased local oxidative stress and levels of free arachidonic acid, lysophospholipids, and non‐esterified fatty acids (Rosenson and Hurt‐Camejo, 2012).
The relevance of sPLA2 isoforms in atherosclerosis has been investigated using knockout and transgenic mice. Transgenic mice expressing the human form of group IIa sPLA2 exhibited significant atherosclerotic lesions even when fed a low‐fat chow diet (Ivandic et al., 1999). LDLr−/− mice overexpressing group V sPLA2 by retrovirus‐mediated gene transfer showed increased atherosclerosis associated with collagen deposition in plaques (Bostrom et al., 2007). Moreover, LDLr−/− chimeric mice deficient in bone marrow group V sPLA2 showed less atherosclerosis compared to control animals (Bostrom et al., 2007). On the contrary, the effect of group X sPLA2 seems to be protective. The overexpression of human group X sPLA2 in murine bone marrow cells of the LDLr−/− chimeric mice leads to the reduction of Th1 response and to a 50% reduction of lesion formation (Ait‐Oufella et al., 2013). All these results suggest different effects of the multiple sPLA2 isotypes and indicate the development of selective inhibitors of PLA2 isoforms as new possible compounds for the treatment of atherosclerosis.
Varespladib is an inhibitor of sPLA2 investigated in several animal models for its anti‐atherosclerotic effect. Treatment of apoE−/− mice with varespladib resulted in the reduction of atherosclerosis development and a more stable plaque phenotype (Fraser et al., 2009; Shaposhnik et al., 2009). In atherosclerotic guinea pigs, treatment with varespladib was not effective in reducing atherosclerosis but decreased cholesterol accumulation in the aorta without changing serum cholesterol levels (Leite et al., 2009).
Lipoprotein‐associated PLA2 (Lp‐PLA2) is an enzyme synthesized in macrophages and which travels in the circulation with LDL‐particles. Although the biology is controversial, several pieces of evidence suggest that Lp‐PLA2 is a pro‐inflammatory enzyme, due to its mediating role in the production of oxidized non‐essential fatty acids and lysophosphatidylcholine (Zalewski and Macphee, 2005), which are thought to be important in the recruitment and retention of inflammatory cells within plaques. Unexpectedly, overexpression of Lp‐PLA2 in apoE−/− mice (Quarck et al., 2001) or in balloon‐denuded rabbits (Turunen et al., 2005) reduced endothelial damage and lesions formation. This anti‐atherogenic effect could, at least in part, be explained by the fact that Lp‐PLA2 hydrolyses PAF, a potent proinflammatory mediator (Talmud and Holmes, 2015).
Darapladib is a relatively selective inhibitor of Lp‐PLA2 (Rosenson and Hurt‐Camejo, 2012). Treatment of diabetic and hypercholesterolemic pigs with darapladib reduced development of advanced coronary atherosclerosis reducing the lyso‐PC content and necrotic core area of the lesion. Moreover, darapladib reduced the expression of several genes associated with macrophage and T lymphocyte activation in pig atherosclerotic vessels (Wilensky et al., 2008), and CCL2, VCAM‐1 and TNF‐α in aortas from apoE−/− mice (Wang et al., 2011).
Observational epidemiology and trial data
Observational epidemiology supports the notion that sPLA2 and LP‐PLA2 might be important pathophysiological pathways. Thus, higher circulating Lp‐PLA2 mass and activity was associated with increased CVD risk (Thompson et al., 2010). In phase 2 trials, darapladib did exactly as predicted; it reduced IL‐6 by 12% and CRP by 13% (Mohler et al., 2008), and also halted the progression of the necrotic core of atherosclerotic plaques (Serruys et al., 2008). In contrast, the phase 3 STABILITY trial (15 828 CHD patients, 3.7 year follow‐up) and the SOLID‐TIMI 52 trial (13 026 ACS patients, 2.5 year follow‐up) disappointingly showed that darapladib did not reduce risk of a composite CVD endpoint (O'Donoghue et al., 2014; White et al., 2014). A similar lack of benefit was reported for varespladib (Nicholls et al., 2014). Interestingly, Mendelian randomization predicted the findings for varespladib would show on benefit, ahead of the publication of the trial results (Holmes et al., 2013).
IL‐12 and IL‐23
Experimental data
IL‐12 and IL‐23 are heterodimeric cytokines that share the subunit p40. The subunit p40 has been detected in foam‐cell‐like regions of the aortic plaque of apoE−/− mice (Lee et al., 1999). IL‐12 is expressed by lymphocytes, activated macrophages and DCs. IL‐12 activates the T‐bet transcription factor, leading to the up‐regulation of IFN‐γ production and polarisation of CD4+ T‐cells to the proinflammatory phenotype Th1 (Teng et al., 2015). Recombinant IL‐12 accelerates the formation of atherosclerotic lesions in apoE−/− mice (Lee et al., 1999), while IL‐12 deficiency (IL‐12−/−/apoE−/−) resulted in reduced atherosclerosis (Davenport and Tipping, 2003). Interestingly, selective inhibition of IL‐12 production in macrophages led to a 50% decrease in aortic lesions in LDLr−/− mice (Zhao et al., 2002). Finally, blockade of IL‐12 by vaccination of LDLr−/− mice resulted in a 60% reduction of atherosclerotic plaque, leading to a stable plaque phenotype (Hauer et al., 2005).
The role of IL‐23 in atherosclerosis is poorly studied, despite the association observed between IL‐23 and disease progression in patients with carotid atherosclerosis. IL‐23 serum levels and the plaque mRNA expression levels were higher in patients with carotid atherosclerosis, compared with healthy patients (Abbas et al., 2015).
The antibodies ustekinumab and briakinumab, which bind to the p40 subunit, were developed for the treatment of psoriasis. Given the arguments set out above, these biological agents might be expected to have a more direct anti‐inflammatory effect than PLA2 inhibitors.
Trial data
Ustekinumab and briakinumab appear efficacious in reducing chronic inflammatory disease symptoms and perhaps reducing CRP (Toedter et al., 2009; Strober et al., 2011), but questions have been raised about safety, with a combined trial meta‐analysis reporting potentially increased major adverse cardiac events (OR = 4.23, 95% CI: 1.07–16.75, P = 0.04) (Tzellos et al., 2013). The confidence intervals around this estimate are very large, and the nuances of quantifying the effect size lie in how the statistics for small event numbers in study arms are handled (sometimes no CVD events occurred).
The question remains as to whether these findings have any relevance for the inflammatory hypothesis of CVD. They certainly do illustrate the complex biology underlying atherogenesis and that an intervention that reduces inflammatory biomarkers cannot be presumed to be beneficial without hard endpoint data to support the findings (Table 1).
Table 1.
