

ABSTRACT
A novel archaeal lytic virus targeting species of the genus Methanosarcina was isolated using Methanosarcina mazei strain Gö1 as the host. Due to its spherical morphology, the virus was designated Methanosarcina spherical virus (MetSV). Molecular analysis demonstrated that MetSV contains double-stranded linear DNA with a genome size of 10,567 bp containing 22 open reading frames (ORFs), all oriented in the same direction. Functions were predicted for some of these ORFs, i.e., such as DNA polymerase, ATPase, and DNA-binding protein as well as envelope (structural) protein. MetSV-derived spacers in CRISPR loci were detected in several published Methanosarcina draft genomes using bioinformatic tools, revealing a potential protospacer-adjacent motif (PAM) motif (TTA/T). Transcription and expression of several predicted viral ORFs were validated by reverse transcription-PCR (RT-PCR), PAGE analysis, and liquid chromatography-mass spectrometry (LC-MS)-based proteomics. Analysis of core lipids by atmospheric pressure chemical ionization (APCI) mass spectrometry showed that MetSV and Methanosarcina mazei both contain archaeol and glycerol dialkyl glycerol tetraether without a cyclopentane moiety (GDGT-0). The MetSV host range is limited to Methanosarcina strains growing as single cells (M. mazei, Methanosarcina barkeri and Methanosarcina soligelidi). In contrast, strains growing as sarcina-like aggregates were apparently protected from infection. Heterogeneity related to morphology phases in M. mazei cultures allowed acquisition of resistance to MetSV after challenge by growing cultures as sarcina-like aggregates. CRISPR/Cas-mediated resistance was excluded since neither of the two CRISPR arrays showed MetSV-derived spacer acquisition. Based on these findings, we propose that changing the morphology from single cells to sarcina-like aggregates upon rearrangement of the envelope structure prevents infection and subsequent lysis by MetSV.
IMPORTANCE Methanoarchaea are among the most abundant organisms on the planet since they are present in high numbers in major anaerobic environments. They convert various carbon sources, e.g., acetate, methylamines, or methanol, to methane and carbon dioxide; thus, they have a significant impact on the emission of major greenhouse gases. Today, very little is known about viruses specifically infecting methanoarchaea that most probably impact the abundance of methanoarchaea in microbial consortia. Here, we characterize the first identified Methanosarcina-infecting virus (MetSV) and show a mechanism for acquiring resistance against MetSV. Based on our results, we propose that growth as sarcina-like aggregates prevents infection and subsequent lysis. These findings allow new insights into the virus-host relationship in methanogenic community structures, their dynamics, and their phase heterogeneity. Moreover, the availability of a specific virus provides new possibilities to deepen our knowledge of the defense mechanisms of potential hosts and offers tools for genetic manipulation.
KEYWORDS: Methanosarcina mazei strain Gö1, virus, methanoarchaea, CRISPR/Cas, virus for methanoarchaea
INTRODUCTION
Since the first archaeal viruses were described in the 1970s, more than 100 have been reported (recently reviewed in reference 1). In addition to viruses infecting crenarchaea or halophilic archaea, only very few viruses, proviruses, and virus-like particles infecting methanoarchaea have been discovered thus far (reviewed in references 2 and 3). Two of them, ψΜ1 and ψΜ2, have been described infecting Methanothermobacter marburgensis (formerly known as Methanobacterium thermoautotrophicum). Both viruses consist of a polyhedral head and a tail (4–6) and contain linear double-stranded DNA (dsDNA) with genome sizes of approximately 27 kb and 26.3 kb, respectively. Moreover, ψΜ2 is a deletion derivative of ψΜ1 that originated by homologous recombination under laboratory conditions. Furthermore, a defective provirus derivative with approximately 70% identity to the ψM2 genome has been identified in the genome of Methanothermobacter wolfeii (7). Two morphologically similar viruses, i.e., ΦF1 and ΦF3, have been isolated from an anaerobic digester. These viruses also consist of a polyhedral head and a tail, but they differ in genome size, genome organization, and host specificity. Virus ΦF1 comprises a linear dsDNA with a size of approximately 85 ± 5 kb and a broad host range. Virus ΦF3 contains dsDNA organized in a circular or terminally redundant linear genome with a size of approximately 36 ± 2 kb and is only able to infect Methanobacterium thermoformicicum. Sequence analysis revealed no genome similarities between ΦF1 and ΦF3 (8).
