

Abstract
Receptor for advanced glycation end products (RAGE) plays a role in inflammatory reactions. The soluble form of RAGE (sRAGE) acts as a decoy to inhibit interactions of RAGE with advanced glycation end products such as High mobility group box 1 (HMGB1). We have demonstrated that HMGB1 directs Th17 skewing by regulating dendritic cell (DC) functions in a previous study. However, the protective effects of HMGB1 blockade with sRAGE in the development of neutrophilic asthma remain unclear. Here, we showed that allergen challenge decreased expression of sRAGE in a murine model of neutrophilic asthma, correlating well with neutrophil counts and interleukin (IL)-17 production. When HMGB1 signalling was blocked by intratracheal administration of sRAGE before sensitisation, HMGB1 expression, neutrophilic inflammation, and Th17-type responses were reduced significantly. Anti-asthma effects of sRAGE were achieved by inhibition of RAGE and IL-23 expression in airway CD11c+ antigen-presenting cells. Finally, we showed that sRAGE inhibited Th17 polarisation induced by recombinant HMGB1 (rHMGB1)-activated dendritic cells (DCs) in vitro. Adoptive transfer of rHMGB1-activated DCs was sufficient to restore airway inflammation, whereas transfer of rHMGB1 plus sRAGE-activated DCs significantly reduced neutrophilic inflammation. Thus, sRAGE prevents Th17-mediated airway inflammation in neutrophilic asthma at least partly by blocking HMGB1/RAGE signalling in DCs.
Introduction
Allergic asthma is one of the most common airway diseases worldwide1. Asthma is generally considered as an airway inflammatory disease characterised by obstruction due to the stimulation of environmental antigens. Despite the progress in therapies and determining the mechanism of pathogenesis in recent years, clinical control of asthma remains poor, which is mainly attributed to the high incidence of recurrence, exacerbation, and therapy resistance2. Increasing evidence suggests that asthma is a heterogeneous disorder regulated by distinct molecular mechanisms. T helper type (Th) 2 cells have been described in the pathogenesis of eosinophilic asthma3, whereas Th17 cells are mainly involved in severe asthma in which neutrophils contribute more than eosinophils to the inflammation. Although Th17 cells are involved in the development of steroid-refractory and neutrophilic asthma4, the underlying molecular mechanism responsible for the dysregulated immune responses mediated by Th17 cells still remains unclear. Therefore, it is of great importance to perform further studies regarding the molecular mechanism of Th17 polarisation in neutrophilic asthma.
Dendritic cells (DCs) are considered as the most important antigen-presenting cells (APCs) that are responsible for cytokine polarisation and expansion of Th2/Th17 subsets5. Many lines of evidence shows that cytokines interleukin (IL)-23 and IL-6 are required for DC-mediated Th17 cell differentiation and maintain cytokine expression in effector Th17 cells6–8. High mobility group box 1 (HMGB1) is a chromatin-associated protein. The function of HMGB1 is complicated and related to its cellular localisation9. In the nucleus, HMGB1 binds to DNA and aids transcription and DNA repair by directly binding to DNA and altering its conformation10. Nevertheless, HMGB1 can be released into the extracellular environment in response to certain stimulations, such as endotoxin11, and plays a critical role in the development of various diseases such as asthma, diabetes, neurodegeneration, and cancer12–15. Some studies postulate that increased HMGB1 enhances Th17 cell polarisation in chronic asthma, acute allograft rejection, and chronic infectious diseases16–18. Our previous study has demonstrated that HMGB1 directs Th17 skewing by regulating DC functions via IL-23 secretion, and blocking HMGB1 by an antibody may benefit attenuation of neutrophilic airway inflammation in asthma19.
Receptor for advanced glycation end products (RAGE), which is an important endogenous pattern recognition receptor, is a central cell surface receptor for HMGB1. RAGE has also been implicated in mediating the cytokine activity of HMGB1, which is a late inflammatory mediator of sepsis, and is involved in the host response to injury, infection, and inflammation20. RAGE exists as both membrane-bound and soluble receptors. Soluble RAGE (sRAGE) is generated through proteolytic cleavage of the extracellular domain of the cell surface receptor or through alternative RNA splicing21,22. Classically, sRAGE functions as a natural antagonist of RAGE signalling, because it sequesters RAGE ligands and inhibits RAGE-dependent cellular responses23,24. Previous studies have found that systemic sRAGE decreases significantly in subjects with neutrophilic asthma25, and correlates with the clinical severity of bronchial asthma and functionally in asthmatic children26. However, the molecular mechanisms of sRAGE deficiency in neutrophilic asthma are not yet fully understood. In this study, we evaluated whether sRAGE blocks HMBG1 signalling in DCs and subsequently contributes to alleviating Th17 polarisation in neutrophilic asthma, indicating that the HMBG1-RAGE axis might play a significant role in the development of neutrophilic asthma.
Results
Levels of sRAGE decrease significantly in bronchoalveolar lavage fluid (BALF) of asthmatic mice
Using a mouse model of neutrophilic asthma induced by ovalbumin (OVA) plus lipopolysaccharide (LPS) sensitisation followed by three sequential daily OVA challenges (Fig. 1A), we investigated the functional relevance of sRAGE levels and allergic Th17 responses in the disease. As shown in Fig. 2A, there was a significant decrease of sRAGE levels in BALF from mice sensitised with OVA plus LPS compared with phosphate-buffered saline (PBS)-sensitised controls. Moreover, a decrease of sRAGE in OVA plus LPS-sensitised mice but not in PBS-sensitised mice, correlated well with a rise in BALF neutrophils and IL-17 levels (Fig. 2B,C), suggesting a strong association of sRAGE with the pathogenesis of experimental neutrophilic asthma.
Figure 1.

(A) Protocols for the OVA and LPS-induced murine model of neutrophilic asthma and experimental intervention. Mice were intranasally sensitised with OVA plus LPS on days 0, 1, 2, and 7 and then challenged with OVA on days 14, 15, 21, and 22. sRAGE or MSA was intranasally administered at 30 min before each OVA challenge. One day after the final challenge, the mice were sacrificed for further analyses. (B) Protocols for establishment of the murine model of asthma by the transfer of DCs and experimental intervention. Mice were intratracheally sensitised with PBS/DCs, HMGB1/OVA-DCs, or HMGB1 + sRAGE/OVA-DCs on day 0 and then challenged with OVA on days 10, 11, and 12. One day after the final challenge, the mice were sacrificed for further analyses.
Figure 2.

