

ABSTRACT
Acute kidney injury (AKI) is a leading cause of chronic kidney disease. Proximal tubules are considered to be the primary origin of pathogenic inflammatory cytokines in AKI. However, it remains unclear whether other cell types, including collecting duct (CD) cells, participate in inflammatory processes. The transcription factor GATA2 is specifically expressed in CD cells and maintains their cellular identity. To explore the pathophysiological function of GATA2 in AKI, we generated renal tubular cell-specific Gata2 deletion (G2CKO) mice and examined their susceptibility to ischemia reperfusion injury (IRI). Notably, G2CKO mice exhibited less severe kidney damage, with reduced granulomacrophagic infiltration upon IRI. Transcriptome analysis revealed that a series of inflammatory cytokine genes were downregulated in GATA2-deficient CD cells, suggesting that GATA2 induces inflammatory cytokine expression in diseased kidney CD cells. Through high-throughput chemical library screening, we identified a potent GATA inhibitor. The chemical reduces cytokine production in CD cells and protects the mouse kidney from IRI. These results revealed a novel pathological mechanism of renal IRI, namely, that CD cells produce inflammatory cytokines and promote IRI progression. In injured kidney CD cells, GATA2 exerts a proinflammatory function by upregulating inflammatory cytokine gene expression. GATA2 can therefore be considered a therapeutic target for AKI.
KEYWORDS: Gata2, kidney, acute kidney disease, inflammation
INTRODUCTION
Acute kidney injury (AKI) is a complex clinical entity characterized by acute renal dysfunction (1). Renal ischemia reperfusion and the resultant tubulointerstitial injury (here referred to as ischemia reperfusion injury [IRI]) are the leading causes of AKI and subsequent progression of chronic kidney disease (CKD) (2–4). The disease emerges in a number of clinical contexts, including renal transplantation, systemic circulatory shock, and vascular surgery (5). The pathological features of AKI include renal tubular cell damage, which results in apoptosis and necrosis (4). In particular, it has been shown that highly metabolically active nephron segments, i.e., the proximal tubules and thick ascending limbs, are the tissues most susceptible to IRI-induced cellular damage (6). Inflammation represents an important pathological component of AKI pathogenesis, exacerbating tubulointerstitial injury during the latter phase of the disease (4). Despite recent progress in advanced critical care medicine, AKI is still associated with high morbidity and mortality (7, 8). Therefore, elucidation of AKI's etiological mechanisms and the development of effective therapeutics are high priorities.
GATA2 is a zinc finger-containing transcription factor that plays crucial roles in diverse developmental programs. GATA2 has important functions in hematopoiesis, including the maintenance of hematopoietic stem cells and the differentiation of multiple hematopoietic lineage cells (9–13). In addition, we previously reported that GATA2 is highly expressed in urogenital primordial tissue in early embryogenesis and plays a crucial role in renal development (14–17). GATA2 expression also persists in the mature kidney. Specifically, GATA2 is robustly expressed in the renal collecting duct (CD), which plays a pivotal role in urinary concentration (18). By examining renal tubular cell-specific Gata2 deletion (G2CKO) mice, we demonstrated that GATA2 participates in urinary volume control by maintaining aquaporin 2 (Aqp2) gene expression, which contributes to water reabsorption from urine in the adult kidney (19).
It is known that various inflammatory cytokines and chemokines are released by the damaged proximal tubular epithelium, evoking the infiltration of macrophages and neutrophils (20). These inflammatory cells subsequently promote further kidney lesions and interstitial fibrosis (21). A number of previous studies have been focused on the pathogenic contribution of damaged proximal tubules (3, 22), while the contributions of other parts of diseased kidney tubules have been largely overlooked. Our group has been paying special attention to the functional contribution of GATA2 to this cell type. While GATA2 maintains the fundamental cellular function of CD cells, whether the lack of GATA2 affects the disease status of AKI remains to be elucidated.
To elucidate the pathophysiological function of GATA2 in AKI, we describe here the use of genetic and pharmacological approaches in IRI model mice. Deletion of the Gata2 gene diminishes the inflammatory cytokine levels in CD cells of the diseased kidney, rendering the mice resistant to IRI. Of note, treatment with a novel GATA inhibitor has therapeutic efficacy for IRI. Thus, we demonstrate a novel mechanism underlying the pathogenesis of renal IRI, i.e., that renal CD cells serve as a major source of inflammatory cytokines during IRI. GATA2 promotes the production of a set of inflammatory cytokines in injured CD cells and thereby contributes to disease progression. Our results provide novel insight into the proinflammatory function of GATA2 in the diseased kidney.
RESULTS
G2CKO mice are resistant to renal IRI.
To address the pathophysiological function of GATA2 in kidney disease progression, we deleted the Gata2 gene specifically in the renal tubular cells by using Pax8-rtTA::tetO-Cre mice (G2CKO mice). We subjected G2CKO and control Gata2flox/flox littermates (here referred to as control mice) to an established IRI procedure at 8 weeks of age after 4 weeks of doxycycline (Dox) feeding (Fig. 1A). Of note, tubular-cell-specific GATA2 deletion markedly improved kidney pathology measures in renal IRI. G2CKO kidneys showed an almost normal histological appearance both 1 and 2 days after unilateral renal IRI, and the renal tubules appeared largely intact (Fig. 1B, a and c). In contrast, control mice exhibited robust coagulative necrotic lesions in the cortical and outer medullary regions (Fig. 1B, b and d, asterisks). Morphometric analysis showed that the percentage of damaged tubular area was significantly lower in the G2CKO mouse kidney than in the control mouse kidney 1 and 2 days after unilateral renal ischemia reperfusion (Fig. 1C).
FIG 1.
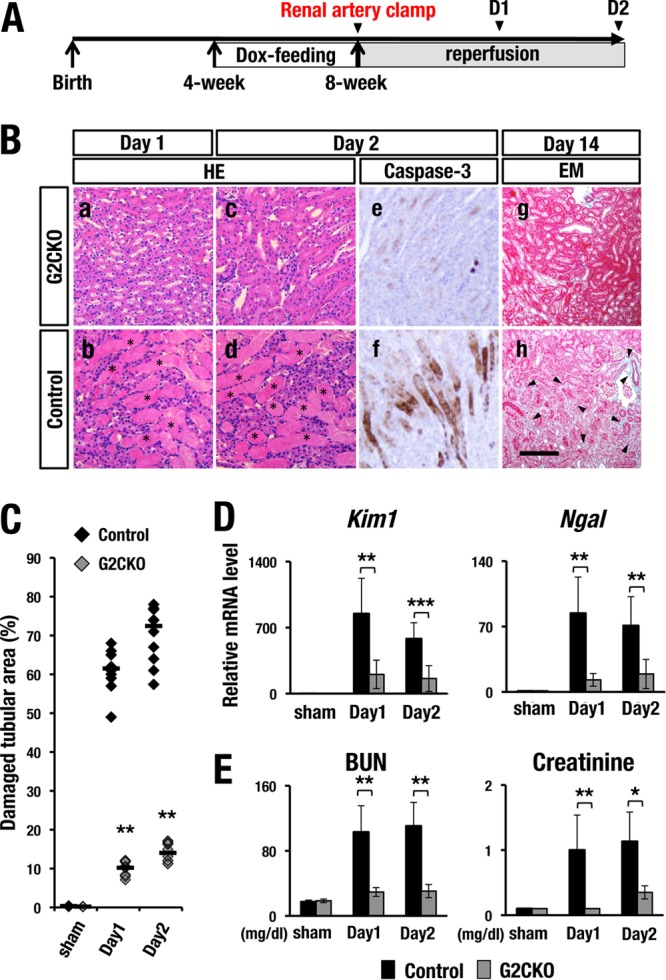
G2CKO mice are resistant to renal IRI. (A) Experimental time course of Dox feeding and IRI induction. Transient renal artery clamping was performed at 8 weeks of age. The mice were subjected to analysis 1 day, 2 days, or 14 days after IR. (B, a to d) H&E staining of the kidney at 1 day and 2 days after unilateral renal IR. Note that control mice showed robust coagulative necrotic lesions in the cortical and outer medullary regions (b and d, asterisks), while G2CKO mice showed lower levels of necrosis (a and c). (e and f) Immunohistochemical analyses with an anti-cleaved-caspase-3 antibody. Note that control mice (f) showed a number of anti-cleaved-caspase-3-positive apoptotic cells in the proximal tubules 2 days after the IR. In contrast, G2CKO mice (e) showed very few apoptotic cells in the IRI kidney. (g and h) Elastica-Masson (EM) staining of IRI kidneys from G2CKO (g) and control (h) mice 14 days after unilateral IR (day 14). Control mouse kidneys showed robust tubular injury, while G2CKO kidneys showed well-preserved renal tubules. Scale bar, 200 μm. (C) Quantitative analysis of damaged tubular area in H&E-stained kidney sections. Percentages of damaged tubular area were significantly decreased in the G2CKO kidneys 1 and 2 days after the unilateral IR compared with control mouse kidneys. The data points represent individual values from each section. The bars indicate the median of each group. (D and E) The expression levels of tubular injury biomarkers (D), as well as BUN and serum creatinine levels (E), 1 and 2 days after IR and 2 days after the sham operation in the kidneys of G2CKO and control mice. Note that the levels of tubular injury biomarkers, BUN, and serum creatinine were maintained at a lower level after IRI in G2CKO mice than in control mice. The data are presented as means ± SD (n = 5 in each group of mice). The statistical significance is indicated (***, P < 0.005; **, P < 0.01; *, P < 0.05).
