

Abstract
Background and Purpose
The side effects of cyclooxygenase‐2 (COX‐2) inhibitors on the cardiovascular system could be associated with reduced prostaglandin (PG)I2 synthesis. Microsomal PGE synthase‐1 (mPGES‐1) catalyses the formation of PGE2 from COX‐derived PGH2. This enzyme is induced under inflammatory conditions and constitutes an attractive target for novel anti‐inflammatory drugs. However, it is not known whether mPGES‐1 inhibitors could be devoid of cardiovascular side effects. The aim of this study was to compare, in vitro, the effects of mPGES‐1 and COX‐2 inhibitors on vascular tone in human blood vessels.
Experimental Approach
The vascular tone and prostanoid release from internal mammary artery (IMA) and saphenous vein (SV) incubated for 30 min with inhibitors of mPGES‐1 or COX‐2 were investigated under normal and inflammatory conditions.
Key Results
In inflammatory conditions, mPGES‐1 and COX‐2 proteins were more expressed, and increased levels of PGE2 and PGI2 were released. COX‐2 and NOS inhibitors increased noradrenaline induced vascular contractions in IMA under inflammatory conditions while no effect was observed in SV. Interestingly, the mPGES‐1 inhibitor significantly reduced (30–40%) noradrenaline‐induced contractions in both vessels. This effect was reversed by an IP (PGI2 receptor) antagonist but not modified by NOS inhibition. Moreover, PGI2 release was increased with the mPGES‐1 inhibitor and decreased with the COX‐2 inhibitor, while both inhibitors reduced PGE2 release.
Conclusions and Implications
In contrast to COX‐2 inhibition, inhibition of mPGES‐1 reduced vasoconstriction by increasing PGI2 synthesis. Targeting mPGES‐1 could provide a lower risk of cardiovascular side effects, compared with those of the COX‐2 inhibitors.
Linked Articles
This article is part of a themed section on Targeting Inflammation to Reduce Cardiovascular Disease Risk. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.22/issuetoc and http://onlinelibrary.wiley.com/doi/10.1111/bcp.v82.4/issuetoc
Abbreviations
- C3
compound 3, 1‐(1‐isopropyl‐5,6‐dimethyl‐1H‐ benzoimidazol‐2‐yl)‐piperidine‐4‐carboxylic acid cyclopentylamide
- COXIB
selective COX‐2 inhibitor
- IMA
internal mammary artery
- mPGES‐1
microsomal PGE synthase‐1
- NSAIDs
non‐steroidal anti‐inflammatory drugs
- SMC
smooth muscle cell
- SV
saphenous vein
Introduction
The effects of selective cyclooxygenase‐2 (COX‐2) inhibitors (COXIB) or non‐steroidal anti‐inflammatory drugs (NSAIDs) are mediated through the inhibition of COX‐2 enzyme (Marnett, 2009), which mostly decreases prostaglandin E2 (PGE2) production at inflammatory sites (Ibuki et al., 2003; Alvarez‐Soria et al., 2008). However, the gastrointestinal side effects of NSAIDs and the cardiovascular side effects associated with both COXIBs and some NSAIDs (such as ibuprofen and diclofenac) limit their use (Bhala et al., 2013; McGettigan and Henry, 2013). Blood pressure and thrombosis are strongly regulated by prostacyclin (PGI2), PGE2 and thromboxane (Tx)A2 synthesized through the COX pathway (Norel, 2007). PGI2 induces vasodilatation and inhibits platelet aggregation via the activation of IP receptors and adenylate cyclase (Reid and Kinsella, 2015). Therefore, the deleterious cardiovascular events induced by COXIBs and NSAIDs have been linked to a decreased level of PGI2. In addition, recent studies have shown that deletion of the COX‐2 gene or treatment with COXIBs in mice lead to vascular dysfunction by decreasing NO release (Yu et al., 2012) or increasing the endogenous NOS inhibitor, asymmetric dimethylarginine and production at a renal level (Ahmetaj‐Shala et al., 2015). For these reasons, new drugs that only suppress synthesis of pro‐inflammatory PGE2 without reducing PGI2 or NO synthesis could be effective in the treatment of inflammatory diseases without increasing cardiovascular risks.
PGE2 controls the vascular tone by inducing contraction (via EP3 receptors) in internal mammary arteries (IMA) or relaxation (via EP4 receptors) in saphenous veins (SV) (Foudi et al., 2011). Three PGE synthase (PGES) isoforms specifically catalyse the final step of PGE2 biosynthesis from PGH2. Among these, microsomal (m)PGES‐1 is the major contributor of PGE2 synthesis in human vascular smooth muscle cells (SMC). This enzyme is constitutively present at a low level or strongly expressed and co‐induced with COX‐2 in inflammatory conditions (Jakobsson et al., 1999; Camacho et al., 2007; Gomez et al., 2013).
PGE2 has been described as a key mediator of pain and inflammation in many animal studies. Reduced pain hypersensitivity, fever and inflammation have been observed in mice deficient for mPGES‐1 (Engblom et al., 2003; Xu et al., 2008) similarly to ablation of COX‐2 (Myers et al., 2000). In addition, mPGES‐1 knockout mice did not exhibit increased thrombogenesis or increased blood pressure, in contrast with COX‐2 knockout mice (Cheng et al., 2006; Wang et al., 2011; Chen et al., 2013). Therefore, mPGES‐1 inhibitors could be safer novel anti‐inflammatory agents by reducing only PGE2 levels in inflammatory diseases. Consequently, during the past few years, novel mPGES‐1 inhibitors [such as MF63, MK886, PF‐4693627, YS121, LY3023703 and compound 3 (C3)] have been developed (Leclerc et al., 2013; Jin et al., 2015; Koeberle and Werz, 2015). However, the potential cardiovascular risks of the mPGES‐1 inhibitors have not been thoroughly evaluated in in vitro models.
For this reason, the aim of our study was to determine and compare the in vitro effects of an mPGES‐1 inhibitor (C3) and COXIBs on the vascular tone of human vessels such as IMA and SV. The mechanisms underlying these effects associated with PGI2 release were analysed in these vessels with or without inflammation.
Methods
Human vascular preparations
This study was approved by the Institutional Review Board of the Istanbul University Institute of Cardiology and the Ethics Committee of INSERM (the French National Institute for Health and Medical Research). These tissues are considered as surgical waste in accordance with French ethical laws (L.1211‐3‐L.1211‐9). All experiments with human subjects were performed in accordance with the Helsinki Declaration. The study was performed on isolated segments of human IMA and SV, with intact endothelium obtained from patients (IMA: 27 males and 8 females aged 67 ± 2; SV: 35 males and 12 females aged 64 ± 2) who had undergone coronary artery bypass surgery. The vascular preparations have been used either after organ culture (18 h incubation) or in the next hour following surgery without any treatment: ‘Normal (0 h)’ conditions.
Organ cultures
The IMA and SV were dissected free from connective tissue, cut into rings of 2–4 mm width and placed immediately into 12‐well plates containing RPMI supplemented with PSA (penicillin, 1000 IU·mL−1; streptomycin, 100 μg·mL−1; amphotericin, 0.25 μg·mL−1). In addition, two conditions were tested: in the presence or absence of both IL‐1β (100 ng·mL−1) and LPS (100 μg·mL−1), named ‘Inflammation (18 h)’ or ‘Normal (18 h)’ respectively. The volume of the culture medium was adjusted to 1 mL for 70 mg of tissue. All tissue incubations were done at 37°C in a humidified atmosphere of 5% CO2 in air using a culture incubator. After 18 h incubation, different protocols were performed for each sample. One part of the samples was set up in organ bath system for vascular reactivity studies. The second part of samples from the same patient was frozen −80°C for Western blot analysis. Organ culture and organ bath solutions were also kept at −80°C for prostanoid measurements.