Levels of sRAGE decrease during asthmatic airway inflammation in the mouse model of neutrophilic asthma. (A) sRAGE levels analysed by ELISA in BALF of OVA plus LPS- or PBS-sensitised mice. (B) Negative correlation between the levels of sRAGE and neutrophils (r = 0.838), and (C) relatively lower correlation between sRAGE and IL-17 levels (r = 0.785) in BALF from OVA plus LPS-sensitised mice. Data are the mean ± SEM; n = 8 mice in each group.
sRAGE inhibits HMGB1 expression in lung tissue
Because HMGB1 has been identified as a crucial advanced glycation end product (AGE) that is involved in many inflammatory diseases, we considered that sRAGE may ameliorate airway inflammation via blockade of HMGB1 signalling. Using an HMGB1 immunostaining technique, we found that HMGB1 expression in lung tissue was elevated significantly in OVA and LPS-sensitised mice. However, this effect was inhibited by administration of sRAGE before sensitisation (Fig. 3A), suggesting that sRAGE has an inhibitory effect on HMGB1 expression. HMGB1 protein and mRNA levels in lung tissue were also measured by western blotting and quantitative PCR, respectively. The results showed that HMGB1 mRNA and protein levels were significantly decreased in sRAGE-treated mice compared with mouse serum albumin (MSA)-treated mice (Supplementary Fig. S1).
Figure 3.

Local administration of sRAGE down-regulates HMGB1 expression in the lung. (A) Representative images of immunohistochemistry for HMGB1 in lung tissue from mice in Control (a), Asthma (b), MSA-treated (c), and sRAGE-treated groups (d). (B) The average optical density in the histogram represents HMGB1 expression. Values represent the means ± SEM (n = 5–6). *P < 0.05 and **P < 0.01 compared with the Asthma group.
To eliminate the possibility of sRAGE masking the HMGB1 antibody epitope, two doses of sRAGE (200 or 400 ng/ml) were directly added to the lung homogenates of asthmatic mice, and then HMGB1 expression was evaluated using the HMGB1 antibody for western blotting. The results showed that the addition of sRAGE to lung homogenates had no effect on HMGB1 expression (Supplementary Fig. S1B), suggesting that sRAGE does not mask the HMGB1 antibody epitope.
Local treatment with sRAGE reduces neutrophilic airway inflammation
Because the decreased expression of sRAGE in asthmatic mice was closely related to neutrophil infiltration and IL-17 production, we next determined whether administration of sRAGE decreased neutrophilic inflammation and Th17 cytokine production in asthmatic mice. To evaluate the effects of sRAGE in allergen-primed airway inflammation, we locally administrated sRAGE or MSA intranasally before OVA plus LPS sensitisation. As expected, there was a substantial increase in total bronchoalveolar (BAL) cells and neutrophils in OVA plus LPS-sensitised mice upon subsequent OVA challenge, but almost no eosinophils, which is one of the characteristics of neutrophilic bronchial asthma. However, pretreatment with sRAGE led to a significant decrease in the number of total BAL cells as well as neutrophils and macrophages in comparison with MSA-treated mice (Fig. 4A).
Figure 4.

Local administration of sRAGE inhibits neutrophilic airway inflammation. (A) sRAGE inhibited the infiltration of neutrophils and macrophages into BALF. BALF was collected at 24 h after the last OVA challenge. The numbers of total and differential cell counts in the BALF of mice in Control, Asthma, MSA-treated, and sRAGE-treated groups were analysed by counting. Values represent the means ± SEM (n = 5–6). *P < 0.05 and **P < 0.01 compared with the Asthma group. (B) Representative H&E and PAS staining of lung tissues. H&E-stained lung tissues (upper panel) from mice in Control (a), Asthma (b), MSA-treated (c), and sRAGE-treated groups (d). PAS-stained lung tissues (lower panel) from mice in Control (e), Asthma (f), MSA-treated (g), and sRAGE-treated groups (h). (C) Inflammation scores were decreased in sRAGE-treated mice. Values represent the means ± SEM (n = 5–6). *P < 0 05 and **P < 0 01 compared with the Asthma group. (D) The proportion of PAS-positive areas along the airways area was decreased in sRAGE-treated mice. Values represent the means ± SEM (n = 5–6). **P < 0 01 compared with the Asthma group.
To further characterise the effects of sRAGE on OVA-induced airway inflammation, we compared lung histology in various groups of mice. As shown in haematoxylin and eosin (H&E)-stained sections (Fig. 4B), OVA plus LPS-sensitised and OVA challenged mice showed increased numbers of inflammatory cells in peribronchial and perivascular regions, which were rarely detected in PBS-sensitised mice. In contrast, sRAGE administration significantly alleviated airway inflammation because we observed little peribronchiolar and perivascular infiltrates in the airways (Fig. 4B). Histological examination also showed decreased periodic acid-Schiff (PAS)-positive mucus-containing goblet cells in sRAGE-treated mice compared with those in MSA-treated mice (Fig. 4B). Collectively, local administration of sRAGE before sensitisation to OVA plus LPS in the mouse model of neutrophilic asthma substantially lessened the severity of neutrophilic airway inflammation.
We also evaluated whether administration of sRAGE before allergen challenge decreased neutrophilic inflammation in asthmatic mice. Sensitised animals treated with sRAGE before allergen challenge exhibited no effects on the appearance of inflammatory cell infiltration in lung tissue (Supplementary Fig. S2). These data provide evidence for the beneficial role of sRAGE in the early phase of neutrophilic asthma.
sRAGE inhibits the development of airway hyperreactivity (AHR)
To assess the effect of sRAGE on AHR, we evaluated airway responsiveness to methacholine (MCh) inhalation. After OVA challenge, AHR was significantly higher in OVA plus LPS-sensitised mice upon subsequent OVA challenge. As expected, administration of sRAGE before airway sensitisation significantly inhibited the development of AHR (Fig. 5).
Figure 5.