The progression of renal IRI is known to be associated with an increased number of caspase-3-positive apoptotic tubular cells (23). Surprisingly, G2CKO mouse kidneys rarely showed apoptotic cells (Fig. 1B, e), whereas control mice showed large numbers of apoptotic tubular cells in the outer medulla and cortical region on day 2 (Fig. 1B, f).
We next used Elastica-Masson staining to examine renal fibrosis 2 weeks after IRI. Most of the tubules of G2CKO mice were well preserved, with intense red Elastica-Masson staining (Fig. 1B, g), while control mouse kidneys exhibited robust tubular injury and weak Elastica-Masson staining (Fig. 1B, h, arrowheads). These results demonstrate that G2CKO mice were markedly resistant to kidney IRI.
Renal function of G2CKO mice is maintained after IRI.
Kim1 (kidney injury molecule 1) and Ngal (neutrophil gelatinase-associated lipocalin) are widely used biomarkers for renal tubular injury, and it has been shown that the expression of these markers is induced upon AKI and faithfully reflects disease severity (24, 25). Consistent with the histological findings, the expression levels of Kim1 and Ngal mRNAs were significantly lower in the G2CKO kidney than in control mouse kidneys both 1 and 2 days after IRI (Fig. 1D). Neither Kim1 nor Nga1 was induced by the sham operation (Fig. 1D).
We also examined the effects of IRI on renal function. To this end, we quantified blood urine nitrogen (BUN) and serum creatinine levels, which are two major indicators of decreased renal function and are indicative of decreased clearance of these waste products (4). We found that BUN and serum creatinine were maintained at low levels in G2CKO mice on days 1 and 2 after the induction of bilateral IRI. In contrast, the levels of both of these indicators were significantly increased in control mice (Fig. 1E). Both BUN and creatinine were not increased by the sham operation. These results support our hypothesis that G2CKO mice are resistant to kidney IRI.
Inflammatory cytokine gene expression is dependent on GATA2.
To address the mechanistic basis of IRI resistance in the G2CKO kidney, we employed microarray analysis data using mRNA from purified CD cells of G2CKO and normal wild-type mice (GSE52448) (18). We analyzed the microarray data using Ingenuity Pathway Analysis (IPA) software and annotated the biological pathways represented by the gene expression signature. A number of affected genes were found in G2CKO-CD cells, which were classified into the following three gene ontology terms with high statistical significance: cell-to-cell signaling, inflammatory response, and cell-mediated immune response (Fig. 2A).
FIG 2.
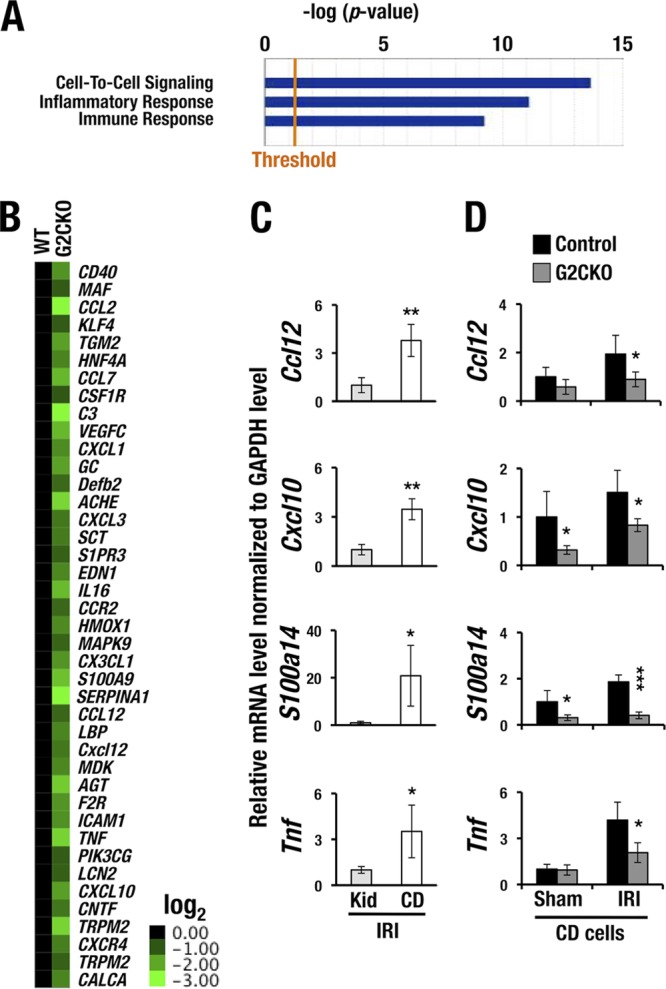
Inflammation-related genes are downregulated in the collecting duct cells of G2CKO mice. (A) Biological pathway annotation based on IPA. The gene signatures that were affected in G2CKO-CD cells were classified into the indicated pathways. (B) Forty-one inflammation-related genes encoding several chemokines and cytokines were reduced more than 2-fold in G2CKO-CD cells compared with wild-type (WT) control CD cells. The mRNA expression levels in the WT samples were set to 1 in the heat map. (C) Ccl12, Cxcl10, S100a14, and Tnf mRNA are more strongly expressed in the DBA-sorted CD cells than in whole kidney cells 1.5 h after the induction of IRI. The data are derived from the CD cells and whole kidney (Kid) cells of three WT mice. The statistical significance of differences between the whole-kidney cell samples and CD cell samples is indicated (**, P < 0.01; *, P < 0.05). (D) Transcript levels of the 4 cytokine genes were significantly lower in G2CKO-CD cells than in control CD cells under the sham or IRI treatment. The statistical significance of differences between the G2CKO and the control CD cells is indicated (3 mice/group; ***, P < 0.005; *, P < 0.05).
Of the influenced genes, the expression levels of 41 inflammation-related genes encoding a series of chemokines and cytokines were reduced more than 2-fold in the G2CKO-CD cells in comparison with CD cells of normal wild-type mice (Fig. 2B). Based on these results, we selected 4 representative chemokine and cytokine genes, namely, Ccl12 (chemokine [C-C motif] ligand 12), Cxcl10 (chemokine [C-X-C motif] ligand 10), S100a14, and Tnf (tumor necrosis factor). All of these genes are known to participate in the pathogenesis of AKI (26, 27). The mRNA levels of these cytokines were assessed in CD cells, as well as in whole-kidney samples from normal mice, following IRI. We found that Ccl12, Cxcl10, S100a14, and Tnf mRNAs were 3- to 20-fold more abundantly expressed in CD cells than those in the whole kidney 1.5 h after the induction of IRI (Fig. 2C). These results indicate that the cytokines are produced predominantly in CD cells of the injured kidney.
We next examined the mRNA levels of the cytokines in CD cells collected from the G2CKO and control mice 1.5 h following IRI or a sham operation. The expression levels of all four genes were significantly decreased in the G2CKO-CD cells compared with those in control mouse CD cells after IRI (Fig. 2D). Cxcl10 and S100a14 mRNA levels in the G2CKO-CD cells were lower than those from control mice subjected to the sham operation (Fig. 2D). Collectively, these results demonstrate that the IRI-induced expression of these inflammatory cytokine mRNAs depends on the presence of GATA2 in renal CD cells.
Reduced infiltration of inflammatory cells in the G2CKO kidney.
Given the reduction of inflammatory cytokine levels in the G2CKO-CD cells, we hypothesized that inflammatory cell infiltration could be repressed in the G2CKO mouse kidneys. To address this hypothesis, we examined the infiltrating inflammatory cell population in the kidney 2 days after IRI. We prepared single-cell suspensions of the whole kidney and stained the cells with anti-Gr-1 and anti-CD11b antibodies. Thereafter, the stained cells were analyzed by flow cytometry. Of note, we found that the Gr-1+ CD11b+ double-positive granulomacrophage population in G2CKO kidneys was much smaller than in control mouse kidneys (4.07% ± 2.58% versus 9.82% ± 2.93%) (Fig. 3A and B, bottom). The granulomacrophage populations were comparable between the G2CKO and control mice in the sham-operated kidney (1.69% ± 0.98% versus 1.74% ± 0.74%) (Fig. 3A and B, top). These results indicate that the G2CKO kidney recruits a lower number of inflammatory cells than the control mouse kidney upon IRI.
FIG 3.
