

ABSTRACT
Oral mucosa is one of the main target tissues of the human pathogen herpes simplex virus 1 (HSV-1). How the virus overcomes the protective epithelial barriers and penetrates the tissue to reach its receptors and initiate infection is still unclear. Here, we established an ex vivo infection assay with human oral mucosa that allows viral entry studies in a natural target tissue. The focus was on the susceptibility of keratinocytes in the epithelium and the characterization of cellular receptors that mediate viral entry. Upon ex vivo infection of gingiva or vestibular mucosa, we observed that intact human mucosa samples were protected from viral invasion. In contrast, the basal layer of the oral epithelium was efficiently invaded once the connective tissue and the basement membrane were removed. Later during infection, HSV-1 spread from basal keratinocytes to upper layers, demonstrating the susceptibility of the stratified squamous epithelium to HSV-1. The analysis of potential receptors revealed nectin-1 on most mucosal keratinocytes, whereas herpesvirus entry mediator (HVEM) was found only on a subpopulation of cells, suggesting that nectin-1 acts as primary receptor for HSV-1 in human oral mucosa. To mimic the supposed entry route of HSV-1 via microlesions in vivo, we mechanically wounded the mucosa prior to infection. While we observed a limited number of infected keratinocytes in some wounded mucosa samples, other samples showed no infected cells. Thus, we conclude that mechanical wounding of mucosa is insufficient for the virus to efficiently overcome epithelial barriers and to make entry-mediating receptors accessible.
IMPORTANCE To invade the target tissue of its human host during primary infection, herpes simplex virus (HSV) must overcome the epithelial barriers of mucosa, skin, or cornea. For most viruses, the mechanisms underlying the invasion into the target tissues of their host organism are still open. Here, we established an ex vivo infection model of human oral mucosa to explore how HSV can enter its target tissue. Our results demonstrate that intact mucosa samples and even compromised tissue allow only very limited access of HSV to keratinocytes. Detailed understanding of barrier functions is an essential precondition to unravel how HSV bypasses the barriers and approaches its receptors in tissue and why it is beneficial for the virus to use a cell-cell adhesion molecule, such as nectin-1, as a receptor.
KEYWORDS: HSV-1, HVEM, epithelial barrier, keratinocytes, Langerhans cells, mechanical wounding, nectin-1, oral human mucosa, viral invasion
INTRODUCTION
The human oral cavity is one of the primary portals of entry for herpes simplex virus 1 (HSV-1). Primary infection usually occurs during childhood and results in viral replication at mucosal surfaces followed by latent infection of the trigeminal ganglion, which represents the largest human reservoir of HSV-1 (1). Reactivation can lead to recurrent herpes labialis, mucosal ulcers, or asymptomatic excretion of HSV-1 into the oral cavity (2). Previous studies showed the presence of specific HSV-1 antigens in human oral mucosa and demonstrated the sensitivity of oral mucosa explants to HSV-1 infection (3–5). In addition, the susceptibility of human respiratory mucosa to HSV-1 was observed upon infection of human nasal explants (6). However, it is still not understood how HSV-1 invades human mucosa during primary infection or penetrates mucosa after reactivation to reach its receptors. The plausible assumption is that mucosal breaches help the virus to bypass epithelial barriers to initiate infection upon primary infection. The oral mucosa shows a number of adaptations to withstand the multiple challenges in the oral cavity such as mechanical forces and the continual exposure to bacterial commensals as well as pathogens. Next to strong physical barriers of the oral epithelia, an immunologic barrier of antimicrobial peptides, chemokines, and cytokines limits infection and inflammation (7, 8). Nonetheless, HSV-1 can penetrate the oral epithelium in vivo. If nectin-1 acts as major receptor, it is an intriguing question how the virus gains access to this cell-cell adhesion molecule, which interacts with the viral envelope glycoprotein gD to mediate fusion with cellular membranes (9).
We established an ex vivo infection model to investigate the cellular components that determine HSV-1 susceptibility in tissue. Using murine epidermal sheets, we characterized this infection assay and demonstrated the involvement of nectin-1 and herpesvirus entry mediator (HVEM) as cellular receptors for successful HSV-1 entry (10–12). From our studies in wounded murine skin and in in vitro keratinocyte differentiation models, we conclude that functional tight junctions (TJs) can act as major physical barriers that prevent the accessibility of the receptors for HSV-1 invasion into tissue (13). Here, we adapted the experimental conditions of ex vivo infection to human oral mucosa, which allows the viral invasion process in the natural target tissue to be studied. Oral mucosa is remarkably different from other mucosae and shows a structure similar to skin. It consists of a stratified multilayer of squamous cells with a basement membrane and underlying connective tissue (termed lamina propria) (14) (Fig. 1). The oral epithelium may be either para/ortho-keratinized or nonkeratinized (Fig. 1) and in both cases exhibits lipid-based permeability barriers in the superficial layer (15). The rather thick epithelial layer of oral mucosa (Fig. 2a and 3a) represents a particularly strong physical barrier that protects the oral cavity against surface abrasions and the invasion of pathogens. The epithelium is characterized by a relatively high rate of cellular turnover, which strongly contributes to an effective barrier function (16).
FIG 1.
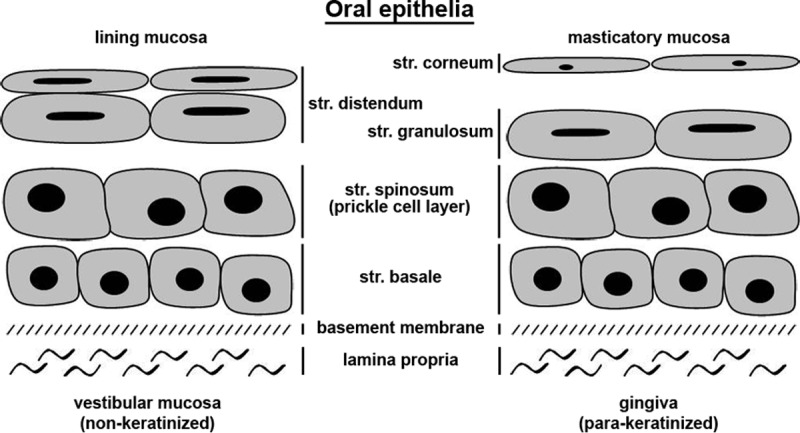
Structure of human oral epithelia. Shown is a scheme illustrating the cell layers of lining and masticatory mucosa. str., stratum.