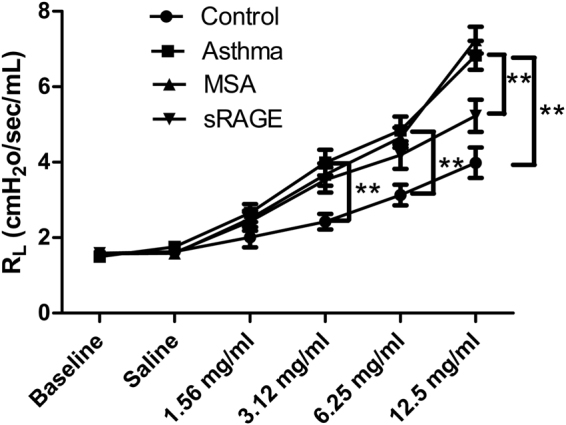
Local administration of sRAGE decreases AHR. Changes in lung resistance (RL) in response to increasing doses of methacholine represents OVA-induced AHR and were measured at 48 h after the final challenge in mice from Control, Asthma, MSA-treated, and sRAGE-treated groups. Values represent the means ± SEM (n = 5–6). **P < 0.01 compared with the Asthma group.
sRAGE suppresses local Th17 skewing in vivo
To analyse the immune response in airways, cytokine levels were measured in BALF. As shown in Fig. 6A, local administration of sRAGE resulted in a small reduction in the levels of IL-17 and IL-23 (Th17-associated cytokines). In contrast, there were no significant differences in the levels of IL-4 (a Th2-associated cytokine) or interferon (IFN)-γ (a Th1-associated cytokine) compared with mice that received MSA.
Figure 6.

Local administration of sRAGE suppresses the Th17 response. (A) sRAGE decreased Th17 cytokine production after OVA challenge. BALFs were collected at 24 h after the last OVA challenge. The levels of IL-23, IL-17A, IL-4, and IFN-γ in the BALF of mice from Control, Asthma, MSA-treated, and sRAGE-treated groups were assessed by ELISAs. Values represent the means ± SEM (n = 5). *P < 0.05 and **P < 0.01 compared with the Asthma group. (B) sRAGE down-regulated the count of Th17 cells in the lung. Representative results of the percentage of Th17 cells in CD3+-gated lung cells from mice in Control (a), Asthma (b), MSA-treated (c), and sRAGE-treated groups (d) were analysed by flow cytometry. (C) Histogram showing the absolute counts of Th17 cells among the lung cells of mice from the various groups. Values represent the means ± SEM (n = 5). *P < 0.05 and **P < 0.01 compared with the Asthma group.
Because administration of sRAGE inhibited the IL-17A level in BALF, we investigated the possibility that this inhibition was mediated through the decreased production of Th17 cells in the lung. As shown in Fig. 6B, OVA plus LPS-sensitised mice exhibited a dramatic increase of Th17 cell production in their lung tissue, whereas administration of sRAGE significantly down-regulated the production of Th17 cells in lung tissue. These results suggested that local administration of sRAGE suppresses Th17 skewing in the mouse model of neutrophilic asthma.
sRAGE decreases RAGE and IL-23 expression in CD11c+ APCs of lung tissue
Because DCs possess the potential to induce Th cell polarisation and play a vital role in sensitisation to the development of asthma, we investigated whether pulmonary DCs are involved in the anti-inflammatory effects of sRAGE in Th17 polarisation. We first assessed RAGE expression in lung DCs, a prerequisite for initiating the immune response to AGE signalling in airways. As shown in Fig. 7A, pretreatment with sRAGE induced a remarkable reduction in RAGE expression by pulmonary DCs in terms of mean fluorescence intensity (MFI) compared with MSA-treated mice (Fig. 7B), suggesting that RAGE expression in lung DCs was substantially affected by the inhibition of HMGB1 signalling with sRAGE.
Figure 7.

Local administration of sRAGE inhibits RAGE and IL-23 expression by lung CD11c+ APCs. (A) sRAGE inhibited RAGE expression in CD11c+ APCs. Representative findings of RAGE expression in CD11c+ APCs of mice from Control, Asthma, MSA-treated, and sRAGE-treated groups were analysed by flow cytometry. (B) Mean fluorescence intensity (MFI) showing the expression of RAGE in CD11c+ APCs of mice from the various groups. Values represent the means ± SEM (n = 5). *P < 0.05 and **P < 0.01 compared with the Asthma group. (C) sRAGE inhibited the percentage of IL-23+ CD11c+ APCs among low density lung cells. Representative results of the percentage of IL-23+ CD11c+ APCs in mice from Control (a), Asthma (b), MSA-treated (c), and sRAGE-treated groups (d) were analysed by flow cytometry. (D) Histogram showing the absolute counts of IL-23+ CD11c+ APCs in mice from the various groups. Values represent the means ± SEM (n = 5). *P < 0.05 and **P < 0.01 compared with the Asthma group.
To eliminate the possibility of sRAGE masking the RAGE antibody epitope, sRAGE was directly added to lung cell suspensions from asthmatic mice, and then RAGE expression in DCs was evaluated using the RAGE antibody for flow cytometric analysis. The results showed that administration of sRAGE had no effect on RAGE expression in DCs (Supplementary Fig. S2), suggesting that sRAGE did not mask the RAGE antibody epitope.
Additionally, we further assessed the polarising ability of DCs in generating Th17 cells. Consistent with the reduced level of IL-23 in BALF, administration of sRAGE significantly decreased IL-23 production by lung CD11c+ APCs, which is an important Th17-polarised cytokine that promotes the differentiation and proliferation of Th17 cells (Fig. 7C,D). These results demonstrated that sRAGE decreased the number of IL-23+ CD11c+ APCs, thus inhibiting development of the Th17-mediated immune response in the lungs.
sRAGE inhibits recombinant HMGB1 (rHMGB1)-activated bone marrow-derived DCs (BMDCs) inducing Th17 responses in vitro
Our previous study showed that rHMGB1-activated BMDCs have the potential to induce efficient Th17 polarisation via endogenous production of IL-2319. Considering the role of HMGB1 as the key instigator of adaptive immune responses for BMDC-mediated Th17 polarisation, blockade of HMGB1 signalling might represent a major mechanism of sRAGE-mediated immune regulatory effects. To examine the contribution of sRAGE to Th17 cell development induced by rHMGB1-activated BMDCs, Th17 polarisation in vitro was analysed by co-culture of OVA-pulsed BMDCs and CD4+ T cells from OVA-sensitised mice in the absence or presence of rHMGB1 and/or sRAGE. As a result, rHMGB1-activated BMDCs significantly enhanced secreted IL-17A levels in the culture supernatants (Fig. 8A) and increased the percentage of IL-17+ CD4+ T cells (Fig. 8B). In contrast, high concentrations of sRAGE (200 or 400 ng/ml) significantly down-regulated the expression of IL-17 induced by rHMGB1-stimulated BMDCs, suggesting that sRAGE suppressed the Th17 response induced by rHMGB1-stimulated BMDCs.
Figure 8.