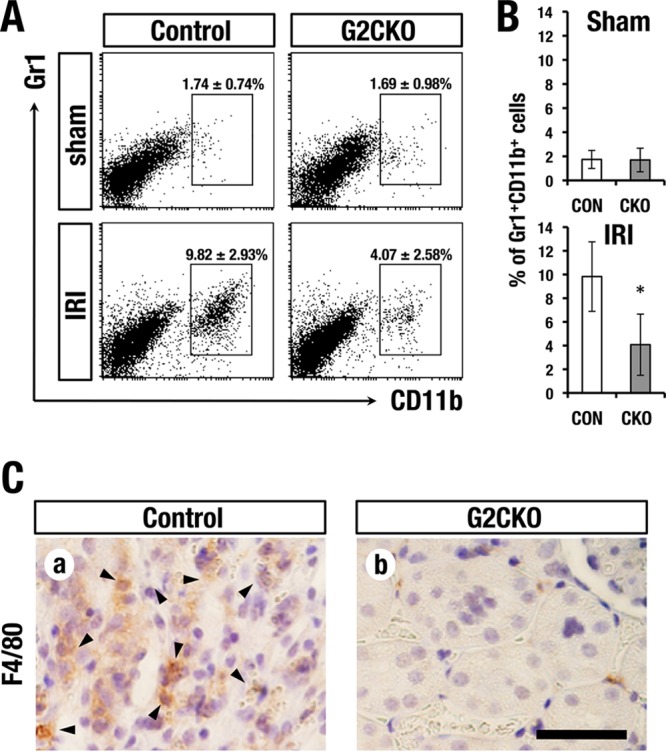
Lower counts of infiltrating inflammatory cells in G2CKO kidney after IRI. (A) Flow cytometry analysis of Gr1+ CD11b+ infiltrating inflammatory cells in the kidney 2 days after IRI. (B) Quantification of infiltrating inflammatory cells in G2CKO and control (Con) mouse kidneys. The data are presented as means ± SD (n = 3 for each group). Statistical significance of differences is indicated (*, P < 0.05). (C) Immunohistochemistry using anti-F4/80 antibody 2 days after renal IRI. Note that the control mouse kidney showed robust infiltration of F4/80-positive macrophages (arrowheads), while the G2CKO kidney showed lower levels of macrophage infiltration than the control mouse (scale bar, 100 μm).
To histologically assess the infiltrating macrophage population, we stained paraffin-embedded sections of IRI kidneys with an anti-F4/80 antibody. Consistent with the flow-cytometry analyses, the G2CKO kidney showed a reduced number of F4/80-positive infiltrating macrophages, while the control mouse kidney contained abundant macrophages (Fig. 3C, a and b, arrowheads). These results indicate that the G2CKO kidney acquires resistance to inflammatory cell infiltration after IRI.
Forced GATA2 expression accelerates inflammatory cytokine induction in a CD cell line.
To further clarify the function of GATA2 in cytokine gene regulation, we focused on Tnf and Cxcl10, two genes that encode representative cytokines involved in AKI (28). We stably introduced a GATA2 expression plasmid into a mouse inner medullary collecting duct (mIMCD) cell line, which is referred to here as GATA2-mIMCD (18). GATA2-mIMCD cells express 4-fold more abundant exogenous GATA2 protein than endogenous GATA2 (18). Utilizing GATA2-mIMCD cells, we tested the effects of forced GATA2 expression on inflammatory cytokine gene induction by lipopolysaccharide (LPS) treatment. The induction of both cytokine genes was significantly higher in GATA2-mIMCD cells than in control mIMCD cells upon LPS challenge (Fig. 4). The expression levels of these inflammatory cytokine genes upon phosphate-buffered saline (PBS) treatment were not significantly different between GATA2-mIMCD and control mIMCD cells (Fig. 4). These results indicate that GATA2 positively regulates the LPS-induced expression of Tnf and Cxcl10 genes in mIMCD cells.
FIG 4.
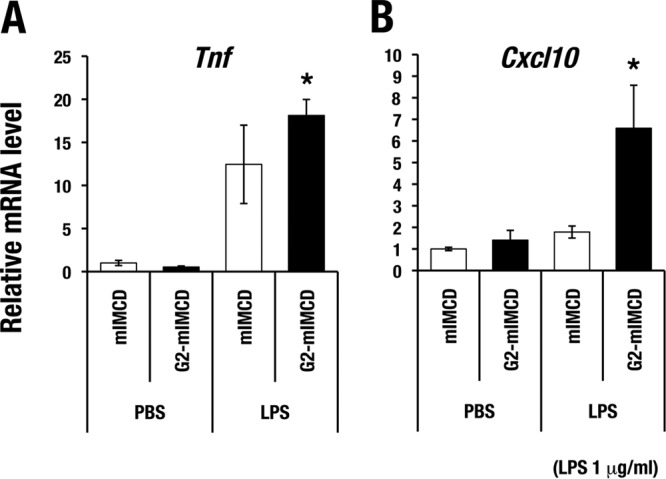
GATA2 enhances LPS-induced cytokine gene expression in mIMCD cells. LPS (1 μg/ml)-induced expression of Tnf (A) and Cxcl10 (B) was higher in mIMCD cells that were stably transfected with GATA2 (G2-mIMCD) than in mock-transfected control cells (mIMCD). The mRNA level of each gene was normalized to the level of GAPDH (glyceraldehyde-3-phosphate dehydrogenase). The data are shown as means ± SD from three independent experiments. Statistically significant differences are indicated (*, P < 0.05).
High-throughput screening of GATA inhibitors.
Given the significant resistance to kidney injury in G2CKO mice, we next addressed whether pharmacological inhibition of GATA2 attenuates kidney injury. Prior to this analysis, we reassessed a known GATA inhibitor, K-7174 (29, 30). While K-7174 has been reported to inhibit the activity of GATA factors in multiple cell types, a recent study suggested that K-7174 has potential cytotoxicity due to proteasome-inhibitory activity (31). Considering this disadvantage of K-7174, we decided to screen and identify novel GATA factor inhibitors by means of high-throughput screening (HTS) of a chemical compound library. To this end, we developed a reporter assay system using the NLucP reporter gene, in which the expression of a NanoLuc reporter is directed by a β-globin locus control region (LCR) that contains multiple GATA-binding sites (Fig. 5A). NanoLuc is a modified luciferase protein that is unstable and has a 20-min half-life (32). Therefore, luciferase activity is predicted to decrease rapidly when cells are exposed to chemicals that suppress GATA factor function. We transduced the NLucP reporter construct into K-562 cells, a human erythroleukemia cell line, and established 3 stable clones (number 3, number 6, and number 27) that showed sufficient levels of luciferase activity (here referred to as K562-NLucP cells) (Fig. 5B). We found that the luciferase activities of these three K562-NLucP clones all decreased upon exposure to K-7174 (Fig. 5B). We chose clone number 3 for the screening assays, as it showed the highest reporter activity (Fig. 5B).
FIG 5.
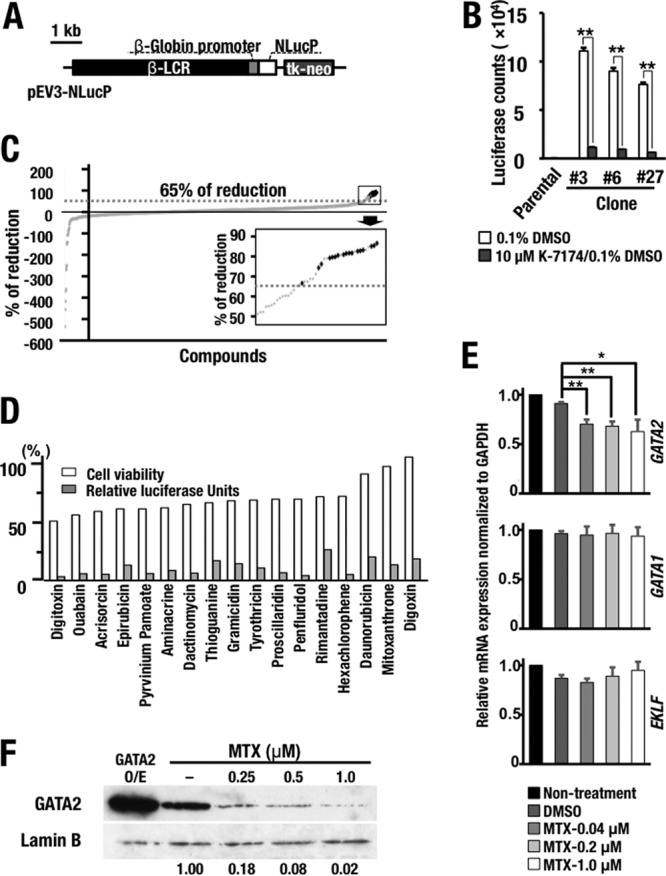
High-throughput screening identifying GATA factor inhibitors. (A) Schematic diagram of the NanoLuc (NLucP) reporter construct. (B) Luciferase activity in the K562-NLucP reporter cell lines. Four thousand cells of three K562-NLucP cell lines (number 3, number 6, and number 27) carrying the NanoLuc reporter construct were exposed to 0.1% DMSO solvent or 10 μM Κ-7174 for 24 h. The results are shown as means and SD, with the experiments performed in triplicate (**, P < 0.01). Parental K562 cells served as a control. (C) Primary chemical library screening data for 1,165 compounds in the off-patent drug library of Keio University. The data are presented as the percent reduction relative to the value obtained for the 0.1% DMSO-treated control. The inset, a magnified view of the boxed area on the upper right, shows the top 29 compounds, which were subjected to the second screening. The dotted lines indicate the 65% reduction level. The 17 hit compounds are indicated by black dots. (D) Validation of the hit compounds. Shown are the percent luciferase activity and cell viability compared with the value for the 0.1% DMSO-treated control after 28-hour exposure to 10 μM each compound. (E) Expression levels of GATA1, GATA2, and EKLF mRNAs in K562 cells treated with MTX. Note that MTX treatment predominantly diminished the GATA2 mRNA level, while the treatment did not affect the GATA1 and EKLF mRNA levels in K562 cells. The statistical significance is indicated (**, P < 0.01; *, P < 0.05). (F) MTX treatment diminishes the GATA2 protein expression level in HEK293T cells in a dose-dependent manner. The far left lane contained HEK293T cells overexpressing (O/E) GATA2 employed as a control.