FIG 2.

HSV-1 enters gingival epithelia. (a) Hematoxylin and eosin (HE)-stained section of gingival mucosa visualizes the stratified epithelium (e) and the underlying lamina propria (lp). (b and c) Epithelial sheets from gingival mucosa were separated from the lamina propria by dispase II and infected with HSV-1 at 100 PFU/cell. (b) Immunostainings of sections show ICP0-expressing cells (green) in the basal layer at 3 h p.i. and viral spreading to the upper layers at 6 h p.i. with DAPI (blue) as nuclear counterstain. (c) Infected whole mounts showing the basal keratinocyte layer were stained for ICP0 (green) and keratin 14 (red). ICP0-expressing cells were mostly found in clusters. A 2-fold magnification of the boxed area is added. Single immunofluorescence is shown. Bars represent 50 μm in panel a and 100 μm in panels b and c.
FIG 3.

HSV-1 enters vestibular epithelia. (a) HE-stained section of epithelium from vestibular mucosa separated from the lamina propria by dispase II. (b) EM analysis of vestibular epithelia shows cell-cell contacts in the basal layer and a magnification of a desmosome. (c) Sections of infected vestibular epithelia (100 PFU/cell) stained for ICP0 (green) visualize viral spreading from basal to upper layers with DAPI (blue) as nuclear counterstain. (d) Sections of infected vestibular epithelia were costained for ICP0 (green) and CD1a (red) to visualize the distribution of Langerhans cells (LCs). No preferred infection of LCs was detected at 16 h p.i. The white line indicates the apical mucosal surface. Single immunofluorescence (c) and overlays (d) are shown. Bars represent 50 μm in panel a, 0.1 μm in panel b, and 50 μm in panels c and d.
We studied HSV-1 entry into human gingiva comprising an epithelium with a cornified layer (para-keratinized) and representing the masticatory mucosa, which is subject to various mechanical forces in the oral cavity (Fig. 1). In addition, we analyzed viral entry into vestibular mucosa, which belongs to the lining mucosa and exhibits a noncornified (nonkeratinized) epithelium representing a more permeable and flexible surface layer than the gingival epithelium (Fig. 1).
Our studies indicate that oral epithelia are very susceptible to HSV-1 once the lamina propria and the basement membrane are removed prior to ex vivo infection. Efficient entry into the basal keratinocyte layer was followed by viral spreading into the prickle cell layers, irrespective whether the epithelia were prepared from gingiva or vestibular mucosa. In contrast, no invasion was observed from the apical surfaces of the epithelia, and only a few infected cells were detected after wounding of mucosa samples by scalpel cuts. Thus, we conclude that mechanical wounding per se allows only minor access to the receptors that mediate cellular entry of HSV-1; this suggests that further disruption of mechanical barriers is needed, which precedes efficient viral invasion.
RESULTS
HSV-1 can enter the oral epithelium via the basal keratinocyte layer.
To address the susceptibility of human oral mucosa to HSV-1 and to characterize the physical barrier function of mucosa, we performed ex vivo infection studies. The samples of oral mucosa were derived from either gingiva or vestibular mucosa, which differ in their structure according to the functional demands in the oral cavity. After submerging complete mucosa samples from at least 7 patients in virus suspension for 3, 6, or 24 h postinfection (p.i.), we observed no infected cells in either gingiva samples with a para-keratinized epithelium (data not shown) or in vestibular mucosa samples with a nonkeratinized epithelium (Fig. 1; see Fig. 7b). Based on our observation in murine skin that infected keratinocytes are only detectable when the dermis is removed from the epidermis prior to ex vivo infection (11, 12, 17), we prepared epithelial sheets from oral mucosa samples to analyze susceptibility to HSV-1 in the explant cultures. After separation from the lamina propria by dispase II treatment, the epithelia were submerged in virus suspension, and infected keratinocytes were determined by visualizing expression of the viral protein ICP0 at various times p.i. As ICP0 is expressed once the viral genome is released into the nucleus, visualization of ICP0 allows the determination of successful entry into individual cells (17, 18). ICP0 staining demonstrated that keratinocytes in the basal layer of the oral epithelia prepared from either gingiva or vestibular mucosa were already infected at 3 h p.i. (Fig. 2b and 3c). Moreover, the number of infected cells increased by this time point when the epithelial sheets were preincubated for 16 h in medium prior to infection, which correlated with flattening of the basal keratinocytes (data not shown). To exclude morphological changes and putative tissue damage during preincubation, further infection studies were performed immediately after preparation of the oral epithelial sheets.
FIG 7.

No efficient HSV-1 entry after wounding of mucosa samples. (a) Gingiva samples completely cut with a scalpel were infected (100 PFU/cell) for 6 h or preincubated in medium for 22 h prior to infection for 6 h. Immunostainings visualize some infected cells at 6 h p.i. and an increased number of ICP0-expressing cells after preincubation at the sample edges of the epithelia. Nuclear counterstaining with DAPI (blue) illustrates elongated nuclei after preincubation. Four-fold magnifications of boxed areas are added. (b) TUNEL-positive cells with DAPI as nuclear counterstain are shown at the wound edges at 22 h + 6 h p.i. and at the apical surfaces in comparison to 6 h p.i. (c) Gingiva samples were slit with a scalpel and infected with HSV-1 at 100 PFU/cell for 24 h. Staining for ICP0 shows infected cells at the wound edges with DAPI as nuclear counterstain. A 2-fold magnification of the boxed area is added. (d) Vestibular mucosa samples slit with a scalpel were infected (100 PFU/cell) for 48 h. Immunostainings visualize no ICP0-expressing cells, and differential interference contrast (DIC) shows tissue morphology. (e) TUNEL-positive cells are shown at the apical surface and the wound edges at 48 h p.i. (f) Intact vestibular mucosa samples were infected (100 PFU/cell) for 24 h. Immunostainings visualize no ICP0-expressing cells. (g) TUNEL-positive cells are visible at the apical surface. (h) The virus titer was determined after infection (50 PFU/cell) of intact or wounded vestibular mucosa at 3 (n = 4), 48 (n = 4), and 72 (n = 2) h p.i. The white line indicates the apical mucosal surface. Confocal projection (c) and overlays of immunofluorescence (a, b, d, e, f, and g) are shown. Bar, 100 μm.
