

ABSTRACT
Many HIV-1-infected patients evolve broadly neutralizing antibodies (bnAbs). This evolutionary process typically takes several years and is poorly understood as selection taking place in germinal centers occurs on the basis of antibody affinity. B cells with the highest-affinity receptors tend to acquire the most antigen from the follicular dendritic cell (FDC) network and present the highest density of cognate peptides to follicular helper T (Tfh) cells, which provide survival signals to the B cell. bnAbs are therefore expected to evolve only when the B cell lineage evolving breadth is consistently capturing and presenting more peptides to Tfh cells than other lineages of more specific B cells. Here we develop mathematical models of Tfh cells in germinal centers to explicitly define the mechanisms of selection in this complex evolutionary process. Our results suggest that broadly reactive B cells presenting a high density of peptides bound to major histocompatibility complex class II molecules (pMHC) are readily outcompeted by B cells responding to lineages of HIV-1 that transiently dominate the within host viral population. Conversely, if broadly reactive B cells acquire a large variety of several HIV-1 proteins from the FDC network and present a high diversity of several pMHC, they can be rescued by a large fraction of the Tfh cell repertoire in the germinal center. Under such circumstances the evolution of bnAbs is much more consistent. Increasing either the magnitude of the Tfh cell response or the breadth of the Tfh cell repertoire markedly facilitates the evolution of bnAbs. Because both the magnitude and breadth can be increased by vaccination with several HIV-1 proteins, this calls for experimental testing.
IMPORTANCE Many HIV-infected patients slowly evolve antibodies that can neutralize a large variety of viruses. Such broadly neutralizing antibodies (bnAbs) could in the future become therapeutic agents. bnAbs appear very late, and patients are typically not protected by them. At the moment, we fail to understand why this takes so long and how the immune system selects for broadly neutralizing capacity. Typically, antibodies are selected based on affinity and not on breadth. We developed mathematical models to study two different mechanisms by which the immune system can select for broadly neutralizing capacity. One of these is based upon the repertoire of different follicular helper T (Tfh) cells in germinal centers. We suggest that broadly reactive B cells may interact with a larger fraction of this repertoire and demonstrate that this would select for bnAbs. Intriguingly, this suggests that broadening the Tfh cell repertoire by vaccination may speed up the evolution of bnAbs.
KEYWORDS: HIV/AIDS, affinity maturation, broadly neutralizing antibodies, mathematical modeling
INTRODUCTION
Many human immunodeficiency virus type 1 (HIV-1)-infected patients evolve broadly neutralizing antibodies (bnAbs) that can effectively neutralize a large subset of phenotypically different HIV-1 variants (1). Treatment with bnAbs may prevent infection upon challenge (2–4), transiently reduce the viral load during chronic infection (5–7), and enhance humoral activity (8). Most of the antibodies in the serum of HIV-infected patients are nonneutralizing, and the small subset of antibodies with significant neutralizing capacity tends to bind epitopes in Env that are relatively conserved because these sites are of functional importance for the virus when it enters target cells (9). One good example of this type of epitope is the CD4-binding site. The mere fact that neutralizing antibodies bind relatively conserved epitopes helps to explain their broad reactivity to many variants of HIV-1. The germ line ancestors of the bnAbs typically fail to recognize the concealed and conserved epitopes that their progeny target after years of evolution (10). The evolution of a bnAb takes many mutations in both the CDR3 and the framework regions of the antibody variable region genes (11).
Most bnAbs have a number of unusual features: they carry a large number of somatic mutations, i.e., up to 30% of the heavy V-chain nucleotides (9); they appear only after several years; and they tend to have long heavy-chain CDR3 regions. The long CDR3 regions would enable them to penetrate deep into conformationally concealed and/or heavily glycosylated sites to contact functional and conserved residues located deep in the structure of Env (12–14). Somatic hypermutation only infrequently increases the length of the CDR3 regions of bnAbs (15), and most of the CDR3 lengths of bnAbs observed in HIV-1-infected patients are therefore determined by the original recombination event at the stage when the naive B cell developed (13). Importantly, this means that the long evolutionary process whereby B cells in a single patient learn to produce bnAbs traces back to relatively rare ancestors with particular VDJ rearrangements having long CDR3 regions (16). Hence, the capacity to become broadly neutralizing is earned by a slow and long sequence of accumulating point mutations and requires repeated rounds of selection of the fittest mutant. Because the process of affinity maturation in germinal centers is known to select for affinity and has no mechanism to select for neutralizing activity, it remains an open question as to how the rare B cells producing bnAbs ultimately become selected.
After the infection of a new host, HIV-1 diversifies by mutation and recombination into a genetically diverse population that is often called a quasispecies. Since the viral population during the chronic infection phase is typically not composed of a single “master sequence” with a cloud of mutants around it but, rather, consists of various lineages, each having numerous mutants, it seems more natural to view the total viral population as a set of quasispecies. We will here refer to each of these as a viral lineage. Particular lineages (or haplotypes) increase in frequency when they carry the best combination of immune escapes from T and B cell responses (17). Immune escape mutations in viral B cell epitopes are expected to reduce the affinity of their cognate antibodies. Although the epitopes targeted by neutralizing antibodies (nAbs) tend to contain relatively conserved amino acid residues, there is strong selection pressure on the virus to mutate these sites, and escape mutations impeding the binding of early nAbs rapidly increase in frequency (18). Thus, the antigenic evolution of HIV-1 within each host provides ample substrate for an ongoing evolution of antibody specificities to better recognize the predominant form of the epitope that the virus is evolving (19).
B cell selection in germinal centers (GCs) involves proliferation and mutation in the dark zone and subsequent selection in the light zone, with some fraction of the selected B cells recycling to the dark zone for additional rounds of proliferation and mutation (20). To be able to proliferate, B cells need to be stimulated by antigen, and the degree of clonal expansion depends on the availability of antigen and the relative affinity of the B cell receptor (BCR) for the antigen (20). In the light zone, B cells acquire antigenic proteins by pulling them off the follicular dendritic cell (FDC) network (21–23). These proteins are internalized and processed into peptides that get presented on MHC molecules (pMHC) to CD4+ follicular helper T cells (Tfh cells). Tfh cells specific for the pMHC expressed by the B cell can positively select such a B cell, i.e., provide a rescue signal (20, 24–27). Thus, germinal centers select for affinity, based on the efficiency of acquiring antigen, and for T-B cell collaboration, based on the likelihood of receiving a rescue signal from Tfh cells.
The important role that Tfh cells play in the evolution of bnAbs has been demonstrated in several studies comparing the frequency and breadth of Tfh cells in simian immunodeficiency virus (SIV)- or simian-human immunodeficiency virus (SHIV)-infected macaques and in HIV-infected patients, with and without bnAbs (28–32). Tfh cell frequencies tend to be higher in patients with bnAbs (28, 30–32). For chronic SHIV infections, Yamamoto et al. (30) found that the frequency of Env-specific IgG+ GC B cells correlates positively with the frequency of Env-specific Tfh cells, suggesting that Env-specific Tfh cells help Env-specific IgG+ GC B cells. Ranasinghe et al. (31) report that the breadth of the Tfh cell responses to HIV Env and Gag are higher in viremic controller patients with bnAbs than in controller patients lacking bnAbs. Since Env-specific Tfh cell responses can be induced by vaccination in rhesus macaques (33) and since well-designed mosaic vaccines can enhance CD4+ T responses (34–36), we urgently need to understand the consequences of increasing Tfh cell numbers and/or breadth on the evolution of bnAbs (28–32).
The selection of B cells in germinal centers operates on the relative affinities of the competing clones for the most prevalent antigen (37). In the absence of competition, low-affinity B cells become selected (38), and B cells with low- or high-affinity BCRs generate GCs with similar kinetics growing to similar sizes (39). The size of an individual GC is closely correlated with the availability of follicular T cell help in that GC (40), suggesting that rescue signals by Tfh cells limit the selection of B cells in the light zone. T cells preferentially form synapses with B cells with the highest surface pMHC density (41, 42), and B cells with higher cognate pMHC surface densities tend to monopolize T cell help, physically interfering with a T cell's ability to interact with low-affinity B cells (25). This seems to be a form of “winner-takes-all” selection (43) whereby the probability of becoming rescued is not determined by the affinity, or even the amount of pMHC, but by the relative amounts of pMHC presented by the B cells present at the border of the T and B cell regions in the germinal center (20, 25, 26). Interestingly, the amount of pMHC presented by a low-affinity B cell is essentially unaffected by the absence or presence of high-affinity competitors (25), and low-affinity B cells tend to be retained in GCs (44, 45). Thus, the BCR captures antigen from FDCs and mediates its internalization and processing in proportion to its affinity (21, 46), essentially mapping BCR affinity onto surface pMHC density. It has indeed been shown that germinal centers are populated by Tfh cells with different antigen specificities (30, 47) and with frequencies reflecting those of conventional CD4+ T cells (48).
Germinal centers develop a few days after infection and typically contract after a few weeks when the antigen is cleared. It is unclear whether GCs also contract during chronic infections, but numerous GCs can be found in patients chronically infected with HIV-1 (49), and the number of germinal centers per lymph node increases during chronic SHIV infection in rhesus macaques (50). GCs in HIV-infected patients are abnormal because as HIV-1 infects Tfh cells (51–55) both Tfh cells and B cells expand (49, 56), and lymph nodes become hyperplastic and undergo gradual remodeling (57–59). Despite the fact that Tfh cells are expanded, they could still be limiting B cell survival because in the GCs of chronically HIV-infected patients, the B cells expand much more than the Tfh cells (49), a large fraction of the Tfh cells is dysfunctional (60, 61), and the number of GC B cells is correlated positively with the number of GC Tfh cells (49). Mature GCs are open to invasion by B cells that were not present during the initiation of the GC reaction (62–64) and could therefore continue to support the evolution of higher affinities. Germinal centers are also open to migrating Tfh cells recognizing different pMHC, and individual germinal centers contain polyclonal Tfh cell responses (47). Additionally, ongoing GCs can be reused for different antigens provided that T cell help for the novel antigen is available (63). One could therefore envision that the selection for bnAbs takes place in chronic GCs displaying novel viruses on their FDC network. Although the FDC network in germinal centers of HIV-1 infected patients contains a large amount of virus (65, 66), the availability of antigen for single B cells could still be limiting if the frequency of viruses expressing their cognate epitopes is low.
Since germinal centers select for affinity and not for neutralizing capacity and since viruses evolving mutated Env proteins will also evolve novel epitopes triggering novel—and typically nonneutralizing—immune responses from naive B cells, leading to new rounds of affinity maturation among their progeny, a major open question is how germinal centers manage to select for bnAbs in HIV-1-infected patients. A necessary requirement seems to be that bnAbs recognize a larger fraction of all the virions in the body than other antibodies. Recognizing a large fraction of the various viral quasispecies can be brought about by evolving poly- or cross-reactive antibodies that also bind mutated forms of the original epitope (11, 67) or by evolving antibodies focusing on the concealed and most conserved residues around the original epitope (12, 13, 19).
Previous mathematical models for the evolution of bnAbs have either ignored Tfh cells (68) or have made the simplifying assumption that the density of Tfh cells is proportional to the concentration of antigen (69–71). Since we consider a repertoire of Tfh cell clonotypes and vary the mechanism whereby Tfh cells rescue germinal center B cells, we make other simplifying assumptions in order to develop a manageable model with different antigen specificities. We model the chronic phase of HIV-1 infection and consider one lineage of B cells evolving bnAbs in a single germinal center and adapting to a nonevolving, but diverse, set of viral lineages. The only temporal variation of the viral quasispecies that we allow for are novel viral variants forming a viral lineage that transiently dominates the pool of virus in the germinal center (e.g., a viral sweep). We first consider a single population of Tfh cells (by ignoring their antigen specificity) and find that a mechanism that is solely based upon the B cell surface density of pMHC presented to Tfh cells is sensitive to the frequency at which novel viral lineages sweep through the viral population. At early stages, when the B cell lineage has yet to evolve broad reactivity, B cells from other lineages that respond specifically to these sweeping viral lineages tend to outcompete the cells of the lineage evolving bnAbs, akin to earlier results of Luo and Perelson (68). Only at late stages would the bnAbs of the main B cell lineage acquire sufficient broad reactivity to also recognize most of the variants sweeping through the viral population and become insensitive to viral sweeps. Second, we subdivide the germinal center Tfh cells into clonotypes specific for different pMHC and find that a rescue mechanism that is based upon the B cell surface diversity of pMHC presented to cognate Tfh cells is largely insensitive to viral sweeps. This is because a new sweeping viral lineage that is transiently dominant is expected to contain fewer functional Tfh cell epitopes than the variety of strains present in all viral lineages recognized by broadly reactive B cells. Selection mechanisms based upon density or diversity of pMHC therefore lead to qualitatively different predictions, and hence it is important to consider the breadth of the Tfh cell repertoire for understanding the evolution of bnAbs.