Summary of studies investigating the effect of key inflammation‐related interventions on the risk for CVD and events
| Target | Intervention | Observational studies (low weighted evidence of causal inference) | Mendelian randomization studies (intermediate weighted evidence of causal inference) | Randomized trials (strong weighted evidence of causal inference) |
|---|---|---|---|---|
| TNF‐α | Adalimumab | ↑ Circulating TNF‐α ↑ CVD risk | NA | ↑Infections (Singh et al., 2011) |
| Infliximab | Biological use ↓risk of CVD (Greenberg et al., 2011) | ? CVD risk | ||
| Etanercept | Biological use ↓NT‐proBNP (Peters et al., 2010) | |||
| IL‐6R | Tocilizumab | ↑ Circulating IL‐6 ↑CVD risk (Sarwar et al., 2012; Interleukin‐6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium, 2012) |
IL‐6R SNPs rs7529229 and rs2228145 (Sarwar et al.,
2012) ↓CRP ↓risk of CHD |
↑LDL‐C, ↓lipoprotein(a), ↓fibrinogen, ↓D‐dimer; ↔ small LDL, ↔ oxidized LDL, (McInnes et al.,
2015) ? CVD risk |
| IL‐12/23 p40 | Ustekinumab | NA | NA | ↓CRP (Toedter et al., 2009) |
| Briakinumab | ↑CVD (Tzellos et al., 2013 ) | |||
| IL‐1β | Canakinumab | NA | IL‐1Ra SNPs rs6743376 and rs1542176 (Interleukin 1 Genetics Consortium, 2015) | Ongoing (https://clinicaltrials.gov/ct2/show/NCT01327846) |
| IL‐1R | Anakinra (rIL‐1RA) |
↓IL‐6; ↓CRP; ↑risk of CHD |
||
| Lp‐PLA2 | Darapladib | ↑Lp‐PLA2 mass and activity ↑CVD risk (Thompson et al., 2010) |
Several SNPs including rs1051931 (Casas et al.,
2010) ↔ risk of CVD |
↓IL‐6; ↓CRP (Mohler et al.,
2008) ↔ risk of CVD (O'Donoghue et al., 2014; White et al., 2014) |
| sPLA2 | Varespladib | ↑sPLA2 circulating concentration ↑CVD risk (Boekholdt et al., 2005) |
SNP rs11573156 (Holmes et al.,
2013) ↔ risk of CVD |
↑risk of CVD (Nicholls et al., 2014) |
| Multiple | Methotrexate | Methotrexate use ↓risk of CVD (Micha et al., 2011) | NA | Ongoing (https://clinicaltrials.gov/ct2/show/NCT01594333) |
NA, not applicable (note this may not mean there are no published studies, but that studies are comparatively small, prone to bias, or inconclusive); ↑, increase; ↓, decrease; ↔, unchanged; TC, total cholesterol; CAD, coronary artery disease; rIL‐1RA, recombinant IL‐1 receptor antagonist.
Ongoing RCTs
The inflammatory hypothesis of CVD has, so far, never been directly tested in RCTs. Two major RCTs, powered for reduction in composite CVD endpoints will now formally test the inflammatory hypothesis in a secondary prevention setting. The Cardiovascular Inflammation Reduction Trial (CIRT: https://clinicaltrials.gov/ct2/show/NCT01594333) uses a methotrexate‐based intervention and the Canakinumab Anti‐inflammatory Thrombosis Outcomes Study uses a monoclonal antibody against IL‐β (CANTOS: https://clinicaltrials.gov/ct2/show/NCT01327846).
Methotrexate experimental data
From a scientific perspective, the mechanisms by which methotrexate exerts anti‐inflammatory effects still need to be fully elucidated. Originally developed as an antifolate drug for the treatment of cancer, methotrexate inhibits cell division. It also shows a range of anti‐inflammatory mechanisms, independent of its antifolate activity, such as inhibition of T‐cell proliferation by affecting purine and pyrimidine metabolism, reduction of intracellular glutathione levels leading to reduced immune cell accumulation at inflammatory sites, and increased release of anti‐inflammatory adenosine (Cronstein, 2005). In TNF‐α‐stimulated human ECs, methotrexate down‐regulates pro‐inflammatory genes, such as those for TNF‐α, IL‐1β, CXCL2 and the toll‐like receptor 2, and up‐regulates the anti‐inflammatory TGF‐β1 gene (Bulgarelli et al., 2012). Incubation with methotrexate prevents the conversion of lipid‐loaded THP‐1 cells into foam cells (Reiss et al., 2008). Following treatment with methotrexate, adipose tissue from obese mice produced less proinflammatory (TNF‐α, IL‐6, leptin) and more anti‐inflammatory mediators (adiponectin and IL‐10) associated with reduced macrophage infiltration and inflammation (DeOliveira et al., 2012). In addition, methotrexate down‐regulates the expression of adhesion molecules (ICAM‐1, E‐selectin, VCAM‐1) in human biopsies rich in inflammatory infiltrate (Dahlman‐Ghozlan et al., 2004) and circulating levels of IL‐6 in psoriatic patients (Elango et al., 2012). All these activities support a potential effect of methotrexate in the treatment of atherosclerosis. Indeed, methotrexate (4 mg·kg−1) intravenously injected four times a week for 30 days has been shown to reduce by 75% atherosclerosis formation in rabbits (Bulgarelli et al., 2012). In addition, administration of methotrexate alone or in combination with etoposide carried in lipid nanoemulsion reduced macrophage, MMP‐9 and lesional content of proinflammatory cytokines, again in the atherosclerotic rabbit model (Bulgarelli et al., 2013; Leite et al., 2015).
CIRT trial
CIRT randomizes low dose methotrexate (15–20 mg·week−1) plus folate (1 mg, 6 days week−1), versus placebo plus folate design. Participants include patients who have had a previous MI or multi‐vessel coronary artery disease, have type 2 diabetes and/or metabolic syndrome, and are therefore high‐risk, secondary prevention patients. As an intervention, methotrexate has several features to recommend it, including a long historical safety profile and very low cost. It should be noted that CIRT is powered to detect a 25% risk reduction in the methotrexate group; a considerable risk reduction against a background of gold standard secondary prevention therapies. If methotrexate fails to lower risk to this level, CIRT may not exclude the inflammatory hypothesis of CVD. The anticipated primary completion date of CIRT is presently the end of 2018.
IL‐1 and CVD experimental data
IL‐1 is the first identified interleukin and affects virtually all cells and organs. It is the major pathogenic mediators of inflammatory and immune diseases (Garlanda et al., 2013; Schett et al., 2016). IL‐1α and IL‐1β share the same receptor (IL‐1R) and the same downstream signalling pathway. Instead, the IL‐1R antagonist (IL‐1RA) serves as a decoy receptor, inhibiting the effects of IL‐1. Both IL‐1α and IL‐1β are produced as precursors and activated by enzymic cleavage (Dinarello, 2011). IL‐1α mediates the early phases of sterile inflammation, whereas IL‐1β is produced as an inactive precursor from tissue‐resident macrophages and monocytes and is activated by caspase‐1. The system is also regulated upstream by the cleavage of procaspase‐1 by the NLRP3 inflammasome (Dinarello, 2011; Garlanda et al., 2013). Neutrophils can also trigger IL‐1β response independently of caspase‐1 and inflammasome activation (Schett et al., 2016).
The IL‐1 pathway seems to be an important player in atherosclerosis. IL‐1α and/or β induce the expression of ICAM‐1, E‐selectin and VCAM‐1 in HUVEC (Aziz and Wakefield, 1996), increasing adhesion of leukocytes (Bevilacqua et al., 1985), leading to local amplification of innate and adaptive immunity (Garlanda et al., 2013). IL‐1RA−/− C57BL/6J mice fed a high cholesterol and cholate diet, developed foam cell lesions, whereas LDLr−/− mice crossed with transgenic mice expressing high levels of murine sIL‐1RA, showed less atherosclerosis (Devlin et al., 2002). The administration of human recombinant IL‐1RA in apoE−/− mice also reduced plaque formation (Elhage et al., 1998). On the contrary, apoE−/− mice lacking IL‐1 receptor type I unexpectedly showed a vulnerable plaque phenotype including reduced SMC and collagen plaque content (Alexander et al., 2012).
The specific role of the two IL‐1α and IL‐1β isoforms in atherosclerosis development is still under debate. Cholesterol crystals and oxLDL have been identified as endogenous triggers of the NLRP3 inflammasome, inducing the secretion of the active form of IL‐1β by plaque macrophages (Duewell et al., 2010; Rajamäki et al., 2010). This pathway is attractive as a potential explanation linking the phenotypes of elevated cholesterol, vascular inflammation and oxidative stress. IL‐1α and β have also been reported to enhance the expression of matrix enzymes (Schett et al., 2016). In addition, deletion of IL‐1β (Kirii et al., 2003) or the use of monoclonal antibodies against IL‐1β (Bhaskar et al., 2011) inhibited the development of atherosclerosis in apoE−/− mice. These findings may suggest a primary role for IL‐1β in the development of atherosclerosis. However, apoE−/− mice lacking inflammasomes develop normal atherosclerotic lesions (Menu et al., 2011) and, more importantly, fatty acid‐induced mitochondrial uncoupling abolished IL‐1β secretion, which turned the cholesterol crystal‐elicited response towards selective production of IL‐1α, as a potent inducer of vascular inflammation (Freigang et al., 2013). This may be evidence of redundancy but could also suggest that IL‐1α could be targeted in patients with CVD. In summary, whether blockade of IL‐1β alone is sufficient to down‐regulate vascular inflammation still remains to be determined.