Vascular reactivity studies
After the incubation period previously described, inflammatory conditions, IMA and SV preparations (cut as rings) were set up in 10 mL organ baths containing Tyrode's solution (concentration mM): NaCl 139.2, KCl 2.7, CaCl2 1.8, MgCl2 0.49, NaHCO3 11.9, NaH2PO4 0.4, glucose 5.5, gassed with 5% CO2 and 95% O2 at 37°C and pH 7.4. Each ring was initially stretched to an optimal load (IMA: 1.0–1.5 g; SV: 1.5–2.0 g). Changes in force were recorded by isometric force displacement transducer (Narco F‐60). Rings were equilibrated for 90 min with bath fluid changes taking place every 10 min.
After the equilibration period, the viability (contractility) of the vessel specimens was checked with KCl (40 mM) stimulation and the preparations were washed until the initial resting tone was reestablished. Thereafter, the vessels were contracted with increasing concentrations of noradrenaline (0.01–100 μM) in a cumulative manner to establish the concentration–response relationship. When a maximal effect (Emax) was obtained, the preparations were washed with Tyrode's solution until they returned to the resting basal tone. Subsequently, these preparations were incubated for 30 min with the following pharmacological treatments: the COX‐2‐selective inhibitors (DuP‐697, 1 μM or DFU, 1 μM), the PGI2 receptor (IP) antagonist [CAY10441 (RO1138452), 1 μM], the PGE2 receptor (EP4) antagonist (GW627368X, 1 μM), the mPGES‐1 inhibitor (C3, 10 μM) (Leclerc et al., 2013) and the NOS inhibitor (L‐NOARG, nitroarginine; 100 μM). Some preparations were incubated without any compounds for 30 min and served as a time control. After this 30 min incubation period, a second noradrenaline concentration–response curve was obtained, after which the organ bath solution was stored at −80°C for prostanoid measurements. Each preparation was used for one protocol, comprising two concentration–response curves of noradrenaline, separated with an incubation period. In order to compare the effect of different compounds between cultured under inflammatory (18 h) conditions and fresh vessels, these protocols have been performed with IMA and SV samples ‘Normal (0 h)’.
Western blot analysis
Following incubation ‘Normal (18 h)’ or ‘Inflammatory (18 h) conditions’, IMA and SV samples were homogenized under liquid nitrogen, using a porcelain mortar. The homogenates were resuspended in RIPA solution [Tris–HCl buffer (in mM): Tris: 50, pH: 8; NaCl: 150; EDTA: 5; Triton X‐100: 1%; sodium desoxycholate 1%; SDS 0.1%] at 4°C (1 mL·100 mg−1 of tissue) with a protease inhibitor cocktail. The homogenates were centrifuged at 4000 x g for 20 min, at 4°C. The supernatants were assayed for protein concentration using a bicinchoninic acid (BCA) protein assay kit. Samples containing 50 μg of protein were loaded on SDS‐PAGE. Proteins were transferred to nitrocellulose membranes. The membranes were subsequently blocked for 1 h in TBS, 0.1% Tween 20 and 5% non‐fat dry milk and incubated overnight at 4°C with an anti‐COX‐2 or anti‐mPGES‐1 monoclonal antibody diluted to 1:250 and 1:1000, respectively, in TBS/0.1% Tween‐20. Subsequently, the membranes were incubated with alkaline phosphatase‐conjugated goat anti‐mouse or rabbit secondary antibody. Bands were visualized using enhanced chemiluminescent (ECL) plus kit. For quantification, the film was scanned and the integrated optical density of the bands was estimated with Scion image (Scion Corporation, NIH, Frederick, MD, USA) and normalized to β‐actin.
Prostanoid measurements
The supernatants of organ culture in the presence or absence of inflammatory conditions were collected after 18 h. In addition, the organ bath solutions after 30 min incubation with the COXIB (DuP‐697, 1 μM) and the mPGES‐1 inhibitor (C3, 10 μM) with or without noradrenaline (100 μM) stimulation were also harvested. The concentrations of PGE2 and 6‐keto‐PGF1α (a stable metabolite of PGI2) were measured in both supernatants using an enzyme immunoassay (EIA) kit according to the manufacturer's instructions. The prostanoid concentrations in supernatants were expressed as pg or ng·(mg tissue wet weight) −1. Technical replicates were used to ensure the reliability of single values.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All results obtained from different patients (n) were expressed as means ± SEM. The first concentration–response curve induced by noradrenaline was expressed in absolute terms, as mN, but the second concentration–response curve induced by noradrenaline was expressed as % of the Emax of the first curve. Where possible, using SigmaPlot version 12.0 (Systat Software, Point Richmond, CA, USA), a four‐parameter logistic equation of the form
was fitted to data (E) obtained from each organ bath protocol to provide estimates of the maximal contraction (Emax) induced by noradrenaline [A], the half‐maximum effective concentration values (EC50), and Hill slope (nH) parameters. The pEC50 values were calculated. Statistical analysis was performed by Student's t‐test (paired data derived from the same patient or unpaired) or repeated‐measures (RM) two‐way ANOVA and Bonferroni's correction for multiple comparisons post hoc tests. If there was more than one preparation (same condition and protocol) for one patient, the results were averaged before statistical analysis. Values of P<0.05 indicated significant differences between means. Statistical analyses were performed using SigmaStat version 3.5 (Systat Software, Point Richmond, CA, USA).
Materials
The protease inhibitor cocktail, IL‐1β, LPS, noradrenaline, KCl, antibiotics and antimycotics were purchased from Sigma–Aldrich (St. Louis, MO, USA). CAY10441, DuP‐697, DFU, GW627368X, U46619, iloprost, PGE2 and 6‐keto‐PGF1α EIA kits were obtained from Cayman Chemical (Ann Arbor, MI, USA). RPMI was obtained from Gibco Invitrogen (Paisley, UK). BCA protein assay kit was from Thermo (Rockford, USA). Nitrocellulose membranes and ECL Plus system were obtained from Amersham Biosciences (Buckinghamshire, UK). C3 [1‐(1‐isopropyl‐5,6‐dimethyl‐1H‐ benzoimidazol‐2‐yl)‐piperidine‐4‐carboxylic acid cyclopentylamide] was a generous gift from NovaSAID AB and Dr Per‐Johan Jakobsson (Karolinska Institutet, Stockholm, Sweden). Antibodies against mPGES‐1 and COX‐2 were from Oxford Bio Therapeutics (Oxford, UK) and Santa Cruz Biotechnology (Santa Cruz, CA, USA) respectively.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
Vasoconstriction induced by noradrenaline in a model of inflammation
The contraction of samples of human IMA and SV induced by noradrenaline was significantly decreased under ‘Inflammatory (18 h) conditions’ compared with ‘Normal (0 h)’ or ‘Normal (18 h)’ conditions. The values of pEC50 were lower after incubation (18 h) either under normal or inflammatory conditions, compared with ‘Normal (0 h)’ conditions (Figure 1; Table 1). On the other hand, there was no difference in the KCl‐induced contractions between normal and inflammatory conditions in both vessels (Supporting Information Figure S1).
Figure 1.

Vascular reactivity induced by noradrenaline in IMA and SV following incubation under ‘Normal (0 h)’ or ‘Inflammatory (18 h)’ conditions. Contractions obtained from first noradrenaline (NAdr) concentration–response curve are expressed in mN. * P < 0.05, significantly different; two‐way ANOVA. Values are means ± SEM derived from (n) different patients (see Table 1 for pEC50, Emax values and statistics).