Models with a single population of Tfh cells that select B cells on the basis of the presented density of pMHC predict that increasing the number of Tfh cells by vaccination could speed up the evolution of bnAbs, and this is in good agreement with data showing higher Tfh cell frequencies in patients having bnAbs (30–32). Our results show that this evolutionary process is sensitive to transient viral lineages sweeping through the quasispecies. Importantly, our second model with various Tfh cell clonotypes selected on the basis of the diversity of pMHC presented by GC B cells can account for data demonstrating that the antigen specificity of Tfh cells matters (31, 72) and predicts that the impact of concomitantly increasing the breadth of the Tfh cell repertoire should be even larger as the evolution of bnAbs would not be compromised by viral sweeps. Since vaccination strategies increasing HIV-specific CD4+ T cell numbers can also be detrimental because HIV-1 preferentially infects HIV-1-specific CD4+ T cells (73, 74), this predicted beneficial effect of diversifying the Tfh cell repertoire calls for experiments adding several CD4+ T cell epitopes to current vaccines designed to generate bnAbs (33, 75).
MATERIALS AND METHODS
We develop several mathematical models for a germinal center harboring one B cell lineage consisting of different B cell clones responding to a particular functional site on Env, e.g., the CD4-binding site. The same germinal center also harbors several small Tfh cell clones, each specific for a particular HLA class II-restricted epitope from the virus. We consider the chronic phase of the infection and assume that the virus has already diversified into a diverse set of lineages. For simplicity, we ignore further evolution of viral diversity by defining a fixed number, U, of viral lineages present at equal frequencies (Fig. 1a). A viral lineage is here defined as all viruses expressing the same unique variant of the B cell epitope under consideration, e.g., the CD4-binding site, and sharing a particular set of functional T cell epitopes (Fig. 1c). Because all viral lineages are simultaneously present at equal frequencies, we are studying a best-case scenario for the evolution of bnAbs. We allow for some temporal variation of the viral quasispecies by randomly introducing novel viral lineages that transiently dominate the virus pool (Fig. 1b). These viral sweeps represent viral lineages having mutated the CD4-binding site such that they escape recognition by at least some of the clones in the B cell lineage evolving bnAbs.
FIG 1.
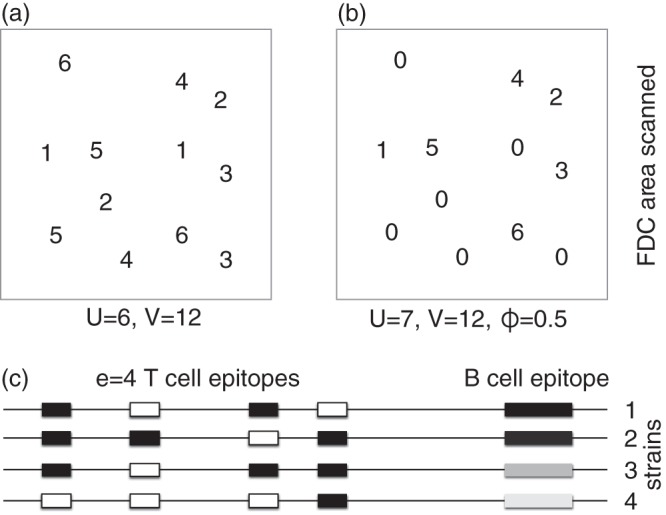
Cartoons explaining the structure of the models. (a) The area on the FDC network scanned by a single B cell contains a total of V = 12 virions of which U = 6 are unique (the digits refer to viral lineage numbers, and each lineage here consists of two strains). The most specific B cells, B1, can bind only the B cell epitope defining one of the viral lineages (say lineage 1), has a specificity f(1) = 1/U, and is expected to capture protein from f(1)V = V/U = 2 virions. B cells of the next class, B2, can bind all strains from two of the viral lineages (say 1 and 2), have a specificity f(2) = 2/U, and would bind f(2)V = 2V/U = 4 virions and so on. The most broadly reactive B cells can bind viruses from all lineages, i.e., f(6) = 6/U = 1, and are expected to capture and present protein from all V = 12 virions in the area. (b) A transiently dominant viral lineage, here number 0, occupies ϕ = 0.5 of the FDC area (replacing one virion of each lineage). The most specific B cells, B1, are now expected to bind at least (1 − ϕ)f(1)V = 1 virions and will bind virus from lineage 0 with the same probability f(1) = 1/6 that they would bind other viruses [in which case they would bind (1 − ϕ)f(1)V + 6 virions]. The broadest reactive B cells, having f(6) = 1, will still bind all 12 virions. (c) Viral lineages have one unique B cell epitope (indicated by the gray bars at the right hand side of each line) and carry e T cell epitopes elsewhere in their sequence (the horizontal line). T cell epitopes can be functional (black) or have escaped MHC binding (open boxes) with probability μ. A B cell capturing virus from all four lineages depicted in panel c would present pMHC for all four T cell epitopes, whereas a B cell processing virus from lineage 1 can only be rescued by only two of the four Tfh cell clonotypes. Note that the number of unique viral lineages captured by a Bi cell is defined as f(i)U = i.
We order the clones in the B cell lineage by the probability that they recognize variants of the B cell epitope. Consider a situation where B cells scan a particular area of the FDC network and attempt to bind all viruses in this area depending on their affinity to the epitope in Env on each virus (Fig. 1). The first clone, B1, is the most specific and recognizes the lowest fraction, f(1), of viral lineages. The second, B2, binds a larger fraction, f(2), than B1, and so on [i.e., f(i + 1) > f(i) for all i]. Since all the clones belong to one B cell lineage, we let Bi+1 cells appear by somatic hypermutation from clone i; i.e., we consider the case where B cells gradually evolve the capacity to capture more variants of the epitope. This evolution of breadth could be due either to evolving novel B cell receptors that better penetrate the glycosylated sites that are concealing a conserved B cell epitope (12–14) or to the evolution of more flexible B cell receptors that bind more variants of this epitope (11, 67). In both cases, Bi+1 cells capture more virus than Bi cells. We initiate the germinal center reaction with a single nonmutated progenitor cell of the B cell lineage and add a subscript 0 to indicate that this cell has undergone zero divisions; i.e., the initial condition is defined as B1,0 = 1. Considering only one progenitor cell implies that this cell has a sufficiently high-affinity for its cognate B cell epitope to outcompete all other B cells not belonging to this B cell lineage (which could be specific for the same or for other epitopes on Env). Thus, B cells not belonging to the B cell lineage are initially ignored, and the germinal center is founded by a progenitor cell of the B cell lineage, which subsequently develops breadth through somatic mutation.
B cells proliferate at per capita rate p as centroblasts, and we use a second index, j, to denote the number of divisions they have completed since their last rescue; i.e., Bi,j is the number of B cells recognizing a fraction f(i) of the epitope variants and having completed j divisions since their last productive interaction with a Tfh cell (Fig. 2). We assume that, after an average of n divisions, B cells become centrocytes that need to interact with a Tfh cell to prevent rapid cell death by apoptosis (at rate dB). A B cell that forms a conjugate with a Tfh cell appears as a “rescued” B cell, Ri, upon the dissociation of the conjugate (at rate kd). The Tfh cell then becomes available to form other conjugates. Rescued B cells may recycle into another round of proliferation, with probability r/(r + dR), or leave the germinal center at rate dR. Note that n here represents the average number of divisions that B cells complete in the dark zone before they migrate to the light zone to interact with Tfh cells (Fig. 2). There will probably be some stochastic variation in the number of divisions individual B cells complete before requiring a rescue signal, but only the average is considered here. Because the precursor frequency of naive B cells with long heavy-chain CDR3 regions that are able to develop breadth is low, simulations start with a single progenitor cell of the B lineage in its most specific class, B1,0 = 1.
FIG 2.
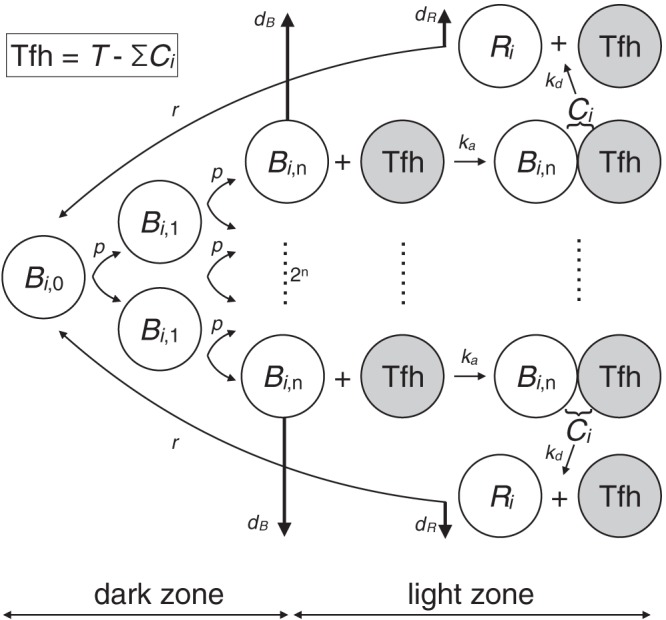
Cartoon explaining the differential equations and their parameters. The vertical heavy arrows denote loss of cells, either by rapid apoptosis at rate dB or by leaving the germinal center at rate dR. Note that a free Tfh cell (gray circle) is written as F in the equations of the density model and as Fl in the diversity model. Further, the association coefficient ka is multiplied with f(i) in the density model and with g(i) in the diversity model.
For reasons of simplicity, we assume that the fraction of viral variants that a B cell of type i, Bi, is able to bind is a simple linear function, f(i) = i/U, where U is the maximum number of unique viral lineages in the area of the FDC network scanned by the B cell. Since we consider only this one area, U is the total number of variants of the epitope under consideration. Further, we let V be the total number of virions in this area. Since we typically consider the case where the viral population is diverse and contains a large number of variants of the epitope, we treat f(i) as the expected fraction of viruses that can be captured by a Bi cell (Fig. 1a); i.e., a Bi cell is expected to capture and present protein from f(i)V virions, where we have chosen a scanning area sufficiently small that the ability of the B cells to capture and present protein is not saturated. Further, we assume a B cell that captures and presents protein from more virions is more likely to interact with Tfh cells and be rescued.
We start with a model considering one dominant T cell epitope that is recognized by one cognate Tfh cell clonotype in the germinal center. A Bi cell forms a conjugate with a free Tfh cell of this clonotype at a maximum rate kaF, where ka is a mass action association rate constant, and F is the number of free cognate Tfh cells. The actual conjugation rate is proportional to the density of pMHC on the B cell surface, and this density is assumed to be proportional to the amount of viral protein, f(i)V, captured from the FDC network. Scaling the total number of virions to V = 1 [by adjusting the association rate ka; i.e., ka = ka′/V when the conjugation rate is written as ka′f(i)VF], we obtain that Bi cells bind free cognate Tfh cells at a rate kaf(i)F. Because the total number of cognate Tfh cells, T, in the germinal center is limiting, we distinguish between conjugated Tfh cells, Ci, and free Tfh cells, F, where the total number of cognate Tfh cells is defined as
Summarizing, we write for the density model:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
where Bi,j is the number of B cells that have undergone j divisions since their last rescue and which recognize a fraction f(i) = i/U of the many variants of the epitope under consideration, with i = 1,2,…,m specificity classes [we typically consider U = 25 viral lineages and evolutionary time periods where m never exceeds U, i.e., f(i) < 1]. T is the total number of cognate Tfh cells in the germinal center, Ci is the number of Bi-Tfh cell conjugates, and Ri is the number of rescued B cells of type i. The total number of B cells of each type is defined as
Finally, in several simulations we consider novel lineages of the virus that appear randomly, which transiently dominate the viral population in the germinal center (and do not overlap in time). The average frequency ϕ of a novel dominant lineage is chosen randomly between 0.5 and 1 from a uniform distribution, and we let the dominant viral lineage be present for approximately 2 months (i.e., for a time period drawn from a Gaussian distribution with a mean and standard deviation of 60 and 10 days, respectively). Whenever a dominant viral lineage is present, we use (1 − ϕ)f(i)V for the expected number of virions bound by Bi cells and use f(i) to define the probability that Bi cells bind the ϕV virions of this particular dominant lineage (see the legend of Fig. 1). These probabilities are drawn each time a novel dominant viral lineage appears. The novel virus is expected to trigger a transient response from B cells not belonging to the B cell lineage. These other B cells obey the same mathematical model and are classified as B0 cells (Fig. 1b). We set the precursor frequency of normal naive B cells at 100-fold larger than that of naive B cells with long CDR3 regions; i.e., we chose B0,0 = 100 cells as the initial condition for the other clone each time a novel dominant viral lineage appeared. In humans, with more than a 1,000 germinal centers (76) and a total of about 1011 B cells, seeding each germinal center with an average of 100 naive cognate B cells would correspond to a reasonable precursor frequency of 10−6 (this remains a rough calculation since the number of germinal centers in chronically HIV-infected patients is not known). Summarizing, B cells of clone Bi capture a fraction, (1 − ϕ)f(i), of the current viral lineages and will be able to capture virus from a sweeping lineage with probability f(i), whereas B cells of clone B0 will capture a fraction ϕ of the virions in the FDC area (Fig. 1b).