CANTOS trial and epidemiological data of the IL‐1 pathway
Despite this optimism, Mendelian randomization data have provided some controversial findings. There are no established SNPs that can act as proxies for circulating IL‐β in Mendelian randomization studies. This is at least partly due to the lack of an assay sensitive enough to measure IL‐1β in healthy people. However, one recent study investigating genetic variants of IL‐1RA reported that variants associated with higher concentrations of IL‐1RA had lower concentrations of CRP (suggesting a true anti‐inflammatory effect), but were also puzzlingly associated with increased CHD (Interleukin 1 Genetics Consortium, 2015). As described previously, the IL‐1 cytokine superfamily signalling system and its regulation are complex (Herder and Donath, 2015). However, this study, and the widespread post hoc explanations of the data, really lays bare our ignorance of the pathways that underlie the inflammatory causes of CVD.
The randomized design in CANTOS, compares three arms of the IL‐1β blocker canakinumab (50, 150 and 300 mg administered subcutaneously every 3 months) to placebo. Participants in the trial are those who have experienced a recent MI and have a circulating hsCRP of >2 mg·L−1. CANTOS is powered for a 20% risk reduction in a composite CVD endpoint in any active arm compared to placebo, and combining doses will further improve power. The anticipated primary completion date is in late 2017 with presentation estimated for September.
Conclusions and proposed next steps
This review highlights the conflict between observational epidemiology and animal models on the one hand, and disappointing Phase III trial results on the other. This conflict remains a major issue, and is one the main difficulties for the inflammatory hypothesis of CVD. There are important debates, outwith the scope of this review, on how to make animal models more relevant to human disease (Libby et al., 2011), and also whether surrogate markers of CVD risk are truly useful to assess causality (Weintraub et al., 2015).
Conduct of RCTs is a critical step in translating a wealth of biological information into tangible benefits for patients. If successful, the CANTOS and CIRT trials may provide the rationale for using anti‐cytokine‐based and anti‐inflammatory therapies for secondary prevention of atherosclerosis‐related CVD and may start a new era in the treatment of chronic vascular disorders. If unsuccessful, these trials will not conclusively disprove the inflammatory hypothesis of atherosclerosis, but might make conducting further trials in this area much more challenging.
Inflammation contributes to atherogenesis and disease development, and therefore, several other or combined anti‐inflammatory treatments may have the potential for preventing cardiovascular events. Importantly, evaluation of risks as well as benefits must drive the development of anti‐inflammatory treatments in CVD. Atherosclerosis is a life‐long process, and it is, therefore, unlikely that the present generation of systemic anti‐inflammatory drugs will ever be prescribed in a primary prevention setting, particularly given great gains in risk reduction in recent years with better treatments of blood pressure, cholesterol and population lowering of smoking rates via smoking bans.
From a biological perspective, several fundamental questions still need to be addressed. For instance, is atherosclerosis in humans a systemic or a local (vascular) immune disease? Are tertiary lymphoid organs in the adventitial connective tissue important in human pathology? The answers to this questions will pave the way for the design of more atherosclerosis‐specific treatments targeting directly vascular (rather than systemic) immune mechanisms for therapeutic utility and potentially reducing the risk of systemic immune suppression. Existing data highlight the complex nature of the immune system, and different signalling pathways may play different roles at different the stages of the pathology. Therefore, we may need different immunomodulatory treatments to affect disease initiation, progression, and/or plaque destabilisation and rupture. Targeting and inhibiting immune‐inflammatory response(s) may be crucial at the onset of the disease. On the contrary, enhancing atheroprotective immunity by expansion of regulatory T‐cells may be the best future therapeutic strategy in secondary prevention. Vaccination approaches have also been successful in experimental models. However, translation of these findings in clinical practice has only just started (Shah et al., 2014; Kimura et al., 2015).
Any new treatment will require robust safety evaluation and testing in randomized cardiovascular outcome trials well before potential adoption in clinical practice. As these studies progress, we will learn more about whether mechanisms of vascular inflammation are indeed viable diagnostic, prognostic and therapeutic targets in atherosclerosis.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Conflict of interest
Naveed Sattar has consulted for Amgen, Sanofi and is an investigator in the CANTOS trial. He was also on the steering committee for ENTRACTE. The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgements
This work was supported by the British Heart Foundation grants PG/12/81/29897 and RE/13/5/30177; the European Commission Marie Skłodowska‐Curie Individual Fellowships 661369; the EPSRC grant EP/L014165/1; and the Tenovus Scotland PROJECT S15/24. This article is also based upon work from the COST Action BM1404 Mye‐EUNITER (www.mye‐euniter.eu), supported by COST (European Cooperation in Science and Technology) part of the EU Framework Program Horizon 2020.
Welsh, P. , Grassia, G. , Botha, S. , Sattar, N. , and Maffia, P. (2017) Targeting inflammation to reduce cardiovascular disease risk: a realistic clinical prospect?. British Journal of Pharmacology, 174: 3898–3913. doi: 10.1111/bph.13818.
Contributor Information
Paul Welsh, Email: paul.welsh@glasgow.ac.uk.
Pasquale Maffia, Email: pasquale.maffia@glasgow.ac.uk.
References
- Abbas A, Gregersen I, Holm S, Daissormont I, Bjerkeli V, Krohg‐Sørensen K et al. (2015). Interleukin 23 levels are increased in carotid atherosclerosis: possible role for the interleukin 23/interleukin 17 axis. Stroke 46: 793–799. [DOI] [PubMed] [Google Scholar]
- Agca R, Heslinga SC, Rollefstad S, Heslinga M, McInnes IB, Peters MJ et al. (2017). EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann Rheum Dis 76: 17–28. [DOI] [PubMed] [Google Scholar]
- Ait‐Oufella H, Herbin O, Lahoute C, Coatrieux C, Loyer X, Joffre J et al. (2013). Group X secreted phospholipase A2 limits the development of atherosclerosis in LDL receptor‐null mice. Arterioscler Thromb Vasc Biol 33: 466–473. [DOI] [PubMed] [Google Scholar]
- Albert MA, Danielson E, Rifai N, Ridker PM, for the PRINCE Investigators (2001). Effect of statin therapy on C‐reactive protein levels. JAMA 286: 64–70. [DOI] [PubMed] [Google Scholar]
- Alexander MR, Moehle CW, Johnson JL, Yang Z, Lee JK, Jackson CL et al. (2012). Genetic inactivation of IL‐1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J Clin Invest 122: 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015c). The concise guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen NN, Jess T (2014). Risk of cardiovascular disease in inflammatory bowel disease. World J Gastrointest Pathophysiol 5: 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz KE, Wakefield D (1996). Modulation of endothelial cell expression of ICAM‐1, E‐selectin, and VCAM‐1 by beta‐estradiol, progesterone, and dexamethasone. Cell Immunol 167: 79–85. [DOI] [PubMed] [Google Scholar]
- Babelova A, Sedding DG, Brandes RP (2013). Anti‐atherosclerotic mechanisms of statin therapy. Curr Opin Pharmacol 13: 260–264. [DOI] [PubMed] [Google Scholar]