We adjusted the screening system for 384-well plates and conducted an HTS assay using the off-patent drug library of Keio University. After the first screening, we found that 29 out of 1,165 compounds lowered the NanoLuc reporter activity to less than 65% of that under the dimethyl sulfoxide (DMSO) control conditions (Fig. 5C, boxed area at upper right). We selected these 29 compounds and conducted the second screening using a 96-well plate, ultimately identifying 17 compounds that reproducibly reduced luciferase activity (Fig. 5D, gray bars). All 17 compounds maintained cellular viability at more than 50% (Fig. 5D, white bars).
GATA inhibitor suppresses LPS-induced cytokine gene expression.
Of the 17 chemicals, we chose mitoxantrone (MTX) for further analysis. This compound reduced luciferase reporter activity to 17% of control levels without exhibiting apparent cytotoxicity (cellular viability was maintained at 98% with the dose tested for the luciferase assay) (Fig. 5D). To address the mechanism through which MTX reduces GATA factor activity, we first examined the influence of MTX on the expression levels of GATA1, GATA2, and EKLF, all of which are known to regulate β-globin LCR (33). We found that GATA2 mRNA expression was decreased by MTX treatment, while that of GATA1 and EKLF was largely maintained in K562 cells (Fig. 5E). Furthermore, the expression level of GATA2 protein was significantly decreased in human embryonic kidney (HEK) 293T cells in an MTX dose-dependent manner (Fig. 5F). These results thus indicate that MTX specifically reduces the mRNA and protein expression levels of GATA2 and that MTX acts as an inhibitor of GATA2.
We next examined the consequences of MTX treatment for LPS-induced cytokine gene expression in mIMCD cells and found that simultaneous MTX treatment significantly diminished the LPS-induced expression of Tnf and Cxcl10 (Fig. 6A and B). The LPS-induced upregulation of Tnf and Cxcl10 was inhibited in an MTX dose-dependent manner, with effects observed at relatively low doses. Aqp4 is a water channel expressed on the basolateral membrane of CD cells, and Aqp4 gene expression did not change much in the CD cells of G2CKO mice (18). We found that the expression level of Aqp4 was not significantly affected by MTX treatment regardless of LPS stimulation (Fig. 6C). These results indicate that, in very good agreement with the genetic loss-of-function assessments, pharmacological inhibition of GATA activity also diminishes LPS-induced cytokine gene expression in mIMCD cells.
FIG 6.
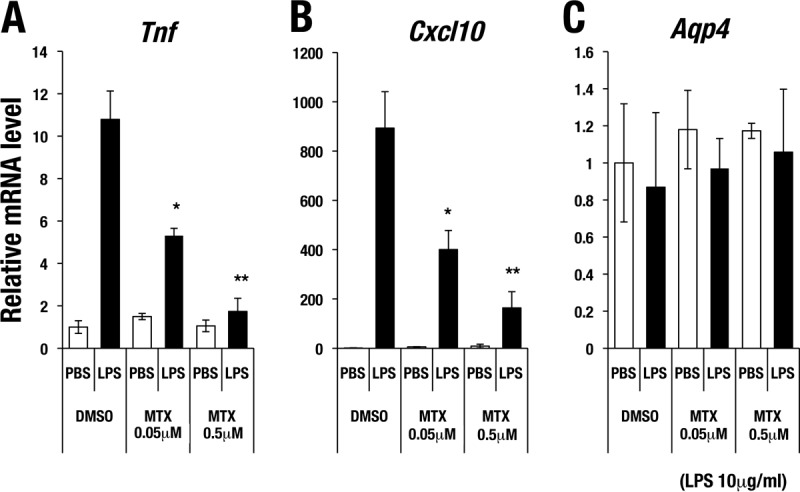
Mitoxantrone attenuates LPS-induced cytokine gene expression in mIMCD cells. (A and B) LPS (10 μg/ml)-induced expression of Tnf (A) and Cxcl10 (B) was suppressed with MTX treatment in a dose-dependent manner. (C) Aqp4 expression was not significantly affected by MTX treatment regardless of LPS stimulation. The data are shown as means ± SD from three independent experiments. Statistically significant differences are indicated (*, P < 0.05; **, P < 0.01).
A GATA inhibitor ameliorates kidney damage after renal IRI.
We next examined whether MTX is protective against IRI in the kidney. To this end, we intraperitoneally injected incremental doses of MTX (1 mg/kg of body weight and 2 mg/kg) or saline into wild-type C57BL/6J male mice daily for 5 consecutive days (Fig. 7A). One day after the final MTX administration (day 6), a unilateral IRI procedure (i.e., 30-min clamping of the renal artery) was performed (Fig. 7A). The injured kidney was subjected to histological analysis 1 day later (day 7). We found that MTX pretreatment efficiently prevented IRI-induced tubular necrosis (Fig. 7B). The percentage of damaged tubular area was significantly improved by MTX pretreatment in a dose-dependent manner (Fig. 7C). Furthermore, MTX pretreatment significantly reduced Kim1 and Tnf mRNA expression levels in the injured kidney (Fig. 7D).
FIG 7.
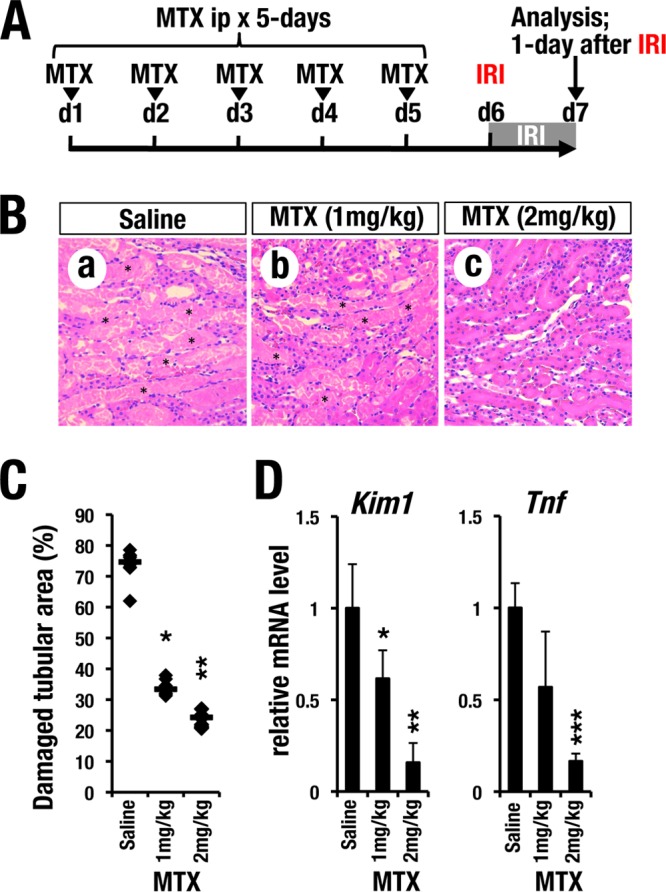
GATA inhibitor pretreatment protects kidneys from ischemia reperfusion injury. (A) Time course of MTX pretreatment. Each concentration of MTX (1 mg/kg or 2 mg/kg) or saline was administered for 5 consecutive days (d) before placement of the transient renal artery clamp. (B) H&E staining of kidneys from mice administered saline or MTX 1 day after reperfusion. Saline-treated control mice (a) showed robust tubular necrosis after renal IR (asterisks). Preadministration of MTX (b and c) prevented acute tubular necrosis in an MTX dose-dependent manner. (C) Quantitative analysis of damaged tubular areas in mouse kidneys. The damaged tubular area was significantly decreased by the MTX pretreatment. Each data point represents an individual value from each section. The bars indicate the median of each group. (D) Relative mRNA expression levels of Kim1 and Tnf in the whole-kidney sample 1 day after clamping. MTX treatment reduced the expression levels of Kim1 and Tnf in an MTX dose-dependent manner. The data are presented as means and SD (n = 5 in each group of mice). Statistically significant differences are indicated (***, P < 0.001; **, P < 0.01; *, P < 0.05).
We next tested the therapeutic efficacy of MTX after the induction of IRI. For this purpose, we performed unilateral IRI surgery using wild-type C57BL/6J mice prior to MTX treatment. Thereafter, the mice were intraperitoneally administered MTX (2 mg/kg) for 3 consecutive days (Fig. 8A). One day after the final MTX administration (day 4), we found that MTX-treated mice showed a lower degree of tubular damage than did saline-treated control mice (Fig. 8B). The percentage of damaged tubular area was significantly improved by MTX posttreatment (Fig. 8C). The induced mRNA levels of Kim1 and Tnf in the kidney were also diminished in MTX-treated mice (Fig. 8D). These data clearly demonstrate that MTX treatment before or after IRI induction is therapeutically effective for IRI-induced kidney injury in mice.
