

ABSTRACT
The human papillomavirus (HPV) E6 oncoproteins recruit the cellular ubiquitin ligase E6AP/UBE3A to target cellular substrates for proteasome-mediated degradation, and one consequence of this activity is the E6 stimulation of E6AP autoubiquitination and degradation. Recent studies identified an autism-linked mutation within E6AP at T485, which was identified as a protein kinase A phosphoacceptor site and which could directly regulate E6AP ubiquitin ligase activity. In this study, we have analyzed how T485-mediated regulation of E6AP might affect E6 targeting of some of its known substrates. We show that modulation of T485 has no effect on the ability of E6 to direct either p53 or Dlg for degradation. Furthermore, T485 regulation has no effect on HPV-16 or HPV-31 E6-induced autodegradation of E6AP but does affect HPV-18 E6-induced autodegradation of E6AP. In cells derived from cervical cancers, we find low levels of both phosphorylated and nonphosphorylated E6AP in the nucleus. However, ablation of E6 results in a dramatic accumulation of phospho-E6AP in the cytoplasm, whereas nonphosphorylated E6AP accumulates primarily in the nucleus. Interestingly, E6AP phosphorylation at T485 confers association with 14-3-3 proteins, and this interaction seems to be important, in part, for the ability of E6 to recruit phospho-E6AP into the nucleus. These results demonstrate that HPV E6 overrides the normal phosphoregulation of E6AP, both in terms of its enzymatic activity and its subcellular distribution.
IMPORTANCE Recent reports demonstrate the importance of phosphoregulation of E6AP for its normal enzymatic activity. Here, we show that HPV E6 is capable of overriding this regulation and can promote degradation of p53 and Dlg regardless of the phosphorylation status of E6AP. Furthermore, E6 interaction with E6AP also significantly alters how E6AP is subject to autodegradation and suggests that this is not a simple stimulation of an already-existing activity but rather a redirection of E6AP activity toward itself. Furthermore, E6-mediated regulation of the subcellular distribution of phospho-E6AP appears to be dependent, in part, upon the 14-3-3 family of proteins.
KEYWORDS: HPV E6, E6AP, phosphorylation
INTRODUCTION
Human papillomaviruses (HPVs) are the causative agents of a number of human cancers, with cervical cancer being the most prevalent (1). There are multiple HPV types capable of infecting either mucosal or cutaneous tissues (2), but only a small subset are associated with the development of human cancers. In the case of cervical cancer, 13 different HPV types have been defined as cancer causing, with the most important being HPV-16 and HPV-18, which together account for approximately 80% of all cervical malignancies (3). Tumor development requires the combined action of two viral oncoproteins, E6 and E7. These two proteins subvert a large number of critical cellular control pathways, which can ultimately result in the development of malignancy. Most importantly, both viral oncoproteins continue to be retained and expressed in the cervical tumor-derived cell lines and tissues many years after immortalization and full transformation (4, 5). Indeed, abolition of E6 or E7 expression results in a cessation of transformed cell growth and induction of either senescence or apoptosis (6, 7). Therefore, both viral oncoproteins represent excellent targets for therapeutic intervention in HPV-induced malignancy.
Understanding the mechanisms by which E6 and E7 subvert cell cycle control and apoptotic pathways is the cornerstone of understanding how these viruses induce malignancy. A major function of E7 is to target cellular proteins that are critical for regulating normal cell cycle progression. This includes members of the pRb family of pocket proteins, as well as cyclins and cyclin-dependent kinase inhibitors, among many others (8). The E6 oncoprotein exerts strong antiapoptotic and growth-promoting activities, which are also achieved through a plethora of protein-protein interactions. These include targeting the tumor suppressor p53 (9), the proapoptotic protein Bak (10), and a number of PDZ (PSD95/Dlg/ZO) domain-containing cell polarity regulators, including Dlg, Scribble, and MAGI-1 (11). A striking feature of many E6 functions is its ability to recruit a cellular ubiquitin ligase, E6AP, which it then redirects to induce the ubiquitin proteasome-mediated degradation of many of its target proteins, including p53 (12, 13). Interestingly, E6 also appears to require association with E6AP for maintaining its own stability, and this appears to be a common feature found in many of the alpha group HPV E6 oncoproteins, regardless of the tumor risk of the HPV type (14).
Much emphasis recently has been placed on understanding how the activities of E6 and E7 are controlled through posttranslational modifications during different phases of the viral life cycle or in different phases of the cell cycle. For example, E7 is subject to phosphorylation by CKII, which appears to regulate a number of E7's activities (15–17). Likewise, certain HPV E6 oncoproteins are phosphorylated within the PDZ binding motif (PBM), which generates a novel interacting motif, thereby adding to the multifunctionality of this specific protein-interacting module (18, 19).
Recent studies have also highlighted the potential for phosphorylation-dependent regulation of the ubiquitin ligase activity of E6AP (20). While perturbation of E6AP function has long been known to be associated with the development of Angelman syndrome, these recent studies have also linked a single point mutation within E6AP to the development of autism in affected individuals. Intriguingly, this residue of E6AP appears to be a phosphoacceptor site for protein kinase A (PKA), with phosphorylated forms being catalytically inactive, while the nonphosphorylated forms of the protein exert increased levels of ubiquitin ligase activity (20). Interestingly, these nonphosphorylatable mutants appeared to be less stable, owing to increased levels of autoubiquitination activity (20).