Diversity of Tfh cell epitopes.
Next, consider an alternative model where we consider Tfh cells with different specificities, i.e., several Tfh cell clonotypes, Tl, where l = 1,2,…,e epitopes. In this diversity model we are more concerned with the diversity of Env molecules captured by a B cell and less by the amount of protein collected. B cells targeting conserved epitopes or B cells having polyreactive BCRs should capture more viral variants from the FDC network than specific B cells. Hence, they would process a larger variety of HIV-1 proteins and present a larger repertoire of pMHC on their cell surfaces (but possibly at a lower density than specific B cells). As a consequence, they can be rescued by a larger fraction of the Tfh cell repertoire present in their germinal center.
The diversity of T cell epitopes that a B cell is expected to present depends on what they pull off the FDC network and which viral proteins they process and present on class II MHC (77). HIV-1 is small enough to be internalized by clathrin-mediated endocytosis (78), and if B cells were to pull entire virions off the FDC network, they could in principle present T cell epitopes from any viral protein. For a much larger virus, Sette et al. (77) provide evidence for a tight linkage between the protein specificities of CD4+ T cells and B cells. This linkage could persist for small virions when the protein targeted by the B cell receptor is protected from intracellular degradation (79), so its peptides could preferentially be presented on class II MHC (77). The fact that the frequency of Gag-specific Tfh cells better correlates with the presence of bnAbs than with the frequency of Env-specific Tfh cells (31) does suggest that GC B cells are presenting HIV Gag epitopes to Tfh cells. Studies of other small pathogens such as influenza virus, vaccinia virus, and hepatitis B virus also indicate that protein specificity does not need to be shared by helper CD4+ T cells and B cells (80–84). Finally, rhesus macaques immunized with a vector expressing SIV Gag/Pol rapidly develop Env antibodies, despite the absence of Env in the vaccine (85). Thus, the T cell epitope could be a small peptide sample from any HIV-1 protein and will typically be unrelated to the B cell epitope under consideration (Fig. 1c). Note that a Tfh cell can rescue only B cells that have captured at least one viral protein lacking immune escape mutations in its cognate T cell epitope.
Defining μl as the frequency of viral immune escape mutations in Tfh cell epitope l, a B cell presenting peptides from only one viral lineage has a chance of 1 − μl to present a functional cognate peptide to a Tfh cell from clone l (Fig. 1c). A Bi cell having extracted proteins from f(i)U = i distinct viral lineages can then be rescued by this Tfh cell with likelihood 1−μli, where μli is the probability that all i extracted viral lineages have an escape mutation in epitope l (see the legend of Fig. 1c). We therefore define g(i, l) = 1−μli as the probability that a Bi cell presenting several pMHC of one particular T cell epitope, l, which were gathered from i different viral lineages, presents at least one functional form of the epitope to a cognate Tfh cell. First, note that the probability of becoming rescued by a particular Tfh cell is not dependent on B cell breadth, i, for all epitopes l lacking escape mutations; i.e., when μl = 0, the probability g(i, l) = 1. Second, B cells cannot be rescued by Tfh cells specific for those epitopes that have fully escaped, i.e., g(i, l) = 0 for μl = 1. Finally, because both B0 cells, which are specific for a transiently dominant viral lineage, and B1 cells, which are the most specific cells of the B cell lineage evolving bnAbs, are defined to extract proteins from a single viral lineage, we define
| (7) |
Since we no longer consider the densities of pMHC in the diversity model, we can ignore the fact the transiently dominant viral lineage is present at a higher frequency, ϕ, than the other viral lineages. By equation 7, we even make the simplifying assumption that B cells of the lineage evolving bnAbs never bind the transiently dominant viral lineage. Since this slows down their evolution of neutralizing capacity and since we find better evolution of bnAbs in the diversity model, this is a conservative assumption.
Now let e be the total number of different T cell epitopes in the virus, and assume that there exist clones of Tfh cells that recognize each of these epitopes. Calling the set of cells recognizing the same epitope a clonotype, there will be e cognate clonotypes of Tfh cells in the germinal center. We assume Bi cells form conjugates with a free cognate Tfh cell from each clonotype, l, at a rate kag(i, l)Fl, where ka remains the association rate, g(i, l) is the probability of rescue (see equation 7), and Fl is the number of free Tfh cells of clone l. We write our new model by keeping equations 1 and 2 and replacing equations 3 to 6 with the following:
| (8) |
| (9) |
| (10) |
| (11) |
where Tl is the total number of cells per Tfh cell clonotype, for l = 1,2,…,e.
The typical number of Tfh cell epitopes, e, in an HIV-infected patient is expected to be relatively small. Ranasinghe et al. (31) typically found five Tfh cell epitopes in Gag (maximally eight in their set of patients) and typically two (maximally four) Tfh cell epitopes in Env in patients having evolved bnAbs. pMHC epitopes from Gag could be overrepresented because Gag is an abundant, conserved, and preferred protein (72, 86). Extrapolating these findings to the other HIV-1 proteins suggests that patients should typically have less than e = 25 Tfh cell epitopes. This estimate resembles the numbers of CD8+ T cell epitopes that are typically found in HIV-infected patients (87, 88) and the number of dominant T cell epitopes found in HLA-transgenic mice infected with vaccinia virus (89). The number of helper T cell epitopes will depend on the HLA haplotype of the patient and could vary markedly between patients (90). Early in the infection most of these T cell epitopes should not be mutated (90) since most patients are infected by HLA-disparate donors (91). Since the virus evolves mutations in the MHC anchor residues of the epitopes over the course of the infection (87, 92, 93), the total number of epitopes, e, should decrease over time, which intensifies the selection between B cells for Tfh cell-mediated rescue.
Although we are interested in the effects of the breadth of the Tfh cell repertoire, we first make a mean-field assumption. This assumption will make the density and diversity models very similar, which helps to identify the major mechanistic difference between selecting for the density or diversity of pMHC. Whenever the different Tfh cell clonotypes have the same size, Tl, and the viral quasispecies has the same fraction of immune escape mutants, μl, per epitope, this mean-field model will be identical to the full model. Thus, let Tl = T/e be the total number of Tfh cells per clone. Since we are assuming that all Tfh cell clones behave similarly, we replace the sum over l in equation 8 with the term g(i)F, where g(i) = 1 − μi remains the chance that a B cell having gathered proteins from i viral lineages presents at least one nonescaped form of the T cell epitope, μ is the average of the μl values (Fig. 3b), and
is the total number of free Tfh cells. Thus, the mean-field model can be described with the equations of the density model [i.e., equations 1 to 6] by just replacing f(i) = i/U with g(i) = 1 − μf(i)U = 1 − μi, for i = 1,2,…,m (Fig. 3). Importantly, the interpretation of the g(i) function remains very different, as the specific B0 cells can present only pMHC from the dominant viral lineage (see equation 7), and each of its T cell epitopes is expected to be functional with probability g(0) = 1 − μ; we similarly define
| (12) |
(Fig. 3a). Thus, after any B cell somatic mutation allowing them to bind more than one viral lineage, B cells in the lineage evolving bnAbs are more likely to be rescued than specific B cells, i.e., g(i) > g(1) = g(0) for i = 2,3,…,m. Surprisingly, replacing f(i) by equation 12 markedly changes the dependence of the evolution of bnAbs on the frequency of novel variants sweeping through the viral population (see Results).
FIG 3.

The probability that a B cell having gathered proteins form various viral lineages presents a functional cognate epitope to a cognate Tfh cell. Panel a depicts the function g(i) defined by equation 12 as a function of the number of B cell mutations i, which here reflects the number of viral lineages presented by a Bi cell [i.e., f(i)U = i]. The special case g(0) = 1 − μ denotes the probability with which B0 cells present a functional cognate epitope derived from a transient dominant viral lineage (which is the same as the probability with which nonmutated B1 cells present Tfh cell epitopes) (see equation 12). Note that μ = 0.5 means that in this Tfh cell epitope, half of the viral lineages carry an escape mutation in the Tfh cell epitope. We also depict g(i) for μ = 0.25 and μ = 0.75 because μ increases from 0 to 0.75 in the data shown in Fig. 6. Panel b illustrates how the fraction of escape mutations averaged over all epitopes increases over time (for a representative example). The dashed lines represents five arbitrary escape mutations (μl, for l = 1,…,5) slowly evolving to fixation. The heavy line illustrates how the average increases over time (in the data shown in Fig. 6 we simplify this by letting μ increase from 0 to 0.75 in a linear manner). Panel c depicts how the normalized sum in the rescue probability of equation 8 hardly depends on the breadth e of the Tfh cell response (see the text). Panel d illustrates that the presence of a single wild-type Tfh cell epitope, i.e., setting μe = 0 and 0 < μl < 1, for l = 1,2,…,l − 1, markedly reduces the differences in the rescue probabilities of the various Bi clonotypes when the number of epitopes, e, is small.
Finally, we analyze and simulate the full diversity model to study how the breadth of Tfh cell epitopes, e, affects the evolution of bnAbs, by drawing random values for the immune escapes, μl, and Tfh cell densities, Tl. Summarizing, the basic idea is that a broadly reactive B cell will capture a larger diversity of each HIV protein than specific B cells and is hence more likely to present at least one wild-type form of the pMHC upon the encounter of a cognate Tfh cell. The parameter μl should be interpreted as the average frequency of immune escape mutants in a particular T cell epitope (Fig. 3b), which is used to define the probability that a B cell presents at least one functional epitope to a particular Tfh cell. Likewise, in the mean-field model, μ does not reflect the fraction of escaped Tfh cell epitopes but the frequency of escapes in a single epitope. Since B cells cannot be rescued by Tfh cells specific for epitopes that have been lost completely from the viral quasispecies, i.e., epitopes l for which μl = 1, and because all B cell lineages have equal chance to be rescued by a Tfh cell specific for an epitope that has no immune escape mutations (μl = 0), the main effect of the breadth, e, of the Tfh cell repertoire is to allow for a variety of Tfh cell epitopes selecting for broadly reactive B cells. In the mean-field model, we consider an average epitope making such a difference by restricting 0 < μ <1.
Parameter values.
We are considering a human lymph node, and most parameter values are discussed in the legend of Fig. 4. For the somatic mutation of B cells, we consider only mutants that bind an increased fraction, f(i), of the virus lineages in the quasispecies. This is done in a stepwise manner as in the affinity maturation model of Kepler and Perelson (94, 95), and B cells binding a fraction f(i + 1) of the viral lineages evolve from the clone binding a fraction f(i) of them. Since we consider only such successful mutations, it is difficult to estimate the effective mutation rate, and we define Bi/106 as the daily probability that one cell is added to population Bi+1,0. Since the maximum clone size in our model is about 105 cells (Fig. 4), large clones generate single cell mutants with an increased breadth approximately once every 10 days. In the density model we use U = 25 unique viral variants, so we need 25 somatic mutations to approach full neutralizing capacity. In the diversity model we will increase μ, 0 ≤ μ < 1, over time to model the evolution of immune escapes. The model was implemented in the C programming language using the variable time step Runge Kutta integrator ODEINT provided by Press et al. (96), and the code is available upon request.
FIG 4.

The behavior of a single clone of B cells in the density model (equations 1 to 6) for a clone recognizing all viruses, i.e., f(1) = 1 in panel a, and a clone recognizing 10% of the virions, i.e., f(1) = 1/10 in panel b. We assume that B cells perform five divisions in the dark zone before moving to the light zone where they can be rescued by a Tfh cell. Since conjugates of B cells and Tfh cells are short-lived (117, 118), we set kd, the dissociation rate, such that these complexes have an expected life span of 3 min. Apoptosis of expanded B cells that fail to get rescued by Tfh cells is a fast process, and we set dB such that B1,5 cells have an expected life span of 2 h. The association rate, ka, is set such that a B cell presenting a high density of pMHC associates with any of the 100 Tfh cells in 2 min. B cells are allowed to perform two divisions per day, and rescued B cells leave the germinal center in about 2 h. Ten percent of the rescued B cells recycle to perform another round of expansion. With these parameters, i.e., T = 100 cells, dB = dR = 12 day−1, kd = 480 day−1, ka = 7.2 cell−1 day−1, p = 2 day−1, r = 1.2 day−1, and n = 5 divisions, the maximal size of a germinal center is B1 = 7.8 × 104 cells (panel a). A clone recognizing 10% of the virions (panel b) expands somewhat more slowly and approaches a slightly lower steady state with far fewer free Tfh cells.
RESULTS
Density model.