- Bellosta S, Ferri N, Arnaboldi L, Bernini F, Paoletti R, Corsini A (2000). Pleiotropic effects of statins in atherosclerosis and diabetes. Diabetes Care 23 (Suppl 2): B72–B78. [PubMed] [Google Scholar]
- Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MA Jr (1985). Interleukin‐1 activation of vascular endothelium. Effects on procoagulant activity and leukocyte adhesion. Am J Pathol 121: 394–403. [PMC free article] [PubMed] [Google Scholar]
- Bhaskar V, Yin J, Mirza AM, Phan D, Vanegas S, Issafras H et al. (2011). Monoclonal antibodies targeting IL‐1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in Apolipoprotein E‐deficient mice. Atherosclerosis 216: 313–320. [DOI] [PubMed] [Google Scholar]
- Blom DJ, Hala T, Bolognese M, Lillestol MJ, Toth PD, Burgess L et al., DESCARTES Investigators (2014). A 52‐week placebo‐controlled trial of evolocumab in hyperlipidemia. N Engl J Med 370: 1809–1819. [DOI] [PubMed] [Google Scholar]
- Boekholdt SM, Keller TT, Wareham NJ, Luben R, Bingham SA, Day NE et al. (2005). Serum levels of type II secretory phospholipase A2 and the risk of future coronary artery disease in apparently healthy men and women: the EPIC‐Norfolk prospective population study. Arterioscler Thromb Vasc Biol 25: 839–846. [DOI] [PubMed] [Google Scholar]
- Boekholdt SM, Meuwese MC, Day NE, Luben R, Welch A, Wareham NJ et al. (2006). Plasma concentrations of ascorbic acid and C‐reactive protein, and risk of future coronary artery disease, in apparently healthy men and women: the EPIC‐Norfolk prospective population study. Br J Nutr 96: 516–522. [PubMed] [Google Scholar]
- Bostrom MA, Boyanovsky BB, Jordan CT, Wadsworth MP, Taatjes DJ, de Beer FC et al. (2007). Group v secretory phospholipase A2 promotes atherosclerosis: evidence from genetically altered mice. Arterioscler Thromb Vasc Biol 27: 600–606. [DOI] [PubMed] [Google Scholar]
- Brånén L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S (2004). Inhibition of tumor necrosis factor‐alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol 24: 2137–2142. [DOI] [PubMed] [Google Scholar]
- Bulgarelli A, Leite AC Jr, Dias AA, Maranhão RC (2013). Anti‐atherogenic effects of methotrexate carried by a lipid nanoemulsion that binds to LDL receptors in cholesterol‐fed rabbits. Cardiovasc Drugs Ther 27: 531–539. [DOI] [PubMed] [Google Scholar]
- Bulgarelli A, Martins Dias AA, Caramelli B, Maranhão RC (2012). Treatment with methotrexate inhibits atherogenesis in cholesterol‐fed rabbits. J Cardiovasc Pharmacol 59: 308–314. [DOI] [PubMed] [Google Scholar]
- Burke JE, Dennis EA (2009). Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res 50 (Suppl): S237–S242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai A, Zhou Y, Li L (2015). Rho‐GTPase and atherosclerosis: pleiotropic effects of statins. J Am Heart Assoc 4: pii: e002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon CP, Cariou B, Blom D, McKenney JM, Lorenzato C, Pordy R et al., ODYSSEY COMBO II Investigators (2015). Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: the ODYSSEY COMBO II randomized controlled trial. Eur Heart J 36: 1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas JP, Ninio E, Panayiotou A, Palmen J, Cooper JA, Ricketts SL et al. (2010). PLA2G7 genotype, lipoprotein‐associated phospholipase A2 activity, and coronary heart disease risk in 10 494 cases and 15 624 controls of European Ancestry. Circulation 121: 2284–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholesterol Treatment Trialists' (CTT) Collaborators , Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J et al. (2012). The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta‐analysis of individual data from 27 randomised trials. Lancet 380: 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins R, Reith C, Emberson J, Armitage J, Baigent C, Blackwell L et al. (2016). Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 388: 2532–2561. [DOI] [PubMed] [Google Scholar]
- Cronstein BN (2005). Low‐dose methotrexate: a mainstay in the treatment of rheumatoid arthritis. Pharmacol Rev 57: 163–172. [DOI] [PubMed] [Google Scholar]
- Dahlman‐Ghozlan K, Ortonne JP, Heilborn JD, Stephansson E (2004). Altered tissue expression pattern of cell adhesion molecules, ICAM‐1, E‐selectin and VCAM‐1, in bullous pemphigoid during methotrexate therapy. Exp Dermatol 13: 65–69. [DOI] [PubMed] [Google Scholar]
- Danesh J, Kaptoge S, Mann AG, Sarwar N, Wood A, Angleman SB et al. (2008). Long‐term interleukin‐6 levels and subsequent risk of coronary heart disease: two new prospective studies and a systematic review. PLoS Med 5: e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport P, Tipping PG (2003). The role of interleukin‐4 and interleukin‐12 in the progression of atherosclerosis in apolipoprotein E‐deficient mice. Am J Pathol 163: 1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey Smith G, Hemani G (2014). Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet 23: R89–R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson JM, LuValle PA, Zoia O, Quaglino D Jr, Giro M (1997). Ascorbate differentially regulates elastin and collagen biosynthesis in vascular smooth muscle cells and skin fibroblasts by pretranslational mechanisms. J Biol Chem 272: 345–352. [DOI] [PubMed] [Google Scholar]
- DeOliveira CC, Acedo SC, Gotardo EM, Carvalho Pde O, Rocha T, Pedrazzoli J Jr et al. (2012). Effects of methotrexate on inflammatory alterations induced by obesity: an in vivo and in vitro study. Mol Cell Endocrinol 361: 92–98. [DOI] [PubMed] [Google Scholar]
- Devlin CM, Kuriakose G, Hirsch E, Tabas I (2002). Genetic alterations of IL‐1 receptor antagonist in mice affect plasma cholesterol level and foam cell lesion size. Proc Natl Acad Sci U S A 99: 6280–6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA (2011). Interleukin‐1 in the pathogenesis and treatment of inflammatory diseases. Blood 117: 3720–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464: 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duivenvoorden R, Tang J, Cormode DP, Mieszawska AJ, Izquierdo‐Garcia D, Ozcan C et al. (2014). A statin‐loaded reconstituted high‐density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun 5: 3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhardt SU, Habersberger J, Murphy A, Chen YC, Woollard KJ, Bassler N et al. (2009). Dissociation of pentameric to monomeric C‐reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ Res 105: 128–137. [DOI] [PubMed] [Google Scholar]
- Elango T, Dayalan H, Subramanian S, Gnanaraj P, Malligarjunan H (2012). Serum interleukin‐6 levels in response to methotrexate treatment in psoriatic patients. Clin Chim Acta 413: 1652–1656. [DOI] [PubMed] [Google Scholar]
- Elhage R, Maret A, Pieraggi MT, Thiers JC, Arnal JF, Bayard F (1998). Differential effects of interleukin‐1 receptor antagonist and tumor necrosis factor binding protein on fatty‐streak formation in apolipoprotein E‐deficient mice. Circulation 97: 242–244. [DOI] [PubMed] [Google Scholar]
- Ettehad D, Emdin CA, Kiran A, Anderson SG, Callender T, Emberson J et al. (2016). Blood pressure lowering for prevention of cardiovascular disease and death: a systematic review and meta‐analysis. Lancet 387: 957–967. [DOI] [PubMed] [Google Scholar]
- Flood C, Gustafsson M, Pitas RE, Arnaboldi L, Walzem RL, Borén J (2004). Molecular mechanism for changes in proteoglycan binding on compositional changes of the core and the surface of low‐density lipoprotein‐containing human apolipoprotein B100. Arterioscler Thromb Vasc Biol 24: 564–570. [DOI] [PubMed] [Google Scholar]
- Fraser H, Hislop C, Christie RM, Rick HL, Reidy CA, Chouinard ML et al. (2009). Varespladib (A‐002), a secretory phospholipase A2 inhibitor, reduces atherosclerosis and aneurysm formation in ApoE−/− mice. J Cardiovasc Pharmacol 53: 60–65. [DOI] [PubMed] [Google Scholar]