Since PKA has also been implicated in the regulation of E6 function (18), we were interested in determining whether posttranslational modification of E6AP could have any impact on the ability of E6 to recruit and use E6AP for the degradation of its target proteins. In this study, we show that phosphoregulation of E6AP has little effect upon the ability of E6 to target its normal substrates for proteasome-mediated degradation but can affect E6-stimulated autodegradation of E6AP.
RESULTS
Phosphorylation of E6AP at T485 affects E6AP degradation of Ring1B.
Recent studies demonstrated that PKA phosphorylation of E6AP at position T485 resulted in decreased enzymatic activity, while a nonphosphorylatable mutant of E6AP appeared to stimulate increased levels of ubiquitin-mediated degradation of its substrates (20). In order to confirm the PKA phosphoacceptor site in E6AP, the wild type and T485A mutant (phospho-dead) of E6AP were expressed as glutathione S-transferase (GST) fusion proteins, purified, and then subjected to in vitro phosphorylation with the catalytic subunit of PKA in the presence of radiolabeled ATP. As can be seen in Fig. 1A, the phosphorylation of the E6AP T485A mutant is greatly decreased, confirming that the T485 residue is a major PKA phosphoacceptor site.
FIG 1.
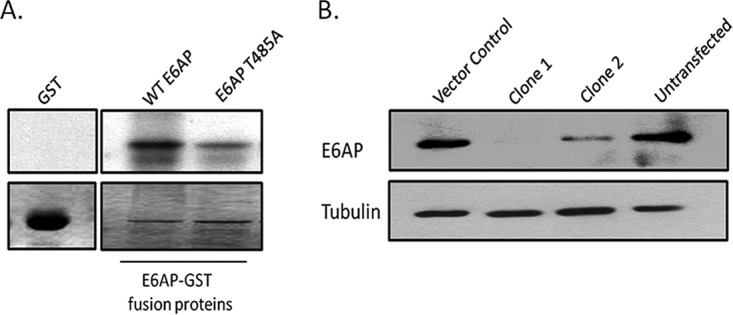
PKA phosphoacceptor site resides mainly at T485 residue of E6AP. (A) The purified GST fusion proteins were incubated with PKA and [γ-32P]ATP. Proteins were then subjected to SDS-PAGE and autoradiographic analysis. (Upper) Autoradiogram of different in vitro-phosphorylated wild-type and mutant E6AP-GST fusion proteins. (Lower) Coomassie blue-stained gel. (B) Western blot analysis against E6AP demonstrating the efficiency of gRNAs in targeting E6AP.
In order to begin to understand the role of phosphoregulation of E6AP in the context of HPV E6-induced degradation of its target proteins, we first wanted to generate a stable cell line where the endogenously expressed E6AP had been ablated. To do this, we performed genome editing of HEK293 cells in which the endogenous E6AP was knocked out by using the CRISPR-Cas9 system. Following selection, single-cell cloning, and initial screening by DNA sequencing, the cells were then further analyzed by Western blotting for any residual levels of E6AP. As can be seen from Fig. 1B, clone 1 was completely deficient for full-length E6AP and has a stop codon at residue 406, and this clone was chosen for all subsequent analyses.
Previous studies had analyzed the effect of phosphoregulation of E6AP upon degradation of HHR23A (20). However, we were first interested in determining whether mutation of the T485 phosphoacceptor site on E6AP could modulate the targeting of another previously described substrate of E6AP. To do this we analyzed Ring1B, which, in previous studies, has been shown to be degraded by E6AP (21, 22). The E6AP-null HEK293 cells were transfected with plasmids expressing Ring1B, together with wild-type E6AP, the T485A phosphodestroyed mutant or the T485E phosphomimic mutant. After 24 h the cells were harvested and proteins analyzed by Western blotting. The results shown in Fig. 2A demonstrate that E6AP alone was capable of inducing degradation of Ring1B, which was further increased in the presence of the T485A mutant, which supports previous studies indicating increased degradation capability with the T485A mutant (20). Surprisingly, the phosphomimic T485E mutant consistently retained some degradation activity in these assays. In order to determine whether E6AP could increase the ubiquitination of Ring1B, we transfected the E6AP-null HEK293 cells with FLAG-tagged Ring1B expression plasmid, together with the different E6AP mutants and hemagglutinin (HA)-tagged ubiquitin expression plasmids. After 24 h the cells were harvested and immunoprecipitated with anti-FLAG-conjugated agarose beads. The ubiquitinated Ring1B was then detected by Western blotting using the anti-HA-horseradish peroxidase (HRP) antibody. Figure 2B shows that there is weak polyubiquitination of Ring1B in the absence of E6AP, but this increases dramatically in the presence of wild-type E6AP. There is a slight further increase in ubiquitination of Ring1B in the presence of the E6AP T485A mutant, while the T485E mutant only weakly ubiquitinates Ring1B. Taken together, these results, using a different substrate of E6AP, largely support previous studies, which indicate that mutation of the T485 phosphoacceptor site can have major effects upon E6AP enzymatic activity (20).
FIG 2.

Regulation of E6AP through T485 affects degradation of the E6AP's normal substrate, Ring1B. (A) E6AP-null HEK293 cells were transfected with the indicated plasmids, and after 24 h the cells were harvested and protein levels analyzed by Western blotting (WB). β-Galactosidase (β-Gal) acted as a control for transfection efficiency. WT, wild type. (B) Cells were transfected with the indicated expression plasmids, and after 24 h they were harvested and subjected to immunoprecipitation (IP) with anti-FLAG-conjugated agarose beads. Polyubiquitinated Ring1B was then detected by Western blotting with anti-HA-HRP antibody. (Lower) Input levels of Ring1B used in the immunoprecipitation.
