

Abstract
The real-time monitoring of specific analytes in situ in the living body would greatly advance our understanding of physiology and the development of personalized medicine. Because they are continuous (wash-free and reagentless) and are able to work in complex media (e.g., undiluted serum), electrochemical aptamer-based (E-AB) sensors are promising candidates to fill this role. E-AB sensors suffer, however, from often-severe baseline drift when deployed in undiluted whole blood either in vitro or in vivo. We demonstrate that cell-membrane-mimicking phosphatidylcholine (PC)-terminated monolayers improve the performance of E-AB sensors, reducing the baseline drift from around 70% to just a few percent after several hours in flowing whole blood in vitro. With this improvement comes the ability to deploy E-AB sensors directly in situ in the veins of live animals, achieving micromolar precision over many hours without the use of physical barriers or active drift-correction algorithms.
Keywords: aptamers, biomimetic surfaces, electrochemical sensors, in vivo measurements, membrane monolayers
Broadcast live
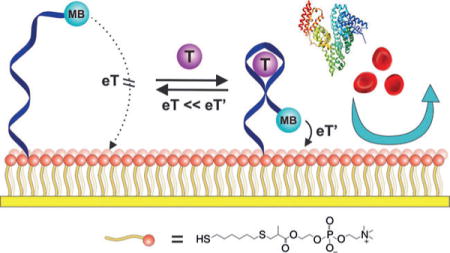
A biomimetic surface employing phosphatidylcholine head groups (red spheres) greatly improves the baseline stability of electrochemical aptamer-based (E-AB) sensors in whole blood and in live rats, reducing the baseline drift from 70% to less than 10% under these challenging conditions. MB=methylene blue, T=target, dark blue ribbon=aptamer, yellow strip= electrode, eT=electron transfer.
The ability to monitor specific molecules in real time in the living body would vastly improve our understanding of and ability to detect, monitor, and treat disease. Such a technology, for example, could provide the high-precision, patient-specific pharmacokinetic information needed to guide the delivery of “the right drug, at the right dose, and at the right time,” and thus help to achieve the promise of personalized medicine.[1] The development of sensors supporting such measurements, however, remains challenging. First, of course, to support this goal, sensors must achieve clinically relevant specificity, precision, and detection limits. Second, they must also operate continuously without requiring sample preparation, batch processing (such as washing steps), or the addition of exogenous reagents. Finally, they must resist or be insensitive to biofouling, that is, the harmful accumulation of proteins and blood cells on the sensor surface.
Towards the goal of real-time measurements in complex sample matrices, we[2] and others[3] have developed electrochemical aptamer-based (E-AB) biosensors (Figure 1), a versatile sensing platform that supports the continuous, real-time measurement of specific molecular analytes.[4] E-AB sensors comprise an aptamer “probe” that is attached at one end to an interrogating electrode using a self-assembled monolayer (SAM), and modified at the other end with a redox-active “reporter”. The binding of an analyte to the aptamer alters the efficiency with which the reporter approaches the electrode (as a result of either binding-induced conformational change or the steric bulk of the target),[5] thereby producing an easily measured change in current when the sensor is interrogated using square wave voltammetry.
Figure 1.
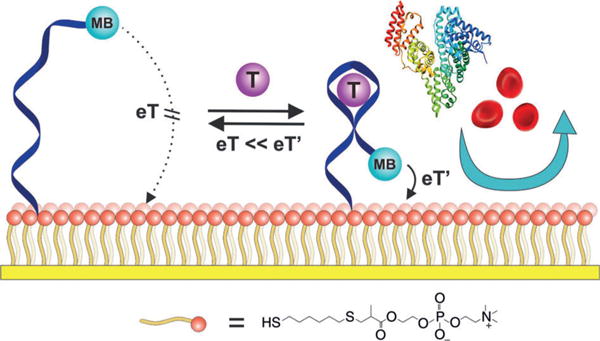
E-AB sensors comprise an electrode-bound, redox-reporter-modified aptamer (dark blue ribbon) that undergoes a binding-induced conformational change on binding to the target (purple sphere). This conformational change alters the positioning of the reporter (methylene blue, MB) relative to the electrode (yellow strip), thereby producing a target-dependent change in current when the sensor is interrogated by square wave voltammetry. Biomimetic monolayers incorporating phosphatidylcholine head-groups (orange spheres) can be used to largely eliminate the signal drift that appears when using these sensors in whole blood.