To assess the integrity of the epithelia at the time of infection, we performed electron microscopy (EM) studies. After preparation of epithelia from vestibular mucosa, we found that basal keratinocytes were loosely connected by extensive cellular extensions and that desmosomes were mostly intact (Fig. 3b), whereas the stratum spinosum was characterized by tightly packed cells (data not shown). These results confirmed the loss of the basement membrane and the hemidesmosomes, which attach the epithelium to the basement membrane; the loss, in turn, might induce the plasma membrane extensions observed at the bottom of the basal cells, suggesting highly motile membrane protrusions (Fig. 3b). Thus, we conclude that HSV-1 had direct contact with an intact basal keratinocyte layer when incubated with epithelial sheets immediately after removal of the lamina propria.
Comparison of infected epithelial sheets revealed no obvious difference in the number of infected cells at 3 h p.i., depending on whether the epithelia were prepared from gingiva (n = 8) or vestibular mucosa (n = 8) (Fig. 2b and 3c). The efficiency of infection, however, varied from patient to patient: while samples from 9 patients exhibited a sparsely infected basal layer at 3 h p.i., samples from 7 patients showed a high number of infected basal cells. These variations might be related to the extremely high rate of cellular turnover in the epithelium, which, in principle, supports the barrier function of the epithelium and can vary from one tissue sample to the other. The varying cellular turnover might affect the amount and availability of receptors so that susceptibility to infection could be reduced if fewer receptors are available on dividing and migrating cells.
When the surface of the basal layer was analyzed by preparing whole mounts, ICP0 expression was often detected in clusters of basal keratinocytes at 3 h p.i. (Fig. 2c). Nearly all basal cells expressed ICP0 at 6 h p.i., demonstrating that all basal keratinocytes are generally accessible for HSV-1, some only in a delayed manner. Spreading to upper layers could be detected already at 6 h p.i. and continued at longer infection times (Fig. 2b and 3c). Overall, these results indicate efficient susceptibility of the stratified epithelium to HSV-1.
We next investigated whether Langerhans cells (LCs) represent a preferred target cell type in the epithelium. Immunity to HSV is initiated by skin dendritic cells that take up the virus; in human skin, HSV infects epidermal LCs that penetrate the dermis, undergo apoptosis, and are taken up by dermal dendritic cells (19). Our interest is based on the observation that epidermal LCs can elongate their dendrites to penetrate cellular keratinocyte junctions, which in turn, might allow circumvention of epidermal barriers during pathogen uptake (20). As the highest density of epithelial LCs was detected within the vestibular mucosa (21), we stained infected epithelia from vestibular mucosa with an antibody against the LC-specific surface antigen CD1a (22), confirming a high number of LCs throughout the epithelium. Costaining of ICP0 at 16 h p.i. revealed no preferred infection of LCs, suggesting that keratinocytes instead represent the initial targets, at least under these experimental conditions (Fig. 3d).
To investigate whether the efficiency of infection was delayed in tissue compared to cells in culture, we determined the time course of ICP0 expression in infected primary human oral keratinocytes. At early times after infection, ICP0 localizes in nuclear foci, but it relocalizes to the cytoplasm later during infection (17). When primary cells were infected at 20 PFU/cell, nuclear ICP0 staining was detected at 2 h p.i., while most cells showed cytoplasmic ICP0 at 4 h p.i. (Fig. 4). As cytoplasmic ICP0 was often present in the basal keratinocytes of the epithelium at 3 h p.i. (100 PFU/cell), we conclude that HSV-1 can enter oral epithelia very efficiently once the lamina propria is removed.
FIG 4.
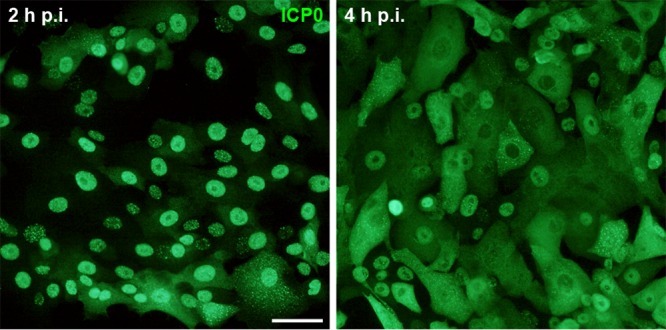
HSV-1 infection of primary oral keratinocytes. Human primary oral keratinocytes were infected with HSV-1 at 20 PFU/cell. Nuclear ICP0 staining (green) at 2 h p.i. followed by cytoplasmic ICP0 at 4 h p.i. illustrates the course of early infection. Bar, 25 μm.
Nectin-1 and HVEM are expressed on human oral keratinocytes.
To address whether the susceptibility of mucosal epithelia correlates with the presence of the putative HSV-1 receptors nectin-1 and HVEM, we investigated receptor expression in epithelia from vestibular mucosa. Nectin-1 and HVEM were present in the oral epithelium and primary oral keratinocytes, respectively, as shown by reverse transcription-PCR (RT-PCR) (Fig. 5a). The surface expression and distribution of the receptors on oral keratinocytes in tissue were analyzed by flow cytometric analysis. After dissociation of epithelial sheets from vestibular mucosa of 4 patients, staining with the anti-human nectin-1 monoclonal antibody CK41 revealed nectin-1 on approximately 90% of the analyzed cells (Fig. 5b and c). In general, the analyzed cell population included at least 93% live cells (data not shown). We then dissociated epithelial sheets from 3 patients by use of an enzyme-free cell dissociation solution (CDS), which is needed to detect HVEM on keratinocytes (10). After staining with the anti-human HVEM polyclonal antibody R140, the HVEM-expressing population represented approximately 40% of the analyzed cells (Fig. 5b and c). Taken together, nectin-1 was present on most oral keratinocytes, while HVEM was detected on the surface of a subpopulation of cells, which suggests nectin-1 as major receptor on oral epithelial cells of vestibular mucosa, although additional receptors cannot be excluded.
FIG 5.