HPV E6 overrides the normal phosphocontrol of E6AP at T485.
Considering that E6AP is recruited by E6 for many of its activities, we were next interested in determining whether the phosphomimic T485E or phosphodestroying T485A amino acid substitution in E6AP could affect the ability of HPV E6 to direct the degradation of p53. To do this, the E6AP-null HEK 293 cells were cotransfected with either HPV-16 E6, HPV-18 E6, or HPV-31 E6, together with p53 and the wild-type or mutant E6AP expression plasmids. After 24 h the cells were harvested and the protein levels analyzed by Western blotting. The results shown in Fig. 3 demonstrate that p53 is not degraded by E6 if E6AP is absent, and this is in agreement with many other previously published studies (13). Cotransfection of wild-type E6AP with E6 promotes p53 degradation, and there is a concomitant increase in the levels of E6 expression, which is also consistent with previous observations showing that E6AP is required for maintaining E6 stability (23). Most interestingly, however, when we cotransfect either the T485A or the T485E mutants, p53 appears to be degraded with an efficiency similar to that observed with the wild-type E6AP. These results suggest that phosphorylation of T485 has little effect upon the ability of either HPV-16 E6, HPV-18 E6, or HPV-31 E6 to utilize E6AP for the degradation of p53.
FIG 3.

E6AP regulation at T485 has no effect on HPV E6 degradation of p53. E6AP-null HEK293 cells were transfected with plasmids expressing E6AP and p53 as indicated, plus plasmids expressing either HPV-16 E6 (A), HPV-18 E6 (B), or HPV-31 E6 (C). After 24 h the cells were harvested and protein levels analyzed by Western blotting. β-Galactosidase acted as a control for transfection efficiency in all assays.
It is also clear from this analysis that there are apparent differences in how the E6AP mutants are degraded by the different HPV E6 proteins, with HPV-16 E6 and HPV-31 E6 targeting the wild type and the two mutants with similar efficiency, while HPV-18 E6 appears incapable of degrading the T485A and T485E mutants. In order to confirm this, the assay was repeated without the presence of exogenously added substrate, and the results obtained are shown in Fig. 4, where it can be seen that HPV-16 E6 effectively degrades wild-type E6AP and the T485A and T485E mutants, while HPV-18 E6 is largely defective in degrading the T485A and T485E mutants. These results suggest that the autodegradation activity of the T485E mutant can be reactivated by HPV-16 E6 and HPV-31 E6 but not by HPV-18 E6. In contrast, the T485A mutant, which has intrinsically more autoubiquitination activity (20), can be further degraded by HPV-16 E6 and HPV-31 E6 but not by HPV-18 E6, indicating intriguing differences in how these viral oncoproteins redirect E6AP activity.
FIG 4.

Different HPV E6s uncouple normal PKA phosphoregulation of E6AP. E6AP-null HEK293 cells were transfected with plasmids expressing E6AP plus plasmids expressing either HPV-16 E6 (A) or HPV-18 E6 (B). After 24 h the cells were harvested and protein levels analyzed by Western blotting. β-Galactosidase acted as a control for transfection efficiency in all assays. (Bottom) Statistical quantification done using Student's t test. Values shown are means from at least 3 independent experiments; standard errors of the means are shown. **, P < 0.005; ns, not significant.
We then proceeded to investigate whether another HPV E6 substrate was similarly unaffected by T485 phosphoregulation. To do this we analyzed Dlg, which is a PDZ domain-containing substrate of HPV E6 (24). The E6AP-null HEK293 cells were transfected with a Dlg expression plasmid, together with the different E6AP expression constructs and HPV-16 E6. After 24 h the cells were harvested and the protein levels analyzed by Western blotting. The results shown in Fig. 5 demonstrate that Dlg degradation by E6 is also unaffected by either the T485A or T485E amino acid substitution.
FIG 5.
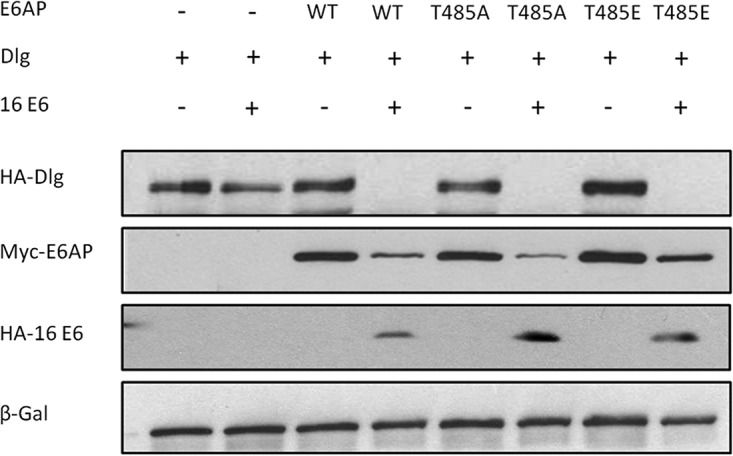
E6AP phosphorylation at T485 has no effect on E6 degradation of Dlg. E6AP-null HEK293 cells were transfected with the indicated plasmids, and after 24 h the cells were harvested and protein levels analyzed by Western blotting. β-Galactosidase acted as a control for transfection efficiency. n = 3.
HPV E6 recruits phospho-E6AP to the nucleus in a 14-3-3-dependent manner.