The general behavior of the models is illustrated by depicting the immune response of the density model with a single clone of B cells responding to a fixed amount of virus (Fig. 4). Figure 4a depicts a clone (B10) starting with a single cell, B1,0 = 1, recognizing all of the virus, f(1) = 1 [where f(i) is a fraction of the epitope variants i], which expands rapidly by performing n = 5 divisions in a few days. Most of its progeny are rescued because the Tfh cells are not yet limiting, i.e., kd f(i)F ≫ dB (where kd is the dissociation rate, F is the number of free cognate Tfh cells, and dB is the rate of B cell death by apoptosis) (Fig. 2). A small fraction, i.e., r/(r + dR) ≃ 0.09, of the cells, having completed five divisions, recycles to the dark zone to complete another round of clonal expansion (Fig. 2). After a few weeks the free Tfh cell density becomes limiting, i.e., F → 1 cell after 3 weeks because at steady state about 99% of the 100 Tfh cells are bound in conjugates, and the B cell clone size approaches a steady state of B1 = 7.8 × 104 cells (where B1 is the first B cell clone) (Fig. 4a). At the steady state, most of the cells are dividing centroblasts, i.e., B1,j cells (where j is the number of B cell divisions since the last interaction with a Tfh cell). B cells recognizing 10% of the virions on the FDC network, e.g., f(1) = 1/10, take about a week longer to approach a slightly smaller maximal clone size and approach a steady state with about 90% of the Tfh cells in conjugates (compare Fig. 4a and b).
Evolution of bnAbs in the density model.
In the density model bnAbs can evolve only if broadly reactive B cells, on average, capture more viral protein from FDC-bound virus than other B cells. This is not necessarily expected to happen because a broadly reactive B cell need not bind more virions on the FDC network than a B cell specifically binding a currently dominant lineage of the virus; i.e., broadly reactive B cells may bind many viral variants but not necessarily the most virus. The evolution of bnAbs could be helped by averaging over time because broadly reactive B cells are more likely to remain activated when the current dominant viral lineage is replaced by a novel dominant lineage, while specific B cells are expected to decline when the frequency of their cognate viral lineage declines. These arguments require that the frequency distribution of the several viral lineages be fairly even as B cells specific for the most dominant lineage within the viral population should not capture more antigen than B cells binding various subdominant lineages. Interestingly, the frequency distribution of the major viral lineages (or haplotypes) broadens slowly over time (17), which could be a reason why bnAbs evolve late.
We first study this scenario with the density model. For simplicity, consider a viral population with a fixed number of lineages present at equal frequencies and study the evolution of neutralizing capacity over the first 2,000 days after infection (Fig. 5a). The first B cell clone, B1, binds the lowest fraction, f(1), of all virions in a small area of the FDC network (Fig. 1a) and approaches its maximal size after about a month. These progenitors generate mutants and are replaced by the second clone, B2, after about 3 months (Fig. 5a). This clone-by-clone replacement continues, with f(i) increasing, and at day 2,000 clone number 12 is taking over. Running 100 simulations and scoring the maximum clone number obtained, we summarize the outcome of these evolutionary simulations by the black line in Fig. 5c, depicting the number of cases at which each level of neutralization was achieved. In all 100 cases bnAbs evolve, and they typically accumulate 11 to 13 mutations. This variation seems too little to account for the observed variation in the evolution of bnAbs in different HIV-1-infected patients (1).
FIG 5.

Evolution of bnAbs in the density model [i.e., using f(i) = i/U in equations 1 to 6]. The gray scale in panels a and b depict the Bi densities on a logarithmic scale (where black is the highest density and white indicates low or zero density). The upper row depicts the various B0 clones. In panel a there are no sweeps, and the B cell lineage accumulates 12 mutations after 2,000 days; in panel b, sweeps occur every 100 days, and the B cell lineage accumulates only 4 mutations. Panel c depicts the maximum clone number attained in 100 simulations with sweeps occurring never, or every 50, 100, 200, or 400 days. Parameters are as defined in the legend of Fig. 4, and U = 25 unique viral lineages and m = 20 B cell clones.
Viral sweeps in the density model.
The fact that bnAbs readily evolve in the density model was to be expected because the different viral lineages remain present at similar densities, and broadly reactive B cells therefore always do better than specific B cells [i.e., f(i + 1) > f(i)]. The evolution of bnAbs is much more difficult when a limited number of viral lineages dominate the quasispecies (68). There is good evidence that viral lineages replace each other by selective sweeps due to immune selection (87, 92, 93), and hence B cells recognizing epitopes expressed by transiently dominant viral lineages could capture much more antigen than their current competitors that are broadly reactive (68). We model this here by randomly introducing novel viral lineages that are specifically recognized by an additional B cell clone, B0, of another B cell lineage (called autologous B cells in Luo and Perelson [68]).
Consider a virus lineage that transiently appears at a high frequency, 0.5 < ϕ < 1 (where ϕ is average frequency), in the viral population (Fig. 1b) and which triggers a response from a novel specific clone of B0 cells [i.e., f(0) = 0 for all the other viral lineages]. The probability that the existing Bi cells of the lineage evolving bnAbs will bind the dominant viral lineage should be given by the function f(i), whereas the expected fraction of the other (1 − ϕ)V (where V is the total number of virions) nondominant viruses that they should still be able to capture reduces to (1 − ϕ)f(i)V (Fig. 1b). Since a dominant viral lineage is likely to be missed by B cells in the evolving bnAb lineages having a low f(i), this tends to reduce the amount of pMHC they are expected to present. The novel B0 cells are expected to bind a large number of virions (i.e., ϕV) (Fig. 1b) and therefore tend to outcompete the Bi cells that have just started to develop broad neutralizing capacity (i.e., Bi cells in the lower classes). More broadly reactive Bi clones are also more likely to bind the dominant viral lineage, and, if they do, are expected to bind a large fraction, ϕ + (1 − ϕ)f(i), of all viruses. On average, broadly reactive B cells will therefore be less affected by the specific B cell response to dominant viral lineages sweeping through the population. Thus, we expect that transient sweeps through the viral population will initially slow down the evolution of bnAbs and that this effect will peter out over time.
The effect of transient sweeps through the viral population is studied in Fig. 5b and c. The average frequency of the sweeping variants, ϕ, is drawn uniformly between 0.5 and 1, and we let the dominant viral lineage be present for approximately 2 months. Setting ϕ = 0 afterwards causes the specific B0 cells to decline rapidly. When dominant viral lineages appear frequently they strongly affect the evolution of bnAbs (Fig. 5b, where the first row denotes the various B0 clones). We quantify the effect of dominant viral lineages by performing 100 simulations and recording the highest clone number that is obtained and observe that the presence of transiently dominant viral lineages can markedly hamper the evolution of bnAbs (Fig. 5c, colored lines). Thus, the evolution of bnAbs is not a robust property of the density model, confirming that this model requires the strong assumption that the viral lineages should be present at relatively similar frequencies. Little is known about the frequency distribution of the viral haplotypes or about the frequency of the transiently dominant sweeps, but one could argue that the variation between the cases shown in Fig. 5c can explain why bnAbs evolve in only a subset of HIV-1-infected individuals (1).
Mean-field diversity model.
Although the diversity model is built upon a repertoire of Tfh cell clonotypes and although the density model considers only one Tfh cell population, the two models differ only in the functions f(i) and g(i), representing the probability that a recycling B cell is rescued by a Tfh cell, after making the mean-field assumption explained in Materials and Methods. Hence, the behavior of a single clone of B cells in the diversity model is identical to that of the density model shown in Fig. 4 [replacing f(1) with g(1) = 1 and g(1) = 0.1 in Fig. 4a and b, respectively]. In the density model, f(i) is the probability that a B cell binds a particular variant of the epitope, whereas in the diversity model g(i) is the probability that a B cell presents a functional pMHC to a cognate Tfh cell in the same germinal center. The latter depends on the frequency of immune escape mutants, μ, in a Tfh cell epitope, which will turn out to be important for the effect of sweeps through the viral population.
In the absence of sweeps the evolution of bnAbs is much slower in the diversity model than in the density model as the g(i) function saturates (Fig. 3a), which decreases the selective advantage of mutant B cells with a broader reactivity, whereas the f(i) function is linear. Since both functions have a phenomenological nature, this difference need not reflect a true biological difference. For instance, HIV-1 tends to escape from the T cell responses in its host, and the parameter μ should increase over time because for each T cell epitope that is under selection, the frequency of escape variants is expected to increase over time (Fig. 3b). Additionally, if cognate Tfh cells were limiting the bnAb responses and if bnAb responses were to impede viral replication, there would be strong selection pressure for the virus to evolve immune escapes in their Tfh cell epitopes (90). Since the value of μ influences the shape of the g(i) function (Fig. 3a), we also study the evolution of bnAbs in simulations starting without immune escapes in the Tfh cell epitopes, i.e., μ = 0, and linearly increase it to μ = 0.75 at day 500 (and keeping it at μ = 0.75 for the next 1,500 days) (Fig. 6c and d). This slows down the early evolution of bnAbs but speeds up the late evolution of B cells with broader neutralizing breadth [until the g(i) function starts to saturate again] (Fig. 3a).
FIG 6.

Evolution of bnAbs in the diversity model [i.e., replacing f(i) with equation 12] in (equations 1 to 6). Panels a and b depict examples for μ = 0.5, and panels c and d depict simulations where μ values increase linearly from μ = 0 at day 0 to μ = 0.75 at day 500. Panels b and d show the same simulations in the presence of sweeps occurring, on average, every 50 days. Parameters are as defined in the legend of Fig. 5, with μ = 0.5 (panels a and b) or 0≤ μ ≤0.75 (panels c and d).
(i) Viral sweeps in the mean-field diversity model.
Sweeps work completely differently in the diversity model because the evolving bnAbs in the B cell lineage will only miss the e(1 − μ) functional T cell epitopes (where e represents the number of Tfh cell epitopes, or breadth) expected to be present in all strains of the dominant viral lineage. The specific B0 cells responding to the dominant viral lineage can only present pMHC from the strains in this lineage, which are expected to be functional with probability g(0) = 1 − μ, whereas Bi cells in the lineage evolving bnAbs are expected to present at least the g(i) = (1 − μi) functional epitopes present in the other viral lineages (see equation 12). Thus, the cells of the lineage evolving bnAbs are not expected to be outcompeted by the B0 cells (Fig. 6). Early in infection, when μ is small, the difference in the rescue rates, g(0) and g(i), will be small since all B cell clones should present a similar number of functional epitopes to a particular Tfh cell. Later, when several T cell epitopes have escaped, Bi cells capturing protein from more than just one viral lineage, i > 1, should have a selective advantage over the B0 cells responding to the novel dominant viral lineage (Fig. 6b and d). Note that the B0 clones are rapidly outcompeted in the examples shown in Fig. 6 (i.e., the B1 bands are darker and broader than the B0 bands) but that they would expand perfectly well in germinal centers lacking the lineage evolving bnAbs as the probability of B0 clones becoming rescued is identical to that of the progenitors of the B cell lineage [i.e., g(0) = g(1) in equation 12], which are expanding vigorously initially.
(ii) Number of Tfh cells.
Searching for interventions that would speed up the evolution of bnAbs, some theoretical studies have argued that it would be beneficial to use broad vaccines composed of numerous lineages of HIV-1 (68), whereas others have argued that it would be more beneficial to provide sequential immunization with single strains (69, 97). In our current models broad reactivity evolves by slow and sequential mutations, despite the fact that we start during the chronic phase of the infection with a diverse viral population (Fig. 5a and 6a). Since the selection for broad reactivity in our models is brought about by competition for cognate Tfh cells, we studied the effect increasing the number of Tfh cells, T. Increasing the number of Tfh cells is known to increase the size of germinal centers (40), and we find that doubling T doubles the steady-state number of B cells in our models (data not shown). Importantly, this markedly speeds up the evolution of bnAbs because when B cell clone sizes become larger, it takes less time for mutants to appear (Fig. 7a and b), which is in good agreement with recent observations finding higher frequencies of Tfh cells in patients or monkeys with bnAbs (28–32). These findings suggest that the evolution of bnAbs can be improved by vaccination strategies increasing the frequencies of CD4+ Tfh cells (33).
FIG 7.

Evolution of bnAbs in the density and diversity models for different numbers of cognate Tfh cells in the germinal center (in the absence of sweeps). Parameters are as defined in the legend of Fig. 5 with U = 25 (panel a) and as defined in the legend of Fig. 6 with 0 ≤ μ ≤ 0.75 (panel b).
Full diversity model: breadth of the cognate Tfh repertoire.
The breadth of the CD4 T cell response to HIV-1 and the rate at which the breadth declines due to the evolution of immune escapes will depend on the individual's HLA (90, 98) as some HLA molecules can present more peptides than average from conserved HIV-1 proteins. Additionally, due to differences in HLAs some patients will have mounted larger Tfh cell responses than others. Interestingly, this may also help to explain the variation in the evolution of bnAbs in the population of patients (1, 99) because HLA polymorphisms accounting for different Tfh cell responses in different individuals are expected to lead to variations in the evolution of bnAbs.