- Freigang S, Ampenberger F, Weiss A, Kanneganti TD, Iwakura Y, Hersberger M et al. (2013). Fatty acid‐induced mitochondrial uncoupling elicits inflammasome‐independent IL‐1α and sterile vascular inflammation in atherosclerosis. Nat Immunol 14: 1045–1053. [DOI] [PubMed] [Google Scholar]
- Fulgoni VL 3rd, Keast DR, Bailey RL, Dwyer J (2011). Foods, fortificants, and supplements: where do Americans get their nutrients? J Nutr 141: 1847–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K (2006). Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L‐selectin dependent. J Exp Med 203: 1273–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkina E, Ley K (2009). Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol 27: 165–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlanda C, Dinarello CA, Mantovani A (2013). The interleukin‐1 family: back to the future. Immunity 39: 1003–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassia G, Maddaluno M, Guglielmotti A, Mangano G, Biondi G, Maffia P et al. (2009). The anti‐inflammatory agent bindarit inhibits neointima formation in both rats and hyperlipidaemic mice. Cardiovasc Res 84: 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassia G, Maddaluno M, Musilli C, De Stefano D, Carnuccio R, Di Lauro MV et al. (2010). The I{kappa}B kinase inhibitor nuclear factor‐{kappa}B essential modulator‐binding domain peptide for inhibition of injury‐induced neointimal formation. Arterioscler Thromb Vasc Biol 30: 2458–2466. [DOI] [PubMed] [Google Scholar]
- Greenberg JD, Kremer JM, Curtis JR, Hochberg MC, Reed G, Tsao P et al., CORRONA Investigators (2011). Tumour necrosis factor antagonist use and associated risk reduction of cardiovascular events among patients with rheumatoid arthritis. Ann Rheum Dis 70: 576–582. [DOI] [PubMed] [Google Scholar]
- Hansson GK, Libby P, Tabas I (2015). Inflammation and plaque vulnerability. J Intern Med 278: 483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer AD, Uyttenhove C, de Vos P, Stroobant V, Renauld JC, van Berkel TJ et al. (2005). Blockade of interleukin‐12 function by protein vaccination attenuates atherosclerosis. Circulation 112: 1054–1062. [DOI] [PubMed] [Google Scholar]
- Herder C, Donath MY (2015). Interleukin‐1 receptor antagonist: friend or foe to the heart? Lancet Diabetes Endocrinol 3: 228–229. [DOI] [PubMed] [Google Scholar]
- Holmes MV, Simon T, Exeter HJ, Folkersen L, Asselbergs FW, Guardiola M et al. (2013). Secretory phospholipase A(2)‐IIA and cardiovascular disease: a mendelian randomization study. J Am Coll Cardiol 62: 1966–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Mohanta SK, Yin C, Peng L, Ma Z, Srikakulapu P et al. (2015). Artery tertiary lymphoid organs control aorta immunity and protect against atherosclerosis via vascular smooth muscle cell lymphotoxin β receptors. Immunity 42: 1100–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HY, Appel LJ, Croft KD, Miller ERI, Mori TA, Puddey IB (2002). Effects of vitamin C and vitamin E on in vivo lipid peroxidation: results of a randomized controlled trial. Am J Clin Nutr 76: 549–555. [DOI] [PubMed] [Google Scholar]
- Huber SA, Sakkinen P, Conze D, Hardin N, Tracy R (1999). Interleukin‐6 exacerbates early atherosclerosis in mice. Arterioscler Thromb Vasc Biol 19: 2364–2367. [DOI] [PubMed] [Google Scholar]
- Interleukin 1 Genetics Consortium (2015). Cardiometabolic effects of genetic upregulation of the interleukin 1 receptor antagonist: a Mendelian randomisation analysis. Lancet Diabetes Endocrinol 3: 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Interleukin‐6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium , Swerdlow DI, Holmes MV, Kuchenbaecker KB, Engmann JE, Shah T et al. (2012). The interleukin‐6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet 379: 1214–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivandic B, Castellani LW, Wang XP, Qiao JH, Mehrabian M, Navab M et al. (1999). Role of group II secretory phospholipase A2 in atherosclerosis: 1. Increased atherogenesis and altered lipoproteins in transgenic mice expressing group IIa phospholipase A2. Arterioscler Thromb Vasc Biol 19: 1284–1290. [DOI] [PubMed] [Google Scholar]
- Kalliolias GD, Ivashkiv LB (2016). TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol 12: 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaptoge S, Di Angelantonio E, Pennells L, Wood AM, White IR, Gao P et al., Emerging Risk Factors Collaboration (2012). C‐reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med 367: 1310–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Tse K, Sette A, Ley K (2015). Vaccination to modulate atherosclerosis. Autoimmunity 48: 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinlay S (2007). Low‐density lipoprotein‐dependent and ‐independent effects of cholesterol‐lowering therapies on C‐reactive protein. J Am Coll Cardiol 49: 2003–2009. [DOI] [PubMed] [Google Scholar]
- Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y et al. (2003). Lack of interleukin‐1beta decreases the severity of atherosclerosis in ApoE‐deficient mice. Arterioscler Thromb Vasc Biol 23: 656–660. [DOI] [PubMed] [Google Scholar]
- Kleinman Y, Krul ES, Burnes M, Aronson W, Pfleger B, Schonfeld G (1988). Lipolysis of LDL with phospholipase A2 alters the expression of selected apoB‐100 epitopes and the interaction of LDL with cells. J Lipid Res 29: 729–743. [PubMed] [Google Scholar]
- Koltsova EK, Garcia Z, Chodaczek G, Landau M, McArdle S, Scott SR et al. (2012). Dynamic T cell‐APC interactions sustain chronic inflammation in atherosclerosis. J Clin Invest 122: 3114–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ et al. (2000). The HMG‐CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med 6: 1004–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak B, Mulhaupt F, Myit S, Mach F (2000). Statins as a newly recognized type of immunomodulator. Nat Med 6: 1399–1402. [DOI] [PubMed] [Google Scholar]
- Lee TS, Yen HC, Pan CC, Chau LY (1999). The role of interleukin 12 in the development of atherosclerosis in ApoE‐deficient mice. Arterioscler Thromb Vasc Biol 19: 734–742. [DOI] [PubMed] [Google Scholar]
- Leite AC Jr, Solano TV, Tavares ER, Maranhão RC (2015). Use of combined chemotherapy with etoposide and methotrexate, both associated to lipid nanoemulsions for atherosclerosis treatment in cholesterol‐fed rabbits. Cardiovasc Drugs Ther 29: 15–22. [DOI] [PubMed] [Google Scholar]
- Leite JO, Vaishnav U, Puglisi M, Fraser H, Trias J, Fernandez ML (2009). A‐002 (Varespladib), a phospholipase A2 inhibitor, reduces atherosclerosis in guinea pigs. BMC Cardiovasc Disord 9: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R, Prospective Studies Collaboration (2002). Age‐specific relevance of usual blood pressure to vascular mortality: a meta‐analysis of individual data for one million adults in 61 prospective studies. Lancet 360: 1903–1913. [DOI] [PubMed] [Google Scholar]
- Liao JK, Laufs U (2005). Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol 45: 89–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Hansson GK (2015). Inflammation and immunity in diseases of the arterial tree: players and layers. Circ Res 116: 307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Hansson GK (2011). Progress and challenges in translating the biology of atherosclerosis. Nature 473: 317–325. [DOI] [PubMed] [Google Scholar]
- Macritchie N, Grassia G, Sabir SR, Maddaluno M, Welsh P, Sattar N et al. (2012). Plasmacytoid dendritic cells play a key role in promoting atherosclerosis in apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol 32: 2569–2579. [DOI] [PubMed] [Google Scholar]