Having found that HPV E6 can redirect E6AP activity independently of its T485 phosphorylation status, we were interested in investigating how the subcellular distribution of phosphorylated and nonphosphorylated forms of E6AP might appear in cells derived from a cervical cancer, and whether E6 might have any impact upon the subcellular distribution of these different forms of E6AP. In order to do this, we performed a series of immunofluorescence analyses in HeLa cells, which contain HPV-18 E6. The cells were transfected with short interfering RNA (siRNA) E6/E7 to determine whether the viral oncoproteins modulate the distribution of the different forms of E6AP. At the same time, siRNA E6AP was also transfected to verify the specificity of the anti-E6AP antibodies. As can be seen in Fig. 6A, control cells have very low levels of p53 and E6AP. There are also correspondingly very low levels of phospho-E6AP, although interestingly, there does appear to be some variability in the staining for phospho-E6AP, suggesting there is an element of cell cycle control in its phosphorylation. Most interestingly, when cells are transfected with siRNA against E6/E7 (Fig. 6B), there is, as expected, a dramatic increase in the levels of nuclear p53 and E6AP, while the phospho-E6AP expression is restored primarily within the cytoplasmic compartment. These results suggest that phospho-E6AP normally resides within the cytoplasm, while nonphosphorylated E6AP is mostly found in the nucleus. However, in the presence of E6, both forms of E6AP accumulate within the nucleus.
FIG 6.

Phosphoforms of E6AP have distinct subcellular distribution in HeLa cells. (A) HeLa cells were transfected with control siRNA against luciferase, and after 72 h, the cells were incubated for a further 3 h with either DMSO or the proteasome inhibitor CBZ. The cells were then fixed and patterns of protein expression monitored using anti-E6AP, anti-p53, and anti-phospho-T485 E6AP antibodies, respectively. Si Luci, control siRNA against luciferase. (B) HeLa cells were transfected with either siRNA E6/E7 or siRNA E6AP, and after 72 h the cells were fixed and stained for E6AP, p53, and phospho-T485 E6AP. n = 3.
In order to verify the E6-mediated nuclear accumulation of phospho-E6AP, we repeated the immunofluorescence analysis in the presence of the proteasome inhibitor MG132. The results obtained are shown in Fig. 6A (lower), and as can be seen, proteasome inhibition results in a dramatic increase in the amount of both total and phosphorylated E6AP within the nucleus.
Differential subcellular localization of phosphorylated proteins is often mediated through the activity of the 14-3-3 family of proteins (25). As we have also recently shown that E6 can interact with certain 14-3-3 isoforms (18), we were initially interested in investigating whether phosphorylation of E6AP at T485 could confer interaction with 14-3-3 proteins. To do this, we purified wild-type GST.E6AP and GST.E6AP T485A fusion proteins and subjected them to in vitro phosphorylation with purified PKA in the absence of radiolabel and then performed binding assays with recombinant 14-3-3γ. As a positive control we also included HPV-18 E6 in the assays. Figure 7 shows that while there is a strong increase in the ability of phospho-E6 to interact with 14-3-3γ, which is in agreement with previous results (18), phosphorylation of E6AP only results in a very modest increase in interaction with 14-3-3γ, although this is dependent upon an intact phosphoacceptor site at T485. Whether this is a reflection of low levels of phosphorylation of E6AP or intrinsically weak interaction with 14-3-3 remains to be determined.
FIG 7.

Phosphorylation of E6AP at T485 confers its interaction with 14-3-3γ. (Left) PKA and 14-3-3 phosphoconsensus motifs. (Right) Direct interaction assay with purified 14-3-3γ. Purified GST fusion proteins were either untreated or subjected to phosphorylation (indicated as “P”) with PKA in the presence of nonradiolabeled ATP. They were then incubated with purified recombinant 14-3-3γ. (Upper) After extensive washing, the bound protein was detected by Western blotting using anti-14-3-3γ antibody. (Lower) Ponceau staining of the nitrocellulose membrane.
In order to investigate a potential role for 14-3-3 in modulating the pattern of E6AP expression, we performed another series of immunofluorescence analyses in HeLa cells, but in this case, we transfected the cells with a plasmid expressing Difopein, which has been shown previously to block endogenous 14-3-3 proteins from interacting with their substrates (26, 27). As can be seen in Fig. 8A, there is a significant increase in the amount of phospho-E6AP in the cytoplasm following transfection of Difopein. In contrast, Difopein does not alter the subcellular distribution of phospho-E6AP when E6 is also removed (Fig. 8B), where the majority of phosphorylated E6AP is found in the cytoplasm. Taken together, these results indicate that nuclear accumulation of phospho-E6AP in the presence of HPV-18 E6 is in part dependent upon 14-3-3.
FIG 8.

HPV E6 recruits phospho-E6AP to the nucleus in a 14-3-3-dependent manner. (A) HeLa cells were transfected with control siRNA against luciferase, plus Difopein in the right panel, and after 72 h the cells were fixed and patterns of protein expression monitored using anti-phospho-T485 E6AP, anti-p53, and anti-E6AP antibodies, respectively. (B) HeLa cells were transfected with either siRNA E6/E7 (left) or with siRNA E6/E7 plus Difopein (right). After 72 h the cells were fixed and stained for anti-phosphoT485 E6AP, anti-p53, and anti-E6AP antibodies, respectively.
DISCUSSION
The cellular ubiquitin ligase E6AP plays a critical role in many of E6's activities. It is essential for the degradation of certain E6 substrates, it is essential for maintaining E6 protein stability, and it contributes directly to the induction of malignancy in transgenic animals (28–31). Furthermore, loss of its expression in cells derived from cervical cancer induces high levels of apoptosis, and therefore the E6-E6AP interaction remains an attractive target for the development of novel anti-HPV therapeutics. The recent demonstration that E6AP enzymatic activity could be modulated by PKA therefore was particularly relevant for understanding how this might affect E6 function. We show here that phosphorylation of E6AP at T485 is unlikely to directly affect the ability of E6 to target its substrates for proteasome-meditated degradation, although depending on the specific HPV type, modulation of T485 can affect the ability of E6 to further promote E6AP autoubiquitination and degradation.