Table 1.
The effect of inflammation on vascular contraction induced by noradrenaline
| IMA | SV | |||||
|---|---|---|---|---|---|---|
| Condition | pEC50 | Emax (mN) | n | pEC50 | Emax (mN) | n |
| Normal (0 h) | 6.80 ± 0.16 | 13.36 ± 1.21 | 5 | 6.71 ± 0.11 | 101.30 ± 10.83 | 14 |
| Normal (18 h) | 6.27 ± 0.24# | 17.36 ± 4.09 | 6 | 6.29 ± 0.10# | 108.13 ± 8.61 | 15 |
| Inflammation (18 h) | 6.26 ± 0.15# | 8.25 ± 1.83*, # | 5 | 6.27 ± 0.06# | 73.31 ± 4.15*, # | 15 |
pEC50 and Emax (maximal contraction expressed as mN) values derived from first concentration–response curve induced by noradrenaline in IMA and SV under ‘Normal (0 h)’, ‘Normal (18 h)’ or ‘Inflammation (18 h)’ conditions.
P < 0.05, significantly different from ‘Normal (18 h)’; Student's t‐test.
Expression of COX‐2 and mPGES‐1 and prostanoid release in a model of inflammation
In inflammatory conditions, expression of COX‐2 and mPGES‐1 protein was significantly increased in IMA, compared with ‘Normal (18 h)’ conditions (Figure 2A–C). In SV, only COX‐2 expression was increased in inflammatory conditions as the increase in mPGES‐1 expression in inflammatory conditions did not reach statistical significance. In addition, concentrations of 6‐keto‐PGF1α (the stable metabolite of PGI2) and PGE2 in culture supernatants were significantly higher under inflammatory conditions, compared with normal conditions, in both IMA and SV (Figure 2D, E). The expression of mPGES‐1 and COX‐2 and the production of PGE2 were greater in SV than in the IMA, in both conditions (Figure 2).
Figure 2.

The expression of COX‐2 and mPGES‐1 and release of 6‐keto‐PGF1α (stable metabolite of PGI2) and PGE2 in IMA or SV following 18 h incubation under ‘Normal’ or ‘Inflam’ (inflammatory) conditions. A representative image of Western blot is presented (A). Histograms represent Western blot quantification of COX‐2 (B) and mPGES‐1 (C) corresponding bands. Optical density (OD, arbitrary units) was quantified by Scion Image and normalized by actin. The release of 6‐keto‐PGF1α (D) and PGE2 (E) in organ culture supernatant after 18 h incubation were expressed as ng·(mg tissue wet weight) −1. * P < 0.05 significantly different as indicated; Student's t‐test. Values are means ±SEM derived from (n) different patients.
Effects of the mPGES‐1 inhibitor on vasoconstriction induced by noradrenaline
The mPGES‐1 inhibitor (C3, 10 μM) decreased the vascular contractile response and sensitivity (pEC50) to noradrenaline, under normal or inflammatory conditions, in both IMA and SV (Figure 3; Table 2). Inhibition of noradrenaline‐induced contractions following C3 incubation was significantly greater in SV, compared with IMA under inflammatory conditions (Supporting Information Figure S2). The decreased vascular tone and sensitivity to noradrenaline induced by C3 were reversed by co‐incubation with the IP receptor antagonist (CAY10441, 1 μM) in SV and IMA (Figure 3; Table 2). Interestingly, under inflammatory conditions, the whole of the concentration‐response curves induced by noradrenaline after co‐incubation with C3 and CAY10441 were significantly higher in IMA and lower in SV as compared with their respective controls (Figure 3C, D). On the other hand, vascular response after co‐incubation with C3 and the TP receptor antagonist (BAY u3405, 1 μM) was comparable with the response obtained following incubation with C3 alone (Supporting Information Figure S3). C3 significantly decreased the potent vasoconstriction response induced by U46619 (TxA2 analogue) in IMA under inflammatory conditions (Supporting Information Figure S4).
Figure 3.
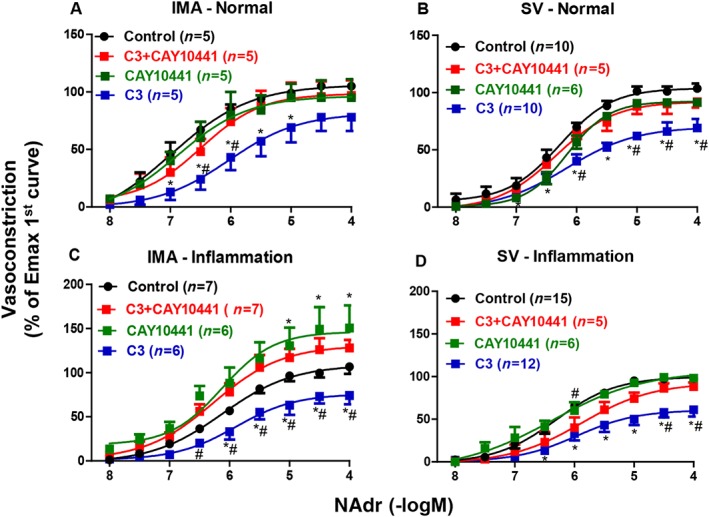
The effects of mPGES‐1 inhibitor (C3, 10 μM, 30 min), IP receptor antagonist (CAY10441, 1 μM, 30 min) and co‐incubation of these treatments on vasoconstriction induced by noradrenaline (NAdr) in IMA and SV. Vessels were used ‘Normal (0 h)’ (A, B) or after incubation under ‘Inflammatory (18 h)’ (C, D) conditions. The contraction was expressed as % of the maximal noradrenaline contraction obtained in the first concentration–response curve. * P < 0.05, significantly different from Control (without any treatment in the organ bath): #P < 0.05, significantly different from C3 + CAY10441; repeated measures two‐way ANOVA.Values are means ± SEM derived from (n) different patients (see Table 2 for pEC50, Emax values and statistics).
Table 2.