As is needed to support continuous, real-time measurements, E-AB sensing is a reagentless and single-step process. Because their signaling mechanism recapitulates the conformation-linked signaling employed by naturally occurring chemo-perception systems, E-AB sensors are also highly selective and thus E-AB sensors perform well even in flowing, undiluted blood serum.[6–8] They suffer, however, from often severe drift when challenged in flowing whole blood.[9,10]
We have previously overcome the problem of drift in whole blood through a combination of physical barriers (membranes or fluid sheaths to prevent cells from approaching the sensor surface) and drift-correction algorithms.[9–11] Membranes, however, increase the sensor bulk and slow sensor response times. And drift-correction algorithms require the collection of additional data at each time point, thereby degrading the time resolution. Motivated by these concerns, we have developed a biomimetic surface treatment that largely eliminates the drift seen in whole blood without invoking physical barriers or drift-correction algorithms, resulting in stable sensor performance even after hours of continuous operation directly in the living body.
A number of monolayer surfaces have been reported to date that resist biofouling,[12–19] one of which, phosphatidylcholine (PC)-terminated SAMs, we have explored here. Our selection of this passivation chemistry was driven by several considerations: 1) PC head-groups are thought to mimic the fouling resistance of eukaryotic cellular membranes by strongly binding water to produce a hydration layer that forms a barrier against protein or cell non-specific adsorption.[20–22] 2) PC-incorporated monolayers can be relatively short, thus rendering them likely to support rapid electron transfer. 3) Consistent with reported claims, we find that a gold surface coated with such a monolayer appears in micrographs to resist bulk fouling by blood cells (Figure S1 in the Supporting Information).
As our first test case, we employed a 28-base DNA aptamer that recognizes the cancer chemotherapeutic agent doxorubicin. We modified this aptamer with a methylene blue (MB) redox reporter on its 3′ end and a six-carbon thiol linker on its 5′ end. We then co-deposited the modified aptamer onto a gold electrode with either the 6-mercapto-1-hexanol (MCH) dilutant, as traditionally employed,[23] or with a phosphatidylcholine alkanethiolate derivative from 2-methacryloyloxyethyl phosphorylcholine (Figure 1) to produce the desired PC-terminated monolayer.
Whereas the MCH- and PC-based sensors both respond robustly when challenged with their target in simple buffers, only the latter perform well in whole blood. For example, the peak currents of both the PC- and MCH-based sensors increase in response to the addition of 10 μm doxorubicin in phosphate-buffered saline (PBS), with a somewhat higher gain for the former (Figure 2A,B), presumably owing to the effects of charged monolayers on the performance of such sensors.[24] In contrast, when challenged in flowing whole blood, the performance of the PC-based sensor is vastly improved relative to that of the MCH-based sensor. For example, whereas the PC-based sensors exhibit a less than 10% loss in peak current over the course of 12 h in flowing whole blood, the signal from MCH-based sensors falls by 70% (Figure 2C, Figure S2). Under these same conditions, the PC-based sensors respond quantitatively to their target in flowing whole blood and return quantitatively (less than 5% baseline drift over 12 h) to their original baseline when the drug is removed (Figure 2D, Figure S3), while the MCH-based sensors exhibit significant drift in both the presence and absence of target.
Figure 2.