Previous studies had shown that the E6AP T485 residue is a phosphoacceptor site for PKA, with the nonphosphorylatable mutant T485A demonstrating increased levels of ubiquitination activity, both with respect to an E6AP target protein, HHR23A, and also with respect to its own autoubiquitination (20). In contrast, a phosphomimic mutation, T485E, apparently has greatly reduced levels of enzymatic activity, again both with respect to a normal substrate and to itself. We initially confirmed these observations in two ways. First, we performed in vitro phosphorylation assays with purified PKA and demonstrated unequivocally that the major PKA phosphoacceptor site on E6AP was at T485. Second, we analyzed how the T485A and T485E mutants would behave with respect to a different E6AP substrate, Ring1B. In agreement with previous studies, we found increased levels of Ring1B degradation and a modest increase in ubiquitination in the presence of the T485A mutant, but reduced levels of enzymatic activity with the T485E mutation. It should also be noted that minor differences from previously published studies could be a reflection of the fact that all our current analyses have been performed in cells in which E6AP expression was stably ablated by CRISPR/Cas9, while previous studies analyzed the E6AP mutants in the context of low levels of endogenously expressed wild-type E6AP, thus potentially complicating interpretation.
We were then interested in determining whether the same regulation of E6AP, reported for itself and its normal substrates, also applied to substrates that were targeted as a result of the interaction with E6. We analyzed two very different targets, p53 and Dlg, each of which interacts with E6 through completely different mechanisms. In both cases we found a striking similarity in that the phosphomodulation of T485 appeared to have no effect upon the capacity of either HPV-16 or HPV-18 E6 to degrade either of these cellular proteins. Similar results were also obtained with HPV-31 E6, indicating that this also holds true for E6 from multiple HPV types. This suggests that E6 recruitment of E6AP overrides the normal regulatory mechanisms that are in place to control E6AP activity.
Most interestingly, other aspects of the E6-E6AP interaction do appear to be affected by the phosphorylation status of T485. While previous analyses had shown that T485A was active for autodegradation and T485E was defective, we find that, depending upon the HPV type, these activities of E6AP are affected differently. Thus, HPV-16 E6 and HPV-31 E6 can efficiently target both the wild-type E6AP and the T485E mutant for degradation, indicating that HPV-16 E6 and HPV-31 E6 can promote the autodegradation of E6AP regardless of the phosphostatus at T485. Interestingly, HPV-16 E6 and HPV-31 E6 also seem capable of further augmenting the autodegradation activity of the T485A mutant. In contrast, HPV-18 E6 can efficiently target WT E6AP but is defective with respect to the T485A mutation, indicating that HPV-18 E6 cannot further stimulate the already highly active autodegradatory activity. In addition, despite T485E being very susceptible to HPV-16 and HPV-31 E6-induced degradation, only a weak activity is seen with HPV-18 E6. This indicates that while HPV-18 E6 can still redirect an apparently inactive T458E mutant to target an E6 substrate, it cannot promote T485E autodegradation. Taken together, these results demonstrate that different HPV E6 oncoproteins can uncouple E6AP from its normal regulation by PKA, presumably by conferring a structural modification upon E6AP itself, but that there are nonetheless quite marked differences in how different E6 oncoproteins bring this about.
Having found that E6 can significantly alter the biochemical regulation of E6AP, we also wanted to determine whether E6 could have any impact upon the subcellular distribution of phospho-E6AP. This was important, since previous studies had indicated that phospho-E6AP was located primarily in the cytoplasm, whereas E6 is known to recruit E6AP to the nucleus (20, 32, 33). To do this, we performed a series of immunofluorescence analyses on HeLa cells and found that in the presence of HPV-18 E6, E6AP was largely found within the nucleus, regardless of its phosphorylation status. However, when E6 was removed, total E6AP was found within the nucleus and cytoplasm, whereas phospho-E6AP was found almost entirely within the cytoplasm. This suggests that the phosphorylation of E6AP can modulate its subcellular distribution, but this is completely overridden by E6.
Since changes in the subcellular distribution of many phosphorylated proteins are controlled by members of the 14-3-3 family (25), which have also been shown to be important partners of the high-risk HPV E6 oncoproteins (18), we were naturally interested in investigating whether 14-3-3 played any role in the regulation of E6AP subcellular distribution. To do this, we made use of a very well-characterized inhibitor of 14-3-3, Difopein, which blocks 14-3-3 interaction with its target proteins (26). Most interestingly, we found that Difopein only had an effect on the subcellular distribution of the phospho-E6AP in the presence of E6, where inhibition of 14-3-3, while having minimal effects on the total E6AP expression pattern, induced a marked relocalization of phospho-E6AP from the nucleus to the cytoplasm. These results indicate that E6-triggered nuclear accumulation of phospho-E6AP is partly dependent upon the activity of 14-3-3 family members. Obviously this raises a number of important questions about the precise mechanisms by which this occurs, but it is tempting to speculate that optimal recruitment of phospho-E6AP to the nucleus requires the ability of E6 to also recognize 14-3-3 proteins. It is also interesting to speculate as to why E6 might wish to recruit phospho-E6AP to the nucleus. Under normal circumstances, this form of the protein would be inactive, and it is quite possible that, in certain phases of the cell cycle or in differentiation, E6AP becomes highly phosphorylated. Therefore, without the ability of E6 to override this phosphoregulation, it is quite possible that there would be times when E6 would lose much of its function. Hence, the recruitment of an apparently inactive form of the protein to the nucleus and reactivating its ability to degrade E6 substrates would appear to make very good virological sense. Future studies will obviously be required to investigate these aspects further.