FIG 8.
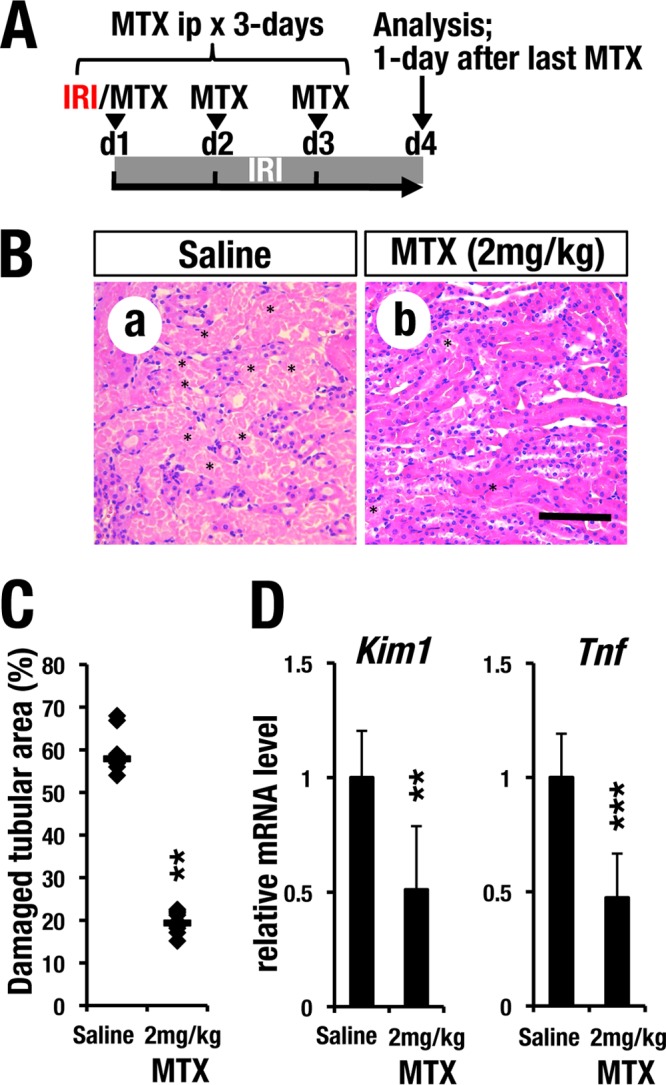
GATA inhibitor posttreatment protects kidneys from ischemia reperfusion injury. (A) Time course of MTX posttreatment. MTX (2 mg/kg) was administered for 3 consecutive days after clamping. (B) H&E staining of the IRI kidney. Note that the postadministration of MTX also prevented tubular necrosis. Scale bar, 200 μm. (C) Quantitative analysis of damaged tubular area. The damaged tubular area was significantly decreased by posttreatment with MTX. The data points represent individual values from each section. The bars indicate the median of each group. (D) Relative mRNA levels of Kim1 and Tnf in IRI kidneys of mice posttreated with MTX. Note that posttreatment with MTX reduced the expression levels of Kim1 and Tnf. The data are shown as means and SD (n = 5 for each group). Statistically significant differences are indicated (***, P < 0.001; **, P < 0.01).
A GATA inhibitor attenuates the inflammatory response after renal IRI.
To examine the mechanistic basis of the observed beneficial effects of MTX in the kidney, we analyzed the inflammatory status of IRI kidneys following treatment with the compound. To this end, we employed the WIM-6 system. This system is a recently developed noninvasive inflammation-monitoring system that makes use of a human interleukin 6 (IL-6) bacterial artificial chromosome (BAC)-driven luciferase reporter transgenic mouse coupled with analysis of a whole-body in vivo imaging system (34). Robust luminescence was detected specifically in the IRI-operated left kidneys of the saline-treated mice 1 day after IRI (Fig. 9A and B, top). Luciferase luminescence gradually decreased 2 days after IRI and thereafter, representing resolution of the acute-phase inflammation (Fig. 9A and C).
FIG 9.
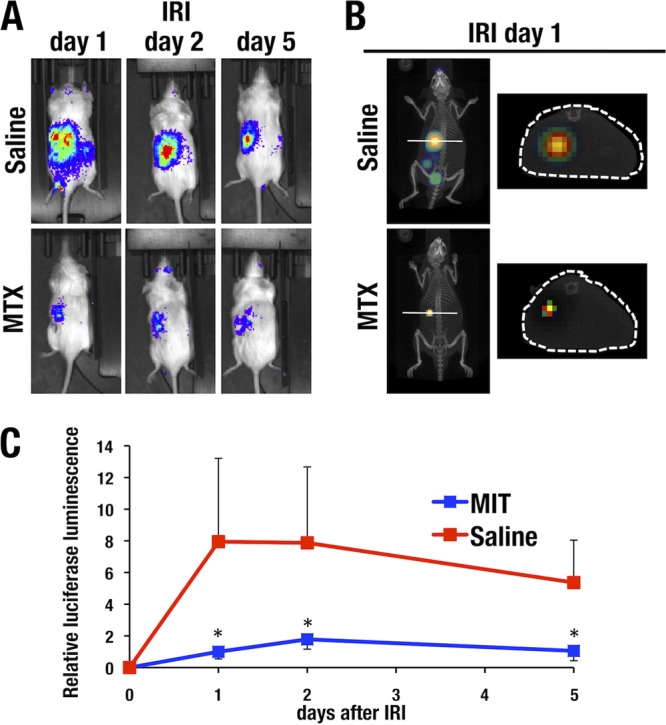
MTX treatment ameliorates renal inflammation after IRI. WIM-6 mice were intraperitoneally administered MTX or saline before induction of left unilateral IRI. The inflammatory status of the kidneys was evaluated 1, 2, and 5 days after IRI using an in vivo imaging system. (A) (Top) Dorsal images of saline-treated control WIM-6 mice showing robust luciferase luminescence in the left kidney after IRI. IRI was performed only for the left kidney. (Bottom) MTX pretreatment reduced luciferase luminescence. (B) (Left) IVIS-CT transverse sections of the lower thoracic level of saline-treated control mice showing strong luciferase luminescence in the left kidney after IRI induction. (Right) MTX pretreatment reduced the luciferase luminescence in the kidney. (C) Quantification of the luminescence data. Robust luciferase luminescence was induced in the saline-treated control mice 1 and 2 days after IRI (red line). Thereafter, the luminescence level gradually decreased. MTX pretreatment dramatically decreased the luciferase luminescence level (blue line). The data are shown as means and SD (n = 3 for each group). Statistically significant differences are indicated (*, P < 0.05).
We next evaluated the efficacy of MTX preadministration for 5 consecutive days before IRI. After IRI (days 1, 2, and 5), the luciferase luminescence from the injured kidney was dramatically attenuated by MTX pretreatment (Fig. 9A). Transverse sections of the lower thoracic level of saline-pretreated control and MTX-pretreated test mice were prepared using in vivo imaging system with computed tomography (IVIS-CT) technology. While strong luciferase luminescence was detected in the left kidney after IRI induction, MTX pretreatment strongly reduced the luminescence (Fig. 9B). The MTX pretreatment reduced luciferase luminescence to 10 to 20% of that observed in saline-pretreated control mice, and this effect was quite reproducible (Fig. 9C). These results show very good agreement with the histological amelioration of tubular injury by MTX treatment.
DISCUSSION
We previously worked to elucidate the physiological function of GATA factors in the kidney, with particular interest on the collecting duct cells (18, 35). In this study, our analyses of GATA2 function in the kidney collecting ducts revealed two previously unidentified findings that led to clarification of a novel pathogenic mechanism underlying acute kidney injury. These findings are summarized in Fig. 10. First, we found that the renal CD cells serve as the major supplier of inflammatory cytokines and play a crucial role in the kidney microenvironment upon the induction of IRI. Second, we found that GATA2 promotes the production of a series of inflammatory cytokines from injured CD cells and contributes to the progression of AKI. In fact, inflammatory cytokine expression in the damaged CD cells of G2CKO mice was significantly reduced, rendering the mice strongly resistant to IRI. To verify our findings, we conducted a high-throughput screening of a chemical library and identified a number of novel GATA inhibitors. Pre- or posttreatment with one of the newly identified inhibitors effectively protected mouse kidneys from IRI. Collectively, this study is the first to demonstrate the proinflammatory function of GATA2 in injured CD cells and suggests that GATA inhibitors may be effective therapeutics for kidney diseases.
FIG 10.