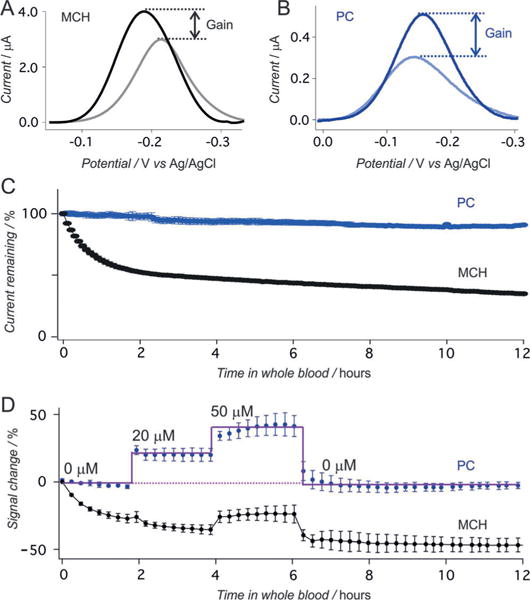
Doxorubicin-detecting E-AB sensors. A) Voltammograms recorded from MCH-based sensors in the absence and presence of target in PBS buffer. The signaling current increases by ca. 35% when the sample is spiked with 10 μm target. B) Under the same conditions a PC-based sensor exhibits higher signal gain, with the current increasing by ca. 65%. C) When challenged in flowing whole blood (no doxorubicin), PC-based sensors (blue) exhibit less than 10% current drift over 12 h, whereas MCH-based sensors (black) lose around two thirds of their original signal. D) The improved drift performance of the PC-based sensors allows real-time measurements in whole blood. Shown are data collected in flowing whole blood samples in vitro spiked at different time points with varying concentrations of doxorubicin. Error bars show the standard deviation of at least three independently fabricated sensors.
The improved performance of PC-monolayer-based sensors also holds for sensors employing aptamers recognizing other targets. To show this, we fabricated sensors employing an aminoglycoside-binding aptamer attached to the electrode via a six-carbon thiol linker. In contrast to the case with the doxorubicin-binding aptamer, we found that the binding constant of the PC-based sensor is poorer than that of the equivalent MCH-based sensor (Figure S4), presumably due to steric blocking caused by the thicker PC monolayer (ca. 17 Å versus ca. 9 Å).[25] To circumvent this problem, we re-fabricated the sensor using an 11-carbon linker (ca. 17 Å in length) on the aptamer and the same PC monolayer. For comparison, we employed an 11-mercapto-1-undecanol (MCU) monolayer in the corresponding “control” sensor. When interrogated using square wave voltammetry in buffer, both sensors exhibit a current increase in response to the addition of the aminoglycoside kanamycin, with the PC-based sensors again exhibiting greater gain (Figure 3A and B).
Figure 3.
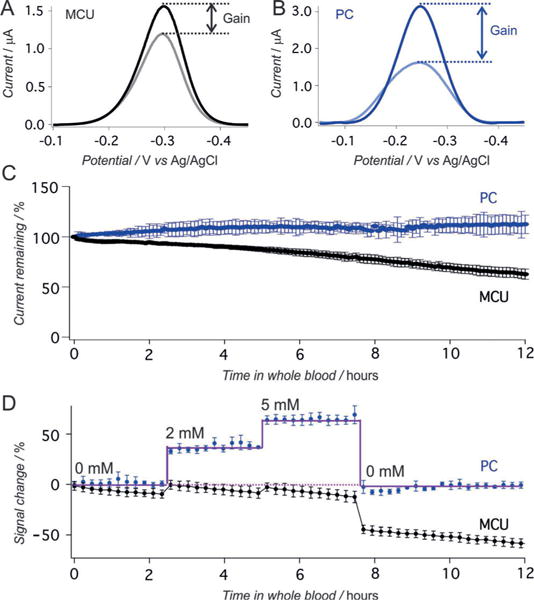
Kanamycin-detecting E-AB sensors. A, B) Higher gain is again obtained for PC-based sensors relative to MCU-based sensors (here both are challenged with 1 mM Kanamycin). C) When challenged in flowing whole blood (no kanamycin), PC-based sensors exhibit less than 10% current drift over 12 h, whereas MCU-based sensors lose half of their original signal. D) Data collected in vitro in flowing blood spiked at different time points with varying concentrations of kanamycin. Error bars show the standard deviation of at least three independently fabricated sensors.