In conclusion, these studies demonstrate that E6 very efficiently overcomes the negative regulation of E6AP activity; however, the precise mechanisms and consequences with respect to the ability of E6 to promote E6AP autoubiquitination vary somewhat between different HPV types, thus implying quite distinct mechanisms by which different HPV E6 oncoproteins redirect the E6AP ubiquitin ligase activity.
MATERIALS AND METHODS
Cell culture and transfection.
The CRISPR/Cas9 guide RNA (gRNA)-mediated E6AP-null HEK 293 cells and HPV-18-positive HeLa cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% bovine fetal serum (FBS), penicillin-streptomycin (100 U/ml), and glutamine (300 μg/ml). Transfection of plasmids was done by using calcium phosphate precipitation. The HeLa cells were transfected with siRNA against appropriate genes by using Lipofectamine RNAiMax transfection reagent as recommended by the manufacturer (Invitrogen). The 14-3-3 inhibitor Difopein was transfected into HeLa cells by using Effectene transfection reagent (Qiagen). The cells were analyzed by immunofluorescence and confocal microscopy 72 h posttransfection.
Plasmid constructs.
pcDNA3 FLAG-p53 (34), pGWI HA-Dlg (35), pGWI HA-18E6, and pGWI HA-16E6 have been described previously (36, 37). HA-tagged 31E6 was subcloned into pGWI vector within compatible HindIII and EcoRI restriction enzyme sites, and pcDNA3 HA-Ub(n) was described previously (38).
HPV-18 E6 and HPV-18 E6 T156E GST fusion proteins were expressed and purified as described previously (18, 19, 37). pGEX-E6AP expression plasmid was described previously (38). pGEX-E6AP T485A was generated by using a GeneArt site-directed mutagenesis kit (Invitrogen). The primer sequences are the following: forward, GTGAACGAAGAATCGCTGTTCTCTACAGC; reverse, GCTGTAGAGAACAGCGATTCTTCGTTCAC.
pCS2-6Myc-Ring1B was a kind gift from Aaron Ciechanover. Ring1B was subcloned into pGWI-FLAG vector by using HindIII and EcoRI restriction enzyme sites. The 14-3-3 inhibitor Myc-Difopein was a kind gift from Haian Fu. Myc-E6AP, Myc-E6AP T485A, and Myc-E6AP T485E were kind gifts from Mark Zylka, USA.
In vitro phosphorylation.
In vitro phosphorylation assays using kinase buffer (25 mM Tris, pH 7.5, 70 mM NaCl, 10 mM MgCl2) containing 2.5μCi [γ-32P]ATP and 25 U of cyclic AMP-dependent protein kinase, catalytic subunit (Promega). The reaction was carried out at 30°C for 20 min. The samples were then washed and analyzed by SDS-PAGE and autoradiography.
In vitro binding assays.
In vitro phosphorylation of the GST fusion proteins was carried out as described above in the presence of 10 μM nonradiolabeled ATP (NEB). After extensive washes, both the phosphorylated and nonphosphorylated GST fusion proteins were incubated with 100 ng of purified human recombinant 14-3-3γ protein (Abcam) at 4°C for 1 h. The samples were then washed and analyzed by SDS-PAGE and Western blotting.
gRNA design and CRISPR/Cas9-mediated gene targeting.
Two different guide RNAs (gRNAs) were designed against the E6AP gene by using DNA2.0 software. The following gRNA sequences were used: gRNA1_F, GTTTCCAGGGGGTCCACTCG; gRNA1_R, CGAGTGGACCCCCTGGAAAC; gRNA2_F, AAGTGGTTTTCGACAATCCA; and gRNA2_R, TGGATTGTCGAAAACCACTT. gRNA1 binds at genomic locus 15:25370913-25370932, and gRNA2 binds at 15:25370857-25370876. The gRNAs were then cloned into pSpCas9(BB)-2A-Puro (PX459) by using the BbsI restriction enzyme site. The Cas9-puro plasmids containing gRNA were then transfected into HEK293 cells, and the single-cell clones were selected by using puromycin. The mutation in E6AP was then verified by picking individual clones and analyzing the relevant region of the E6AP genomic DNA by PCR and DNA sequencing. Verification of the loss of E6AP was performed by Western blotting for E6AP.
Antibodies.
Anti-Myc mouse monoclonal antibody (Santa Cruz Biotechnology), anti-FLAG mouse monoclonal antibody M2 (Sigma), anti-β-galactosidase mouse monoclonal antibody (Promega), monoclonal anti-HA-peroxidase antibody produced in mouse clone HA-7 (Sigma-Aldrich), rabbit 14-3-3γ antibody (Santa Cruz Biotechnology), and appropriate secondary antibodies conjugated to horseradish peroxidase (HRP) (Dako) were used. For immunofluorescence, mouse monoclonal anti-E6AP (BD Transduction Laboratories), rabbit polyclonal anti-p53 (Santa Cruz Biotechnology), and chicken anti-(pT485) E6AP (a kind gift from Mark Zylka, USA) were used. Appropriate Alexa-Fluor secondary antibodies (Life Technologies) were used.
Western blotting.