Nectin-1 and HVEM are expressed in human oral epithelia and primary keratinocytes. (a) RNAs were isolated from epithelial sheets prepared from vestibular mucosa or from primary oral keratinocytes. RT-PCR demonstrates expression of nectin-1 and HVEM; as a control, GAPDH transcripts are shown. (b) Cells were dissociated from vestibular epithelial sheets by TrypLE Select treatment and stained for nectin-1. To investigate surface expression of HVEM, cells were dissociated from epithelial sheets by CDS treatment. Flow cytometric analyses indicate 92% and 40% of cells were positive for nectin-1 and HVEM, respectively. APC-A, allophycocyanin-conjugated antibody; FITC-A, fluorescein isothiocyanate-conjugated antibody. (c) Results from samples from 4 (nectin-1) or 3 (HVEM) different patients are shown as means ± standard deviation (SD), demonstrating that surface expression of HVEM is present on fewer cells than nectin-1.
As a key component of adherens junctions (AJs), nectin-1 is located underneath the tight junctions (TJs) and is expressed throughout the epidermis of human skin (23, 24). When we visualized nectin-1 in comparison to AJ and TJ components in vestibular mucosa, we observed nectin-1 staining mainly at the basal epithelial layer (Fig. 6a). In contrast, the AJ component E-cadherin was visible throughout the epithelium, while the TJ component ZO-1 was detected in the stratum distendum, where E-cadherin was less prominent (Fig. 1 and 6b, c, and d). The prominent ZO-1 localization in the stratum distendum of the vestibular mucosa corresponds to the distribution of the TJ component in the stratum granulosum of human epidermis from adult skin samples (25). We assume that nectin-1 expression is not restricted to the basal layer, but due to a rather weak staining of the basal cells, it might be difficult to detect nectin-1 signals on the larger cells of the upper epithelial layers (Fig. 6a). The weak staining might indicate a rather low and varying amount of nectin-1 per oral keratinocyte in the context of tissue, which in turn might be largest in basal cells.
FIG 6.

Localization of nectin-1, ZO-1, and E-cadherin in vestibular epithelia. (a) Sections of vestibular mucosa were stained for nectin-1 (green) or (b) costained for ZO-1 (red) or (c) E-cadherin (green) and keratin 14 (magenta), respectively, with DAPI (blue) as nuclear counterstain. Nectin-1 is rather weakly detected in the basal layers, while ZO-1 is found in the intermediate and superficial layers and E-cadherin throughout the stratified epithelium. Costaining of the junctional proteins ZO-1 and E-cadherin illustrates their presence in the intermediate layers. Two-fold magnifications of the boxed areas are added. The white line indicates the apical mucosal surface. Single immunofluorescence (a) and confocal projections and merged images (b, c, and d) are shown. Bars represent 25 μm in panel a and 50 μm in panels b, c, and d.
HSV-1 can enter oral human mucosa only sparsely after wounding.
We never observed ICP0-expressing cells on the apical side of infected epithelia from either gingiva or vestibular mucosa samples (Fig. 7f), which is in line with the efficient physical barrier in oral epithelia protecting them against pathogen invasion. When we infected intact gingival mucosa samples for 6 or 12 h, we sometimes observed very few ICP0-expressing cells, which were only at the sample edges (Fig. 7a); this effect was even more pronounced after incubation of the samples in medium for 22 h prior to infection for 6 or 9 h (Fig. 7a). As the presence of these infected cells correlated with elongated nuclei (Fig. 7a) or even dissociation at the sample edges, we assume that the infected cells do not reflect viral penetration into intact cellular layers but into dissociated cells. As control, we visualized apoptotic cells by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining, confirming the viability of most cells at the wound edges after preincubation for 22 h (Fig. 7b). However, an increase of TUNEL-positive cells was observed at the apical surface of preincubated samples compared to samples without preincubation (Fig. 7b). Thus, we hypothesize that the number of apoptotic cells at the apical surface at 24 h p.i. correlated with the time in culture (Fig. 7g).
To investigate whether the physical barrier function of intact mucosa can be overcome by wounding, we applied various protocols to wound the oral mucosa samples immediately prior to ex vivo infection. The small size of the tissue allowed no stripping procedures to remove epithelial layers, and the use of punches was not suitable. Thus, complete mucosa samples were slit with one or multiple scalpel cuts, which just reached the lamina propria. After various scalpel cuts into complete gingiva samples from 3 patients followed by infection, we observed no ICP0-expressing cells at 6 h p.i. (data not shown). At longer times of infection, the extent of dissociation varied strongly from patient to patient. When we analyzed scalpel cuts in complete gingiva with moderate dissociation at 24 h p.i., we found infected cells at the wound edges in the lower part of the epithelium and no penetration from the apical surface (Fig. 7c). In contrast, when we analyzed scalpel cuts in vestibular mucosa from 4 patients that was not completely dissociated, we observed no infected cells at 24 and even 48 h p.i. at the wound edges (Fig. 7d). TUNEL staining confirmed again the viability of most cells at the wound edges at 48 h p.i. (Fig. 7e). Both in wounded gingiva and in vestibular mucosa, we never found infected cells in the lamina propria (Fig. 7a, c, d, and f). The heterogeneity in viral susceptibility at wound edges suggests that the dissociation of the mechanical barriers in scalpel cuts varies strongly.
We also determined production of virus progeny in wounded vestibular mucosa in comparison to intact mucosa samples. Infection of intact mucosa as well as of wounded samples led only to some increase in virus progeny at 48 and 72 h p.i., supporting that wounding did not improve the susceptibility to HSV-1 (Fig. 7h). Assuming that the rate of dissociation advances with increasing incubation time of the mucosa samples in medium, infection is probably limited to areas of disintegrated tissue. Thus, we conclude that wounding overcomes the mechanical barriers for HSV-1 invasion only after distinctive breakup of cellular junctions.
DISCUSSION
The general assumption is that HSV enters its host through breaks in tissue and immediately initiates infection; however, the experimental evidence of how the virus invades human oral or genital mucosa during primary infection is still missing. Here, we addressed the initial steps of HSV-1 invasion into human oral mucosa prior to virus production. Based on an ex vivo infection model with human explants, we analyzed the determinants of viral invasion into individual cells in tissue. One precondition for successful invasion is the presence of cell surface receptors that mediate interaction with viral glycoproteins (26). The major receptors for entry into mouse and human cells are HVEM and the cell adhesion molecule nectin-1 (9, 27). In murine epidermis, we found that HSV-1 entry strongly depends on the presence of nectin-1, while HVEM has a more limited role (10). Here, we demonstrate the surface expression of nectin-1 on most of the cells in the human oral epithelium and the presence of approximately 40% of HVEM-expressing cells. By means of immunofluorescence stainings, we detected nectin-1 only very weakly at the basal epithelial layer, which might be insufficiently reflecting the distribution throughout the epithelium. However, Hung et al. (28) observed nectin-1 also only in the basal layer and stratum spinosum of human gingiva. Future studies are needed to understand the precise distribution of nectin-1 in human epithelia.