The effects of different treatments on vascular contractions induced by noradrenaline
| IMA | SV | ||||||
|---|---|---|---|---|---|---|---|
| Condition | Treatments | pEC50 | Emax (%) | n | pEC50 | Emax (%) | n |
| Normal (0 h) | Control | 6.57 ± 0.31 | 105 ± 2 | 5 | 6.43 ± 0.08 | 103 ± 4 | 10 |
| Normal (0 h) | C3 | 5.75 ± 0.40 * , # | 78 ± 12 | 5 | 6.07 ± 0.13 * , # | 69 ± 8 * , # | 10 |
| Normal (0 h) | C3 + CAY10441 | 6.71 ± 0.18 | 97 ± 13 | 5 | 6.58 ± 0.11 | 92 ± 5 | 5 |
| Normal (0 h) | CAY10441 | 6.04 ± 0.10 | 95 ± 16 | 5 | 6.38 ± 0.12 | 91 ± 2 | 5 |
| Inflam. (18 h) | Control | 6.15 ± 0.18 | 106 ± 8 | 7 | 6.36 ± 0.09# | 98 ± 3 | 15 |
| Inflam. (18 h) | C3 | 5.91 ± 0.08 | 74 ± 10 * , # | 6 | 5.59 ± 0.18 * | 61 ± 8 * , # | 12 |
| Inflam. (18 h) | C3 + CAY10441 | 6.22 ± 0.25 | 128 ± 9 | 7 | 5.82 ± 0.31 | 88 ± 5 | 5 |
| Inflam. (18 h) | CAY10441 | 6.38 ± 0.27 | 150 ± 25 * | 6 | 6.41 ± 0.19 | 98 ± 2 | 5 |
| Inflam. (18 h) | Control | 6.20 ± 0.10 | 103 ± 12 | 5 | 6.35 ± 0.09 | 88 ± 2 | 6 |
| Inflam. (18 h) | DuP‐697 | 6.42 ± 0.18 | 165 ± 22 * | 5 | 6.41 ± 0.11 | 90 ± 2 | 6 |
| Inflam. (18 h) | DFU | nt | nt | – | 6.27 ± 0.09 | 85 ± 3 | 6 |
| Normal (0 h) | Control | 6.68 ± 0.15 | 105 ± 4 | 5 | 6.32 ± 0.12 | 103 ± 4 | 5 |
| Normal (0 h) | L‐NOARG | 6.14 ± 0.22 | 160 ± 20 * | 5 | 6.28 ± 0.15 | 92 ± 5 | 5 |
| Inflam. (18 h) | Control | 6.18 ± 0.04 | 102 ± 2 | 5 | 6.52 ± 0.15 | 89 ± 5 | 5 |
| Inflam. (18 h) | L‐NOARG | 5.82 ± 0.07 | 142 ± 13 * | 6 | 6.37 ± 0.13 | 95 ± 1 | 5 |
pEC50 and Emax (maximal contraction) values derived from the second concentration–response curve induced by NE in IMA or SV under ‘Normal (0 h)’ and ‘Inflammatory (18 h)’ (Inflam.) conditions. Emax are expressed as % of the maximal noradrenaline contraction obtained in the first concentration–response curve. The treatments used are as follows: mPGES‐1 inhibitor (C3, 10 μM), COX‐2 inhibitor (DuP‐697 or DFU, 1 μM), NOS inhibitor (L‐NOARG, 100 μM) and IP receptor antagonist (CAY10441, 1 μM). Values are means ± SEM derived from (n) different patients.
P < 0.05, significantly different from their respective paired Control (without any treatment in organ bath);
P < 0.05, significantly different from C3 + CAY10441; Student's t‐test. nt indicates protocol not tested.
Effects of the IP‐receptor antagonist and COX‐2 inhibitors on vasoconstriction induced by noradrenaline
Under ‘Normal (0 h)’ conditions, the IP receptor antagonist (CAY10441, 1 μM) did not modify the contractions induced by noradrenaline in IMA and SV (Figure 3A, B; Table 2). Under ‘Inflammatory (18 h) conditions’, incubation with CAY10441 or the COX‐2 inhibitor DuP‐697 (1 μM) increased the contractions induced by noradrenaline in IMA while pEC50 values were not changed (Figures 3C and 4A; Table 2). There was no statistical difference between maximal contraction obtained after these treatments under inflammatory condtions (DuP‐697: 15 ± 8 mN, CAY10441: 14 ± 5 mN) and those obtained under normal conditions in IMA (17 ± 4 mN, Table 1). In contrast, these effects of CAY10441 and DuP‐697 were not observed in SV (Figures 3D and 4B; Table 2). For this reason, another COXIB (DFU, 1 μM) and an EP4 receptor antagonist (GW627368X, 1 μM) were tested and found to have no effect on the vascular response to noradrenaline in SV under inflammatory conditions (Figure 4B; Table 2; Supporting Information Table S1).
Figure 4.
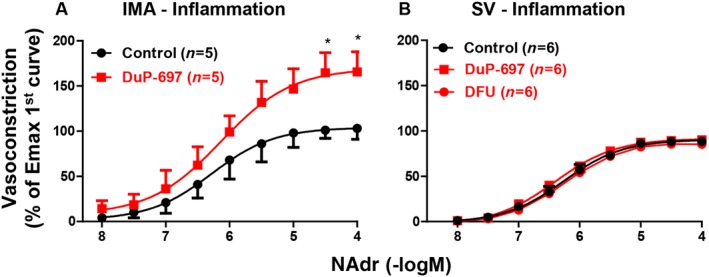
The effects of COX‐2 inhibitors (DuP‐697, DFU 1 μM, 30 min) on vasoconstriction induced by noradrenaline. IMA and SV were used after incubation under ‘Inflammatory (18 h)’ conditions. The contraction was expressed as % of the maximal noradrenaline (NAdr) contraction obtained in the first concentration–response curve. * P < 0.05, significantly different from Control (without any treatment in the organ bath); repeated measures two‐way ANOVA. Values are means ± SEM derived from (n) different patients (see Table 2 for pEC50, Emax values and statistics).
Effects of mPGES‐1 and COX‐2 inhibitors on prostanoid release in inflammatory conditions
After 18 h culture under inflammatory conditions, vascular preparations were set up in an organ bath. The measurements of 6‐keto‐PGF1α (the stable metabolite of PGI2) and PGE2 in organ bath solutions, were performed after 30 min incubation with the respective treatments. The levels of PGE2 and 6‐keto‐PGF1α in the organ bath solutions (without any pharmacological treatment) were increased after noradrenaline (100 μM) stimulation (Figure 5). Interestingly, incubation with the mPGES‐1 inhibitor (C3, 10 μM) increased the basal release of 6‐keto‐PGF1α while the COX‐2 inhibitor (DuP‐697, 1 μM) caused a significant decrease of 6‐keto‐PGF1α concentrations, in IMA and SV (Figure 5A, B). PGE2 release with or without noradrenaline stimulation was significantly decreased in IMA and SV after either C3 or DuP‐697 incubation (Figure 5C, D).
Figure 5.
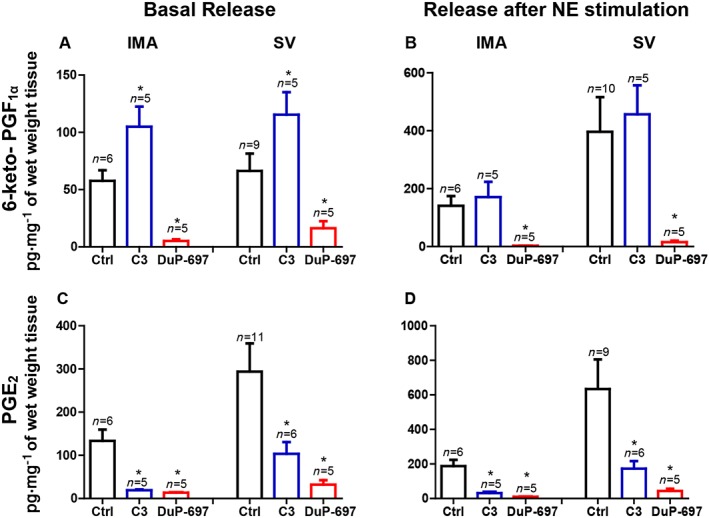
The effects of mPGES‐1 inhibitor (C3, 10 μM, 30 min) and COX‐2 inhibitor (DuP‐697, 1 μM, 30 min) on the release of 6‐keto‐PGF1α (stable metabolite of PGI2; A, B) and PGE2 (C, D) in basal and after noradrenaline (maximal stimulation (100 μM). The production of prostanoids was measured in organ bath solutions containing IMA and SV after incubation under ‘Inflammatory (18 h)’ conditions. The release of prostanoids were expressed as pg·(mg tissue wet weight) −1. * P < 0.05, significantly different from control (Ctrl) of respective vessel; Student's t‐test. Values are means ± SEM derived from (n) different patients.