Total cell extracts were obtained by lysing the cells directly in 2× SDS-PAGE sample buffer. Western blotting and processing were then performed as described previously (23) and developed using the ECL detection system (Amersham).
Ubiquitination assays.
For ubiquitination assays, the relevant plasmids were transfected into E6AP-null HEK293 cells, and after 24 h cell lysates were prepared, followed by immunoprecipitation using anti-FLAG-conjugated agarose beads to pull down ubiquitin-conjugated proteins (Sigma-Aldrich), as described previously (23). The beads were then washed and polyubiquitinated Ring1B protein was detected using Western blotting.
ACKNOWLEDGMENTS
We are most grateful to Jason Yi and Mark Zylka for providing the E6AP expression plasmids and the anti-phospho-E6AP antibody, to Aaron Ciechanover for providing the Ring1B expression plasmid, and to Haian Fu for providing the Difopein expression construct.
We are also grateful to Miranda Thomas and Mike Myers for comments on the manuscript.
J.T. is a recipient of an Arturo Falaschi ICGEB predoctoral fellowship and is registered with the Open University, United Kingdom. This work was supported in part by a research grant from the Associazione Italiana per la Ricerca sul Cancro, project no. 18578.
REFERENCES
- 1.zur Hausen H. 2009. Papillomaviruses in the causation of human cancers–a brief historical account. Virology 384:260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 2.Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA. 2012. The biology and life-cycle of human papillomaviruses. Vaccine 30(Suppl 5):F55–F70. doi: 10.1016/j.vaccine.2012.06.083. [DOI] [PubMed] [Google Scholar]
- 3.Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V, WHO International Agency for Research on Cancer Monograph Working Group. 2009. A review of human carcinogens–part B: biological agents. Lancet Oncol 10:321–322. doi: 10.1016/S1470-2045(09)70096-8. [DOI] [PubMed] [Google Scholar]
- 4.Smotkin D, Wettstein FO. 1986. Transcription of human papillomavirus type 16 early genes in a cervical cancer and a cancer-derived cell line and identification of the E7 protein. Proc Natl Acad Sci U S A 83:4680–4684. doi: 10.1073/pnas.83.13.4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banks L, Spence P, Androphy E, Hubbert N, Matlashewski G, Murray A, Crawford L. 1987. Identification of human papillomavirus type 18 E6 polypeptide in cells derived from human cervical carcinomas. J Gen Virol 68(Part 5):1351–1359. doi: 10.1099/0022-1317-68-5-1351. [DOI] [PubMed] [Google Scholar]
- 6.Butz K, Ristriani T, Hengstermann A, Denk C, Scheffner M, Hoppe-Seyler F. 2003. siRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene 22:5938–5945. doi: 10.1038/sj.onc.1206894. [DOI] [PubMed] [Google Scholar]
- 7.Yoshinouchi M, Yamada T, Kizaki M, Fen J, Koseki T, Ikeda Y, Nishihara T, Yamato K. 2003. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by E6 siRNA. Mol Ther 8:762–768. doi: 10.1016/j.ymthe.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. 2004. Mechanisms of human papillomavirus-induced oncogenesis. J Virol 78:11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 10.Thomas M, Banks L. 1998. Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene 17:2943–2954. doi: 10.1038/sj.onc.1202223. [DOI] [PubMed] [Google Scholar]
- 11.Banks L, Pim D, Thomas M. 2012. Human tumour viruses and the deregulation of cell polarity in cancer. Nat Rev Cancer 12:877–886. doi: 10.1038/nrc3400. [DOI] [PubMed] [Google Scholar]
- 12.Scheffner M, Huibregtse JM, Howley PM. 1994. Identification of a human ubiquitin-conjugating enzyme that mediates the E6-AP-dependent ubiquitination of p53. Proc Natl Acad Sci U S A 91:8797–8801. doi: 10.1073/pnas.91.19.8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 14.Thomas M, Tomaic V, Pim D, Myers MP, Tommasino M, Banks L. 2013. Interactions between E6AP and E6 proteins from alpha and beta HPV types. Virology 435:357–362. doi: 10.1016/j.virol.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Firzlaff JM, Galloway DA, Eisenman RN, Luscher B. 1989. The E7 protein of human papillomavirus type 16 is phosphorylated by casein kinase II. New Biol 1:44–53. [PubMed] [Google Scholar]
- 16.Massimi P, Pim D, Storey A, Banks L. 1996. HPV-16 E7 and adenovirus E1a complex formation with TATA box binding protein is enhanced by casein kinase II phosphorylation. Oncogene 12:2325–2330. [PubMed] [Google Scholar]
- 17.Zine El Abidine A, Tomaic V, Bel Haj Rhouma R, Massimi P, Guizani I, Boubaker S, Ennaifer E, Banks L. 2017. A naturally occurring variant of HPV-16 E7 exerts increased transforming activity through acquisition of an additional phospho-acceptor site. Virology 500:218–225. doi: 10.1016/j.virol.2016.10.023. [DOI] [PubMed] [Google Scholar]