Our ex vivo infection studies revealed efficient HSV-1 invasion into the human oral epithelium via the basal layer under the experimental conditions in which the lamina propria was removed prior to infection. EM studies confirmed that removal of the lamina propria by dispase II treatment also detached the basement membrane from the epithelium, which can provide a barrier toward pathogen invasion (29). Importantly, cell-cell junctions of the basal layer appeared intact, indicating that HSV-1 can reach its receptors once it gains access to plasma membranes that lost contact with underlying structures. Time course experiments demonstrate no preferential virus entry into a subset of basal keratinocytes but penetration into all basal cells in a time-delayed fashion. These findings are in line with our observations in murine epidermis once nectin-1 and/or HVEM are present (10). Entry via the basal layer of the epithelium might reflect invasion upon reactivation and anterograde axonal transport. As most nerves terminate within the lamina propria, and only a few endings occur between epithial cells (14), we assume that in vivo HSV-1 is either shed into the connective tissue or reaches basal keratinocytes already after release from nerve endings.
Upon ex vivo infection of epithelial samples, we found virus spreading from the basal to the prickle cell layers. To address the mode of spreading in tissue, we added human anti-HSV serum 1 h p.i. (data not shown). These initial experiments suggest direct cell-to-cell spread of HSV-1 in the oral epithelium as a major route of virus spreading.
Human oral mucosa provides a strong physical barrier that generally protects from pathogen invasion and mechanical injuries (30). As the permeability barrier can vary within the oral cavity, we performed the ex vivo infection studies with gingiva representing the least permeable masticatory mucosa and with vestibular mucosa belonging to the lining mucosa whose areas are most permeable. The variation in the permeability reflects differences in the types of lipid making up the intercellular permeability barrier in the stratum distendum (lining mucosa) or stratum granulosum (masticatory mucosa) (15, 31). We found no infected cells in the apical layers after submerging gingiva as well as vestibular mucosa samples in virus suspension, although minor lesions from sample taking sometimes occurred. Only very few infected cells were observed at the edges of some samples. These observations were made in tissue samples, which were incubated less than 20 h in medium to avoid cell dissociation prior to virus entry. Assuming that the cell-cell junctions in gingiva as well as vestibular mucosa samples were still functional at the time of infection, our results demonstrate that the permeability of the epithelia does not allow the virus to reach its receptors via penetration into intercellular spaces of the stratum distendum (vestibular mucosa) or stratum granulosum (gingiva). Surprisingly, scalpel cuts into the epithelia just prior to infection were still insufficient to allow efficient virus entry. Even when virus production was compared after infection of intact or wounded mucosa samples, we found no enhanced production in wounded samples. Our results indicate that mechanical wounding is insufficient for enhanced virus invasion into human oral mucosa, suggesting that further contributions play a role in virus entry. Under in vivo conditions, the mucosal surfaces are coated with saliva, a relatively mobile fluid with few mucins, limited enzymatic activity, and nearly no proteases, but which contains antimicrobial peptides, which contribute to the barrier function of oral mucosa (7, 32). Furthermore, the biofilm of the oral cavity harbors commensal as well as pathogenic bacteria, whose imbalance can lead to inflammatory responses and bacterial invasion into tissue (33). A plausible assumption is that disturbance of epithelial integrity induced by bacteria facilitates invasion of viral pathogens. Studies supporting a periodontopathic role of herpesviruses emphasize the potential cross talk of various pathogens during disease development (34). In immunocompromised individuals, HSV can cause opportunistic infections. In the case of HIV infection, the increased risk of HSV invasion may be mediated by immune dysfunction; however, recent reports demonstrate HIV-induced impairment of mucosal barrier function (35, 36). For human oral epithelia, Sufiawati and Tugizov (37) suggested that HIV-associated disruption of cellular junctions contributes to HSV-1 infection. Taken together, we hypothesize that HSV-1 invasion into human oral mucosa needs further disruption of intercellular junctions in addition to mechanical wounding of tissue to make receptors such as nectin-1 accessible for virus penetration.
MATERIALS AND METHODS
Preparation of human oral mucosa, isolation of human oral keratinocytes, and viruses.
Samples of human oral mucosa were received from patients undergoing removal of retained wisdom teeth (gingiva) or corrective jaw surgeries (vestibular mucosa). Samples were taken from healthy individuals and from areas of the oral cavity without signs of inflammation (no periodontal pockets, no redness, no swelling). Immediately after surgery, tissue samples were stored in phosphate-buffered saline (PBS) and prepared for infection. After removal of subcutaneous fat tissue, samples were cut into pieces of approximately 0.5 by 0.5 cm. Depending on the size, complete mucosa pieces were either left untreated or were cut with a scalpel prior to infection. The surface of complete mucosa was slit with one or multiple scalpel cuts. Epithelial sheets were prepared from gingival or vestibular mucosa after incubation of the mucosa samples for 2 h at 37°C or overnight at 4°C with 5 mg/ml dispase II (Roche) in PBS, followed by 3 washing steps and gentle removal as an intact sheet from the underlying lamina propria. Epithelial sheets and intact or wounded mucosa samples were incubated in Dulbecco's modified Eagle's medium (DMEM; high glucose, GlutaMAX supplement [Life Technologies]). For gentle cell dissociation, the epithelial sheets were incubated with the basal side on TrypLE Select cell dissociation enzyme (Life Technologies) for 30 min at room temperature. These cell suspensions were used to isolate keratinocytes or extract RNA.
Human primary keratinocytes isolated from gingival epithelia were cultured in the presence of mitomycin C-treated 3T3 fibroblasts (strain J2) as feeder cells at 37°C. Cells were maintained in DMEM (high glucose, GlutaMAX supplement [Life Technologies])–Ham's F-12 (Gibco) (3.5:1.1) containing 10% fetal calf serum (FCS), penicillin (400 IU/ml), streptomycin (50 μg/ml), adenine (1.8 × 10−4 M), glutamine (300 μg/ml), hydrocortisone (0.5 μg/ml), epidermal growth factor (EGF) (10 ng/ml), cholera enterotoxin (10−10 M), insulin (5 μg/ml), and ascorbic acid (0.05 mg/ml).