Increasing the breadth of the Tfh cell repertoire, e, in the full pMHC diversity model could have two different effects. First, greater breadth makes it more likely that there is at least one Tfh cell epitope l that has mutated but has not approached fixation, i.e., at least one epitope l that is polymorphic with 0 < μl < 1, such that B cells encountering a Tfh cell specific for this epitope are selected on the basis of the diversity of the cognate pMHC they are presenting to this Tfh cell. Second, if B cells were to typically encounter several Tfh cells in the light zone (42), great breadth could mean that B0 cells that fail to become rescued by the first Tfh cell they encounter can still become rescued by other Tfh cell clonotypes, e.g., those specific for wild-type epitopes. Although the latter suggests that increasing Tfh cell breadth, e, decreases the differences among the summed probabilities
determining the rate at which Bi cells are rescued (see equation 8), the data in Fig. 3c show that this is not the case. To study the effect of Tfh cell breadth on rescue rates, we draw e random 0 < μl < 1 values from a uniform distribution, and compute the normalized sum
for i = 0,1,…,15 [where g(i, l) is defined by equation 7]. Repeating this 1,000 times for each value of e and depicting the average in Fig. 3c, we see that increasing breadth is not expected to change the relative rescue probabilities of the Bi lineages. Thus, the main effect of the breadth of the Tfh cell repertoire, e, is to increase the likelihood that at least one of the Tfh cell epitopes is polymorphic and makes a difference.
Tfh cell epitopes that evolve complete immune escape, μl = 1, can be removed from the sum in equation 8, which corresponds to a decrease in the number of epitopes e, and we just showed that this fails to affect the rescue probabilities (Fig. 3c). However, the rate of selection for broad B cell reactivity does decrease when some of the Tfh cell epitopes are not mutated. For instance, assuming that one of the epitopes remains functional, e.g., μe = 0, the normalized sum term becomes
and plotting the average of this for e = 1,2,…,5 and i = 0,1,…,15 shows that this decreases the differences between the rescue rates of the Bi lineages (Fig. 3d), especially at low breadth, e.
Finally, we simulated the full diversity model, as defined by equations 8 to 11, to study the effect of Tfh cell breadth on the evolution of bnAbs. This model is identical to the mean-field model when we consider a single Tfh cell clonotype (by setting e = 1; where a clonotype is the set of cells recognizing the same epitope), but for e > 1, the full model depends on Tfh cell breadth, e, whenever we use different densities, Tl, and/or different frequencies of the immune escapes, μl, for the various Tfh cell clonotypes. In the legend of Fig. 8 we describe that both are drawn randomly for each simulation of the full diversity model. To study breadth independently of the Tfh cell density, we normalize the e random T cell densities, Tl, such that the total remains a 100 Tfh cells. As above, we consider two scenarios, one in which the frequencies of the immune escapes, μl, do not change over time (t) and another where the μl(t) increases logistically over time (similar to the dashed lines in Fig. 3b), each with a uniformly drawn rate of increase. Because the various transiently dominant viral lineages should represent different samples from the viral quasispecies, we randomly draw the presence of the immune escapes in the sweeping viral lineage,
from the expected frequencies, μl or μl(t), in the quasispecies. The behavior of the full diversity model is very similar to that of the mean-field diversity model shown in Fig. 6 and is therefore not shown.
FIG 8.

Evolution of bnAbs in the full diversity model for different numbers of cognate Tfh cell clonotypes in the germinal center, in the absence (a and c) and presence (b and d) of sweeps. Each curve is the frequency distribution of a 100 simulations over 2,000 days of chronic HIV infection (as shown in the data of Fig. 5c). For each simulation we draw random densities for the number of cells per Tfh cell clonotype, Tl, from a uniform distribution and normalize these such that the total number of Tfh cells is ∑leTl = 100 cells. In panels a and b, the fraction of immune escapes per Tfh cell epitope, μl, is fixed over time, and for each simulation these values are drawn from a uniform distribution, 0 < μl < 1. In panels c and d, the μl values increase logistically over time, i.e., μl(t) = 1/(1 + e−rl(max[0,t−Δl])[1/μl(0)−1]), with uniformly drawn rates at which they increase, 0 < rl <0.05, uniformly drawn starting times, 0 < Δl < 2,000 days, and fixed starting values, μl(0) = 0.01. Whenever a new transiently dominant viral lineage appears, we randomly draw the presence of the e epitopes in the sweeping viral lineage, μ0l∈{0,1}, from the expected frequencies, μl or μl(t), in the quasispecies. Finally, because g(i, l) = 1 when all μl = 1 (equation 7), which would imply that all B cell lineages are rescued at the same rate, we introduce the first immune escape at day zero (i.e., Δ0 = 0) in panels c and d where the immune escapes are evolving. All other parameters are as defined in the legend of Fig. 5.
These simulations confirm that the B cell lineages evolve broader neutralizing capacity when all Tfh cell epitopes remain polymorphic [compare Fig. 8a, where the μl is fixed at a random value during each simulation, with c, where μl(t) is approximately 0 or 1 most of the time]. They also confirm that increasing the breadth of the Tfh cell clonotypes improves the neutralizing capacity of the B cell lineage (Fig. 8, all panels). The presence or absence of viral sweeps hardly affects the evolution of bnAbs (compare Fig. 8a with b and c with d), implying that the evolution of bnAbs is hardly affected by transiently dominant viral lineages sweeping through the quasispecies. Summarizing, increasing the Tfh cell breadth should increase the rate at which bnAbs are selected because there will be more polymorphic Tfh cell epitopes in the viral quasispecies that can select for bnAbs.
DISCUSSION
We have identified two mechanisms by which cognate Tfh cells in germinal centers can select for a larger breadth of B cell receptors. Broadly reactive B cells are expected to present a larger diversity of Tfh cell epitopes when they capture a larger variety of HIV variants from the FDC network, and they may present these epitopes at a higher density. Although the density and diversity mechanisms are probably occurring simultaneously in germinal centers, we have modeled them separately to identify each of their properties. We find that conventional selection on the basis of the density of pMHC only is very sensitive to transient sweeps through the viral population, whereas evolution on the basis of the diversity of pMHC presented to a repertoire of Tfh cells is hardly sensitive to sweeps. Since HIV-infected patients harbor diverse repertoires of HIV-specific Tfh cells (31, 72), the selection of B cells presenting a high diversity of Tfh cell epitopes could play a major role in the evolution of bnAbs. We have seen that this depends on the diversity and the polymorphism of Tfh cell epitopes in the viral quasispecies, which calls for novel studies delineating the relative roles of these two selection mechanisms during affinity maturation. As long as we have not ruled out the markedly different selection based upon the diversity of the pMHC presented to Tfh cells, we have to remain careful in interpreting the results of models implementing selection on the basis of density only.
bnAbs tend to evolve after years of infection, and there is a large variation among patients in their ability to evolve bnAbs (1). Our modeling has identified a number of mechanisms contributing to this. First, if the selection mechanism for B cell survival in a germinal center is largely based on the density of pMHC, patients with frequent sweeps through the viral population would be expected to evolve antibodies with a limited breadth, as shown in Fig. 5c (which is similar to the results of Luo and Perelson [68] obtained with a very different model allowing for viral evolution). Similarly, the evolution of bnAbs is expected to depend on the frequency distribution of the major haplotypes in the viral quasispecies within a host (17) and would be expected to slowly speed up when this distribution becomes more even over the years. Second, according to both selection mechanisms, patients with a poor HIV-specific CD4 T cell response to HIV-1 should evolve bnAbs more slowly than patients with a large and/or broad HIV-specific CD4 T cell response. These two processes may reinforce each other because the number of T cell immune escapes observed in a patient, which are responsible for a subset of the sweeps, depends on the breadth of the T cell response (87, 100, 101). Deep sequencing of the T and B cell repertoires in HIV-1-infected patients (102) will in the future provide additional insights into the frequency distribution of B and T cell clones.
Interestingly, there may be positive feedback in the evolution of bnAbs as neutralizing antibodies select for novel escape variants and thereby could be diversifying the viral population (103). This positive feedback between bnAbs could also be responsible for the cooperation that was observed between different lineages of bnAbs (104) and could also explain why the evolution of bnAbs tends to be late and variable (1). However, purifying selection can also decrease the diversity of the population, and the virus can escape from a bnAb not only by mutating the conserved B cell epitope but also by mutating the Tfh cell epitopes required for rescuing the B cells producing the bnAb. Studying this would require more elaborate models describing both the evolution of the viral quasispecies and the repertoire of cognate Tfh and B cells. Additionally, it would be interesting to develop models combining the density- and diversity-based selection mechanisms. One could extend the existing spatially explicit models of germinal centers that track cognate T and B cells (26, 43, 105) in this direction, but to complement this with the evolution of a diverse viral quasispecies is challenging and not within the scope of this study.
Affinity mutation in patients chronically infected with HIV-1 is expected to be distributed over many germinal centers. In humans having about 550 (106) or more (76) lymph nodes, each with several germinal centers, 100 to 200 Peyer's patches (107), and many germinal centers in the spleen, one readily expects thousands of active germinal centers in an HIV-1-infected patient. Although little is known about the formation and persistence of germinal centers during chronic infection, the mere fact that B cells in HIV-1-infected patients perform somatic hypermutation and affinity maturation over a time period of several years suggests that their memory B cells either populate existing germinal centers or form new ones to undergo additional rounds of expansion, mutation, and selection. Since our simple model considers only one germinal center, we need to discuss whether our results depend on this simplifying assumption. First, the large pools of virus captured on the FDC networks within the thousands of germinal centers are most likely to be fairly similar as the FDC network appears to bind virus reversibly, allowing exchange of virus particles with the circulation (108–113). Second, mature GCs are open to invasion by higher-affinity B cells that can take over an existing GC reaction (62–64). Although the first germinal centers are probably started by several clones of naive B cells (44), the B cell clones they generate expand and form large clones of circulating memory cells, and the reinitiation of germinal centers by memory B cells becomes less stochastic because the largest B cell clones would contribute the most seeders. Thus, during the chronic stage of the infection, germinal centers are expected to have a similar composition of B cells and viruses, and modeling just one germinal center seems a reasonable assumption.
We have argued that the repertoire of cognate Tfh cells capable of helping and rescuing centrocytes in germinal centers is expected to be narrow because there are a restricted number of CD4 T cell epitopes in a small virus like HIV-1. Because HIV preferentially infects cognate CD4+ T cells and because Tfh cells serve as a major component for HIV-1 infection (51–53), Tfh cells are expected to become a limiting resource during HIV-1 infection (29). We have shown that this should slow down the evolution of bnAbs (Fig. 7). Given the paucity of T cell epitopes in the whole virus, it would be very important to characterize the actual antigens that high-affinity HIV-specific B cells pull off the FDC network and to know whether they present epitopes from genes other than Env to the Tfh cells. For large viruses, Sette et al. (77) provide evidence for a tight linkage between the protein specificities of CD4+ T cells and their cognate B cells. Yamamoto et al. (30) quantified the frequencies of Gag- and Env-specific Tfh cells in SHIV-infected macaques and found that the frequency of Env-specific IgG+ GC B cells correlates positively with the frequency of Env-specific Tfh cells but not with the frequency of Gag-specific Tfh cells. On the other hand, Ranasinghe et al. (31) found that the frequency of Gag-specific Tfh cells better correlates with the presence of bnAbs than with the frequency of Env-specific Tfh cells, which suggests that GC B cells are presenting Gag epitopes to Tfh cells. Finally, note that high-affinity B cells would be able to acquire whole virions from the FDC network only if the binding energy holding their BCR in the cell membrane as well as to the cognate Env epitope is higher than the sum of the energies of the antibodies (or complement molecules) that are binding the virion to the FDC network (114, 115). Hence, what B cells are capturing and subsequently presenting to the Tfh cells may change dynamically as B cell affinity increases by somatic mutation and selection.
The first vaccination experiments testing Tfh cell immunity used a vaccine encoding SIV Env, Rev, Gag, and Nef, which was followed by two boosts with Env protein (33). This vaccination procedure elicited Env-specific Tfh cells (33); other Tfh cell specificities were not tested. Following several low-dose rectal challenges with SIVmac251, all vaccinated macaques ultimately were infected and followed for approximately 40 weeks. Vaccinated monkeys approached an almost 10-fold-lower set point viral load than unvaccinated controls (116). Unfortunately, whether the improved control was due to (broadly neutralizing) antibodies was not tested. Moreover, although our results predict that an initially broad Tfh cell response should speed up the evolution of bnAbs after infection, this need not be beneficial clinically as HIV-infected patients are typically not protected by the bnAbs they evolve naturally. A major final question therefore is whether our modeling results suggest that bnAbs can evolve by vaccination only.
Since the B cells have to accumulate many somatic mutations, evolution of bnAbs will require repeated vaccinations with a vaccine containing many viral variants (68) or with different vaccines, each containing a limited number of strains (69, 97). We have learned from our modeling that the various antigens in a vaccine should be presented on the FDC network at similar densities; otherwise, B cells specific for the dominant antigen in the vaccine(s) tend to be selected. As there should be no transiently dominant antigens due to sweeping viruses during vaccinations aimed at generating sterilizing immunity, a selection mechanism that is based solely upon the density of protein that is internalized and presented to cognate Tfh cells should now be sufficient (although presenting the highest diversity of Tfh cell epitopes remains advantageous). For Tfh cell epitopes located in Env, one could develop different variants of a mosaic vaccine containing many different T cell epitopes in Env (34–36) as our models predict that vaccinations with successively different variants of Env should select for broadly reactive B cells since they will present the highest diversity of the Tfh cell epitopes in Env at the highest density. To include Tfh cell epitopes not contained in Env, one should work with a viral vector that is pulled off from the FDC network in its entirety, such that B cells binding many variants of Env also internalize many copies and/or variants of the other HIV-1 proteins in the vaccine. To select for broad reactivity, different T and B cell epitopes should be inserted in these vaccines. Whether such repeated vaccinations with various vaccines are going to elicit bnAbs will depend on the timing, the diversity, and the variation of the vaccinations and is unfortunately difficult to predict.