- Maddaluno M, Grassia G, Di Lauro MV, Parisi A, Maione F, Cicala C et al. (2012). Bindarit inhibits human coronary artery smooth muscle cell proliferation, migration and phenotypic switching. PLoS One 7: e47464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda N, Hagihara H, Nakata Y, Hiller S, Wilder J, Reddick R (2000). Aortic wall damage in mice unable to synthesize ascorbic acid. Proc Natl Acad Sci U S A 97: 841–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffia P, Zinselmeyer BH, Ialenti A, Kennedy S, Baker AH, McInnes IB et al. (2007). Images in cardiovascular medicine. Multiphoton microscopy for 3‐dimensional imaging of lymphocyte recruitment into apolipoprotein‐E‐deficient mouse carotid artery. Circulation 115: e326–e328. [DOI] [PubMed] [Google Scholar]
- Makheja AN, Bloom S, Muesing R, Simon T, Bailey JM (1989). Anti‐inflammatory drugs in experimental atherosclerosis. 7. Spontaneous atherosclerosis in WHHL rabbits and inhibition by cortisone acetate. Atherosclerosis 76: 155–161. [DOI] [PubMed] [Google Scholar]
- Margaritis M, Channon KM, Antoniades C (2014). Statins as regulators of redox state in the vascular endothelium: beyond lipid lowering. Antioxid Redox Signal 20: 1198–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes IB, Thompson L, Giles JT, Bathon JM, Salmon JE, Beaulieu AD et al. (2015). Effect of interleukin‐6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo‐controlled study. Ann Rheum Dis 74: 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L et al. (2011). Atherosclerosis in ApoE‐deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis 2: e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micha R, Imamura F, Wyler von Ballmoos M, Solomon DH, Hernán MA, Ridker PM et al. (2011). Systematic review and meta‐analysis of methotrexate use and risk of cardiovascular disease. Am J Cardiol 108: 1362–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira E, León B, Barber DF, Jiménez‐Baranda S, Goya I, Almonacid L et al. (2008). Statins induce regulatory T cell recruitment via a CCL1 dependent pathway. J Immunol 181: 3524–3534. [DOI] [PubMed] [Google Scholar]
- Mohler ER 3rd, Ballantyne CM, Davidson MH, Hanefeld M, Ruilope LM, Johnson JL et al., Darapladib Investigators (2008). The effect of darapladib on plasma lipoprotein‐associated phospholipase A2 activity and cardiovascular biomarkers in patients with stable coronary heart disease or coronary heart disease risk equivalent: the results of a multicenter, randomized, double‐blind, placebo‐controlled study. J Am Coll Cardiol 51: 1632–1641. [DOI] [PubMed] [Google Scholar]
- Montezano AC, Dulak‐Lis M, Tsiropoulou S, Harvey A, Briones AM, Touyz RM (2015). Oxidative stress and human hypertension: vascular mechanisms, biomarkers, and novel therapies. Can J Cardiol 31: 631–641. [DOI] [PubMed] [Google Scholar]
- Mulhaupt F, Matter CM, Kwak BR, Pelli G, Veillard NR, Burger F et al. (2003). Statins (HMG‐CoA reductase inhibitors) reduce CD40 expression in human vascular cells. Cardiovasc Res 59: 755–766. [DOI] [PubMed] [Google Scholar]
- Murad S, Grove D, Lindberg KA, Reynolds G, Sivarajah A, Pinnell SR (1981). Regulation of collagen synthesis by ascorbic acid. Proc Natl Acad Sci U S A 78: 2879–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myasoedova E, Crowson CS, Kremers HM, Fitz‐Gibbon PD, Therneau TM, Gabriel SE (2010). Total cholesterol and LDL levels decrease before rheumatoid arthritis. Ann Rheum Dis 69: 1310–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung SK, Ju W, Cho B, Oh SW, Park SM, Koo BK et al., Korean Meta‐Analysis Study Group (2013). Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta‐analysis of randomised controlled trials. BMJ 346: f10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata Y, Maeda N (2002). Vulnerable atherosclerotic plaque morphology in apolipoprotein E‐deficient mice unable to make ascorbic Acid. Circulation 105: 1485–1490. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ, Kastelein JJ, Schwartz GG, Bash D, Rosenson RS, Cavender MA et al., VISTA‐16 Investigators (2014). Varespladib and cardiovascular events in patients with an acute coronary syndrome: the VISTA‐16 randomized clinical trial. JAMA 311: 252–262. [DOI] [PubMed] [Google Scholar]
- O'Donoghue ML, Braunwald E, White HD, Steen DN, Lukas MA, Tarka E et al., for the SOLID‐TIMI 52 Investigators (2014). Effect of darapladib on major coronary events after an acute coronary syndrome: the SOLID‐TIMI 52 randomized clinical trial. JAMA 312: 1006–1015. [DOI] [PubMed] [Google Scholar]
- Ogdie A, Yu Y, Haynes K, Love TJ, Maliha S, Jiang Y et al. (2015). Risk of major cardiovascular events in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a population‐based cohort study. Ann Rheum Dis 74: 326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta H, Wada H, Niwa T, Kirii H, Iwamoto N, Fujii H et al. (2005). Disruption of tumor necrosis factor‐alpha gene diminishes the development of atherosclerosis in ApoE‐deficient mice. Atherosclerosis 180: 11–17. [DOI] [PubMed] [Google Scholar]
- Pauling L (1971). The significance of the evidence about ascorbic acid and the common cold. Proc Natl Acad Sci U S A 68: 2678–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters MJ, Welsh P, McInnes IB, Wolbink G, Dijkmans BA, Sattar N et al. (2010). Tumour necrosis factor {alpha} blockade reduces circulating N‐terminal pro‐brain natriuretic peptide levels in patients with active rheumatoid arthritis: results from a prospective cohort study. Ann Rheum Dis 69: 1281–1285. [DOI] [PubMed] [Google Scholar]
- Qiao H, Bell J, Juliao S, Li L, May JM (2009). Ascorbic acid uptake and regulation of type I collagen synthesis in cultured vascular smooth muscle cells. J Vasc Res 46: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarck R, De Geest B, Stengel D, Mertens A, Lox M, Theilmeier G et al. (2001). Adenovirus‐mediated gene transfer of human platelet‐activating factor‐acetylhydrolase prevents injury‐induced neointima formation and reduces spontaneous atherosclerosis in apolipoprotein E‐deficient mice. Circulation 103: 2495–2500. [DOI] [PubMed] [Google Scholar]
- Rajamäki K, Lappalainen J, Oörni K, Välimäki E, Matikainen S, Kovanen PT et al. (2010). Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One 5: e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastogi S, Rizwani W, Joshi B, Kunigal S, Chellappan SP (2012). TNF‐α response of vascular endothelial and vascular smooth muscle cells involve differential utilization of ASK1 kinase and p73. Cell Death Differ 19: 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss AB, Carsons SE, Anwar K, Rao S, Edelman SD, Zhang H et al. (2008). Atheroprotective effects of methotrexate on reverse cholesterol transport proteins and foam cell transformation in human THP‐1 monocyte/macrophages. Arthritis Rheum 58: 3675–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaie‐Majd A, Prager GW, Bucek RA, Schernthaner GH, Maca T, Kress HG et al. (2003). Simvastatin reduces the expression of adhesion molecules in circulating monocytes from hypercholesterolemic patients. Arterioscler Thromb Vasc Biol 23: 397–403. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ et al., JUPITER Trial Study Group (2009). Reduction in C‐reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet 373: 1175–1182. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E (1999). Long‐term effects of pravastatin on plasma concentration of C‐reactive protein. The Cholesterol and Recurrent Events (CARE) investigators. Circulation 100: 230–235. [DOI] [PubMed] [Google Scholar]
- del Rincón ID, Williams K, Stern MP, Freeman GL, Escalante A (2001). High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum 44: 2737–2745. [DOI] [PubMed] [Google Scholar]