Infection was performed with purified preparations of HSV-1 wild-type strain 17 as described previously (38). Tissue samples were placed in virus suspension at 37°C defining time point zero. The calculation of the virus dose was based on the estimated cell number in the basal layer and the lateral surfaces (1 × 104 cells per mm2). Intact or wounded mucosa and epithelial sheets were infected with HSV-1 at approximately 100 PFU/cell; after incubation for 1 h at 37°C, the virus-containing medium was replaced. Human primary oral keratinocytes were seeded without 3T3 fibroblasts prior to infection at 20 PFU/cell. After infection of intact or wounded vestibular mucosa samples at 50 PFU/cell, the titers of cell-released virus were determined by plaque assays on Vero B4 cells at 3, 48, and 72 h p.i.
Ethics statement.
Human oral mucosa specimens were obtained, after informed consent, from patients undergoing dental surgery at the Department of Oral and Maxillofacial Plastic Surgery, University Hospital of Cologne. Samples were taken in strict accordance with the recommendations of the ethics commission of the Medical Faculty, University of Cologne. The study was approved by the Ethics Commission, Medical Faculty (approval no. 10-205, 15 September 2010).
RNA preparation and RT-PCR.
TRIzol reagent (Life Technologies) was used to isolate RNA from keratinocytes directly after dissociation from vestibular epithelia or from primary oral keratinocytes that were cultivated for 1 day without 3T3 fibroblasts. cDNAs were synthesized using the SuperScript II reverse transcriptase (Life Technologies); PCR was performed with Taq DNA polymerase (Life Technologies) and the following primer pairs: (i) nectin-1 primers (forward, 5′-TTGACCGCATTCTTCCTCCC-3′; reverse, 5′-CCACCAGGACCTTGTCATCC-3′), (ii) HVEM primers (forward, 5′-GTCCAGCGGAAAAGACAGGA-3′; reverse, 5′-TGGACAGCCTCTTTCAGCAG-3′), and (iii) GAPDH (glyceraldehyde-3-phosphate dehydrogenase) primers (forward: 5′-TGATGACATCAAGAAGGTGGTGAAG-3′; reverse: 5′-TCCTTGGAGGCCATGTGGGCCAT-3′).
Immunocytochemistry and antibodies.
For cryosections, complete mucosa or epithelial sheets were embedded in Tissue Tek (Sakura), frozen, and cut into 10-μm cross sections as described previously (12). For hematoxylin and eosin (HE) staining, tissue sections were fixed with 1% formaldehyde and stained for 10 min with hemalum to visualize nuclei followed by counterstaining of the cytoplasm with eosin for 5 min. For immunofluorescence, tissue sections fixed with 1% formaldehyde were stained with mouse anti-ICP0 (11060; 1:60) (39), AF488-conjugated anti-mouse IgG (Life Technologies), and DAPI (4′,6-diamidino-2-phenylindole). In addition, fixed cryosections were blocked with 5% normal goat serum (NGS) and 0.2% Tween 20 in PBS for 3 h at room temperature and costained with mouse anti-E-cadherin (1:400 [BD]) or polyclonal rabbit anti-ZO-1 (1:400 [Life Technologies]) and guinea pig anti-keratin 14 (1:150 [Progen]) at 4°C overnight followed by incubation with the corresponding secondary antibodies and DAPI for 45 min at room temperature. Nectin-1 was visualized in cryosections after fixation with 100% methanol for 10 min at −20°C, blocking with 5% NGS and 0.005% saponin in PBS for 3 h at room temperature, and staining with mouse anti-nectin-1 (CK41; 1:50 in 5% NGS and 0.01% saponin) (40) overnight at 4°C followed by incubation with the corresponding secondary antibodies and DAPI. For detection of DNA fragmentation, tissue sections were fixed with 4% formaldehyde, labeled using the DeadEnd fluorometric TUNEL system (Promega), and counterstained with DAPI.
For whole mounts (17, 41), gingival epithelia were fixed with 3.4% formaldehyde for 2 h, costained with mouse anti-ICP0 (11060; 1:60) (39) and rabbit polyclonal anti-mouse keratin 14 (AF64; 1:10,000 [Covance]) as counterstain of the basal epithelial layer, and visualized with the corresponding secondary antibodies as described previously (12).
Human primary oral keratinocytes were fixed with 2% formaldehyde for 10 min, permeabilized with 0.5% NP-40 for 10 min, and stained for 60 min with mouse anti-ICP0 (11060; 1:60) (39) and then visualized by the corresponding secondary antibody, all at room temperature.
Microscopy was performed using a Leica DM IRB/E microscope linked to a Leica TCS-SP/5 confocal unit or a Zeiss Axiophot. Images were assembled using Photoshop (version CS2; Adobe).
Transmission electron microscopy.
Vestibular epithelia were prepared for EM as described previously (42). Semithin sections were stained with uranyl acetate and lead citrate and analyzed in a Zeiss EM109.
Flow cytometric analysis.
Vestibular epithelial sheets were incubated on TrypLE Select (Life Technologies) or on enzyme-free cell dissociation solution (CDS) (Sigma) and processed as described previously (10). Cell suspensions prepared by TrypLE Select (Life Technologies) were incubated in PBS–5% FCS on ice for 45 min with mouse anti-nectin-1 (CK41; 1:100) (40), and nectin-1 was visualized with anti-mouse IgG-Cy5 (1:100 [Jackson ImmunoResearch Laboratories, Inc.]). Cell suspensions prepared by enzyme-free CDS (Sigma) were kept in PBS–5% FCS and incubated on ice for 45 min with rabbit polyclonal anti-human HVEM (R140; 1:500) (43) followed by visualization of HVEM with anti-rabbit AF488 (1:200 [Life Technologies]). For nectin-1 and HVEM, mouse IgG1 (Life Technologies) and polyclonal rabbit IgG (Abcam) were used as isotype controls. Viable cells were assessed by 7-aminoactinomycin D (7-AAD; 1:40 [BD]) after incubation for 10 min on ice. Samples were analyzed by using a FACSCanto II flow cytometer and FACSDiva (version 6.1.3, BD) with FlowJo (version 7.6.3, Tree Star) software.