Effect of NOS inhibitor on vasoconstriction induced by noradrenaline
Under normal and inflammatory conditions, contractions induced by noradrenaline in IMA (but not in SV) were significantly increased in the presence of the inhibitor of NO synthesis (L‐NOARG, 100 μM; Figure 6A–D; Table 2). These vascular contractions after co‐incubation with C3 and the NOS inhibitor (L‐NOARG, 100 μM) were comparable with those obtained after incubation with C3 alone in IMA under inflammatory conditions (Figure 6C).
Figure 6.
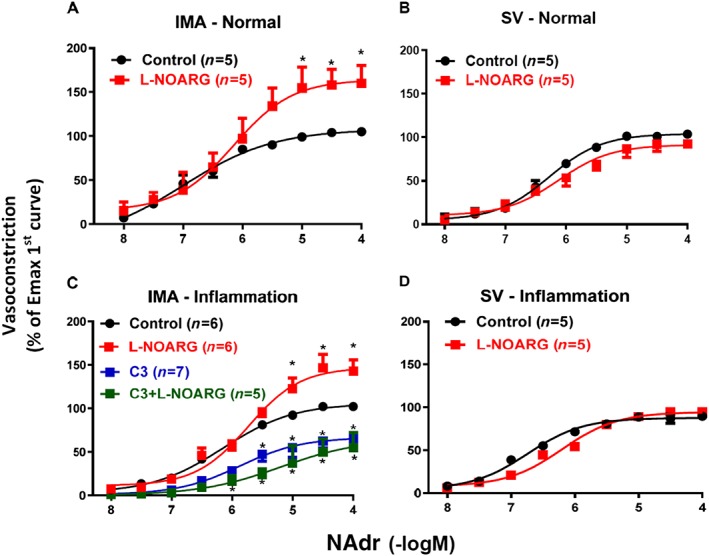
The effect of the NOS inhibitor (L‐NOARG, 100 μM, 30 min) on vasoconstriction induced by noradrenaline (NAdr) in IMA and SV in ‘Normal (0 h)’ or ‘Inflammatory (Inflam. 18 h)’ conditions. The effect of co‐incubation with mPGES‐1 inhibitor (C3, 10 μM, 30 min) and NOS inhibitor (L‐NOARG, 100 μM, 30 min) was tested in IMA under inflammatory conditions. * P < 0.05, significantly different from Control (without any treatment in the organ bath); repeated measures two‐way ANOVA. Values are means ± SEM derived from (n) different patients (see Table 2 for pEC50, Emax values and statistics).
Discussion
COX‐2 inhibitors are effective in the treatment of inflammatory disease by decreasing PGE2 levels. However, their cardiovascular side effects, associated with the reduction of PGI2 production, have limited their use (Grosser et al., 2010). In recent years, the inhibition of mPGES‐1 has gained importance as an alternative to inhibition of COX‐2. Our study focused on the effects of mPGES‐1 inhibitor on human vascular reactivity in inflammatory conditions. The present report shows that in contrast to the COX‐2 inhibitor, the mPGES‐1 inhibitor (C3) reduced the contractions induced by noradrenaline (Figures 3 and 7) by increasing (twofold) PGI2 production (Figures 5 and 7). Furthermore, treatment with this mPGES‐1 inhibitor on human vessels was associated with reduced levels (threefold to fivefold) of PGE2 (Figure 5). These results suggest that C3 could have a potent anti‐inflammatory effect, without the increased cardiovascular risk associated with COX‐2 inhibitors. In addition, our study provides a comparison of the regulation of vascular tone in IMA and SV, induced by selective inhibitors of PGI2, PGE2 and NO pathways (Figure 7).
Figure 7.
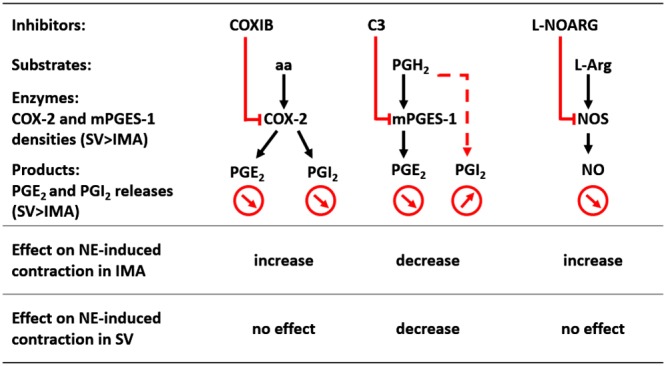
The effects of different inhibitors on vascular tone and their mechanisms of action in human vessels. aa: arachidonic acid, L‐arg: L‐arginine.
Both COX‐2 and mPGES‐1 enzymes are co‐induced by inflammatory stimuli in many cells and are the main enzymes responsible for PGE2 synthesis (Jakobsson et al., 1999). In an earlier study, the induction of COX‐2 enzyme, after 24 h incubation of IMA with IL‐1β and LPS, was accompanied by increased levels of PGE2 and PGI2 (Foudi et al., 2009). These results were confirmed in the present report after 18 h incubation with the same inflammatory stimuli (Figure 2A, B, D, E). In addition, we demonstrated here that the mPGES‐1 enzyme was also induced in inflammatory conditions in IMA (Figure 2A, C). This acute inflammation (either 18 or 24 h incubations with inflammatory stimuli) reduced the contraction induced by noradrenaline (Figure 1, Foudi et al., 2009). This decreased reactivity can be explained, at least in part, by the inflammation‐induced synthesis of PGI2 and its vasodilatory effect. This hypothesis was confirmed by the fact that the decreased contraction under inflammatory conditions was increased after incubation with the IP receptor antagonist (CAY10441) or a COX‐2 inhibitor (DuP‐697, DFU, etoricoxib) (Figures 3C and 4A; Foudi et al., 2009). Both treatments were able to restore the contractions to a level, similar to that observed under normal conditions (Table 1). This increase in vascular tone due to inhibition of PGI2 synthesis induced by COX‐2 inhibitors could be associated with their cardiovascular side effects such as arterial hypertension. As several studies have shown that PGE2 is a key mediator of inflammation, drugs that specifically inhibit PGE2 while sparing other prostanoids would be more specific and possibly safer anti‐inflammatory therapy. From this aspect, mPGES‐1 inhibition represents a promising target for drug development (Wang and FitzGerald, 2010).
In this study, we have chosen C3 as an mPGES‐1 inhibitor because this compound is a selective mPGES‐1 inhibitor in humans and no concentration‐dependent inhibition has been detected against COX‐1, COX‐2, PGIS or haematopoietic PGD synthase, at concentrations up to 50 μM C3 (Leclerc et al., 2013). The novel finding of our study was that C3 attenuated the noradrenaline‐induced vasoconstrictions of human vessels (IMA and SV) by increasing PGI2 production within the vascular wall. This shift towards increased PGI2 synthesis after mPGES‐1 inhibition was confirmed by both measuring the PGI2 metabolite, 6‐keto‐PGF1α, and using a specific IP receptor antagonist in organ bath studies (Figures 3 and 5A). However, the effect of C3 on PGI2 levels after noradrenaline stimulation (100 μM) was not significant (Figure 5B) while a decreased contraction was still measured (Figure 3). The high concentration of PGI2 (threefold to sixfold) produced after noradrenaline stimulation, the metabolism of 6‐keto‐PGF1α and the accumulation of the metabolites in organ bath solutions could account for this discrepancy. The redirection of PGH2 metabolism towards PGI2 has been reported with various mPGES‐1 inhibitors in preparations derived from animals (PF‐9184, MF63, LY3023703, C3) (Mbalaviele et al., 2010; Leclerc et al., 2013; Jin et al., 2015; Chandrasekhar et al., 2016). In addition, our data obtained with C3 are in accordance with higher concentrations of PGI2 observed in mPGES‐1 knockout mice (Cheng et al., 2006; Wang et al., 2006; Raouf et al., 2016a). Moreover, in these mice, blood pressure was not elevated (Cheng et al., 2006) while increased blood pressure was observed in COX‐2 knockout mice (Qi et al., 2002). These in vivo murine data are in agreement with our in vitro human data, which show an increased contraction in the presence of the COX‐2 inhibitor (Figure 4A) and a decreased contraction by the mPGES‐1 inhibitor (Figure 3). The shift in favour of PGI2 production induced by mPGES‐1 inhibitors might be a great improvement in the cardiovascular safety of anti‐inflammatory drugs in comparison to COX‐2 inhibitors.