sRAGE inhibits rHMGB1-stimulated BMDCs inducing Th17 responses in vitro. (A) sRAGE inhibited rHMGB1-stimulated BMDCs to produce IL-23. BMDCs were cultured in medium or stimulated with 500 ng/ml HMGB1 alone or cocultured with graded concentrations of sRAGE (50, 100, 200, 400 ng/ml) for 2 days. IL-23 levels in culture supernatants were measured using an ELISA. Values represent the means ± SEM (n = 5). *P < 0.05 and **P < 0.01 compared with the rHMGB1-stimulated BMDC group. (B, C, and D) sRAGE inhibited Th17 polarisation induced by rHMGB1-stimulated BMDCs. CD4+ T cells from the spleens of OVA-sensitised mice (1 × 105) were cultured alone or cocultured with BMDCs (2.5 × 104) that were treated with or without 500 ng/ml rHMGB1 in the presence or absence of 400 ng/ml sRAGE or MSA. The culture supernatants were analysed for IL-17A using an ELISA (B), and the percentages of IL-17+ CD4+ T cells were analysed by flow cytometry (C). (D) Histogram showing the percentages of IL-17+ CD4+ T cells after the 5 days of coculture. Values represent the means ± SEM (n = 5). *P < 0.05 and **P < 0.01 compared with the rHMGB1-stimulated BMDC group.
sRAGE inhibits the Th17 priming induced by HMGB1-activated BMDCs in vivo
To evaluate the role of sRAGE in HMGB1-activated BMDCs of Th17 polarisation in vivo, we adoptively transferred rHMGB1 or rHMGB1 plus sRAGE-treated DCs into the airways of mice (Fig. 1B). Adoptive transfer of HMGB1-activated DCs resulted in significant neutrophilic inflammation characterised by infiltration of neutrophils into the BALF (Fig. 9A) and Th17/Th1 priming as shown by increases in IL-23, IL-17A, and IFN-γ levels of the BALF (Fig. 9B). However, adoptive transfer of HMGB1 plus sRAGE-treated DCs markedly decreased the neutrophils in the BALF (Fig. 9A) and the Th17 response as shown by decreases in IL-23 and IL-17A levels (Fig. 9B). These data suggested that sRAGE inhibits the capability of HMGB1-activated DCs to induce neutrophilic inflammation and Th17 skewing in asthmatic mice.
Figure 9.

sRAGE impedes the potential of rHMGB1-stimulated BMDCs to prime Th17 responses in vivo. Mice received an intratracheal (i.t.) injection of 2 × 106 PBS-treated, non-pulsed DCs (PBS/DCs), rHMGB1-treated, OVA-pulsed DCs (rHMGB1/OVA-DCs), or rHMGB1 plus sRAGE-treated, OVA-pulsed DCs (rHMGB1 + sRAGE/OVA-DCs) and were then exposed to OVA aerosols on days 10–12. One day after the final challenge, the mice were sacrificed for further analysis. (A) Total and differential cells counts and (B) cytokine profiles were analysed in the BALF. Values represent the means ± SEM (n = 5). *P < 0.05 and **P < 0.01 compared with the Asthma group.
Discussion
Neutrophilic asthma is a prevalent, yet recently described phenotype of asthma, characterised by the mass infiltration of neutrophilic rather than eosinophilic airway inflammation and AHR, which may have an infectious origin27. Th17 immune responses may have a role in the pathogenesis of neutrophilic asthma. However, the nature of the association between Th17 responses and the development of neutrophilic asthma is not understood. DCs have been regarded as critical initiators of allergic inflammation, not only because of their abilities to process and present inhaled antigens, but also their capabilities to activate naïve T cells and even determine the type of immune response28. Some experiments indicate that HMGB1 is involved in local inflammatory responses and contributes to direct DC functions29,30. Therefore, we postulated that HMGB1 acts on DCs to promote the local Th17 response in neutrophilic asthma.
Previous studies have revealed up-regulation of HMGB1 expression in a variety of diseases, and activation of HMGB1/RAGE signalling is a poor prognostic factor for these diseases. For example, HMGB1/RAGE signalling is upregulated in patients with diabetes and atherosclerosis31. Some investigations have reported that reduced sRAGE is associated with neutrophilic airway inflammation in asthma and chronic obstructive pulmonary disease (COPD)25,32, suggesting that sRAGE might have a protective role in inflammatory lung diseases. Here, we confirmed that production of sRAGE was significantly decreased in the mouse model of neutrophilic asthma, and this decrease was well correlated with elevated IL-17 levels and increased neutrophil infiltration, suggesting that sRAGE production was responsible for the development of Th17-biased inflammation. Furthermore, our study showed that administration of sRAGE inhibited HMGB1 expression, airway neutrophilic inflammation, and AHR in the mouse model of neutrophilic asthma, indicating that HMGB1/RAGE signalling is a key regulator of the immune cascade leading to subsequent Th17 polarisation, neutrophilic airway inflammation, and AHR.
RAGE is a pattern recognition receptor that binds AGEs, HMGB1, amyloid β-peptide, and S100 protein. It interacts with diverse endogenous ligands and elicits activation of immune and inflammatory responses, induction of oxidant stress, and tissue remodelling responses33. Recently, several carboxyl-terminal truncated isoforms of RAGE, such as sRAGE and endogenous secretory RAGE (esRAGE), were identified in the lungs of both humans and mice34,35. Because these isoforms lack a transmembrane domain, they are secreted and act as decoy receptors. Indeed, sRAGE has been shown to prevent or reverse RAGE signals in experimental models of diabetic atherosclerosis, wound healing, amyloidosis, and colitis36–39. In addition, Zhang and colleagues reported that sRAGE treatment significantly attenuates the neutrophilic inflammation and pathological changes in the LPS-induced murine model of acute lung injury, suggesting that sRAGE may be secreted as a decoy receptor and contributes to the suppression of excessive inflammatory responses during actual lung injury40. Izushi and colleagues reported that the release of sRAGE has the potential to attenuate the excessive inflammatory response in acute respiratory distress syndrome (ARDS), and supports the notion that RAGE may be a promising candidate as a molecular target for the treatment of ARDS41. Sukkar and colleagues reported that systemic sRAGE was significantly decreased in subjects with neutrophilic asthma or COPD compared with those without airway neutrophilia, and that neutrophilic airway inflammation in asthma and COPD is associated with reduced sRAGE. They proposed that sRAGE deficiency is a primary consequence of excessive neutrophilic inflammation. They also identified sRAGE as a potential biomarker for the prognosis or management of asthmatic or COPD subjects with neutrophilic airway inflammation25. Smith and colleagues reported lower plasma levels of sRAGE in COPD patients during disease exacerbation, which is frequently caused by respiratory bacterial and/or viral infection, compared with patients with stable COPD. These data support the theory that sRAGE sequesters RAGE ligands and limits RAGE signalling, and that correcting deficiencies in sRAGE could represent a therapeutic strategy for neutrophilic asthma and COPD42. Milutinovic and colleagues reported that the absence of RAGE abolishes most assessed measures of pathology, including airway hypersensitivity, eosinophilic inflammation, and airway remodelling in a house dust mite mouse model of asthma43. All these studies indicate that RAGE inhibition or blockage with sRAGE may serve as a promising therapeutic strategy to control airway inflammation. However, its protective effects and the molecular mechanisms of sRAGE in neutrophilic asthma have not been characterised previously.