ACKNOWLEDGMENTS
Portions of this work were performed under the auspices of the U.S. Department of Energy under contract DE-AC52-06NA25396 and were supported by the Center of Nonlinear Studies at Los Alamos National Laboratory, NIH grants R01-AI028433 and R01-OD011095. This paper was partly written at the Santa Fe Institute and during the Quantitative Immunology workshop at the Kavli Institute for Theoretical Physics. The latter was supported in part by the National Science Foundation under grant number NSF PHY11-25915.
We thank Will Fisher for very helpful comments on an early version of the manuscript.
REFERENCES
- 1.Hraber P, Seaman MS, Bailer RT, Mascola JR, Montefiori DC, Korber BT. 2014. Prevalence of broadly neutralizing antibody responses during chronic HIV-1 infection. AIDS 28:163–169. doi: 10.1097/QAD.0000000000000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gautam R, Nishimura Y, Pegu A, Nason MC, Klein F, Gazumyan A, Golijanin J, Buckler-White A, Sadjadpour R, Wang K, Mankoff Z, Schmidt SD, Lifson JD, Mascola JR, Nussenzweig MC, Martin MA. 2016. A single injection of anti-HIV-1 antibodies protects against repeated SHIV challenges. Nature 533:105–109. doi: 10.1038/nature17677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hessell AJ, Jaworski JP, Epson E, Matsuda K, Pandey S, Kahl C, Reed J, Sutton WF, Hammond KB, Cheever TA, Barnette PT, Legasse AW, Planer S, Stanton JJ, Pegu A, Chen X, Wang K, Siess D, Burke D, Park BS, Axthelm MK, Lewis A, Hirsch VM, Graham BS, Mascola JR, Sacha JB, Haigwood NL. 2016. Early short-term treatment with neutralizing human monoclonal antibodies halts SHIV infection in infant macaques. Nat Med 22:362–368. doi: 10.1038/nm.4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu J, Ghneim K, Sok D, Bosche WJ, Li Y, Chipriano E, Berkemeier B, Oswald K, Borducchi E, Cabral C, Peter L, Brinkman A, Shetty M, Jimenez J, Mondesir J, Lee B, Giglio P, Chandrashekar A, Abbink P, Colantonio A, Gittens C, Baker C, Wagner W, Lewis MG, Li W, Sekaly RP, Lifson JD, Burton DR, Barouch DH. 2016. Antibody-mediated protection against SHIV challenge includes systemic clearance of distal virus. Science 353:1045–1049. doi: 10.1126/science.aag0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynch RM, Boritz E, Coates EE, DeZure A, Madden P, Costner P, Enama ME, Plummer S, Holman L, Hendel CS, Gordon I, Casazza J, Conan-Cibotti M, Migueles SA, Tressler R, Bailer RT, McDermott A, Narpala S, O'Dell S, Wolf G, Lifson JD, Freemire BA, Gorelick RJ, Pandey JP, Mohan S, Chomont N, Fromentin R, Chun TW, Fauci AS, Schwartz RM, Koup RA, Douek DC, Hu Z, Capparelli E, Graham BS, Mascola JR, Ledgerwood JE. 2015. Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV-1 infection. Sci Transl Med 7:319ra206. doi: 10.1126/scitranslmed.aad5752. [DOI] [PubMed] [Google Scholar]
- 6.Caskey M, Klein F, Lorenzi JC, Seaman MS, West AP Jr, Buckley N, Kremer G, Nogueira L, Braunschweig M, Scheid JF, Horwitz JA, Shimeliovich I, Ben-Avraham S, Witmer-Pack M, Platten M, Lehmann C, Burke LA, Hawthorne T, Gorelick RJ, Walker BD, Keler T, Gulick RM, Fatkenheuer G, Schlesinger SJ, Nussenzweig MC. 2015. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 522:487–491. doi: 10.1038/nature14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu CL, Murakowski DK, Bournazos S, Schoofs T, Sarkar D, Halper-Stromberg A, Horwitz JA, Nogueira L, Golijanin J, Gazumyan A, Ravetch JV, Caskey M, Chakraborty AK, Nussenzweig MC. 2016. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science 352:1001–1004. doi: 10.1126/science.aaf1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schoofs T, Klein F, Braunschweig M, Kreider EF, Feldmann A, Nogueira L, Oliveira T, Lorenzi JC, Parrish EH, Learn GH, West AP Jr, Bjorkman PJ, Schlesinger SJ, Seaman MS, Czartoski J, McElrath MJ, Pfeifer N, Hahn BH, Caskey M, Nussenzweig MC. 2016. HIV-1 therapy with monoclonal antibody 3BNC117 elicits host immune responses against HIV-1. Science 352:997–1001. doi: 10.1126/science.aaf0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwong PD, Mascola JR. 2012. Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies. Immunity 37:412–425. doi: 10.1016/j.immuni.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoot S, McGuire AT, Cohen KW, Strong RK, Hangartner L, Klein F, Diskin R, Scheid JF, Sather DN, Burton DR, Stamatatos L. 2013. Recombinant HIV envelope proteins fail to engage germline versions of anti-CD4bs bNAbs. PLoS Pathog 9:e1003106. doi: 10.1371/journal.ppat.1003106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein F, Diskin R, Scheid JF, Gaebler C, Mouquet H, Georgiev IS, Pancera M, Zhou T, Incesu RB, Fu BZ, Gnanapragasam PN, Oliveira TY, Seaman MS, Kwong PD, Bjorkman PJ, Nussenzweig MC. 2013. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 153:126–138. doi: 10.1016/j.cell.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwong PD, Doyle ML, Casper DJ, Cicala C, Leavitt SA, Majeed S, Steenbeke TD, Venturi M, Chaiken I, Fung M, Katinger H, Parren PW, Robinson J, Van Ryk D, Wang L, Burton DR, Freire E, Wyatt R, Sodroski J, Hendrickson WA, Arthos J. 2002. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420:678–682. doi: 10.1038/nature01188. [DOI] [PubMed] [Google Scholar]
- 13.Briney BS, Willis JR, Crowe JE Jr. 2012. Human peripheral blood antibodies with long HCDR3s are established primarily at original recombination using a limited subset of germline genes. PLoS One 7:e36750. doi: 10.1371/journal.pone.0036750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein F, Mouquet H, Dosenovic P, Scheid JF, Scharf L, Nussenzweig MC. 2013. Antibodies in HIV-1 vaccine development and therapy. Science 341:1199–1204. doi: 10.1126/science.1241144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson PC, De Bouteiller O, Liu YJ, Potter K, Banchereau J, Capra JD, Pascual V. 1998. Somatic hypermutation introduces insertions and deletions into immunoglobulin V genes. J Exp Med 187:59–70. doi: 10.1084/jem.187.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willis JR, Finn JA, Briney B, Sapparapu G, Singh V, King H, LaBranche CC, Montefiori DC, Meiler J, Crowe JE Jr. 2016. Long antibody HCDR3s from HIV-naive donors presented on a PG9 neutralizing antibody background mediate HIV neutralization. Proc Natl Acad Sci U S A 113:4446–4451. doi: 10.1073/pnas.1518405113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pandit A, De Boer RJ. 2014. Reliable reconstruction of HIV-1 whole genome haplotypes reveals clonal interference and genetic hitchhiking among immune escape variants. Retrovirology 11:56. doi: 10.1186/1742-4690-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bar KJ, Tsao CY, Iyer SS, Decker JM, Yang Y, Bonsignori M, Chen X, Hwang KK, Montefiori DC, Liao HX, Hraber P, Fischer W, Li H, Wang S, Sterrett S, Keele BF, Ganusov VV, Perelson AS, Korber BT, Georgiev I, McLellan JS, Pavlicek JW, Gao F, Haynes BF, Hahn BH, Kwong PD, Shaw GM. 2012. Early low-titer neutralizing antibodies impede HIV-1 replication and select for virus escape. PLoS Pathog 8:e1002721. doi: 10.1371/journal.ppat.1002721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao HX, Lynch R, Zhou T, Gao F, Alam SM, Boyd SD, Fire AZ, Roskin KM, Schramm CA, Zhang Z, Zhu J, Shapiro L, Mullikin JC, Gnanakaran S, Hraber P, Wiehe K, Kelsoe G, Yang G, Xia SM, Montefiori DC, Parks R, Lloyd KE, Scearce RM, Soderberg KA, Cohen M, Kamanga G, Louder MK, Tran LM, Chen Y, Cai F, Chen S, Moquin S, Du X, Joyce MG, Srivatsan S, Zhang B, Zheng A, Shaw GM, Hahn BH, Kepler TB, Korber BT, Kwong PD, Mascola JR, Haynes BF. 2013. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 496:469–476. doi: 10.1038/nature12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Victora GD, Nussenzweig MC. 2012. Germinal centers. Annu Rev Immunol 30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 21.Batista FD, Iber D, Neuberger MS. 2001. B cells acquire antigen from target cells after synapse formation. Nature 411:489–494. doi: 10.1038/35078099. [DOI] [PubMed] [Google Scholar]
- 22.Natkanski E, Lee WY, Mistry B, Casal A, Molloy JE, Tolar P. 2013. B cells use mechanical energy to discriminate antigen affinities. Science 340:1587–1590. doi: 10.1126/science.1237572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nowosad CR, Spillane KM, Tolar P. 2016. Germinal center B cells recognize antigen through a specialized immune synapse architecture. Nat Immunol 17:870–877. doi: 10.1038/ni.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, Nussenzweig MC. 2010. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 143:592–605. doi: 10.1016/j.cell.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwickert TA, Victora GD, Fooksman DR, Kamphorst AO, Mugnier MR, Gitlin AD, Dustin ML, Nussenzweig MC. 2011. A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J Exp Med 208:1243–1252. doi: 10.1084/jem.20102477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer-Hermann M, Mohr E, Pelletier N, Zhang Y, Victora GD, Toellner KM. 2012. A theory of germinal center B cell selection, division, and exit. Cell Rep 2:162–174. doi: 10.1016/j.celrep.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 27.Gitlin AD, Mayer CT, Oliveira TY, Shulman Z, Jones MJ, Koren A, Nussenzweig MC. 2015. T cell help controls the speed of the cell cycle in germinal center B cells. Science 349:643–646. doi: 10.1126/science.aac4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, Su LF, Cubas R, Davis MM, Sette A, Haddad EK, Poignard P, Crotty S. 2013. Human circulating PD-1+ CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity 39:758–769. doi: 10.1016/j.immuni.2013.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boswell KL, Paris R, Boritz E, Ambrozak D, Yamamoto T, Darko S, Wloka K, Wheatley A, Narpala S, McDermott A, Roederer M, Haubrich R, Connors M, Ake J, Douek DC, Kim J, Petrovas C, Koup RA. 2014. Loss of circulating CD4 T cells with B cell helper function during chronic HIV infection. PLoS Pathog 10:e1003853. doi: 10.1371/journal.ppat.1003853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamamoto T, Lynch RM, Gautam R, Matus-Nicodemos R, Schmidt SD, Boswell KL, Darko S, Wong P, Sheng Z, Petrovas C, McDermott AB, Seder RA, Keele BF, Shapiro L, Douek DC, Nishimura Y, Mascola JR, Martin MA, Koup RA. 2015. Quality and quantity of TFH cells are critical for broad antibody development in SHIVAD8 infection. Sci Transl Med 7:298ra120. doi: 10.1126/scitranslmed.aab3964. [DOI] [PubMed] [Google Scholar]