- 18.Boon SS, Banks L. 2013. High-risk human papillomavirus E6 oncoproteins interact with 14-3-3zeta in a PDZ binding motif-dependent manner. J Virol 87:1586–1595. doi: 10.1128/JVI.02074-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boon SS, Tomaic V, Thomas M, Roberts S, Banks L. 2015. Cancer-causing human papillomavirus E6 proteins display major differences in the phospho-regulation of their PDZ interactions. J Virol 89:1579–1586. doi: 10.1128/JVI.01961-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yi JJ, Berrios J, Newbern JM, Snider WD, Philpot BD, Hahn KM, Zylka MJ. 2015. An autism-linked mutation disables phosphorylation control of UBE3A. Cell 162:795–807. doi: 10.1016/j.cell.2015.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mortensen F, Schneider D, Barbic T, Sladewska-Marquardt A, Kuhnle S, Marx A, Scheffner M. 2015. Role of ubiquitin and the HPV E6 oncoprotein in E6AP-mediated ubiquitination. Proc Natl Acad Sci U S A 112:9872–9877. doi: 10.1073/pnas.1505923112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaaroor-Regev D, de Bie P, Scheffner M, Noy T, Shemer R, Heled M, Stein I, Pikarsky E, Ciechanover A. 2010. Regulation of the polycomb protein Ring1B by self-ubiquitination or by E6-AP may have implications to the pathogenesis of Angelman syndrome. Proc Natl Acad Sci U S A 107:6788–6793. doi: 10.1073/pnas.1003108107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomaic V, Pim D, Banks L. 2009. The stability of the human papillomavirus E6 oncoprotein is E6AP dependent. Virology 393:7–10. doi: 10.1016/j.virol.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 24.Gardiol D, Kuhne C, Glaunsinger B, Lee SS, Javier R, Banks L. 1999. Oncogenic human papillomavirus E6 proteins target the discs large tumour suppressor for proteasome-mediated degradation. Oncogene 18:5487–5496. doi: 10.1038/sj.onc.1202920. [DOI] [PubMed] [Google Scholar]
- 25.Muslin AJ, Xing H. 2000. 14-3-3 proteins: regulation of subcellular localization by molecular interference. Cell Signal 12:703–709. doi: 10.1016/S0898-6568(00)00131-5. [DOI] [PubMed] [Google Scholar]
- 26.Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, Thomas LM, Al-Qahtani A, White CA, Park SR, Steinacker P, Li Z, Yates J III, Herron B, Otto M, Zan H, Fu H, Casali P. 2010. 14-3-3 adaptor proteins recruit AID to 5′-AGCT-3′-rich switch regions for class switch recombination. Nat Struct Mol Biol 17:1124–1135. doi: 10.1038/nsmb.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao W, Yang X, Zhou J, Teng Z, Cao L, Zhang X, Fei Z. 2010. Targeting 14-3-3 protein, difopein induces apoptosis of human glioma cells and suppresses tumor growth in mice. Apoptosis 15:230–241. doi: 10.1007/s10495-009-0437-4. [DOI] [PubMed] [Google Scholar]
- 28.Herber R, Liem A, Pitot H, Lambert PF. 1996. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. J Virol 70:1873–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shai A, Nguyen ML, Wagstaff J, Jiang YH, Lambert PF. 2007. HPV16 E6 confers p53-dependent and p53-independent phenotypes in the epidermis of mice deficient for E6AP. Oncogene 26:3321–3328. doi: 10.1038/sj.onc.1210130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shai A, Pitot HC, Lambert PF. 2010. E6-associated protein is required for human papillomavirus type 16 E6 to cause cervical cancer in mice. Cancer Res 70:5064–5073. doi: 10.1158/0008-5472.CAN-09-3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Padash Barmchi M, Gilbert M, Thomas M, Banks L, Zhang B, Auld VJ. 2016. A Drosophila model of HPV E6-induced malignancy reveals essential roles for Magi and the insulin receptor. PLoS Pathog 12:e1005789. doi: 10.1371/journal.ppat.1005789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vaeteewoottacharn K, Chamutpong S, Ponglikitmongkol M, Angeletti PC. 2005. Differential localization of HPV16 E6 splice products with E6-associated protein. Virol J 2:50. doi: 10.1186/1743-422X-2-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daniels PR, Sanders CM, Maitland NJ. 1998. Characterization of the interactions of human papillomavirus type 16 E6 with p53 and E6-associated protein in insect and human cells. J Gen Virol 79(Part 3):489–499. doi: 10.1099/0022-1317-79-3-489. [DOI] [PubMed] [Google Scholar]
- 34.Pim D, Massimi P, Banks L. 1997. Alternatively spliced HPV-18 E6* protein inhibits E6 mediated degradation of p53 and suppresses transformed cell growth. Oncogene 15:257–264. doi: 10.1038/sj.onc.1201202. [DOI] [PubMed] [Google Scholar]
- 35.Gardiol D, Galizzi S, Banks L. 2002. Mutational analysis of the discs large tumour suppressor identifies domains responsible for human papillomavirus type 18 E6-mediated degradation. J Gen Virol 83:283–289. doi: 10.1099/0022-1317-83-2-283. [DOI] [PubMed] [Google Scholar]
- 36.Pim D, Thomas M, Javier R, Gardiol D, Banks L. 2000. HPV E6 targeted degradation of the discs large protein: evidence for the involvement of a novel ubiquitin ligase. Oncogene 19:719–725. doi: 10.1038/sj.onc.1203374. [DOI] [PubMed] [Google Scholar]
- 37.Thomas M, Matlashewski G, Pim D, Banks L. 1996. Induction of apoptosis by p53 is independent of its oligomeric state and can be abolished by HPV-18 E6 through ubiquitin mediated degradation. Oncogene 13:265–273. [PubMed] [Google Scholar]
- 38.Tomaic V, Pim D, Thomas M, Massimi P, Myers MP, Banks L. 2011. Regulation of the human papillomavirus type 18 E6/E6AP ubiquitin ligase complex by the HECT domain-containing protein EDD. J Virol 85:3120–3127. doi: 10.1128/JVI.02004-10. [DOI] [PMC free article] [PubMed] [Google Scholar]