A recent study has demonstrated that isolated aorta preparations derived from mPGES‐1 knockout mice exhibit greater ACh‐induced relaxation than those from the wild‐type mice (Raouf et al., 2016a). As the relaxation of ACh is mostly dependent on NO and PGI2 releases in murine aorta (Shen et al., 2013), the increased ACh relaxation in mPGES‐1 knockout mice (Raouf et al., 2016a) could be due to either increased PGI2 or NO levels. In our study, we tested the possibility for an augmentation of NO levels after mPGES‐1 inhibition. Our results demonstrated that co‐incubation with NOS inhibitor and C3 exhibited similar profile to treatment with C3 alone on vascular tone (Figure 6C). This result suggests that C3 did not affect NO levels.
In addition to the beneficial effects of mPGES‐1 inhibitors described above, the present study could provide new aspects for improving the patency of graft materials by using a mPGES‐1 inhibitor. In this study, we used IMA and SV, which are frequently used as a graft material to bypass stenosed coronary arteries (Goldman et al., 2004). During bypass surgery or the post‐operative period, vascular cells are likely to be exposed to inflammatory conditions, resulting in an increase of prostanoid production. In addition, the PGE2 produced by the vascular wall exacerbates atherothrombosis (Gross et al., 2007) while PGI2 has a reverse effect. All patients are prescribed antiplatelet therapy including aspirin after bypass surgery. Another recent study showed that the inhibition of mPGES‐1 also prevents platelet activation (Raouf et al., 2016b). For all these reasons, we suggest that using an mPGES‐1 inhibitor such as C3 in the post‐operative period could prevent graft spasm, thrombosis or occlusion, not only by increasing PGI2 release but also by decreasing PGE2 levels.
In IMA and SV under inflammatory conditions, mPGES‐1 and COX‐2 expressions were induced and similar reduction of noradrenaline‐induced vasoconstriction was measured (Figures 1 and 2A–C). The mPGES‐1 inhibitor reduced vascular tone to the same extent in both IMA and SV under normal conditions (Figures 3A, B). On the other hand, we have found many discrepancies between these vessels. Under inflammatory conditions, the inhibition of noradrenaline‐induced vasoconstriction by C3 was less pronounced in IMA (Supporting Information Figures S2) and restauration with CAY10441 was significantly greater (twofold) in IMA than SV (Figures 3C, D; Table 2). Moreover, the inhibition of the vasodilatory effects of PGI2, either by COX‐2 inhibition or by antagonism of IP receptors antagonist, induced an increased contraction in IMA but not in SV under inflammatory conditions (Figures 3C, D and 4A, B; Table 2). In addition, we showed that SV is 100‐fold less sensitive than IMA to the relaxation induced by the PGI2 analogue (iloprost, Supporting Information Figure S5 and Table S2) under inflammatory conditions. This could explain the lack of effect of IP receptor antagonism or COX‐2 inhibition on vascular tone in SV. In contrast, these treatments augmented the contractile response to noradrenaline (Figures 3C and 4A) in IMA, which was much more sensitive to the PGI2 analogue (Supporting Information Figure S5 and Table S2). As PGE2 induces vasodilatation via EP4 receptors in SV but not in IMA (Foudi et al., 2011), the effect of the EP4 receptor antagonist on vascular tone was tested only in SV and no effect was found (Supporting Information Table S1).
Like COX‐2 inhibition and antagonism of IP receptors, the inhibition of NOS also regulated the vascular tone differently between IMA and SV. After incubation with the NOS inhibitor, a greater response to noradrenaline under normal and inflammatory conditions was detected in IMA, while the SV responses were not modified (Figure 6A–D; Table 2). The same variation between IMA and SV was observed in endothelium‐denuded vessels after 22 h incubation with LPS (Thorin‐Trescases et al., 1995). This different regulation of vascular tone by NO could be due to the higher eNOS activity described in IMA (Hamilton et al., 1997; Shapira et al., 1999), and consequently greater effect of NOS inhibitor on vascular tone was measured in IMA. Globally, these results suggest that the contractions induced by noradrenaline in IMA were under control of both PGI2 and NO. In contrast, in SV, only the increased level of PGI2 after C3 incubation controlled the vascular tone (Figure 3B, D) and no role for NOS activity was found (Figure 6B, D).
Another difference observed between IMA and SV was that the expression of mPGES‐1, COX‐2 and PGE2 release were greater in SV versus IMA (Figures 2 and 5C, D). A similar result has been described using SMC cultures derived from IMA and SV (Bishop‐Bailey et al., 1998). In addition, this study showed that COX‐2 expression in arterial SMC, but not in venous SMC, was strongly inhibited by exogenously applied PGE2 (Bishop‐Bailey et al., 1998). This negative feedback could explain the lower levels of COX‐2 expression and consequently lower prostanoid productions in IMA presented in Figures 2 and 5. In our results, in spite of these greater enzymic activities in SV, inhibition or antagonism of the PGI2 pathway (even with high doses) was without effect on SV vascular tone. Only the greater effect of mPGES‐1 inhibitor on vascular tone (Supporting Information Figure S2) was associated with higher mPGES‐1 expression in SV versus IMA (Figure 2C).
In conclusion; while mPGES‐1 and COX‐2 inhibitors have similar anti‐inflammatory effect in reducing inflammation‐induced PGE2 levels, they affect PGI2 synthesis and regulation of vascular tone in opposing ways. From our in vitro study, the decreased vasoreactivity to noradrenaline due to increased PGI2 release is the predominant effect induced by mPGES‐1 inhibition either in human artery or vein. This effect is predictive of the absence of cardiovascular side effects of mPGES‐1 inhibition, such as the increased blood pressure observed with COX‐2 inhibitors.
Author contributions
G.O. and X.N. designed the study; G.O., X.N., A.D. and L.B. performed experiments; G.O., X.N. and I.G. analysed the data; C.D. and O.T. obtained human vessel samples and recruitment of patients; P.J.J. provisioned of C3 study protocols; G.O and X.N. wrote the manuscript; I.G., A.D., P.J.J., D.L., S.U.D. and G.T. revised manuscript.
Conflict of interest
Per‐Johan Jakobsson is a member of the board of NovaSAID AB. The other authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Contractions induced by potassium chloride (KCl, 40 mM) in internal mammary artery (IMA) and saphenous vein (SV) preparations after incubation with ‘Normal (18 h) or Inflammatory (18 h)’ conditions.
Figure S2 The effect of mPGES‐1 (microsomal prostaglandin E synthase‐1) inhibitor (C3, 10 μM, 30 min) on the vasoconstriction induced by noradrenaline (NE) in internal mammary artery (IMA) and saphenous vein (SV) in ‘Inflammatory (18 h)’ conditions. The values are expressed as % of differences between first concentration–response curve and second concentration–response curve after C3 incubation. * P < 0.05, significantly different; two‐way ANOVA. Values are means ± s.e.mean derived from (n) different patients.