ACKNOWLEDGMENTS
We are grateful to Frank Meier and Franziska Möller for taking mucosa samples. We thank Daniel Sälzer, Mei-Ju Hsu, Lisa Wirtz, and Maureen Möckel for help with sample preparations, Roger Everett for the antibodies against ICP0, Mario Fabri for the antibodies against CD1a, and Claude Krummenacher for the antibodies against nectin-1 (CK41) or HVEM (R140). In addition, we thank Markus Plomann, Mats Paulsson, and Henrik Dommisch for discussion and advice.
This research was supported by the German Research Foundation (grants KN536/16-3 and KN536/18-1) and the Köln Fortune Program/Faculty of Medicine, University of Cologne.
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Scott DA, Coulter WA, Lamey PJ. 1997. Oral shedding of herpes simplex virus type 1: a review. J Oral Pathol Med 26:441–447. doi: 10.1111/j.1600-0714.1997.tb00012.x. [DOI] [PubMed] [Google Scholar]
- 3.Ehrlich J, Cohen GH, Hochman N. 1983. Specific herpes simplex virus antigen in human gingiva. J Periodontol 54:357–360. doi: 10.1902/jop.1983.54.6.357. [DOI] [PubMed] [Google Scholar]
- 4.Yura Y, Iga H, Terashima K, Yoshida H, Yanagawa T, Hayashi Y, Sato M. 1987. The role of epithelial cell differentiation in the expression of herpes simplex virus type 1 in normal human oral mucosa in culture. Arch Virol 92:41–53. doi: 10.1007/BF01310061. [DOI] [PubMed] [Google Scholar]
- 5.Yura Y, Iga H, Kondo Y, Harada K, Yanagawa T, Yoshida H, Sato M. 1991. Herpes simplex virus type 1 and type 2 infection in human oral mucosa in culture. J Oral Pathol Med 20:68–73. doi: 10.1111/j.1600-0714.1991.tb00892.x. [DOI] [PubMed] [Google Scholar]
- 6.Glorieux S, Bachert C, Favoreel HW, Vandekerckhove AP, Steukers L, Rekecki A, Van den Broeck W, Goossens J, Croubels S, Clayton RF, Nauwynck HJ. 2011. Herpes simplex virus type 1 penetrates the basement membrane in human nasal respiratory mucosa. PLoS One 6:e22160. doi: 10.1371/journal.pone.0022160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dommisch H, Jepsen S. 2015. Diverse functions of defensins and other antimicrobial peptides in periodontal tissues. Periodontol 2000 69:96–110. doi: 10.1111/prd.12093. [DOI] [PubMed] [Google Scholar]
- 8.Groeger SE, Meyle J. 2015. Epithelial barrier and oral bacterial infection. Peridontol 2000 69:46–67. doi: 10.1111/prd.12094. [DOI] [PubMed] [Google Scholar]
- 9.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 10.Petermann P, Thier K, Rahn E, Rixon FJ, Bloch W, Özcelik S, Krummenacher C, Barron MJ, Dixon MJ, Scheu S, Pfeffer K, Knebel-Mörsdorf D. 2015. Entry mechanisms of herpes simplex virus 1 into murine epidermis: involvement of nectin-1 and herpesvirus entry mediator as cellular receptors. J Virol 89:262–274. doi: 10.1128/JVI.02917-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rahn E, Petermann P, Thier K, Bloch W, Morgner J, Wickström SA, Knebel-Mörsdorf D. 2015. Invasion of herpes simplex virus type 1 into murine epidermis: an ex vivo infection study. J Invest Dermatol 135:3009–3016. doi: 10.1038/jid.2015.290. [DOI] [PubMed] [Google Scholar]
- 12.Rahn E, Thier K, Petermann P, Knebel-Mörsdorf D. 2015. Ex vivo infection of murine epidermis with herpes simplex virus type 1. J Vis Exp 102:e53046. doi: 10.3791/53046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rahn E, Thier K, Petermann P, Rübsam M, Staeheli P, Iden S, Niessen CM, Knebel-Mörsdorf D. 2017. Epithelial barriers in murine skin during herpes simplex virus 1 infection: the role of tight junction formation. J Invest Dermatol 137:884–893. doi: 10.1016/j.jid.2016.11.027. [DOI] [PubMed] [Google Scholar]
- 14.Squier C, Brogden K. 2011. Human oral mucosa: development, structure and function. Wiley-Blackwell, Hoboken, NJ. [Google Scholar]
- 15.Squier CA. 1991. The permeability of oral mucosa. Crit Rev Oral Biol Med 2:13–32. doi: 10.1177/10454411910020010301. [DOI] [PubMed] [Google Scholar]
- 16.Rowat JS, Squier CA. 1986. Rates of epithelial cell proliferation in the oral mucosa and skin of the tamarin monkey (Saguinus fuscicollis). J Dent Res 65:1326–1331. doi: 10.1177/00220345860650110901. [DOI] [PubMed] [Google Scholar]
- 17.Petermann P, Haase I, Knebel-Mörsdorf D. 2009. Impact of Rac1 and Cdc42 signaling during early herpes simplex virus type 1 infection of keratinocytes. J Virol 83:9759–9772. doi: 10.1128/JVI.00835-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boutell C, Everett RD. 2013. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol 94:465–481. doi: 10.1099/vir.0.048900-0. [DOI] [PubMed] [Google Scholar]
- 19.Kim M, Truong NR, James V, Bosnjak L, Sandgren KJ, Harman AN, Nasr N, Bertram KM, Olbourne N, Sawleshwarkar S, McKinnon K, Cohen RC, Cunningham AL. 2015. Relay of herpes simplex virus between Langerhans cells and dermal dendritic cells in human skin. PLoS Pathog 13:e1004812. doi: 10.1371/journal.ppat.1004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kubo A, Nagoa K, Yokouchi M, Sasaki H, Amagai M. 2009. External antigen uptake by Langerhans cells with reorganization of epidermal tight junction barriers. J Exp Med 206:2937–2946. doi: 10.1084/jem.20091527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allam JP, Stojanovski G, Friedrichs N, Peng W, Bieber T, Wenzel J, Novak N. 2008. Distribution of Langerhans cells and mast cells within the human oral mucosa: new application sites of allergens in sublingual immunotherapy? Allergy 63:720–727. doi: 10.1111/j.1398-9995.2007.01611.x. [DOI] [PubMed] [Google Scholar]