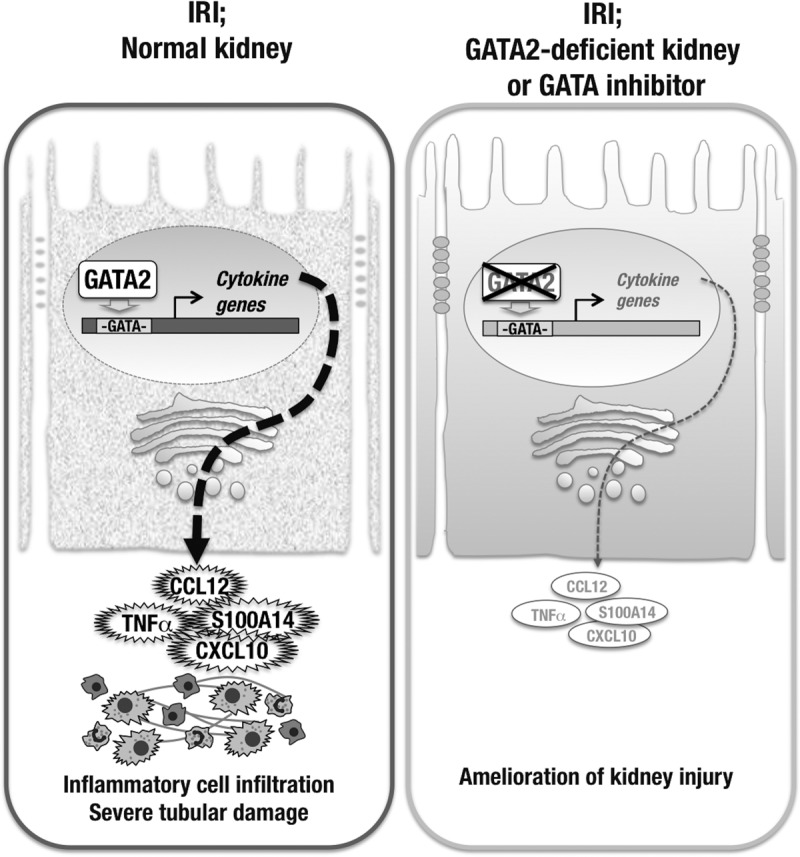
Collecting duct cells play a role in inflammatory cytokine production upon IRI. (Left) GATA2 positively regulates inflammatory cytokine gene expression in CD cells and provokes inflammatory cell infiltration and tubular damage in the IRI kidney. (Right) Conditional knockout or chemical inhibition of GATA2 reduces inflammatory cytokine gene expression in CD cells and protects kidney cells from IRI.
Inflammatory cell infiltration is a key process in the pathogenesis of AKI and the progression of tubulointerstitial injury. However, it remains unclear how the inflammatory processes are initiated and promoted in the kidney upon IRI. Renal collecting ducts primarily contribute to the final adjustment of urinary volume by water reabsorption through Aqp2 (19). In this study, we show evidence that CD cells participate in the inflammation process after IRI. Consistent with this notion, there are several reports suggesting that CD cells change their characteristics under disease conditions. For instance, CD cells undergo an epithelial-mesenchymal transition and transform into pathogenic myofibroblasts upon unilateral ureteral obstruction (UUO) of the kidney (36). It was reported that the injured CD cells in the UUO model produced several chemoattractive proteins (37).
While we showed that a variety of inflammatory cytokines and chemokines are under the regulation of GATA2 in CD cells, it remains unclear whether GATA2 directly regulates these downstream genes. In this regard, it is interesting that chromatin immunoprecipitation sequencing (ChIP-seq) data in the UCSC Genome Browser shows GATA1 binding to putative regulatory sequences of Tnf, Cxcl10, and S100A14 genes in a mouse erythroleukemia cell line (https://genome.ucsc.edu). As GATA1 and GATA2 often share binding sequences in hematopoietic cells (38–40), we surmise that GATA2 also binds to the GATA-binding sequences in the regulatory regions of these cytokine genes in CD cells. Another study using human acute myeloid leukemia cells showed that GATA2 directly transactivates IL1b and Cxcl2 genes and promotes the proliferation of leukemia cells (41). It has also been shown that several cytokine and cell adhesion molecule genes that are involved in leukocyte infiltration are cooperatively regulated by GATA2 and NF-κB (29, 42). Thus, these previous studies and our present results suggest that GATA2 may be a proinflammatory factor in multiple pathogenic contexts.
In the present study, using HTS of a chemical library, we identified MTX as a potent GATA factor inhibitor. MTX was originally identified as a topoisomerase inhibitor that disrupts DNA replication (43), and the compound has been developed as a clinically approved anticancer drug (44). We found that MTX specifically reduces mRNA and protein expression of GATA2, but the underlying mechanism by which MTX suppresses GATA2 expression remains unknown. It will be very important to address this issue in future studies. Similarly, it would be of value to test if low-dose MTX treatment has clinical efficacy for AKI.
GATA2 colocalizes with GATA3, another GATA factor, in renal CD cells (18, 45). We previously found that the Gata2 mRNA levels are much higher than those of Gata3 in renal CD cells (18), and GATA2 plays a more predominant role than GATA3 in Aqp2 gene regulation in this cell type (18). Therefore, GATA2 is likely the primary target of MTX in the kidney. We plan to use CD cell-specific Gata3-deficient mice to examine whether the protein also participates in the regulation of inflammatory cytokine gene expression in CD cells.
In summary, we demonstrated a novel pathological mechanism of renal IRI. Upon IRI induction in the kidney, CD cells robustly produce cytokines and chemokines, which play important roles in initiating immune cell infiltration and promoting inflammation. GATA2 positively regulates the expression of these inflammatory cytokine genes in CD cells and contributes to disease progression. In CD cells with GATA2 deletion, inflammatory cytokine gene expression is attenuated and IRI-induced kidney damage is ameliorated. In agreement with this genetic approach, a GATA factor inhibitor was found to have beneficial effects on renal IRI by reducing inflammatory cytokine gene expression. As AKI is a major risk factor for CKD, the prevention of AKI via GATA factor inhibition would confer a significant clinical benefit in kidney disease patients. This study provides the novel insight that pharmacologically inhibiting GATA2 activity could offer therapeutic protection against AKI-CKD progression.
MATERIALS AND METHODS
Mice.
Renal tubular cell-specific Gata2-deficient mice were characterized previously (18). Briefly, Gata2GFP/flox::Pax8-rtTA::tetO-Cre or Gata2flox/flox::Pax8-rtTA::tetO-Cre mice (4 weeks old) were fed with 1 mg/ml Dox in drinking water for 4 weeks to induce renal tubular cell-specific deletion of Gata2 (referred to as G2CKO mice) (Fig. 1A). Littermate mice (Gata2flox/flox) were similarly treated with Dox and were used as controls. In vivo imaging analysis of hIL6 BAC-luciferase transgenic mice (WIN-6 mice) was carried out as described previously (34). Primer sequence information for genotyping is available upon request.
IRI.
IRI was performed as described previously (23). The renal artery was clamped using a disposable vascular clip (Bear Medic Corporation) for 30 min. These mice were subjected to analysis 1 day, 2 days, and 2 weeks after release of the clip. All of the mice were handled according to the regulations of the standards for use of laboratory animals in Tohoku University. All of the animal procedures followed the guidelines established for the proper conduct of animal experiments by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
Immunostaining, histology, and immunoblotting.
Paraffin-embedded sections were autoclaved in citrate buffer for antigen retrieval. Primary antibodies against caspase-3 (Cell Signaling Technology; catalog number 9661) and F4/80 (Bio-Rad; MCA497GA) were used. A horseradish peroxidase (HRP)-labeled antibody (Dako) and 3,3′-diaminobenzidine (Dako) were used to detect the immunoreactivity. Hematoxylin and eosin (H&E) and Elastica-Masson staining were conducted employing a standard procedure (23). Damaged tubular areas were quantitatively analyzed with the H&E-stained sections as previously described (23, 46). For each kidney, images (magnification, ×200) were taken at 2 selected regions in the medullas from 5 mice of each group. The surface areas of the coagulative necrotic regions were determined using ImageJ software (National Institutes of Health), and the percentages of the damaged tubular area in the injured kidneys were calculated. An immunoblotting assay was performed as described previously (47) using anti-GATA2 (H-6; sc-515178) and anti-lamin B (M-20; sc-6217) antibodies purchased from Santa Cruz Biotechnology.
RT-qPCR.
Total RNA was extracted using Isogen (Nippon Gene). cDNA was synthesized using Superscript III (Invitrogen). Real-time quantitative PCR (RT-qPCR) was performed with an ABI7300 system (Thermo Fisher Scientific) and SYBR green master mix (Toyobo).
Flow cytometry.
Kidneys were digested with Liberase (Roche) to prepare single-cell suspensions, as previously described (18). The remaining red blood cells were removed with Histopaque-1083 (Sigma). The cells were stained with CD11b-fluorescein isothiocyanate (FITC) (M1/70; 11-0112041) and Gr1-allophycocyanin (APC) (RB6-8C5; 17-5931-81) antibodies (eBioscience). Flow cytometry was performed with a FACSCalibur system (BD Biosciences).
Isolation of CD cells.
CD cells were isolated from kidneys as previously described (18). Briefly, kidney single-cell suspensions were incubated with biotin-tagged Dolichos biflorus agglutinin (DBA) (Vector Laboratories), and DBA-labeled CD cells were isolated with M-280 streptavidin beads (Life Technologies).
BUN and creatinine.
Mouse serum was isolated from fresh blood. A Fuji Dri-Chem 7000 V instrument was used to measure BUN and serum creatinine levels.
Microarray and data mining.