IL-17 has been suggested to play a role in Th17 responses. In this study, IL-17 was up-regulated in the BALF of asthmatic mice, and administration of sRAGE actively inhibited the polarisation of Th17 cells and expression of IL-23 in CD11+ APCs. These results suggest that sRAGE is a negative regulator of established allergic asthma and may drive the IL-23/IL-17 axis that in turn controls the Th17 response in neutrophilic asthma. More interestingly, RAGE expression in pulmonary DCs was regulated by sRAGE treatment, implying that negative feedback through HMGB1 signalling might exist to inhibit the development of allergic inflammation.
HMGB1/RAGE signalling not only occurs in DCs but also in other inflammatory cells44. To rule out the direct effects of HMGB1 blockage by sRAGE on other effector cells in allergic inflammation, we performed experiments in which rHMGB1-activated DCs were treated with sRAGE in vitro prior to adoptive transfer into the airways of naive mice. As expected, rHMGB1-activated DCs triggered strong neutrophilic airway inflammation that was accompanied by massive production of Th17 cytokines such as IL-23 and IL-17. However, pretreatment of HMGB1-activated DCs with sRAGE resulted in a significant reduction of neutrophil infiltration as well as IL-23 and IL-17 levels, further illuminating the putative role of HMGB1 signalling in DC-primed allergic diseases and demonstrating the potential of HMGB1 signalling blockade in DCs to control Th17 responses and neutrophilic airway inflammation.
In conclusion, our findings indicate that local administration of sRAGE inhibits Th17-mediated cardinal features of neutrophilic asthma at least partly by blocking HMGB1 signalling in lung DCs. Therefore, it might be worth determining whether decreased levels of sRAGE correlate with the severity of asthma, and further studies on the physiological and pathological significance of sRAGE may shed more light on allergic pathogenesis and reveal a new therapeutic perspective for these diseases.
Methods
Mice
Female 8–10-week-old C57BL/6 mice were obtained from the Animal Center of Jinling Hospital. All mice were bred and maintained under pathogen-free conditions, where the temperature was maintained at 20–22 °C and the humidity was kept at 50–60%. The dark/light cycles were 12 h each. All experiments involving animals and tissue samples were performed according to the guidelines of the National Institutes of Health and Medical School of Nanjing University (Nanjing, China), and all procedures were approved by the Institutional Animal Care and Use Committee of Medical School of Nanjing University.
Protocols for the mouse model and experimental intervention
A murine model of neutrophilic asthma characterised by Th17 cell responses was generated as described previously45. Briefly, mice underwent intranasal sensitisation with 75 μg OVA (grade V; Sigma-Aldrich, St Louis, MO, USA) plus 10 μg LPS (E. coli serotype 026:B6; Sigma-Aldrich) on days 0, 1, 2, and 7, and were then challenged by intranasal instillation of 50 μg OVA alone on days 14, 15, 21, and 22. All mice were analysed at 24 h after the last OVA challenge.
In this study, the mice were divided randomly into four groups (n = 5–6 mice each) as follows: (i) mice sensitised with PBS and challenged with OVA (Control group); (ii) mice sensitised with OVA plus LPS and challenged with OVA (Asthma group); (iii) mice treated with MSA (Sigma-Aldrich) at 30 min before sensitisation to OVA plus LPS and the same challenge with OVA (MSA group); (iv) mice treated with sRAGE (R&D Systems) at 30 min before sensitisation to OVA plus LPS and the same challenge with OVA (sRAGE group). MSA or sRAGE was administered intranasally (200 μg/kg) on days 0, 1, 2, and 7 before sensitisation (Fig. 1A). The dose of sRAGE was predetermined by staining analysis of airway inflammation in mice that received 100–400 μg/kg sRAGE. In some experiments, different dosages of sRAGE (200 or 400 μg/kg) were administered intranasally to the mice at 30 min before the secondary OVA challenges on days 14, 15, 21, and 22. One the day after the final challenge, the mice were sacrificed for histological analysis.
BALF
To assess differential BALF cell counts, lungs were lavaged three times with 0.75 ml Ca2+ and Mg2+-free Hank’s balanced salt solution containing 0.1 mM sodium EDTA. Cells in the BALF samples were centrifuged at 300 g for 5 min to generate a cell pellet and cell-free supernatant that was frozen and stored at −80 °C until analysis by enzyme-linked immunosorbent assays (ELISAs). For differential cell counts, cytocentrifuged preparations were fixed and stained with Diff-Quick (Kokusaishiyaku, Kobe, Japan) and differentiated morphologically by counting 300 cells/slide. The levels of sRAGE, IL-23, IL-17A, IL-4, and IFN-γ in the BALF were determined by ELISAs according to the manufacturer’s instructions (eBioscience, CA, USA).
Histological examination
For histopathological assessment, non-lavaged lobes of the lungs were fixed and embedded in paraffin. Sections (5 µm thick) from all lobes were prepared and stained with H&E for analysis of cellular infiltrates. Mucus-containing goblet cells were detected by staining with PAS. The numbers of PAS-positive goblet cells were determined only in cross-sectional areas of the airway wall. The sections were observed under a microscope at × 200 magnification. Six to eight fields per slide in five to six samples from each group of mice were examined in a blinded manner.
AHR
Airway responsiveness of mice to increasing concentrations of aerosolised MCh was measured as described in detail previously46. After the mice were anesthetised with an intraperitoneal injection of pentobarbital sodium (100 mg/kg), the trachea was cannulated via tracheotomy. The mice were connected to a computer-controlled, small animal ventilator and ventilated with a tidal volume of 10 ml/kg at a frequency of 150 breaths per min using the flexiVent System (SCIREQ, Montreal, Canada). After obtaining baseline measurements, each mouse was challenged with MCh aerosol at increasing concentrations (0, 1.56, 3.12, 6.25, and 12.5 mg/ml), and the lung resistance (RL) to the inhaled MCh was recorded.