- 31.Ranasinghe S, Soghoian DZ, Lindqvist M, Ghebremichael M, Donaghey F, Carrington M, Seaman MS, Kaufmann DE, Walker BD, Porichis F. 2015. HIV-1 antibody neutralization breadth is associated with enhanced HIV-specific CD4+ T cell responses. J Virol 90:2208–2220. doi: 10.1128/JVI.02278-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moody MA, Pedroza-Pacheco I, Vandergrift NA, Chui C, Lloyd KE, Parks R, Soderberg KA, Ogbe AT, Cohen MS, Liao HX, Gao F, McMichael AJ, Montefiori DC, Verkoczy L, Kelsoe G, Huang J, Shea PR, Connors M, Borrow P, Haynes BF. 2016. Immune perturbations in HIV-1—infected individuals who make broadly neutralizing antibodies. Sci Immunol 1:aag0851. doi: 10.1126/sciimmunol.aag0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vargas-Inchaustegui DA, Demers A, Shaw JM, Kang G, Ball D, Tuero I, Musich T, Mohanram V, Demberg T, Karpova TS, Li Q, Robert-Guroff M. 2016. Vaccine induction of lymph node-resident simian immunodeficiency virus Env-specific T follicular helper cells in rhesus macaques. J Immunol 196:1700–1710. doi: 10.4049/jimmunol.1502137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barouch DH, O'Brien KL, Simmons NL, King SL, Abbink P, Maxfield LF, Sun YH, La Porte A, Riggs AM, Lynch DM, Clark SL, Backus K, Perry JR, Seaman MS, Carville A, Mansfield KG, Szinger JJ, Fischer W, Muldoon M, Korber B. 2010. Mosaic HIV-1 vaccines expand the breadth and depth of cellular immune responses in rhesus monkeys. Nat Med 16:319–323. doi: 10.1038/nm.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santra S, Liao HX, Zhang R, Muldoon M, Watson S, Fischer W, Theiler J, Szinger J, Balachandran H, Buzby A, Quinn D, Parks RJ, Tsao CY, Carville A, Mansfield KG, Pavlakis GN, Felber BK, Haynes BF, Korber BT, Letvin NL. 2010. Mosaic vaccines elicit CD8+ T lymphocyte responses that confer enhanced immune coverage of diverse HIV strains in monkeys. Nat Med 16:324–328. doi: 10.1038/nm.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barouch DH, Stephenson KE, Borducchi EN, Smith K, Stanley K, McNally AG, Liu J, Abbink P, Maxfield LF, Seaman MS, Dugast AS, Alter G, Ferguson M, Li W, Earl PL, Moss B, Giorgi EE, Szinger JJ, Eller LA, Billings EA, Rao M, Tovanabutra S, Sanders-Buell E, Weijtens M, Pau MG, Schuitemaker H, Robb ML, Kim JH, Korber BT, Michael NL. 2013. Protective efficacy of a global HIV-1 mosaic vaccine against heterologous SHIV challenges in rhesus monkeys. Cell 155:531–539. doi: 10.1016/j.cell.2013.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mesin L, Ersching J, Victora GD. 2016. Germinal center B cell dynamics. Immunity 45:471–482. doi: 10.1016/j.immuni.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dal Porto JM, Haberman AM, Kelsoe G, Shlomchik MJ. 2002. Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J Exp Med 195:1215–1221. doi: 10.1084/jem.20011550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shih TA, Meffre E, Roederer M, Nussenzweig MC. 2002. Role of BCR affinity in T cell dependent antibody responses in vivo. Nat Immunol 3:570–575. doi: 10.1038/ni803. [DOI] [PubMed] [Google Scholar]
- 40.Rolf J, Bell SE, Kovesdi D, Janas ML, Soond DR, Webb LM, Santinelli S, Saunders T, Hebeis B, Killeen N, Okkenhaug K, Turner M. 2010. Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J Immunol 185:4042–4052. doi: 10.4049/jimmunol.1001730. [DOI] [PubMed] [Google Scholar]
- 41.Depoil D, Zaru R, Guiraud M, Chauveau A, Harriague J, Bismuth G, Utzny C, Muller S, Valitutti S. 2005. Immunological synapses are versatile structures enabling selective T cell polarization. Immunity 22:185–194. doi: 10.1016/j.immuni.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 42.Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, Craft JE, Nussenzweig MC. 2014. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science 345:1058–1062. doi: 10.1126/science.1257861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keşmir C, De Boer RJ. 2003. Spatial models of affinity maturation in germinal centers. J Theor Biol 222:9–22. doi: 10.1016/S0022-5193(03)00010-9. [DOI] [PubMed] [Google Scholar]
- 44.Tas JM, Mesin L, Pasqual G, Targ S, Jacobsen JT, Mano YM, Chen CS, Weill JC, Reynaud CA, Browne EP, Meyer-Hermann M, Victora GD. 2016. Visualizing antibody affinity maturation in germinal centers. Science 351:1048–1054. doi: 10.1126/science.aad3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuraoka M, Schmidt AG, Nojima T, Feng F, Watanabe A, Kitamura D, Harrison SC, Kepler TB, Kelsoe G. 2016. Complex antigens drive permissive clonal selection in germinal centers. Immunity 44:542–552. doi: 10.1016/j.immuni.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Batista FD, Arana E, Barral P, Carrasco YR, Depoil D, Eckl-Dorna J, Fleire S, Howe K, Vehlow A, Weber M, Treanor B. 2007. The role of integrins and coreceptors in refining thresholds for B-cell responses. Immunol Rev 218:197–213. doi: 10.1111/j.1600-065X.2007.00540.x. [DOI] [PubMed] [Google Scholar]
- 47.Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, Victora GD. 2013. T follicular helper cell dynamics in germinal centers. Science 341:673–677. doi: 10.1126/science.1241680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leddon SA, Sant AJ. 2012. The peptide specificity of the endogenous T follicular helper cell repertoire generated after protein immunization. PLoS One 7:e46952. doi: 10.1371/journal.pone.0046952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hey-Nguyen WJ, Xu Y, Pearson CF, Bailey M, Suzuki K, Tantau R, Obeid S, Milner B, Field A, Carr A, Bloch M, Cooper DA, Kelleher AD, Zaunders JJ, Koelsch KK. 2017. Quantification of residual germinal center activity and HIV-1 DNA and RNA levels using fine needle biopsies of lymph nodes during antiretroviral therapy. AIDS Res Hum Retroviruses 33:648–657. doi: 10.1089/aid.2016.0171. [DOI] [PubMed] [Google Scholar]
- 50.Margolin DH, Saunders EF, Bronfin B, De Rosa N, Axthelm MK, Alvarez X, Letvin NL. 2002. High frequency of virus-specific B lymphocytes in germinal centers of simian-human immunodeficiency virus-infected rhesus monkeys. J Virol 76:3965–3973. doi: 10.1128/JVI.76.8.3965-3973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cubas RA, Mudd JC, Savoye AL, Perreau M, Van Grevenynghe J, Metcalf T, Connick E, Meditz A, Freeman GJ, Abesada-Terk G Jr, Jacobson JM, Brooks AD, Crotty S, Estes JD, Pantaleo G, Lederman MM, Haddad EK. 2013. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med 19:494–499. doi: 10.1038/nm.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perreau M, Savoye AL, De Crignis E, Corpataux JM, Cubas R, Haddad EK, De Leval L, Graziosi C, Pantaleo G. 2013. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med 210:143–156. doi: 10.1084/jem.20121932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kohler SL, Pham MN, Folkvord JM, Arends T, Miller SM, Miles B, Meditz AL, McCarter M, Levy DN, Connick E. 2016. Germinal center T follicular helper cells are highly permissive to HIV-1 and alter their phenotype during virus replication. J Immunol 196:2711–2722. doi: 10.4049/jimmunol.1502174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukazawa Y, Lum R, Okoye AA, Park H, Matsuda K, Bae JY, Hagen SI, Shoemaker R, Deleage C, Lucero C, Morcock D, Swanson T, Legasse AW, Axthelm MK, Hesselgesser J, Geleziunas R, Hirsch VM, Edlefsen PT, Piatak M Jr, Estes JD, Lifson JD, Picker LJ. 2015. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med 21:132–139. doi: 10.1038/nm.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Banga R, Procopio FA, Noto A, Pollakis G, Cavassini M, Ohmiti K, Corpataux JM, De Leval L, Pantaleo G, Perreau M. 2016. PD-1+ and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat Med 22:754–761. doi: 10.1038/nm.4113. [DOI] [PubMed] [Google Scholar]
- 56.Lindqvist M, Van Lunzen J, Soghoian DZ, Kuhl BD, Ranasinghe S, Kranias G, Flanders MD, Cutler S, Yudanin N, Muller MI, Davis I, Farber D, Hartjen P, Haag F, Alter G, Schulze zur Wiesch J, Streeck H. 2012. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J Clin Invest 122:3271–3280. doi: 10.1172/JCI64314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pantaleo G, Graziosi C, Demarest JF, Butini L, Montroni M, Fox CH, Orenstein JM, Kotler DP, Fauci AS. 1993. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature 362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- 58.Zeng M, Haase AT, Schacker TW. 2012. Lymphoid tissue structure and HIV-1 infection: life or death for T cells. Trends Immunol 33:306–314. doi: 10.1016/j.it.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 59.Hong JJ, Chang KT, Villinger F. 2016. The dynamics of T and B cells in lymph node during chronic HIV infection: TFH and HIV, unhappy dance partners? Front Immunol 7:522. doi: 10.3389/fimmu.2016.00522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cubas R, Perreau M. 2014. The dysfunction of T follicular helper cells. Curr Opin HIV AIDS 9:485–491. doi: 10.1097/COH.0000000000000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Petrovas C, Koup RA. 2014. T follicular helper cells and HIV/SIV-specific antibody responses. Curr Opin HIV AIDS 9:235–241. doi: 10.1097/COH.0000000000000053. [DOI] [PubMed] [Google Scholar]
- 62.Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, Dustin ML, Nussenzweig MC. 2007. In vivo imaging of germinal centres reveals a dynamic open structure. Nature 446:83–87. doi: 10.1038/nature05573. [DOI] [PubMed] [Google Scholar]
- 63.Schwickert TA, Alabyev B, Manser T, Nussenzweig MC. 2009. Germinal center reutilization by newly activated B cells. J Exp Med 206:2907–2914. doi: 10.1084/jem.20091225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suzuki K, Grigorova I, Phan TG, Kelly LM, Cyster JG. 2009. Visualizing B cell capture of cognate antigen from follicular dendritic cells. J Exp Med 206:1485–1493. doi: 10.1084/jem.20090209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haase AT, Henry K, Zupancic M, Sedgewick G, Faust RA, Melroe H, Cavert W, Gebhard K, Staskus K, Zhang ZQ, Dailey PJ, Balfour HH Jr, Erice A, Perelson AS. 1996. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 274:985–989. doi: 10.1126/science.274.5289.985. [DOI] [PubMed] [Google Scholar]
- 66.Haase AT. 2005. Perils at mucosal front lines for HIV and SIV and their hosts. Nat Rev Immunol 5:783–792. doi: 10.1038/nri1706. [DOI] [PubMed] [Google Scholar]
- 67.Dimitrov JD, Planchais C, Roumenina LT, Vassilev TL, Kaveri SV, Lacroix-Desmazes S. 2013. Antibody polyreactivity in health and disease: statu variabilis. J Immunol 191:993–999. doi: 10.4049/jimmunol.1300880. [DOI] [PubMed] [Google Scholar]
- 68.Luo S, Perelson AS. 2015. Competitive exclusion by autologous antibodies can prevent broad HIV-1 antibodies from arising. Proc Natl Acad Sci U S A 112:11654–11659. doi: 10.1073/pnas.1505207112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang S, Mata-Fink J, Kriegsman B, Hanson M, Irvine DJ, Eisen HN, Burton DR, Wittrup KD, Kardar M, Chakraborty AK. 2015. Manipulating the selection forces during affinity maturation to generate cross-reactive HIV antibodies. Cell 160:785–797. doi: 10.1016/j.cell.2015.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shaffer JS, Moore PL, Kardar M, Chakraborty AK. 2016. Optimal immunization cocktails can promote induction of broadly neutralizing abs against highly mutable pathogens. Proc Natl Acad Sci U S A 113:E7039–E7048. doi: 10.1073/pnas.1614940113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang S. 2017. Optimal sequential immunization can focus antibody responses against diversity loss and distraction. PLoS Comput Biol 13:e1005336. doi: 10.1371/journal.pcbi.1005336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ranasinghe S, Flanders M, Cutler S, Soghoian DZ, Ghebremichael M, Davis I, Lindqvist M, Pereyra F, Walker BD, Heckerman D, Streeck H. 2012. HIV-specific CD4 T cell responses to different viral proteins have discordant associations with viral load and clinical outcome. J Virol 86:277–283. doi: 10.1128/JVI.05577-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, Casazza JP, Kuruppu J, Kunstman K, Wolinsky S, Grossman Z, Dybul M, Oxenius A, Price DA, Connors M, Koup RA. 2002. HIV preferentially infects HIV-specific CD4+ T cells. Nature 417:95–98. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- 74.Brenchley JM, Ruff LE, Casazza JP, Koup RA, Price DA, Douek DC. 2006. Preferential infection shortens the life span of human immunodeficiency virus-specific CD4+ T cells in vivo. J Virol 80:6801–6809. doi: 10.1128/JVI.00070-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jardine JG, Kulp DW, Havenar-Daughton C, Sarkar A, Briney B, Sok D, Sesterhenn F, Ereno-Orbea J, Kalyuzhniy O, Deresa I, Hu X, Spencer S, Jones M, Georgeson E, Adachi Y, Kubitz M, DeCamp AC, Julien JP, Wilson IA, Burton DR, Crotty S, Schief WR. 2016. HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science 351:1458–1463. doi: 10.1126/science.aad9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qatarneh SM, Kiricuta IC, Brahme A, Tiede U, Lind BK. 2006. Three-dimensional atlas of lymph node topography based on the visible human data set. Anat Rec B New Anat 289:98–111. doi: 10.1002/ar.b.20102. [DOI] [PubMed] [Google Scholar]