Figure S3 The effect of mPGES‐1 (microsomal prostaglandin E synthase‐1) inhibitor (C3, 10 μM, 30 min) with or without thromboxane receptor (TP) antagonist (BAY u3405, 1 μM, 30 min) on vasoconstriction induced by norepinephrine (NE) in saphenous vein (SV) in ‘Inflammatory (18 h)’ conditions. The contraction was expressed as % of the maximal noradrenaline contraction obtained in the first concentration–response curve. * P < 0.05, significantly different from control; two‐way ANOVA. Values are means ± SEM derived from (n) different patients.
Figure S4 The effect of mPGES‐1 (microsomal prostaglandin E synthase‐1) inhibitor (C3, 10 μM, 30 min) on vasoconstriction induced by U46619 (TP receptor agonist, 100 nM) in internal mammary artery under ‘Inflammatory (18 h)’ conditions. The contraction was expressed as % of KCl (40 mM) induced contraction. * P < 0.05, significantly different; Student's t‐test. Values are means ± SEM derived from (n = 5) different patients.
Figure S5 The relaxation response induced by iloprost (PGI2 analogue) in internal mammary artery (IMA) and saphenous vein (SV) precontracted with U46619 (TP receptor agonist, 0.1 μM) after incubation under ‘Inflammatory (18 h)’ conditions. Values are means ± s.e.mean derived from (n) different patients (see Table S2 for pEC50, Emax values and statistics).
Table S1 The effect of an EP4 receptor antagonist (GW627368X) on vascular contractions induced by noradrenaline in saphenous vein.
Table S2 Pharmacological values derived from relaxation‐curves induced by iloprost.
Acknowledgements
We thank the Department of Cardiac Surgery at Bichat Hospital Paris, France, and the Department of Cardiovascular Surgery, Aile Hospital, Istanbul, Turkey, for providing tissue.
This work was supported by INSERM (Institut National de la Santé et de la Recherche Médicale). Gulsev Ozen is a recipient of a postgraduate fellowship (BIDEB‐2214) from the Scientific and Technological Research Council of Turkey (TUBITAK).
Ozen, G. , Gomez, I. , Daci, A. , Deschildre, C. , Boubaya, L. , Teskin, O. , Uydeş‐Doğan, B. S. , Jakobsson, P.‐J. , Longrois, D. , Topal, G. , and Norel, X. (2017) Inhibition of microsomal PGE synthase‐1 reduces human vascular tone by increasing PGI2: a safer alternative to COX‐2 inhibition. British Journal of Pharmacology, 174: 4087–4098. doi: 10.1111/bph.13939.
References
- Ahmetaj‐Shala B, Kirkby NS, Knowles R, Al'Yamani M, Mazi S, Wang Z et al. (2015). Evidence that links loss of cyclooxygenase‐2 with increased asymmetric dimethylarginine: novel explanation of cardiovascular side effects associated with anti‐inflammatory drugs. Circulation 131: 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Soria MA, Herrero‐Beaumont G, Moreno‐Rubio J, Calvo E, Santillana J, Egido J et al. (2008). Long‐term NSAID treatment directly decreases COX‐2 and mPGES‐1 production in the articular cartilage of patients with osteoarthritis. Osteoarthritis Cartilage 16: 1484–1493. [DOI] [PubMed] [Google Scholar]
- Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA et al. (2013). Vascular and upper gastrointestinal effects of non‐steroidal anti‐inflammatory drugs: meta‐analyses of individual participant data from randomised trials. Lancet 382: 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop‐Bailey D, Pepper JR, Larkin SW, Mitchell JA (1998). Differential induction of cyclooxygenase‐2 in human arterial and venous smooth muscle: role of endogenous prostanoids. Arterioscler Thromb Vasc Biol 18: 1655–1661. [DOI] [PubMed] [Google Scholar]
- Camacho M, Gerboles E, Escudero JR, Anton R, Garcia‐Moll X, Vila L (2007). Microsomal prostaglandin E synthase‐1, which is not coupled to a particular cyclooxygenase isoenzyme, is essential for prostaglandin E(2) biosynthesis in vascular smooth muscle cells. J Thromb Haemost 5: 1411–1419. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar S, Harvey AK, Yu XP, Chambers MG, Oskins JL, Lin C et al. (2016). Identification and characterization of novel microsomal prostaglandin E synthase‐1 inhibitors for analgesia. J Pharmacol Exp Ther 356: 634–644. [DOI] [PubMed] [Google Scholar]
- Chen L, Yang G, Xu X, Grant G, Lawson JA, Bohlooly YM et al. (2013). Cell selective cardiovascular biology of microsomal prostaglandin E synthase‐1. Circulation 127: 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA (2006). Cyclooxygenases, microsomal prostaglandin E synthase‐1, and cardiovascular function. J Clin Invest 116: 1391–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engblom D, Saha S, Engstrom L, Westman M, Audoly LP, Jakobsson PJ et al. (2003). Microsomal prostaglandin E synthase‐1 is the central switch during immune‐induced pyresis. Nat Neurosci 6: 1137–1138. [DOI] [PubMed] [Google Scholar]
- Foudi N, Louedec L, Cachina T, Brink C, Norel X (2009). Selective cyclooxygenase‐2 inhibition directly increases human vascular reactivity to norepinephrine during acute inflammation. Cardiovasc Res 81: 269–277. [DOI] [PubMed] [Google Scholar]
- Foudi N, Kotelevets L, Gomez I, Louedec L, Longrois D, Chastre E et al. (2011). Differential reactivity of human mammary artery and saphenous vein to prostaglandin E(2): implication for cardiovascular grafts. Br J Pharmacol 163: 826–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman S, Zadina K, Moritz T, Ovitt T, Sethi G, Copeland JG et al. (2004). Long‐term patency of saphenous vein and left internal mammary artery grafts after coronary artery bypass surgery: results from a Department of Veterans Affairs Cooperative Study. J Am Coll Cardiol 44: 2149–2156. [DOI] [PubMed] [Google Scholar]
- Gomez I, Foudi N, Longrois D, Norel X (2013). The role of prostaglandin E2 in human vascular inflammation. Prostaglandins Leukot Essent Fatty Acids 89: 55–63. [DOI] [PubMed] [Google Scholar]
- Gross S, Tilly P, Hentsch D, Vonesch JL, Fabre JE (2007). Vascular wall‐produced prostaglandin E2 exacerbates arterial thrombosis and atherothrombosis through platelet EP3 receptors. J Exp Med 204: 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosser T, Yu Y, Fitzgerald GA (2010). Emotion recollected in tranquility: lessons learned from the COX‐2 saga. Annu Rev Med 61: 17–33. [DOI] [PubMed] [Google Scholar]
- Hamilton CA, Berg G, McIntyre M, McPhaden AR, Reid JL, Dominiczak AF (1997). Effects of nitric oxide and superoxide on relaxation in human artery and vein. Atherosclerosis 133: 77–86. [DOI] [PubMed] [Google Scholar]
- Ibuki T, Matsumura K, Yamazaki Y, Nozaki T, Tanaka Y, Kobayashi S (2003). Cyclooxygenase‐2 is induced in the endothelial cells throughout the central nervous system during carrageenan‐induced hind paw inflammation; its possible role in hyperalgesia. J Neurochem 86: 318–328. [DOI] [PubMed] [Google Scholar]
- Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B (1999). Identification of human prostaglandin E synthase: a microsomal, glutathione‐dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A 96: 7220–7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Smith CL, Hu L, Campanale KM, Stoltz R, Huffman LG Jr et al. (2015). Pharmacodynamic comparison of LY3023703, a novel microsomal prostaglandin E synthase 1 inhibitor, with celecoxib. Clin Pharmacol Ther 99: 274–284. [DOI] [PubMed] [Google Scholar]