Measurement of HMGB1 expression
Immunohistochemical staining of lung tissue was performed to detect the expression of HMGB1 in the lungs as described previously47. In brief, 5 μm paraffin-embedded sections were stained with a primary antibody against HMGB1 (Santa Cruz Biotechnology Inc., CA, USA), which was diluted at 1:50, and a biotinylated secondary antibody that was diluted at 1:500. The bound peroxidase was visualised using the 3,3′-diaminobenzidine method. Ten randomly chosen fields per section were counted under a microscope at × 200 magnification, and the intensity of HMGB1 protein staining was determined as the average optical density using IPP software (Image-Pro Plus 6.0, Media, Cybernetics).
HMGB1 expression was also analysed by western blotting. Lung tissues were homogenised to prepare lung lysates. Protein samples (30 μg) were subjected to 10% SDS-polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes. The membranes were blocked in blocking buffer (5% dry milk powder in TBS plus Tween 20) for 1 h at room temperature. Then, the membranes were incubated with a primary HMGB1 antibody (Santa Cruz Biotechnology Inc.). In some experiments, 200 or 400 ng/ml sRAGE was directly added to the lung homogenates, and HMGB1 expression was evaluated using the HMGB1 antibody by western blotting. HMGB1 mRNA levels were analysed in the lung tissue by quantitative PCR as described previously48.
Culture and treatment of DCs
BMDCs were isolated and cultured in DC medium as described previously49. After 8 days of culture, DCs were enriched using anti-CD11c-coated magnetic microbeads (Miltenyi-Biotec, Auburn, CA, USA) and treated with medium in the absence or presence of rHMGB1 (500 ng/ml) (R&D Systems, Minneapolis, MN, USA) with or without various concentrations of sRAGE (10, 100, 200, or 400 ng/ml) (R&D Systems) for 2 days. Cytokine concentrations in the cell culture supernatants were measured using an IL-23 ELISA kit (R&D Systems). rHMGB1 and sRAGE were tested by Limulus amebocyte lysate (ZhanJiang A&C Biological, China) and considered to be endotoxin free. In addition, rHMGB1 was in the disulphide form in this study.
Coculture of DCs with CD4+ T Cells
CD4+ T cells were highly enriched from the spleens of OVA-sensitised mice using a Mouse CD4+ T cell enrichment kit (Stem Cell Technologies, Vancouver, Canada) following the manufacturer’s instructions50. The OVA-sensitised mouse model was established as described previously30. The purity of isolated CD4+ T cells was greater than 98%. CD4+ T cells (1 × 104 cells/well) were mixed in a 96-well plate with BMDCs (2.5 × 104/well) stimulated with rHMGB1 (500 ng/ml) in the absence or presence of sRAGE (200 ng/ml). OVA (10 μg/mL) was added, and the mixed cells were cultured at 37 °C with 5% CO2. After 5 days of incubation, the culture supernatants were analysed for IL-17A by an ELISA, and the expression of intracellular IL-17 in CD4+ T cells was analysed by flow cytometry.
Flow cytometric analysis
To detect IL-17+ CD4+ T cells in the DC-T cell coculture system, the cells were incubated with 10 μg/ml brefeldin A (eBioscience) for 2 h and then stained for cell surface CD4 by a FITC-anti-CD4 mAb (eBioscience) for 30 min at 4 °C. After incubation in a fixation/permeabilisation solution (eBioscience), the cells were stained for intracellular IL-17 with a PE-anti-IL-17 mAb (eBioscience) for 30 min and analysed using a FACSCalibur flow cytometer (BD Biosciences).
To detect IL-17+ CD4+ T cells in lung tissue, lung cells were obtained according to previously reported methods19. Lung cells (4 × 106/ml) were washed three times in FACS buffer (PBS containing 1% bovine serum albumin and 0.1% sodium azide), incubated with brefeldin A (10 μg/ml) for 2 h, and then stained with surface-specific Abs (anti-CD3-APC and anti-CD4-FITC; eBioscience) for 30 min at 4 °C. For intracellular staining, cells were fixed and permeabilised with the fixation/permeabilisation solution, intracellularly stained with the PE-anti-IL-17 mAb for 30 min, and subsequently analysed using the FACSCalibur flow cytometer.
To detect RAGE and IL-23 expression in CD11C+ APCs, low density lung cells were enriched as described previously19. Briefly, digested lung cells (10 × 106/ml) were filtered through a nylon wool plug and resuspended in high density Percoll (ρ = 1.075 g/ml), overlaid with an equal volume of lower density Percoll (ρ = 1.030 g/ml), and centrifuged at 400 × g for 20 min. Low density lung cells, which were enriched for mononuclear cells, were recovered from the 1.075/1.030 Percoll interface and washed with Hank’s balanced salt solution. Low density lung cells were then stained with a DC-specific Ab (FITC-anti-CD11C mAb, eBioscience) and PE-conjugated goat anti-mouse RAGE mAb (Sigma-Aldrich) for 30 min at 4 °C. For intracellular staining, cells were fixed and permeabilised with the fixation/permeabilisation solution, stained with PE-anti-IL-23 mAb (eBioscience) for 30 min, and subsequently analysed using the FACSCalibur flow cytometer. In some experiments, 200 or 400 ng/ml sRAGE was directly added to lung cell suspensions from asthmatic mice, and RAGE expression in DCs was evaluated using the RAGE antibody by flow cytometry.
Adoptive transfer of DCs
A murine model of asthma through the transfer of BMDCs was established as described previously51. Briefly, BMDCs were enriched and pulsed with OVA overnight (OVA-DCs). OVA-DCs were then injected into naïve mice via the intratracheal route (i.t.). Ten days after intratracheal immunisation, the mice were challenged with OVA (1% w/v in PBS; grade V; Sigma-Aldrich) aerosol during a daily, 30 min challenge on 3 consecutive days. In this experiment, the mice were divided randomly into the three groups (n = 5–6 per group) as follows: (i) mice received an i.t. injection of 2 × 106 PBS-treated and non-pulsed DCs (PBS/DCs); (ii) mice received an i.t. injection of 2 × 106 rHMGB1-treated OVA-DCs (rHMGB1/OVA-DCs); (iii) mice received an i.t. injection of 2 × 106 rHMGB1 plus sRAGE-treated OVA-DCs (rHMGB1 + sRAGE/OVA-DCs). The OVA-DCs were treated with rHMGB1, sRAGE, or control IgG at concentrations of 500, 200, or 200 ng/ml, respectively. All mice were sacrificed at 24 h after the final challenge for further analyses.