The microarray data set was previously described (GSE52448) (18). Ingenuity pathway analysis (Ingenuity System) was performed to classify genes according to their biological functions. A heat map was generated using Cluster 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm) and JAVA Treeview (http://jtreeview.sourceforge.net/).
Chemicals and cell culture.
Compounds in the off-patent drug library of Keio University were subjected to HTS. Mitoxantrone dihydrochloride was purchased from Sigma-Aldrich. K-7174 {N,N′-bis-(E)-[5-(3,4,5-trimethoxy-phenyl)-4-pentenyl] homopiperazine} was generously provided by Kowa Pharmaceutical, Inc. For in vivo animal experiments, mitoxantrone dihydrochloride dissolved in saline was intraperitoneally administered to mice at doses of 1 or 2 mg/kg. Culture of mIMCD and the generation of GATA2-mIMCD cells were as described previously (18).
Generation of the K562-NLucP reporter line for HTS.
K562 cells were purchased from the American Type Culture Collection (ATCC) and maintained in RPMI 1640 (Nacalai) culture medium containing 10% fetal bovine serum (FBS) (Gibco) and 1 U/ml penicillin-streptomycin (Gibco) in a humidified atmosphere at 5% CO2 at 37°C. NanoLuc (NLucP) cDNA was PCR amplified from a pNL1.2 (NLucP) vector (Promega) and introduced into the EcoRI and NotI doubly digested pEV3 vector, which contains a human β-LCR and β-globin promoter (33). The reporter plasmid was transfected into K562 cells using Lipofectamine 2000 (Life Technologies). One day after transfection and thereafter, the cells were cultured in fresh medium containing 100 μg/ml G418 (Gibco). The surviving cells were cloned via a limiting-dilution protocol. The reporter activity of each clone was measured with the Nano-Glo luciferase assay system (Promega) with PheraStar-FS (BMG-Labtech).
Quality validation of the HTS system.
Four thousand cells in a 50-μl volume of medium were plated into each well of a 96-well plate and mixed with 50-μl medium containing 20 μM K-7174 or a 0.2% DMSO vehicle control. After 24 h of incubation at 5% CO2 and 37°C, the luciferase intensities of individual wells were measured using a Flexstation 3 (Molecular Devices). The value of each parameter was calculated as follows: coefficient of variation (CV) = standard deviation (SD)/average; signal/background ratio (S/B) = average of DMSO controls/average of K-7174 samples; signal/noise ratio = (average of DMSO controls − average of K-7174 samples)/SD of K-7174 samples; Z′-factor = 1 − (3 × SD of DMSO controls + 3 × SD of K-7174 samples)/(average of DMSO controls − average of K-7174 samples) (48).
HTS and luciferase assay.
For the first screen, 50 μl of K562-NLucP cells (1.5 × 104 cells/ml) was plated into 384-well plates with a Multidrop Combi dispenser (Thermo Scientific) and subsequently mixed with 10 μl medium containing 60 μM the tested compounds in 0.6% DMSO. For the second screen, 50 μl of K562-NLucP cells (1.5 × 105 cells/ml) was plated into a 96-well plate with a Multidrop Combi dispenser and mixed with 50 μl of medium containing 20 μM compounds that were selected in the first screen in 0.2% DMSO. After the cells were incubated in a CO2 incubator (at 37°C in 5% CO2, 20% O2) for 28 h, aliquots from each well were transferred to a fresh 384-well or 96-well plate and tested for luciferase activity using a Nano-Glo luciferase assay (Promega) with a PheraStar-FS plate reader (BMG-Labtech). The percent reduction in luciferase activity was calculated using the following formula: (1 − luciferase count of sample/luciferase count of 0.1% DMSO control) × 100.
Validation of hit compounds.
Two sets of 96-well plates were prepared in which 50 μl of K562-NLuc2 clone number 3 cells (1 × 105 cells/ml) was mixed with 50 μl of medium containing 20 μM tested compounds in 0.2% DMSO. One plate was used for a cell viability assay, and the other plate was used for a luciferase assay. The cells were incubated at 37°C in 5% CO2 and 20% O2 for 28 h. The cell viability was analyzed using a CellTiter 96 AQueous One solution cell proliferation assay (Promega) by following the manufacturer's instructions.
Statistical analyses.
All the data are presented as means ± SD. Statistical analyses were performed using the unpaired Student t test (two-tailed). In Fig. 1C, 7C, and 8C, data were analyzed using the Mann-Whitney U test.
ACKNOWLEDGMENTS
We thank Tomokazu Souma and Norio Suzuki for technical help and scientific comments. We also thank Kowa Pharmaceutical, Inc., for the generous gift of chemical compounds.
This study was supported by JSPS Kakenhi (grant 16H05147 to T.M. and grant 15H02507 to M.Y.), the Kobayashi Foundation for Cancer Research (T.M.), the Platform Project for Supporting Drug Discovery and Life Science Research funded by AMED (M.Y. and R.S.), and a research grant from the Astellas Foundation for Research on Metabolic Disorders (R.S.).
REFERENCES
- 1.Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A. 2007. Acute kidney injury network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 11:R31. doi: 10.1186/cc5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. 2010. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16:535–543. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferenbach DA, Bonventre JV. 2015. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11:264–276. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basile DP, Bonventre JV, Mehta R, Nangaku M, Unwin R, Rosner MH, Kellum JA, Ronco C, ADQI XIII Work Group . 2016. Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol 27:687–697. doi: 10.1681/ASN.2015030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rewa O, Bagshaw SM. 2014. Acute kidney injury—epidemiology, outcomes and economics. Nat Rev Nephrol 10:193–207. doi: 10.1038/nrneph.2013.282. [DOI] [PubMed] [Google Scholar]
- 6.Chevalier RL. 2016. The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Renal Physiol 311:F145–F161. doi: 10.1152/ajprenal.00164.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amdur RL, Chawla LS, Amodeo S, Kimmel PL, Palant CE. 2009. Outcomes following diagnosis of acute renal failure in U.S. veterans: focus on acute tubular necrosis. Kidney Int 76:1089–1097. doi: 10.1038/ki.2009.332. [DOI] [PubMed] [Google Scholar]
- 8.Ishani A, Xue JL, Himmelfarb J, Eggers PW, Kimmel PL, Molitoris BA, Collins AJ. 2009. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol 20:223–228. doi: 10.1681/ASN.2007080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto M, Ko LJ, Leonard MW, Beug H, Orkin SH, Engel JD. 1990. Activity and tissue-specific expression of the transcription factor NF-E1 multigene family. Genes Dev 4:1650–1662. doi: 10.1101/gad.4.10.1650. [DOI] [PubMed] [Google Scholar]
- 10.Minegishi N, Suzuki N, Yokomizo T, Pan X, Fujimoto T, Takahashi S, Hara T, Miyajima A, Nishikawa S, Yamamoto M. 2003. Expression and domain-specific function of GATA-2 during differentiation of the hematopoietic precursor cells in midgestation mouse embryos. Blood 102:896–905. doi: 10.1182/blood-2002-12-3809. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki N, Ohneda O, Minegishi N, Nishikawa M, Ohta T, Takahashi S, Engel JD, Yamamoto M. 2006. Combinatorial Gata2 and Sca1 expression defines hematopoietic stem cells in the bone marrow niche. Proc Natl Acad Sci U S A 103:2202–2207. doi: 10.1073/pnas.0508928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim KC, Hosoya T, Brandt W, Ku CJ, Hosoya-Ohmura S, Camper SA, Yamamoto M, Engel JD. 2012. Conditional Gata2 inactivation results in HSC loss and lymphatic mispatterning. J Clin Invest 122:3705–3717. doi: 10.1172/JCI61619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moriguchi T, Suzuki M, Yu L, Takai J, Ohneda K, Yamamoto M. 2015. Progenitor stage-specific activity of a cis-acting double GATA motif for Gata1 gene expression. Mol Cell Biol 35:805–815. doi: 10.1128/MCB.01011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Y, Lim KC, Onodera K, Takahashi S, Ohta J, Minegishi N, Tsai FY, Orkin SH, Yamamoto M, Engel JD. 1998. Rescue of the embryonic lethal hematopoietic defect reveals a critical role for GATA-2 in urogenital development. EMBO J 17:6689–6700. doi: 10.1093/emboj/17.22.6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khandekar M, Suzuki N, Lewton J, Yamamoto M, Engel JD. 2004. Multiple, distant Gata2 enhancers specify temporally and tissue-specific patterning in the developing urogenital system. Mol Cell Biol 24:10263–10276. doi: 10.1128/MCB.24.23.10263-10276.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoshino T, Shimizi R, Ohmori S, Nagano M, Pan X, Ohneda O, Khandekar M, Yamamoto M, Lim KC, Engel JD. 2008. Reduced BMP4 abundance in Gata2 hypomorphic mutant mice result in uropathies resembling human CAKUT. Genes Cells 13:159–170. doi: 10.1111/j.1365-2443.2007.01158.x. [DOI] [PubMed] [Google Scholar]