- Koeberle A, Werz O (2015). Perspective of microsomal prostaglandin E2 synthase‐1 as drug target in inflammation‐related disorders. Biochem Pharmacol 98: 1–15. [DOI] [PubMed] [Google Scholar]
- Leclerc P, Idborg H, Spahiu L, Larsson C, Nekhotiaeva N, Wannberg J et al. (2013). Characterization of a human and murine mPGES‐1 inhibitor and comparison to mPGES‐1 genetic deletion in mouse models of inflammation. Prostaglandins Other Lipid Mediat 107: 26–34. [DOI] [PubMed] [Google Scholar]
- Marnett LJ (2009). The COXIB experience: a look in the rearview mirror. Annu Rev Pharmacol Toxicol 49: 265–290. [DOI] [PubMed] [Google Scholar]
- Mbalaviele G, Pauley AM, Shaffer AF, Zweifel BS, Mathialagan S, Mnich SJ et al. (2010). Distinction of microsomal prostaglandin E synthase‐1 (mPGES‐1) inhibition from cyclooxygenase‐2 inhibition in cells using a novel, selective mPGES‐1 inhibitor. Biochem Pharmacol 79: 1445–1454. [DOI] [PubMed] [Google Scholar]
- McGettigan P, Henry D (2013). Use of non‐steroidal anti‐inflammatory drugs that elevate cardiovascular risk: an examination of sales and essential medicines lists in low‐, middle‐, and high‐income countries. PLoS Med 10: e1001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers LK, Kang AH, Postlethwaite AE, Rosloniec EF, Morham SG, Shlopov BV et al. (2000). The genetic ablation of cyclooxygenase 2 prevents the development of autoimmune arthritis. Arthritis Rheum 43: 2687–2693. [DOI] [PubMed] [Google Scholar]
- Norel X (2007). Prostanoid receptors in the human vascular wall. ScientificWorldJournal 7: 1359–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, Hao CM, Langenbach RI, Breyer RM, Redha R, Morrow JD et al. (2002). Opposite effects of cyclooxygenase‐1 and ‐2 activity on the pressor response to angiotensin II. J Clin Invest 110: 788–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raouf J, Kirkby N, Ahmetaj‐Shala B, Liu B, Mazi S, Korotkova M et al. (2016a). MPGES‐1 deletion increases prostacyclin and evades the elevated systemic ADMA associated with COX‐2 inhibitors: relevance to cardiovascular safety of mpges‐1 inhibitors. Ann Rheum Dis 75 (Suppl 1): A11–A12. [Google Scholar]
- Raouf J, Mobarrez F, Larsson K, Jakobsson PJ, Korotkova M (2016b). mPGES‐1 deletion affects platelet functions in mice. Clin Sci (Lond). https://doi.org/10.1042/CS20160463. [DOI] [PubMed] [Google Scholar]
- Reid HM, Kinsella BT (2015). Prostacyclin receptors: transcriptional regulation and novel signalling mechanisms. Prostaglandins Other Lipid Mediat 121 (Pt A): 70–82. [DOI] [PubMed] [Google Scholar]
- Shapira OM, Xu A, Aldea GS, Vita JA, Shemin RJ, Keaney JF Jr (1999). Enhanced nitric oxide‐mediated vascular relaxation in radial artery compared with internal mammary artery or saphenous vein. Circulation 100 (19 Suppl): 322–327. [DOI] [PubMed] [Google Scholar]
- Shen B, Ye CL, Ye KH, Liu JJ (2013). Mechanism underlying enhanced endothelium‐dependent vasodilatation in thoracic aorta of early stage streptozotocin‐induced diabetic mice. Acta Pharmacol Sin 24: 422–428. [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorin‐Trescases N, Hamilton CA, Reid JL, McPherson KL, Jardine E, Berg G et al. (1995). Inducible L‐arginine/nitric oxide pathway in human internal mammary artery and saphenous vein. Am J Physiol 268: 1122–1132. [DOI] [PubMed] [Google Scholar]
- Wang M, FitzGerald GA (2010). Cardiovascular biology of microsomal prostaglandin E synthase‐1. Trends Cardiovasc Med 20: 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Zukas AM, Hui Y, Ricciotti E, Pure E, FitzGerald GA (2006). Deletion of microsomal prostaglandin E synthase‐1 augments prostacyclin and retards atherogenesis. Proc Natl Acad Sci U S A 103: 14507–14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ihida‐Stansbury K, Kothapalli D, Tamby MC, Yu Z, Chen L et al. (2011). Microsomal prostaglandin e2 synthase‐1 modulates the response to vascular injury. Circulation 123: 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Rowland SE, Clark P, Giroux A, Cote B, Guiral S et al. (2008). MF63 [2‐(6‐chloro‐1H‐phenanthro[9,10‐d]imidazol‐2‐yl)‐isophthalonitrile], a selective microsomal prostaglandin E synthase‐1 inhibitor, relieves pyresis and pain in preclinical models of inflammation. J Pharmacol Exp Ther 326: 754–763. [DOI] [PubMed] [Google Scholar]
- Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, Yu Z et al. (2012). Vascular COX‐2 modulates blood pressure and thrombosis in mice. Sci Transl Med 4: 132ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Contractions induced by potassium chloride (KCl, 40 mM) in internal mammary artery (IMA) and saphenous vein (SV) preparations after incubation with ‘Normal (18 h) or Inflammatory (18 h)’ conditions.
Figure S2 The effect of mPGES‐1 (microsomal prostaglandin E synthase‐1) inhibitor (C3, 10 μM, 30 min) on the vasoconstriction induced by noradrenaline (NE) in internal mammary artery (IMA) and saphenous vein (SV) in ‘Inflammatory (18 h)’ conditions. The values are expressed as % of differences between first concentration–response curve and second concentration–response curve after C3 incubation. * P < 0.05, significantly different; two‐way ANOVA. Values are means ± s.e.mean derived from (n) different patients.
Figure S3 The effect of mPGES‐1 (microsomal prostaglandin E synthase‐1) inhibitor (C3, 10 μM, 30 min) with or without thromboxane receptor (TP) antagonist (BAY u3405, 1 μM, 30 min) on vasoconstriction induced by norepinephrine (NE) in saphenous vein (SV) in ‘Inflammatory (18 h)’ conditions. The contraction was expressed as % of the maximal noradrenaline contraction obtained in the first concentration–response curve. * P < 0.05, significantly different from control; two‐way ANOVA. Values are means ± SEM derived from (n) different patients.
Figure S4 The effect of mPGES‐1 (microsomal prostaglandin E synthase‐1) inhibitor (C3, 10 μM, 30 min) on vasoconstriction induced by U46619 (TP receptor agonist, 100 nM) in internal mammary artery under ‘Inflammatory (18 h)’ conditions. The contraction was expressed as % of KCl (40 mM) induced contraction. * P < 0.05, significantly different; Student's t‐test. Values are means ± SEM derived from (n = 5) different patients.
Figure S5 The relaxation response induced by iloprost (PGI2 analogue) in internal mammary artery (IMA) and saphenous vein (SV) precontracted with U46619 (TP receptor agonist, 0.1 μM) after incubation under ‘Inflammatory (18 h)’ conditions. Values are means ± s.e.mean derived from (n) different patients (see Table S2 for pEC50, Emax values and statistics).
Table S1 The effect of an EP4 receptor antagonist (GW627368X) on vascular contractions induced by noradrenaline in saphenous vein.
Table S2 Pharmacological values derived from relaxation‐curves induced by iloprost.