As for the doxorubicin sensor, the performance of the PC-based kanamycin sensor is greatly improved over the MCU-based sensor when challenged in flowing whole blood. For example, whereas the PC-based sensor exhibits less than 10% signal drift over 12 h under these conditions (Figure 3C), the signal from the MCU-based sensors falls by nearly half despite the generally improved stability of this monolayer relative to shorter-chain monolayers.[26] This improved performance likewise holds when the sensors are challenged with their target, with the PC-based sensors responding quantitatively to their target (Figure 3D and Figure S5) and returning to baseline quantitatively upon removal of the target.
Given the improved performance of the PC-based sensors in whole blood in vitro, we were motivated to test their ability to support continuous real-time measurements in vivo in the blood of a living animal. To do so we emplaced the sensors for the detection of doxorubicin in the external jugular vein of an anesthetized Sprague-Dawley rat and injected doxorubicin into the opposite external jugular vein. We have previously found that, under these conditions, even membrane-protected MCH-based sensors suffer from significant drift, exhibiting signal loss on the order of 50% over the course of 3 h (Figure 4A). Historically, we have corrected for this using protective membranes and drift-correction algorithms,[10] which reduces the time resolution (due to reduced diffusion rates through the membrane and the need to collect additional voltammetric data to perform drift correction). The PC-based sensor, however, exhibits excellent in vivo baseline stability without the need for either membranes or drift-correction algorithms (Figure 4B), achieving micromolar precision in the measurement of clinically relevant levels of the drug. Specifically, following three sequential injections, the resulting plot of concentration versus time presents consecutive spikes corresponding to each injection, with maximum doxorubicin concentrations (Cmax) of approximately 30 μm and an effective clearance of 90% of the drug from the circulatory system within 50 min, values that are consistent with our previous studies and other prior reports.[10,27]
Figure 4.
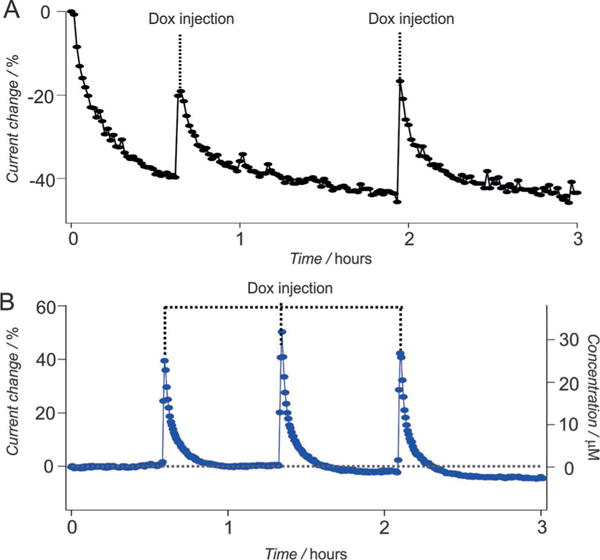
Real-time continuous measurement of doxorubicin (Dox) in living animals. A) When deployed in vivo, conventional MCH-based sensors exhibit significant drift; the sensor shown loses nearly half of its signaling current over a few hours even though it is protected with a microporous membrane[10] (each Dox dose is 2 mg m−2). B) Replacing the MCH monolayer with a PC monolayer largely eliminates this drift; the resulting sensor exhibits less than 5% drift after the same duration in vivo (each Dox dose is 40 mg m−2).