- 22.Fithian E, Kung P, Goldstein G, Rubenfeld M, Fenoglio C, Edelson R. 1981. CD1a: reactivity of Langerhans cells with hybridoma antibody. Proc Natl Acad Sci U S A 78:2541–2544. doi: 10.1073/pnas.78.4.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsushima H, Utani A, Endo H, Matsuura H, Kakuta M, Nakamura Y, Matsuyoshi N, Matsui C, Nakanishi H, Takai Y, Shinkai H. 2003. The expression of nectin-1alpha in normal human skin and various skin tumours. Br J Dermatol 148:755–762. doi: 10.1046/j.1365-2133.2003.05225.x. [DOI] [PubMed] [Google Scholar]
- 24.Takai Y, Ikeda W, Ogita H, Rikitake Y. 2008. The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu Rev Cell Dev Biol 24:309–342. doi: 10.1146/annurev.cellbio.24.110707.175339. [DOI] [PubMed] [Google Scholar]
- 25.Pummi K, Malminen M, Aho H, Karvonen SL, Peltonen J, Peltonen S. 2001. Epidermal tight junctions: ZO-1 and occludin are expressed in mature, developing, and affected skin and in vitro differentiating keratinocytes. J Invest Dermatol 117:1050–1058. doi: 10.1046/j.0022-202x.2001.01493.x. [DOI] [PubMed] [Google Scholar]
- 26.Heldwein EE, Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell Mol Life Sci 65:1653–1668. doi: 10.1007/s00018-008-7570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436. doi: 10.1016/S0092-8674(00)81363-X. [DOI] [PubMed] [Google Scholar]
- 28.Hung SL, Cheng YY, Wang YH, Chang KW, Chen YT. 2002. Expression and roles of herpesvirus entry mediators A and C in cells of oral origin. Oral Microbiol Immunol 17:215–223. doi: 10.1034/j.1399-302X.2002.170403.x. [DOI] [PubMed] [Google Scholar]
- 29.Steukers L, Glorieux S, Vandekerckhove AP, Favoreel HW, Nauwynck HJ. 2012. Diverse microbial interactions with the basement membrane barrier. Trends Microbiol 20:147–155. doi: 10.1016/j.tim.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Squier CA, Kremer MJ. 2001. Biology of oral mucosa and esophagus. J Natl Cancer Inst Monogr 29:7–15. doi: 10.1093/oxfordjournals.jncimonographs.a003443. [DOI] [PubMed] [Google Scholar]
- 31.Squier CA, Wertz PW. 1993. Permeability and the pathophysiology of oral mucosa. Adv Drug Deliv Rev 12:13–24. doi: 10.1016/0169-409X(93)90038-6. [DOI] [Google Scholar]
- 32.Malamud D, Abrams WR, Barber CA, Weissman D, Rehtanz M, Golub E. 2011. Antiviral activities in human saliva. Adv Dent Res 23:34–37. doi: 10.1177/0022034511399282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roberts FA, Darveau RP. 2015. Microbial protection and virulence in periodontal tissue as a function of polymicrobial communities: symbiosis and dysbiosis. Periodontol 2000 69:18–27. doi: 10.1111/prd.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slots J. 2015. Periodontal herpesviruses: prevalence, pathogenicity, systemic risk. Periodontol 2000 69:28–45. doi: 10.1111/prd.12085. [DOI] [PubMed] [Google Scholar]
- 35.Burgener A, McGowan I, Klatt NR. 2015. HIV and mucosal barrier interactions: consequences for transmission and pathogenesis. Curr Opin Immunol 36:22–30. doi: 10.1016/j.coi.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 36.Tugizov S. 2016. Human immunodeficiency virus-associated disruption of mucosal barriers and its role in HIV transmission and pathogenesis of HIV/AIDS disease. Tissue Barriers 4:e1159276. doi: 10.1080/21688370.2016.1159276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sufiawati I, Tugizov SM. 2014. HIV-associated disruption of tight and adherens junctions of oral epithelial cells facilitates HSV-1 infection and spread. PLoS One 9:e88803. doi: 10.1371/journal.pone.0088803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schelhaas M, Jansen M, Haase I, Knebel-Mörsdorf D. 2003. Herpes simplex virus type 1 exhibits a tropism for basal entry in polarized epithelial cells. J Gen Virol 84:2473–2484. doi: 10.1099/vir.0.19226-0. [DOI] [PubMed] [Google Scholar]
- 39.Everett RD, Cross A, Orr A. 1993. A truncated form of herpes simplex virus type 1 immediate-early protein Vmw110 is expressed in a cell type dependent manner. Virology 197:751–756. doi: 10.1006/viro.1993.1651. [DOI] [PubMed] [Google Scholar]
- 40.Krummenacher C, Baribaud I, Ponce de Leon M, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. 2000. Localization of a binding site for herpes simplex virus glycoprotein D on herpesvirus entry mediator C by using antireceptor monoclonal antibodies. J Virol 74:10863–10872. doi: 10.1128/JVI.74.23.10863-10872.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Braun KM, Niemann C, Jensen UB, Sundberg JP, Silva-Vargas V, Watt FM. 2003. Manipulation of stem cell proliferation and lineage commitment: visualisation of label-retaining cells in whole mounts of mouse epidermis. Development 130:5241–5255. doi: 10.1242/dev.00703. [DOI] [PubMed] [Google Scholar]
- 42.Bechtel M, Keller MV, Bloch W, Sasaki T, Boukamp P, Zaucke F, Paulsson M, Nischt R. 2012. Different domains in nidogen-1 and nidogen-2 drive basement membrane formation in skin organotypic cocultures. FASEB J 26:3637–3648. doi: 10.1096/fj.11-194597. [DOI] [PubMed] [Google Scholar]
- 43.Terry-Allison T, Montgomery RI, Whitbeck JC, Xu R, Cohen GH, Eisenberg RJ, Spear PG. 1998. HveA (herpesvirus entry mediator A), a coreceptor for herpes simplex virus entry, also participates in virus-induced cell fusion. J Virol 72:5802–5810. [DOI] [PMC free article] [PubMed] [Google Scholar]