- Robertson J, Peters MJ, McInnes IB, Sattar N (2013). Changes in lipid levels with inflammation and therapy in RA: a maturing paradigm. Nat Rev Rheumatol 9: 513–523. [DOI] [PubMed] [Google Scholar]
- Romano M, Diomede L, Sironi M, Massimiliano L, Sottocorno M, Polentarutti N et al. (2000). Inhibition of monocyte chemotactic protein‐1 synthesis by statins. Lab Invest 80: 1095–1100. [DOI] [PubMed] [Google Scholar]
- Rosenson RS (2010). Phospholipase A2 inhibition and atherosclerotic vascular disease: prospects for targeting secretory and lipoprotein‐associated phospholipase A2 enzymes. Curr Opin Lipidol 21: 473–480. [DOI] [PubMed] [Google Scholar]
- Rosenson RS, Hurt‐Camejo E (2012). Phospholipase A2 enzymes and the risk of atherosclerosis. Eur Heart J 33: 2899–2909. [DOI] [PubMed] [Google Scholar]
- Rössig L, Hoffmann J, Hugel B, Mallat Z, Haase A, Freyssinet JM et al. (2001). Vitamin C inhibits endothelial cell apoptosis in congestive heart failure. Circulation 104: 2182–2187. [DOI] [PubMed] [Google Scholar]
- Roubille C, Richer V, Starnino T, McCourt C, McFarlane A, Fleming P et al. (2015). The effects of tumour necrosis factor inhibitors, methotrexate, non‐steroidal anti‐inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta‐analysis. Ann Rheum Dis 74: 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan C, Leonardi CL, Krueger JG, Kimball AB, Strober BE, Gordon KB et al. (2011). Association between biologic therapies for chronic plaque psoriasis and cardiovascular events: a meta‐analysis of randomized controlled trials. JAMA 306: 864–871. [DOI] [PubMed] [Google Scholar]
- Saeed RW, Peng T, Metz CN (2003). Ascorbic acid blocks the growth inhibitory effect of tumor necrosis factor‐alpha on endothelial cells. Exp Biol Med (Maywood) 228: 855–865. [DOI] [PubMed] [Google Scholar]
- Sage AP, Murphy D, Maffia P, Masters LM, Sabir SR, Baker LL et al. (2014). MHC class II‐restricted antigen presentation by plasmacytoid dendritic cells drives proatherogenic T cell immunity. Circulation 130: 1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarwar N, Butterworth AS, Freitag DF, Gregson J, Willeit P, Gorman DN et al., IL6R Genetics Consortium Emerging Risk Factors Collaboration (2012). Interleukin‐6 receptor pathways in coronary heart disease: a collaborative meta‐analysis of 82 studies. Lancet 379: 1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattar N, McCarey DW, Capell H, McInnes IB (2003). Explaining how “high‐grade” systemic inflammation accelerates vascular risk in rheumatoid arthritis. Circulation 108: 2957–2963. [DOI] [PubMed] [Google Scholar]
- Scalia R, Gooszen ME, Jones SP, Hoffmeyer M, Rimmer DM 3rd, Trocha SD et al. (2001). Simvastatin exerts both anti‐inflammatory and cardioprotective effects in apolipoprotein E‐deficient mice. Circulation 103: 2598–2603. [DOI] [PubMed] [Google Scholar]
- Schett G, Dayer JM, Manger B (2016). Interleukin‐1 function and role in rheumatic disease. Nat Rev Rheumatol 12: 14–24. [DOI] [PubMed] [Google Scholar]
- Schieffer B, Selle T, Hilfiker A, Hilfiker‐Kleiner D, Grote K, Tietge UJ et al. (2004). Impact of interleukin‐6 on plaque development and morphology in experimental atherosclerosis. Circulation 110: 3493–3500. [DOI] [PubMed] [Google Scholar]
- Schuett H, Oestreich R, Waetzig GH, Annema W, Luchtefeld M, Hillmer A et al. (2012). Transsignaling of interleukin‐6 crucially contributes to atherosclerosis in mice. Arterioscler Thromb Vasc Biol 32: 281–290. [DOI] [PubMed] [Google Scholar]
- Serruys PW, García‐García HM, Buszman P, Erne P, Verheye S, Aschermann M et al., Integrated Biomarker and Imaging Study‐2 Investigators (2008). Effects of the direct lipoprotein‐associated phospholipase A(2) inhibitor darapladib on human coronary atherosclerotic plaque. Circulation 118: 1172–1182. [DOI] [PubMed] [Google Scholar]
- Sever PS, Poulter NR, Chang CL, Hingorani A, Thom SA, Hughes AD et al., ASCOT Investigators (2012). Evaluation of C‐reactive protein prior to and on‐treatment as a predictor of benefit from atorvastatin: observations from the Anglo‐Scandinavian Cardiac Outcomes Trial. Eur Heart J 33: 486–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever PS, Poulter NR, Chang CL, Thom SA, Hughes AD, Welsh P et al., ASCOT Investigators (2013). Evaluation of C‐reactive protein before and on‐treatment as a predictor of benefit of atorvastatin: a cohort analysis from the Anglo‐Scandinavian Cardiac Outcomes Trial lipid‐lowering arm. J Am Coll Cardiol 62: 717–729. [DOI] [PubMed] [Google Scholar]
- Shah PK, Chyu KY, Dimayuga PC, Nilsson J (2014). Vaccine for atherosclerosis. J Am Coll Cardiol 64: 2779–2791. [DOI] [PubMed] [Google Scholar]
- Shaposhnik Z, Wang X, Trias J, Fraser H, Lusis AJ (2009). The synergistic inhibition of atherogenesis in apoE−/− mice between pravastatin and the sPLA2 inhibitor varespladib (A‐002). J Lipid Res 50: 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh JA, Wells GA, Christensen R, Tanjong Ghogomu E, Maxwell L, Macdonald JK et al. (2011). Adverse effects of biologics: a network meta‐analysis and Cochrane overview. Cochrane Database Syst Rev 2: CD008794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirtori CR (2014). The pharmacology of statins. Pharmacol Res 88: 3–11. [DOI] [PubMed] [Google Scholar]
- Sniderman AD, Williams K, Contois JH, Monroe HM, McQueen MJ, de Graaf J et al. (2011). A meta‐analysis of low‐density lipoprotein cholesterol, non‐high‐density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ Cardiovasc Qual Outcomes 4: 337–345. [DOI] [PubMed] [Google Scholar]
- Soedamah‐Muthu SS, Livingstone SJ, Charlton‐Menys V, Betteridge DJ, Hitman GA, Neil HA et al., CARDS Investigators (2015). Effect of atorvastatin on C‐reactive protein and benefits for cardiovascular disease in patients with type 2 diabetes: analyses from the Collaborative Atorvastatin Diabetes Trial. Diabetologia 58: 1494–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DH, Reed GW, Kremer JM, Curtis JR, Farkouh ME, Harrold LR et al. (2015). Disease activity in rheumatoid arthritis and the risk of cardiovascular events. Arthritis Rheumatol 67: 1449–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souverein PC, Berard A, Van Staa TP, Cooper C, Egberts AC, Leufkens HG et al. (2004). Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case‐control study. Heart 90: 859–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague AH, Khalil RA (2009). Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol 78: 539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikakulapu P, Hu D, Yin C, Mohanta SK, Bontha SV, Peng L et al. (2016). Artery tertiary lymphoid organs control multilayered territorialized atherosclerosis B‐cell responses in aged ApoE−/− mice. Arterioscler Thromb Vasc Biol 36: 1174–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober BE, Crowley JJ, Yamauchi PS, Olds M, Williams DA (2011). Efficacy and safety results from a phase III, randomized controlled trial comparing the safety and efficacy of briakinumab with etanercept and placebo in patients with moderate to severe chronic plaque psoriasis. Br J Dermatol 165: 661–668. [DOI] [PubMed] [Google Scholar]
- Sukovich DA, Kauser K, Shirley FD, DelVecchio V, Halks‐Miller M, Rubanyi GM (1998). Expression of interleukin‐6 in atherosclerotic lesions of male ApoE‐knockout mice: inhibition by 17beta‐estradiol. Arterioscler Thromb Vasc Biol 18: 1498–1505. [DOI] [PubMed] [Google Scholar]
- Swirski FK, Nahrendorf M, Libby P (2016). Mechanisms of myeloid cell modulation of atherosclerosis. Microbiol Spectr 4: MCHD‐0026‐2015. [DOI] [PubMed] [Google Scholar]
- Talmud PJ, Holmes MV (2015). Deciphering the causal role of sPLA2s and Lp‐PLA2 in coronary heart disease. Arterioscler Thromb Vasc Biol 35: 2281–2289. [DOI] [PubMed] [Google Scholar]