- 77.Sette A, Moutaftsi M, Moyron-Quiroz J, McCausland MM, Davies DH, Johnston RJ, Peters B, Rafii-El-Idrissi Benhnia M, Hoffmann J, Su HP, Singh K, Garboczi DN, Head S, Grey H, Felgner PL, Crotty S. 2008. Selective CD4+ T cell help for antibody responses to a large viral pathogen: deterministic linkage of specificities. Immunity 28:847–858. doi: 10.1016/j.immuni.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McMahon HT, Boucrot E. 2011. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- 79.Watts C. 1997. Capture and processing of exogenous antigens for presentation on MHC molecules. Annu Rev Immunol 15:821–850. doi: 10.1146/annurev.immunol.15.1.821. [DOI] [PubMed] [Google Scholar]
- 80.Lake P, Mitchison NA. 1976. Associative control of the immune response to cell surface antigens. Immunol Commun 5:795–805. doi: 10.3109/08820137609047620. [DOI] [PubMed] [Google Scholar]
- 81.Russell SM, Liew FY. 1979. T cells primed by influenza virion internal components can cooperate in the antibody response to haemagglutinin. Nature 280:147–148. doi: 10.1038/280147a0. [DOI] [PubMed] [Google Scholar]
- 82.Scherle PA, Gerhard W. 1986. Functional analysis of influenza-specific helper T cell clones in vivo. T cells specific for internal viral proteins provide cognate help for B cell responses to hemagglutinin. J Exp Med 164:1114–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Milich DR, McLachlan A, Thornton GB, Hughes JL. 1987. Antibody production to the nucleocapsid and envelope of the hepatitis B virus primed by a single synthetic T cell site. Nature 329:547–549. doi: 10.1038/329547a0. [DOI] [PubMed] [Google Scholar]
- 84.Yin L, Calvo-Calle JM, Cruz J, Newman FK, Frey SE, Ennis FA, Stern LJ. 2013. CD4+ T cells provide intermolecular help to generate robust antibody responses in vaccinia virus-vaccinated humans. J Immunol 190:6023–6033. doi: 10.4049/jimmunol.1202523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nabi G, Genannt Bonsmann MS, Tenbusch M, Gardt O, Barouch DH, Temchura V, Uberla K. 2013. GagPol-specific CD4+ T-cells increase the antibody response to Env by intrastructural help. Retrovirology 10:117. doi: 10.1186/1742-4690-10-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Borghans JA, Mølgaard A, De Boer RJ, Kesmir C. 2007. HLA alleles associated with slow progression to AIDS truly prefer to present HIV-1 p24. PLoS One 2:e920. doi: 10.1371/journal.pone.0000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu MK, Hawkins N, Ritchie AJ, Ganusov VV, Whale V, Brackenridge S, Li H, Pavlicek JW, Cai F, Rose-Abrahams M, Treurnicht F, Hraber P, Riou C, Gray C, Ferrari G, Tanner R, Ping LH, Anderson JA, Swanstrom R, Cohen M, Karim SS, Haynes B, Borrow P, Perelson AS, Shaw GM, Hahn BH, Williamson C, Korber BT, Gao F, Self S, McMichael A, Goonetilleke N. 2013. Vertical T cell immunodominance and epitope entropy determine HIV-1 escape. J Clin Invest 123:380–393. doi: 10.1172/JCI65330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Turnbull EL, Wong M, Wang S, Wei X, Jones NA, Conrod KE, Aldam D, Turner J, Pellegrino P, Keele BF, Williams I, Shaw GM, Borrow P. 2009. Kinetics of expansion of epitope-specific T cell responses during primary HIV-1 infection. J Immunol 182:7131–7145. doi: 10.4049/jimmunol.0803658. [DOI] [PubMed] [Google Scholar]
- 89.Assarsson E, Greenbaum JA, Sundstrom M, Schaffer L, Hammond JA, Pasquetto V, Oseroff C, Hendrickson RC, Lefkowitz EJ, Tscharke DC, Sidney J, Grey HM, Head SR, Peters B, Sette A. 2008. Kinetic analysis of a complete poxvirus transcriptome reveals an immediate-early class of genes. Proc Natl Acad Sci U S A 105:2140–2145. doi: 10.1073/pnas.0711573105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Erdmann N, Du VY, Carlson J, Schaefer M, Jureka A, Sterrett S, Yue L, Dilernia D, Lakhi S, Tang J, Sidney J, Gilmour J, Allen S, Hunter E, Heath S, Bansal A, Goepfert PA. 2015. HLA class-II associated HIV polymorphisms predict escape from CD4+ T cell responses. PLoS Pathog 11:e1005111. doi: 10.1371/journal.ppat.1005111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.van Dorp CH, Van Boven M, De Boer RJ. 2014. Immuno-epidemiological modeling of HIV-1 predicts high heritability of the set-point virus load, while selection for CTL escape dominates virulence evolution. PLoS Comput Biol 10:e1003899. doi: 10.1371/journal.pcbi.1003899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fischer W, Ganusov VV, Giorgi EE, Hraber PT, Keele BF, Leitner T, Han CS, Gleasner CD, Green L, Lo CC, Nag A, Wallstrom TC, Wang S, McMichael AJ, Haynes BF, Hahn BH, Perelson AS, Borrow P, Shaw GM, Bhattacharya T, Korber BT. 2010. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One 5:e12303. doi: 10.1371/journal.pone.0012303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ganusov VV, Goonetilleke N, Liu MK, Ferrari G, Shaw GM, McMichael AJ, Borrow P, Korber BT, Perelson AS. 2011. Fitness costs and diversity of the cytotoxic T lymphocyte (CTL) response determine the rate of CTL escape during acute and chronic phases of HIV infection. J Virol 85:10518–10528. doi: 10.1128/JVI.00655-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kepler TB, Perelson AS. 1995. Modeling and optimization of populations subject to time-dependent mutation. Proc Natl Acad Sci U S A 92:8219–8223. doi: 10.1073/pnas.92.18.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kepler TB, Perelson AS. 1993. Somatic hypermutation in B cells: an optimal control treatment. J theor Biol 164:37–64. doi: 10.1006/jtbi.1993.1139. [DOI] [PubMed] [Google Scholar]
- 96.Press WH, Plannery BP, Teukolsky SA, Vetterling WT. 1988. Numerical recipes in C. The art of scientific computing. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 97.Malherbe DC, Doria-Rose NA, Misher L, Beckett T, Puryear WB, Schuman JT, Kraft Z, O'Malley J, Mori M, Srivastava I, Barnett S, Stamatatos L, Haigwood NL. 2011. Sequential immunization with a subtype B HIV-1 envelope quasispecies partially mimics the in vivo development of neutralizing antibodies. J Virol 85:5262–5274. doi: 10.1128/JVI.02419-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Streeck H. 2016. Designing optimal HIV-vaccine T-cell responses. Curr Opin HIV AIDS 11:593–600. doi: 10.1097/COH.0000000000000313. [DOI] [PubMed] [Google Scholar]
- 99.Hraber P, Korber BT, Lapedes AS, Bailer RT, Seaman MS, Gao H, Greene KM, McCutchan F, Williamson C, Kim JH, Tovanabutra S, Hahn BH, Swanstrom R, Thomson MM, Gao F, Harris L, Giorgi E, Hengartner N, Bhattacharya T, Mascola JR, Montefiori DC. 2014. Impact of clade, geography, and age of the epidemic on HIV-1 neutralization by antibodies. J Virol 88:12623–12643. doi: 10.1128/JVI.01705-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.van Deutekom HW, Wijnker G, De Boer RJ. 2013. The rate of immune escape vanishes when multiple immune responses control an HIV infection. J Immunol 191:3277–3286. doi: 10.4049/jimmunol.1300962. [DOI] [PubMed] [Google Scholar]
- 101.Gadhamsetty S, Coorens T, De Boer RJ. 2016. Notwithstanding circumstantial alibis, cytotoxic T cells can be major killers of HIV-1-infected cells. J Virol 90:7066–7083. doi: 10.1128/JVI.00306-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Heather JM, Best K, Oakes T, Gray ER, Roe JK, Thomas N, Friedman N, Noursadeghi M, Chain B. 2015. Dynamic perturbations of the T-cell receptor repertoire in chronic HIV infection and following antiretroviral therapy. Front Immunol 6:644. doi: 10.3389/fimmu.2015.00644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Frost SD, Wrin T, Smith DM, Kosakovsky Pond SL, Liu Y, Paxinos E, Chappey C, Galovich J, Beauchaine J, Petropoulos CJ, Little SJ, Richman DD. 2005. Neutralizing antibody responses drive the evolution of human immunodeficiency virus type 1 envelope during recent HIV infection. Proc Natl Acad Sci U S A 102:18514–18519. doi: 10.1073/pnas.0504658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gao F, Bonsignori M, Liao HX, Kumar A, Xia SM, Lu X, Cai F, Hwang KK, Song H, Zhou T, Lynch RM, Alam SM, Moody MA, Ferrari G, Berrong M, Kelsoe G, Shaw GM, Hahn BH, Montefiori DC, Kamanga G, Cohen MS, Hraber P, Kwong PD, Korber BT, Mascola JR, Kepler TB, Haynes BF. 2014. Cooperation of B cell lineages in induction of HIV-1-broadly neutralizing antibodies. Cell 158:481–491. doi: 10.1016/j.cell.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Y, Meyer-Hermann M, George LA, Figge MT, Khan M, Goodall M, Young SP, Reynolds A, Falciani F, Waisman A, Notley CA, Ehrenstein MR, Kosco-Vilbois M, Toellner KM. 2013. Germinal center B cells govern their own fate via antibody feedback. J Exp Med 210:457–464. doi: 10.1084/jem.20120150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Textor J, Henrickson SE, Mandl JN, Von Andrian UH, Westermann J, De Boer RJ, Beltman JB. 2014. Random migration and signal integration promote rapid and robust T cell recruitment. PLoS Comput Biol 10:e1003752. doi: 10.1371/journal.pcbi.1003752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cornes JS. 1965. Number, size, and distribution of Peyer's patches in the human small intestine. Part II. The development of Peyer's patches. Gut 6:230–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hlavacek WS, Wofsy C, Perelson AS. 1999. Dissociation of HIV-1 from follicular dendritic cells during HAART: mathematical analysis. Proc Natl Acad Sci U S A 96:14681–14686. doi: 10.1073/pnas.96.26.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hlavacek WS, Stilianakis NI, Notermans DW, Danner SA, Perelson AS. 2000. Influence of follicular dendritic cells on decay of HIV during antiretroviral therapy. Proc Natl Acad Sci U S A 97:10966–10971. doi: 10.1073/pnas.190065897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schacker T, Little S, Connick E, Gebhard-Mitchell K, Zhang ZQ, Krieger J, Pryor J, Havlir D, Wong JK, Richman D, Corey L, Haase AT. 2000. Rapid accumulation of human immunodeficiency virus (HIV) in lymphatic tissue reservoirs during acute and early HIV infection: implications for timing of antiretroviral therapy. J Infect Dis 181:354–357. doi: 10.1086/315178. [DOI] [PubMed] [Google Scholar]
- 111.Müller V, Marée AFM, De Boer RJ. 2001. Release of virus from lymphoid tissue affects human immunodeficiency virus type 1 and hepatitis C virus kinetics in the blood. J Virol 75:2597–2603. doi: 10.1128/JVI.75.6.2597-2603.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Keele BF, Tazi L, Gartner S, Liu Y, Burgon TB, Estes JD, Thacker TC, Crandall KA, McArthur JC, Burton GF. 2008. Characterization of the follicular dendritic cell reservoir of human immunodeficiency virus type 1. J Virol 82:5548–5561. doi: 10.1128/JVI.00124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.De Boer RJ, Ribeiro RM, Perelson AS. 2010. Current estimates for HIV-1 production imply rapid viral clearance in lymphoid tissues. PLoS Comput Biol 6:e1000906. doi: 10.1371/journal.pcbi.1000906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Oprea M, Perelson AS. 1997. Somatic mutation leads to efficient affinity maturation when centrocytes recycle back to centroblasts. J Immunol 158:5155–5162. [PubMed] [Google Scholar]
- 115.Oprea M, Perelson AS. 1996. Exploring the mechanisms of primary antibody responses to T cell-dependent antigens. J Theor Biol 181:215–236. doi: 10.1006/jtbi.1996.0127. [DOI] [PubMed] [Google Scholar]
- 116.Tuero I, Mohanram V, Musich T, Miller L, Vargas-Inchaustegui DA, Demberg T, Venzon D, Kalisz I, Kalyanaraman VS, Pal R, Ferrari MG, LaBranche C, Montefiori DC, Rao M, Vaccari M, Franchini G, Barnett SW, Robert-Guroff M. 2015. Mucosal B cells are associated with delayed SIV acquisition in vaccinated female but not male rhesus macaques following SIVmac251 rectal challenge. PLoS Pathog 11:e1005101. doi: 10.1371/journal.ppat.1005101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Allen CD, Okada T, Tang HL, Cyster JG. 2007. Imaging of germinal center selection events during affinity maturation. Science 315:528–531. doi: 10.1126/science.1136736. [DOI] [PubMed] [Google Scholar]
- 118.Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, Haberman AM. 2011. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity 34:947–960. doi: 10.1016/j.immuni.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]