- 17.Ainoya K, Moriguchi T, Ohmori S, Souma T, Takai J, Morita M, Chandler KJ, Mortlock DP, Shimizi R, Engel JD, Lim KC, Yamamoto M. 2012. UG4 enhancer-driven GATA-2 and bone morphogenetic protein 4 complementation remedies the CAKUT phenotype in Gata2 hypomorphic mutant mice. Mol Cell Biol 32:2312–2322. doi: 10.1128/MCB.06699-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu L, Moriguchi T, Souma T, Takai J, Satoh H, Morito N, Engel JD, Yamamoto M. 2014. GATA2 regulates body water homeostasis through maintaining aquaporin 2 expression in renal collecting ducts. Mol Cell Biol 34:1929–1941. doi: 10.1128/MCB.01659-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fushimi K, Uchida S, Hara Y, Hirata Y, Marumo F, Sasaki S. 1993. Cloning and expression of apical membrane water channel of rat kidney collecting tubule. Nature 361:549–552. doi: 10.1038/361549a0. [DOI] [PubMed] [Google Scholar]
- 20.Sean Eardley K, Cockwell P. 2005. Macrophages and progressive tubulointerstitial disease. Kidney Int 68:437–455. doi: 10.1111/j.1523-1755.2005.00422.x. [DOI] [PubMed] [Google Scholar]
- 21.Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti VS, Ichimura T, Humphreys BD, Bonventre JV. 2012. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 82:172–183. doi: 10.1038/ki.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takaori K, Nakamura J, Yamamoto S, Nakata H, Sato Y, Takase M, Nameta M, Yamamoto T, Economides AN, Kohno K, Haga H, Sharma K, Yanagita M. 2016. Severity and frequency of proximal tubule injury determines renal prognosis. J Am Soc Nephrol 27:2393–2406. doi: 10.1681/ASN.2015060647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nezu M, Souma T, Yu L, Suzuki T, Saigusa D, Ito S, Suzuki N, Yamamoto M. 2017. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int 91:387–401. doi: 10.1016/j.kint.2016.08.023. [DOI] [PubMed] [Google Scholar]
- 24.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. 2002. Kidney injury molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int 62:237–244. doi: 10.1046/j.1523-1755.2002.00433.x. [DOI] [PubMed] [Google Scholar]
- 25.Chakraborty S, Kaur S, Guha S, Batra SK. 2012. The multifaceted roles of neutrophil gelatinase associated lipocalin (ngal) in inflammation and cancer. Biochim Biophys Acta 1826:129–169. doi: 10.1016/j.bbcan.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chung AC, Lan HY. 2011. Chemokines in renal injury. J Am Soc Nephrol 22:802–809. doi: 10.1681/ASN.2010050510. [DOI] [PubMed] [Google Scholar]
- 27.Akcay A, Nguyen Q, Edelstein CL. 2009. Mediators of inflammation in acute kidney injury. Mediators Inflamm 2009:137072. doi: 10.1155/2009/137072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petrovic-Djergovic D, Popovic M, Chittiprol S, Cortado H, Ransom RF, Partida-Sánchez S. 2015. CXCL10 induces the recruitment of monocyte-derived macrophages into kidney, which aggravate puromycin aminonucleoside nephrosis. Clin Exp Immunol 180:305–315. doi: 10.1111/cei.12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Umetani M, Nakao H, Doi T, Iwasaki A, Ohtaka M, Nagoya T, Mataki C, Hamakubo T, Kodama T. 2000. A novel cell adhesion inhibitor, K-7174, reduces the endothelial VCAM-1 induction by inflammatory cytokines, acting through the regulation of GATA. Biochem Biophys Res Commun 272:370–374. doi: 10.1006/bbrc.2000.2784. [DOI] [PubMed] [Google Scholar]
- 30.Imagawa S, Nakano Y, Obara N, Suzuki N, Doi T, Kodama T, Nagasawa T, Yamamoto M. 2003. A GATA-specific inhibitor (K-7174) rescues anemia induced by IL-1beta, TNF-alpha, or L-NMMA. FASEB J 17:1742–1744. [DOI] [PubMed] [Google Scholar]
- 31.Kikuchi J, Yamada S, Koyama D, Wada T, Nobuyoshi M, Izumi T, Akutsu M, Kano Y, Furukawa Y. 2013. The novel orally active proteasome inhibitor K-7174 exerts anti-myeloma activity in vitro and in vivo by down-regulating the expression of class I histone deacetylases. J Biol Chem 288:25593–25602. doi: 10.1074/jbc.M113.480574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV. 2012. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol 7:1848–1857. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whyatt D, Lindeboom F, Karis A, Ferreira R, Milot E, Hendriks R, de Bruijn M, Langeveld A, Gribnau J, Grosveld F, Philipsen S. 2000. An intrinsic but cell-nonautonomous defect in GATA-1-overexpressing mouse erythroid cells. Nature 406:519–524. doi: 10.1038/35020086. [DOI] [PubMed] [Google Scholar]
- 34.Hayashi M, Takai J, Yu L, Motohashi H, Moriguchi T, Yamamoto M. 2015. Whole-body in vivo monitoring of inflammatory diseases exploiting human interleukin 6-luciferase transgenic mice. Mol Cell Biol 35:3590–3601. doi: 10.1128/MCB.00506-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moriguchi T, Yu L, Otsuki A, Ainoya K, Lim KC, Yamamoto M, Engel JD. 2016. Gata3 hypomorphic mutant mice rescued with a yeast artificial chromosome transgene suffer a glomerular mesangial cell defect. Mol Cell Biol 36:2272–2281. doi: 10.1128/MCB.00173-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Butt MJ, Tarantal AF, Jimenez DF, Matsell DG. 2007. Collecting duct epithelial-mesenchymal transition in fetal urinary tract obstruction. Kidney Int 72:936–944. doi: 10.1038/sj.ki.5002457. [DOI] [PubMed] [Google Scholar]
- 37.Fujiu K, Manabe I, Nagai R. 2011. Renal collecting duct epithelial cells regulate inflammation in tubulointerstitial damage in mice. J Clin Invest 121:3425–3441. doi: 10.1172/JCI57582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suzuki M, Kobayashi-Osaki M, Tsutsumi S, Pan X, Ohmori S, Takai J, Moriguchi T, Ohneda O, Ohneda K, Shimizu R, Kanki Y, Kodama T, Aburatani H, Yamamoto M. 2013. GATA factor switching from GATA2 to GATA1 contributes to erythroid differentiation. Genes Cells 18:921–933. doi: 10.1111/gtc.12086. [DOI] [PubMed] [Google Scholar]
- 39.Moriguchi T, Yu L, Takai J, Hayashi M, Satoh H, Suzuki M, Ohneda K, Yamamoto M. 2015. The human GATA1 gene retains a 5′ insulator that maintains chromosomal architecture and GATA1 expression levels in splenic erythroblasts. Mol Cell Biol 35:1825–1837. doi: 10.1128/MCB.00011-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu L, Takai J, Otsuki A, Katsuoka F, Suzuki M, Katayama S, Nezu M, Engel JD, Moriguchi T, Yamamoto M. 2017. Derepression of the DNA methylation machinery of Gata1 gene triggers the differentiation cue for erythropoiesis. Mol Cell Biol 37:e00592-16. doi: 10.1128/MCB.00592-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katsumura KR, Ong IM, DeVilbiss AW, Sanalkumar R, Bresnick EH. 2016. GATA factor-dependent positive-feedback circuit in acute myeloid leukemia cells. Cell Rep 16:2428–2441. doi: 10.1016/j.celrep.2016.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen GY, Sakuma K, Kannagi R. 2008. Significance of NF-kappaB/GATA axis in tumor necrosis factor-alpha-induced expression of 6-sulfated cell recognition glycans in human T-lymphocytes. J Biol Chem 283:34563–34570. doi: 10.1074/jbc.M804271200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazerski J, Martelli S, Borowski E. 1998. The geometry of intercalation complex of antitumor mitoxantrone and ametantrone with DNA: molecular dynamics simulations. Acta Biochim Pol 45:1–11. [PubMed] [Google Scholar]
- 44.Parker C, Waters R, Leighton C, Hancock J, Sutton R, Moorman AV, Ancliff P, Morgan M, Masurekar A, Goulden N, Green N, Révész T, Darbyshire P, Love S, Saha V. 2010. Effect of mitoxantrone on outcome of children with first relapse of acute lymphoblastic leukaemia (ALL R3): an open-label randomised trial. Lancet 376:2009–2017. doi: 10.1016/S0140-6736(10)62002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hasegawa SL, Moriguchi T, Rao A, Kuroha T, Engel JD, Lim KC. 2007. Dosage-dependent rescue of definitive nephrogenesis by a distant Gata3 enhancer. Dev Biol 301:568–577. doi: 10.1016/j.ydbio.2006.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J, Vemuri C, Palekar RU, Gaut JP, Goette M, Hu L, Cui G, Zhang H, Wickline SA. 2015. Antithrombin nanoparticles improve kidney reperfusion and protect kidney function after ischemia-reperfusion injury. Am J Physiol Renal Physiol 308:F765–F773. doi: 10.1152/ajprenal.00457.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hasegawa A, Kaneko H, Ishihara D, Nakamura M, Watanabe A, Yamamoto M, Trainor CD, Shimizu R. 2016. GATA1 binding kinetics on conformation-specific binding sites elicit differential transcriptional regulation. Mol Cell Biol 36:2151–2167. doi: 10.1128/MCB.00017-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]