Statistical analysis
Data are expressed as means ± standard error of the mean (SEM). Differences between groups were analysed by SPSS for windows (version 16.0) using the unpaired, two-tailed, parametric Student t-test or one-way analysis of variance followed by Dunnett’s multiple comparison tests. P-values of less than 0.05 were considered as statistically significant.
Electronic supplementary material
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81270079 and 81570025). The authors are grateful to Dr. Huahao Shen (The Second Affiliated Hospital School of Medicine of Zhejiang University, Hangzhou, China) for reviewing this manuscript and giving constructive suggestions, and a special thank you to the members of the integrated Lee Lab Group (Mayo Clinic Scottsdale) for their valuable help and encouragement when Dr. Fang Zhang studied in the Lee Laboratory. We thank Mitchell Arico from Liwen Bianji, Edanz Group China, for editing the English text of a draft of this manuscript.
Author Contributions
F.Z., X.S., G.H., X.F.X., E.H.C. and Y.S. performed the experiments; F.Z. and Y.S. designed the study; F.Z. wrote the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-14667-4.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Fang Zhang, Email: zhangfanglab@163.com.
Yong Song, Email: yong_song6310@yahoo.com.
References
- 1.Sears MR. Trends in the prevalence of asthma. Chest. 2014;145:219–225. doi: 10.1378/chest.13-2059. [DOI] [PubMed] [Google Scholar]
- 2.Price D, Fletcher M, van der Molen T. Asthma control and management in 8,000 European patients: the REcognise Asthma and LInk to Symptoms and Experience (REALISE) survey. NPJ primary care respiratory medicine. 2014;24:14009. doi: 10.1038/npjpcrm.2014.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wardlaw, A. J. et al. New insights into the relationship between airway inflammation and asthma. Clin Sci (Lond)103, 201–211, https://doi.org/10.1042/ (2002). [DOI] [PubMed]
- 4.Agache I, Ciobanu C, Agache C, Anghel M. Increased serum IL-17 is an independent risk factor for severe asthma. Respir Med. 2010;104:1131–1137. doi: 10.1016/j.rmed.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 5.Plantinga M, Hammad H, Lambrecht BN. Origin and functional specializations of DC subsets in the lung. Eur J Immunol. 2010;40:2112–2118. doi: 10.1002/eji.201040562. [DOI] [PubMed] [Google Scholar]
- 6.Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. 2012;181:8–18. doi: 10.1016/j.ajpath.2012.03.044. [DOI] [PubMed] [Google Scholar]
- 7.Shainheit MG, et al. Dendritic cell IL-23 and IL-1 production in response to schistosome eggs induces Th17 cells in a mouse strain prone to severe immunopathology. J Immunol. 2008;181:8559–8567. doi: 10.4049/jimmunol.181.12.8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sonderegger I, et al. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med. 2008;205:2281–2294. doi: 10.1084/jem.20071119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muller S, Ronfani L, Bianchi ME. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med. 2004;255:332–343. doi: 10.1111/j.1365-2796.2003.01296.x. [DOI] [PubMed] [Google Scholar]
- 10.Osmanov T, Ugrinova I, Pasheva E. The chaperone like function of the nonhistone protein HMGB1. Biochem Biophys Res Commun. 2013;432:231–235. doi: 10.1016/j.bbrc.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 11.Huang W, Tang Y, Li L. HMGB1, a potent proinflammatory cytokine in sepsis. Cytokine. 2010;51:119–126. doi: 10.1016/j.cyto.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 12.Shim EJ, et al. The role of high-mobility group box-1 (HMGB1) in the pathogenesis of asthma. Clin Exp Allergy. 2012;42:958–965. doi: 10.1111/j.1365-2222.2012.03998.x. [DOI] [PubMed] [Google Scholar]
- 13.Zhang S, Zhong J, Yang P, Gong F, Wang CY. HMGB1, an innate alarmin, in the pathogenesis of type 1 diabetes. Int J Clin Exp Pathol. 2009;3:24–38. [PMC free article] [PubMed] [Google Scholar]
- 14.Fang P, Schachner M, Shen YQ. HMGB1 in development and diseases of the central nervous system. Mol Neurobiol. 2012;45:499–506. doi: 10.1007/s12035-012-8264-y. [DOI] [PubMed] [Google Scholar]
- 15.Gnanasekar M, et al. HMGB1: A Promising Therapeutic Target for Prostate Cancer. Prostate cancer. 2013;2013:157103. doi: 10.1155/2013/157103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee CC, et al. Inhibition of high-mobility group box 1 in lung reduced airway inflammation and remodeling in a mouse model of chronic asthma. Biochem Pharmacol. 2013;86:940–949. doi: 10.1016/j.bcp.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 17.Duan L, et al. High-mobility group box 1 promotes early acute allograft rejection by enhancing IL-6-dependent Th17 alloreactive response. Lab Invest. 2011;91:43–53. doi: 10.1038/labinvest.2010.141. [DOI] [PubMed] [Google Scholar]
- 18.Mu HH, Nourian MM, Jiang HH, Tran JW, Cole BC. Mycoplasma superantigen initiates a TLR4-dependent Th17 cascade that enhances arthritis after blocking B7-1 in Mycoplasma arthritidis-infected mice. Cell Microbiol. 2014;16:896–911. doi: 10.1111/cmi.12247. [DOI] [PubMed] [Google Scholar]
- 19.Zhang F, et al. Anti-HMGB1 neutralizing antibody ameliorates neutrophilic airway inflammation by suppressing dendritic cell-mediated Th17 polarization. Mediators Inflamm. 2014;2014:257930. doi: 10.1155/2014/257930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie J, Mendez JD, Mendez-Valenzuela V, Aguilar-Hernandez MM. Cellular signalling of the receptor for advanced glycation end products (RAGE) Cell Signal. 2013;25:2185–2197. doi: 10.1016/j.cellsig.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 21.Sterenczak KA, et al. Cloning, characterisation, and comparative quantitative expression analyses of receptor for advanced glycation end products (RAGE) transcript forms. Gene. 2009;434:35–42. doi: 10.1016/j.gene.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, et al. Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J Biol Chem. 2008;283:35507–35516. doi: 10.1074/jbc.M806948200. [DOI] [PubMed] [Google Scholar]