Herein, we have shown that a PC-terminated SAM that mimics the surface of cell membranes improves the performance of E-AB sensors deployed in flowing whole blood both in vitro and in vivo in our live animal model. Given these results, we believe that the biomimetic approach proposed here will benefit a wide variety of electrochemical sensor architectures with the objective of achieving real-time continuous monitoring of pharmacokinetics to provide patients with personalized medicine.
Supplementary Material
Acknowledgments
The biosensor work at UCSB was carried out at the Institute for Collaborative Biotechnologies (supported by the Army Research Office, Grant W911NF-09-0001 and the National Institutes of Health, Grant R01AI107936) and with partial support from the W. M. Keck Foundation. L. H. was partially supported by Swiss National Science Foundation with an “Early Postdoc Mobility fellowship”. P. D.-D. is partially supported by Fonds de recherche du Quebec—Nature et Technologies and Natural Sciences and Engineering Research Council with a postdoctoral fellowship. N. A.-C. is supported by the Otis Williams Postdoctoral Fellowship of the Santa Barbara foundation. J. S. is supported by the National Cancer Institute of the National Institutes of Health (NRSA F31CA183385). The authors thank Dr. Gabriel Ortega Quintanilla for help with Matlab script for data processing.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: http://dx.doi.org/10.1002/anie.201700748.
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Dr. Hui Li, Department of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA 93106 (USA)
Dr. Philippe Dauphin-Ducharme, Department of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA 93106 (USA)
Dr. Netzahualcóyotl Arroyo-Currás, Department of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA 93106 (USA)
Claire H. Tran, Department of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA 93106 (USA)
Dr. Philip A. Vieira, Department of Psychology, California State University, Dominguez Hills, 1000 E. Victoria Ave., Carson, CA 90747 (USA)
Dr. Shaoguang Li, Department of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA 93106 (USA)
Christina Shin, Department of Psychological and Brain Sciences, University of California Santa Barbara, Santa Barbara, CA 93106 (USA); The Neuroscience Research Institute and Department of Molecular, Cellular and Developmental Biology, University of California Santa Barbara, Santa Barbara, CA 93106 (USA).
Jacob Somerson, Interdepartmental Program, Biomolecular Science and Engineering, University of California Santa Barbara, Santa Barbara, CA 93106 (USA).
Tod E. Kippin, Department of Psychological and Brain Sciences, University of California Santa Barbara, Santa Barbara, CA 93106 (USA) The Neuroscience Research Institute and Department of Molecular, Cellular and Developmental Biology, University of California Santa Barbara, Santa Barbara, CA 93106 (USA).
Kevin W. Plaxco, Department of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA 93106 (USA) Center for Bioengineering, University of California Santa Barbara, Santa Barbara, CA 93106 (USA).
References
- 1.Hamburg MA, Collins FS. N Engl J Med. 2010;363:301–304. doi: 10.1056/NEJMp1006304. [DOI] [PubMed] [Google Scholar]
- 2.Cash KJ, Ricci F, Plaxco KW. J Am Chem Soc. 2009;131:6955–6957. doi: 10.1021/ja9011595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zuo X, Song S, Zhang J, Pan D, Wang L, Fan C. J Am Chem Soc. 2007;129:1042–1043. doi: 10.1021/ja067024b. [DOI] [PubMed] [Google Scholar]