- Tay C, Liu YH, Hosseini H, Kanellakis P, Cao A, Peter K et al. (2016). B‐cell‐specific depletion of tumour necrosis factor alpha inhibits atherosclerosis development and plaque vulnerability to rupture by reducing cell death and inflammation. Cardiovasc Res 111: 385–397. [DOI] [PubMed] [Google Scholar]
- Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM et al. (2015). IL‐12 and IL‐23 cytokines: from discovery to targeted therapies for immune‐mediated inflammatory diseases. Nat Med 21: 719–729. [DOI] [PubMed] [Google Scholar]
- Thompson A, Gao P, Orfei L, Watson S, Di Angelantonio E, Kaptoge S et al., Lp‐PLA(2) Studies Collaboration (2010). Lipoprotein‐associated phospholipase A(2) and risk of coronary disease, stroke, and mortality: collaborative analysis of 32 prospective studies. Lancet 375: 1536–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tits LJ, Stienstra R, van Lent PL, Netea MG, Joosten LA, Stalenhoef AF (2011). Oxidized LDL enhances pro‐inflammatory responses of alternatively activated M2 macrophages: a crucial role for Krüppel‐like factor 2. Atherosclerosis 214: 345–349. [DOI] [PubMed] [Google Scholar]
- Toedter GP, Blank M, Lang Y, Chen D, Sandborn WJ, de Villiers WJ (2009). Relationship of C‐reactive protein with clinical response after therapy with ustekinumab in Crohn's disease. Am J Gastroenterol 104: 2768–2773. [DOI] [PubMed] [Google Scholar]
- Turunen P, Puhakka H, Rutanen J, Hiltunen MO, Heikura T, Gruchala M et al. (2005). Intravascular adenovirus‐mediated lipoprotein‐associated phospholipase A2 gene transfer reduces neointima formation in balloon‐denuded rabbit aorta. Atherosclerosis 179: 27–33. [DOI] [PubMed] [Google Scholar]
- Tzellos T, Kyrgidis A, Zouboulis CC (2013). Re‐evaluation of the risk for major adverse cardiovascular events in patients treated with anti‐IL‐12/23 biological agents for chronic plaque psoriasis: a meta‐analysis of randomized controlled trials. J Eur Acad Dermatol Venereol 27: 622–627. [DOI] [PubMed] [Google Scholar]
- Walker BR (2007). Glucocorticoids and cardiovascular disease. Eur J Endocrinol 157: 545–559. [DOI] [PubMed] [Google Scholar]
- Wang WY, Zhang J, Wu WY, Li J, Ma YL, Chen WH et al. (2011). Inhibition of lipoprotein‐associated phospholipase A2 amelioratesb inflammation and decreases atherosclerotic plaque formation in ApoE‐deficient mice. PLoS One 6: e23425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XL, Zhou YL, Sun W, Li L (2016). Rosuvastatin attenuates CD40L‐induced downregulation of extracellular matrix production in human aortic smooth muscle cells via TRAF6‐JNK‐NF‐κB pathway. PLoS One 11: e0153919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson C, Whittaker S, Smith N, Vora AJ, Dumonde DC, Brown KA (1996). IL‐6 acts on endothelial cells to preferentially increase their adherence for lymphocytes. Clin Exp Immunol 105: 112–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub WS, Lüscher TF, Pocock S (2015). The perils of surrogate endpoints. Eur Heart J 36: 2212–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh P, Doolin O, Willeit P, Packard C, Macfarlane P, Cobbe S et al. (2013). N‐terminal pro‐B‐type natriuretic peptide and the prediction of primary cardiovascular events: results from 15‐year follow‐up of WOSCOPS. Eur Heart J 34: 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh P, Hart C, Papacosta O, Preiss D, McConnachie A, Murray H et al. (2016a). Prediction of cardiovascular disease risk by cardiac biomarkers in 2 United Kingdom cohort studies: does utility depend on risk thresholds for treatment? Hypertension 67: 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh P, Murray HM, Ford I, Trompet S, de Craen AJ, Jukema JW et al., PROSPER Study Group (2011). Circulating interleukin‐10 and risk of cardiovascular events: a prospective study in the elderly at risk. Arterioscler Thromb Vasc Biol 31: 2338–2344. [DOI] [PubMed] [Google Scholar]
- Welsh P, Tuckwell K, McInnes IB, Sattar N (2016b). Effect of IL‐6 receptor blockade on high‐sensitivity troponin T and NT‐proBNP in rheumatoid arthritis. Atherosclerosis 254: 167–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng TC, Yang YH, Lin SJ, Tai SH (2010). A systematic review and meta‐analysis on the therapeutic equivalence of statins. J Clin Pharm Ther 35: 139–151. [DOI] [PubMed] [Google Scholar]
- Wensley F, Gao P, Burgess S, Kaptoge S, Di Angelantonio E, Shah T et al., C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC) (2011). Association between C reactive protein and coronary heart disease: mendelian randomisation analysis based on individual participant data. BMJ 342: d548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White HD, Held C, Stewart R, Tarka E, Brown R, Davies RY et al., STABILITY Investigators (2014). Darapladib for preventing ischemic events in stable coronary heart disease. N Engl J Med 370: 1702–1711. [DOI] [PubMed] [Google Scholar]
- Wilensky RL, Shi Y, Mohler ER 3rd, Hamamdzic D, Burgert ME, Li J et al. (2008). Inhibition of lipoprotein‐associated phospholipase A2 reduces complex coronary atherosclerotic plaque development. Nat Med 14: 1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willeit P, Thompson A, Aspelund T, Rumley A, Eiriksdottir G, Lowe G et al. (2013). Hemostatic factors and risk of coronary heart disease in general populations: new prospective study and updated meta‐analyses. PLoS One 8: e55175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis GC (1953). An experimental study of intimal ground substance in atherosclerosis. Can Med Assoc J 69: 17–22. [PMC free article] [PubMed] [Google Scholar]
- Wong B, Lumma WC, Smith AM, Sisko JT, Wright SD, Cai TQ (2001). Statins suppress THP‐1 cell migration and secretion of matrix metalloproteinase 9 by inhibiting geranylgeranylation. J Leukoc Biol 69: 959–962. [PubMed] [Google Scholar]
- Woodward M, Rumley A, Welsh P, MacMahon S, Lowe G (2007). A comparison of the associations between seven hemostatic or inflammatory variables and coronary heart disease. J Thromb Haemost 5: 1795–1800. [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO) (2011). Global status report on noncommunicable diseases 2010. ISBN: 978 92 4 156422 9.
- Xanthoulea S, Gijbels MJ, van der Made I, Mujcic H, Thelen M, Vergouwe MN et al. (2008). P55 tumour necrosis factor receptor in bone marrow‐derived cells promotes atherosclerosis development in low‐density lipoprotein receptor knock‐out mice. Cardiovasc Res 80: 309–318. [DOI] [PubMed] [Google Scholar]
- Ye Y, Li J, Yuan Z (2013). Effect of antioxidant vitamin supplementation on cardiovascular outcomes: a meta‐analysis of randomized controlled trials. PLoS One 8: e56803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Song H (2008). Antioxidant vitamins intake and the risk of coronary heart disease: meta‐analysis of cohort studies. Eur J Cardiovasc Prev Rehabil 15: 26–34. [DOI] [PubMed] [Google Scholar]
- Yilmaz A, Reiss C, Weng A, Cicha I, Stumpf C, Steinkasserer A et al. (2006). Differential effects of statins on relevant functions of human monocyte‐derived dendritic cells. J Leukoc Biol 79: 529–538. [DOI] [PubMed] [Google Scholar]
- Youssef S, Stüve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM et al. (2002). The HMG‐CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature 420: 78–84. [DOI] [PubMed] [Google Scholar]
- Zalewski A, Macphee C (2005). Role of lipoprotein‐associated phospholipase A2 in atherosclerosis: biology, epidemiology, and possible therapeutic target. Arterioscler Thromb Vasc Biol 25: 923–931. [DOI] [PubMed] [Google Scholar]
- Zhao L, Cuff CA, Moss E, Wille U, Cyrus T, Klein EA et al. (2002). Selective interleukin‐12 synthesis defect in 12/15‐lipoxygenase‐deficient macrophages associated with reduced atherosclerosis in a mouse model of familial hypercholesterolemia. J Biol Chem 277: 35350–35356. [DOI] [PubMed] [Google Scholar]