- 23.Lalla E, et al. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J Clin Invest. 2000;105:1117–1124. doi: 10.1172/JCI8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng S, et al. Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury to the liver in mice. Hepatology. 2004;39:422–432. doi: 10.1002/hep.20045. [DOI] [PubMed] [Google Scholar]
- 25.Sukkar MB, et al. Soluble RAGE is deficient in neutrophilic asthma and COPD. Eur Respir J. 2012;39:721–729. doi: 10.1183/09031936.00022011. [DOI] [PubMed] [Google Scholar]
- 26.El-Seify MY, Fouda EM, Nabih ES. Serum level of soluble receptor for advanced glycation end products in asthmatic children and its correlation to severity and pulmonary functions. Clin Lab. 2014;60:957–962. doi: 10.7754/Clin.Lab.2013.130418. [DOI] [PubMed] [Google Scholar]
- 27.Nakagome K, Matsushita S, Nagata M. Neutrophilic inflammation in severe asthma. Int Arch Allergy Immunol. 2012;158(Suppl 1):96–102. doi: 10.1159/000337801. [DOI] [PubMed] [Google Scholar]
- 28.Lambrecht BN, Hammad H. Lung dendritic cells in respiratory viral infection and asthma: from protection to immunopathology. Annu Rev Immunol. 2012;30:243–270. doi: 10.1146/annurev-immunol-020711-075021. [DOI] [PubMed] [Google Scholar]
- 29.Bianchi ME, Manfredi AA. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev. 2007;220:35–46. doi: 10.1111/j.1600-065X.2007.00574.x. [DOI] [PubMed] [Google Scholar]
- 30.Zhang F, Huang G, Hu B, Qian GS, Song Y. Recombinant HMGB1 A box protein inhibits Th17 responses in mice with neutrophilic asthma by suppressing dendritic cell-mediated Th17 polarization. Int Immunopharmacol. 2015;24:110–118. doi: 10.1016/j.intimp.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 31.Koulis C, de Haan JB, Allen TJ. Novel pathways and therapies in experimental diabetic atherosclerosis. Expert review of cardiovascular therapy. 2012;10:323–335. doi: 10.1586/erc.12.13. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, et al. Changes of HMGB1 and sRAGE during the recovery of COPD exacerbation. J Thorac Dis. 2014;6:734–741. doi: 10.3978/j.issn.2072-1439.2014.04.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mosquera JA. Role of the receptor for advanced glycation end products (RAGE) in inflammation. Invest Clin. 2010;51:257–268. [PubMed] [Google Scholar]
- 34.Santilli F, Vazzana N, Bucciarelli LG, Davi G. Soluble forms of RAGE in human diseases: clinical and therapeutical implications. Curr Med Chem. 2009;16:940–952. doi: 10.2174/092986709787581888. [DOI] [PubMed] [Google Scholar]
- 35.Kalea AZ, et al. Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J. 2009;23:1766–1774. doi: 10.1096/fj.08-117739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wendt T, et al. RAGE modulates vascular inflammation and atherosclerosis in a murine model of type 2 diabetes. Atherosclerosis. 2006;185:70–77. doi: 10.1016/j.atherosclerosis.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 37.Goova MT, et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol. 2001;159:513–525. doi: 10.1016/S0002-9440(10)61723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emanuele E, et al. Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch Neurol. 2005;62:1734–1736. doi: 10.1001/archneur.62.11.1734. [DOI] [PubMed] [Google Scholar]
- 39.Meijer B, et al. Total soluble and endogenous secretory receptor for advanced glycation endproducts (RAGE) in IBD. J Crohns Colitis. 2014;8:513–520. doi: 10.1016/j.crohns.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, et al. Role of soluble receptor for advanced glycation end products on endotoxin-induced lung injury. Am J Respir Crit Care Med. 2008;178:356–362. doi: 10.1164/rccm.200707-1069OC. [DOI] [PubMed] [Google Scholar]
- 41.Izushi Y, et al. Soluble form of the receptor for advanced glycation end-products attenuates inflammatory pathogenesis in a rat model of lipopolysaccharide-induced lung injury. J Pharmacol Sci. 2016;130:226–234. doi: 10.1016/j.jphs.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Smith DJ, et al. Reduced soluble receptor for advanced glycation end-products in COPD. Eur Respir J. 2011;37:516–522. doi: 10.1183/09031936.00029310. [DOI] [PubMed] [Google Scholar]
- 43.Milutinovic PS, Alcorn JF, Englert JM, Crum LT, Oury TD. The receptor for advanced glycation end products is a central mediator of asthma pathogenesis. Am J Pathol. 2012;181:1215–1225. doi: 10.1016/j.ajpath.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 45.Kim YS, et al. Distinct roles of vascular endothelial growth factor receptor-1- and receptor-2-mediated signaling in T cell priming and Th17 polarization to lipopolysaccharide-containing allergens in the lung. J Immunol. 2010;185:5648–5655. doi: 10.4049/jimmunol.1001713. [DOI] [PubMed] [Google Scholar]
- 46.Liu JN, et al. The role of autophagy in allergic inflammation: a new target for severe asthma. Exp Mol Med. 2016;48:e243. doi: 10.1038/emm.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ebina M, et al. Gradual increase of high mobility group protein b1 in the lungs after the onset of acute exacerbation of idiopathic pulmonary fibrosis. Pulmonary medicine. 2011;2011:916486. doi: 10.1155/2011/916486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang J, et al. TLR4-HMGB1-, MyD88- and TRIF-dependent signaling in mouse intestinal ischemia/reperfusion injury. World J Gastroenterol. 2015;21:8314–8325. doi: 10.3748/wjg.v21.i27.8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang F, Huang G, Hu B, Song Y, Shi Y. A soluble thymic stromal lymphopoietin (TSLP) antagonist, TSLPR-immunoglobulin, reduces the severity of allergic disease by regulating pulmonary dendritic cells. Clin Exp Immunol. 2011;164:256–264. doi: 10.1111/j.1365-2249.2011.04328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quan J, Tan PH, MacDonald A, Friend PJ. Manipulation of indoleamine 2,3-dioxygenase (IDO) for clinical transplantation: promises and challenges. Expert Opin Biol Ther. 2008;8:1705–1719. doi: 10.1517/14712598.8.11.1705. [DOI] [PubMed] [Google Scholar]
- 51.Masten BJ, Olson GK, Kusewitt DF, Lipscomb MF. Flt3 ligand preferentially increases the number of functionally active myeloid dendritic cells in the lungs of mice. J Immunol. 2004;172:4077–4083. doi: 10.4049/jimmunol.172.7.4077. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.