- 4.Lubin AA, Plaxco KW. Acc Chem Res. 2010;43:496–505. doi: 10.1021/ar900165x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao Y, Uzawa T, White RJ, Demartini D, Plaxco KW. Electroanalysis. 2009;21:1267–1271. doi: 10.1002/elan.200804564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferapontova EE, Olsen EM, Gothelf KV. J Am Chem Soc. 2008;130:4256–4258. doi: 10.1021/ja711326b. [DOI] [PubMed] [Google Scholar]
- 7.Lubin AA, Lai RY, Baker BR, Heeger AJ, Plaxco KW. Anal Chem. 2006;78:5671–5677. doi: 10.1021/ac0601819. [DOI] [PubMed] [Google Scholar]
- 8.Vallée-Bélisle A, Ricci F, Uzawa T, Xia F, Plaxco KW. J Am Chem Soc. 2012;134:15197–15200. doi: 10.1021/ja305720w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferguson BS, Hoggarth DA, Maliniak D, Ploense K, White RJ, Woodward N, Hsieh K, Bonham AJ, Eisenstein M, Kippin TE, Plaxco KW, Soh HT. Sci Transl Med. 2013;5 doi: 10.1126/scitranslmed.3007095. 213ra165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arroyo-Currás N, Somerson J, Vieira PA, Ploense KL, Kippin TE, Plaxco KW. Proc Natl Acad Sci USA. 2017;114:645–650. doi: 10.1073/pnas.1613458114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Arroyo-Currás N, Kang D, Ricci F, Plaxco KW. J Am Chem Soc. 2016;138:15809–15812. doi: 10.1021/jacs.6b08671. [DOI] [PubMed] [Google Scholar]
- 12.Hu Y, Liang B, Fang L, Ma G, Yang G, Zhu Q, Chen S, Ye X. Langmuir. 2016;32:11763–11770. doi: 10.1021/acs.langmuir.6b03016. [DOI] [PubMed] [Google Scholar]
- 13.Wu J, Campuzano S, Halford C, Haake DA, Wang J. Anal Chem. 2010;82:8830–8837. doi: 10.1021/ac101474k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blaszykowski C, Sheikh S, Thompson M. Chem Soc Rev. 2012;41:5599–5612. doi: 10.1039/c2cs35170f. [DOI] [PubMed] [Google Scholar]
- 15.Vermette P, Meagher L. Colloids Surf B. 2003;28:153–198. [Google Scholar]
- 16.Ishihara K, Fukumoto K, Iwasaki Y, Nakabayashi N. Biomaterials. 1999;20:1545–1551. doi: 10.1016/s0142-9612(99)00052-6. [DOI] [PubMed] [Google Scholar]
- 17.Chen S, Zheng J, Li L, Jiang S. J Am Chem Soc. 2005;127:14473–14478. doi: 10.1021/ja054169u. [DOI] [PubMed] [Google Scholar]
- 18.Laughlin RG. Langmuir. 1991;7:842–847. [Google Scholar]
- 19.Goda T, Tabata M, Sanjoh M, Uchimura M, Iwasaki Y, Miyahara Y. Chem Commun. 2013;49:8683–8685. doi: 10.1039/c3cc44357d. [DOI] [PubMed] [Google Scholar]
- 20.Chen S, Li L, Zhao C, Zheng J. Polymer. 2010;51:5283–5293. [Google Scholar]
- 21.Nakaya T, Li YJ. Prog Polym Sci. 1999;24:143–181. [Google Scholar]
- 22.Schlenoff JB. Langmuir. 2014;30:9625–9636. doi: 10.1021/la500057j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao Y, Lai RY, Plaxco KW. Nat Protoc. 2007;2:2875–2880. doi: 10.1038/nprot.2007.413. [DOI] [PubMed] [Google Scholar]
- 24.Ricci F, Zari N, Caprio F, Recine S, Amine A, Moscone D, Palleschi G, Plaxco KW. Bioelectrochemistry. 2009;76:208–213. doi: 10.1016/j.bioelechem.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel DJ, Suri AK. Rev Mol Biotechnol. 2000;74:39–60. doi: 10.1016/s1389-0352(99)00003-3. [DOI] [PubMed] [Google Scholar]
- 26.Lai RY, Seferos DS, Heeger AJ, Bazan GC, Plaxco KW. Langmuir. 2006;22:10796–10800. doi: 10.1021/la0611817. [DOI] [PubMed] [Google Scholar]
- 27.Ahmed S, Kishikawa N, Ohyama K, Wada M, Nakashima K, Kuroda N. Talanta. 2009;78:94–100. doi: 10.1016/j.talanta.2008.10.043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.