A few virus-like particles (VLPs) have been reported which infect methanoarchaea. The first VLPs associated with Methanococcus voltae A3 were described by Wood et al. They detected extrachromosomal closed circular DNA and particles which appeared similar to viruses. Interestingly, an integrated chromosomal copy was verified; however, neither infectivity nor inducibility of the VLPs could be demonstrated (9). Sequence analyses revealed an integrated copy of the A3 VLP in the genome of M. voltae A3 inside a provirus (MVV). It appears that the A3 VLP was integrated into the preintegrated proviral genome, which might have led to the inactivation of the provirus (10). When Methanofollis aquaemaris sp. nov. is in the late exponential phase of growth, VLP release by budding has been demonstrated, indicating the presence of a provirus, which was named MMV (11). Further VLPs have been detected that are able to infect the archaeal genus Methanosaeta in the deep and anoxic sediments of the volcanic Lake Pavin (France) (12) and in Methanosaeta-dominated methanogenic reactors (13). In addition, several proviruses were identified by genome sequence analyses in strains of Methanobrevibacter ruminantium, Methanobrevibacter smithii, and Methanococcus jannaschii (14–17).
Here, we report the isolation and characterization of a new lytic archaeal virus infecting Methanosarcina strains, which now allows new insights into resistance formation in Methanosarcina. In addition, these findings offer enormous potential for analyzing the function of CRISPR/Cas systems in Methanosarcina strains and the development of additional genetic tools.
RESULTS
Isolation and morphological characterization of virus MetSV.
The Methanosarcina spherical virus (MetSV) was enriched and isolated from anaerobic sewage sludge collected in the digestion tower of a municipal wastewater treatment plant (Göttingen, Germany) by serially challenging Methanosarcina mazei DSM 3647 growing in liquid cultures with solutions of filtered sludge. Transmission electron microscopy of negatively stained virus preparations revealed a spherical shape with a diameter of 56.6 ± 1.9 nm and a blackberry-like envelope of the virus (Fig. 1). Disrupted virus particles (i.e., ghost particles without DNA) were typically filled with the dark staining solution. These damaged particles exhibited a unique thick structure of the coat envelope. The MetSV virus exhibits an overall morphotype similar to that of the previously reported Pyrobaculum spherical virus (PSV) infecting Pyrobaculum or Thermoproteus (18); however, MetSV has a significantly smaller particle diameter than PSV (with a diameter of approximately 100 nm), and no helical nucleoparticles were detected. Due to the overall morphological appearance of the virus, it was designated Methanosarcina spherical virus (MetSV).
FIG 1.
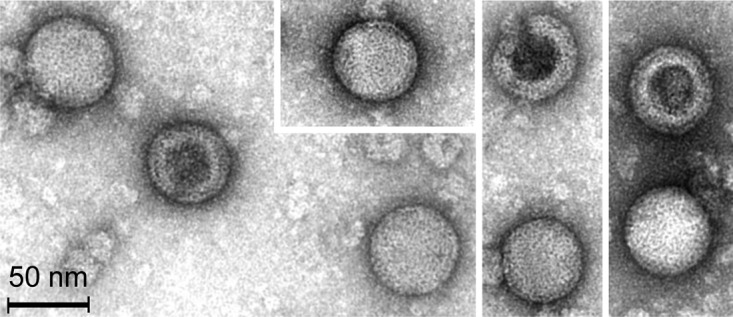
Transmission electron micrograph of five intact and three empty (negatively stained) MetSV particles.
The linear genome of MetSV.
DNA from MetSV was isolated as described in the Materials and Methods section. Subsequent treatment with different restriction endonucleases demonstrated a linear, double-stranded DNA (dsDNA) with an approximate length of 11 kb (data not shown). The DNA was sequenced using a high-throughput shotgun sequencing approach (using 454 sequencing technology) and in parallel by subcloning, followed by Sanger sequencing. The size of the MetSV genome was determined to be 10,567 bp, which was in agreement with the endonuclease restriction analyses. Both ends of the linear genome were flanked by inverted terminal repeats (ITRs) 59 nucleotides (nt) long. The G+C content was determined to be 38.46%, which is in a range similar to the G+C content of the host DNA (41.5%) (19). The MetSV genome contains 22 open reading frames (ORFs) encoding potential proteins ranging from 3,630 Da to 36,700 Da. These include 11 long ORFs encoding proteins with a predicted size of approximately >90 amino acids (aa) (Fig. 2) and 11 small ORFs (sORFs) encoding proteins with a size of ≤90 aa. Promoter elements typical for Methanosarcina (TATA and BRE boxes) were identified for several ORFs (Table 1). All predicted ORFs were located on the same DNA strand, resulting in a consistent direction of transcription (Fig. 2). Of the 22 ORFs, 15 showed very low or no similarities to sequences in public databases, while only 4 ORFs showed homologies to proteins with an assigned function. ORF7 showed homology to DNA polymerase B (37% local identity [67 aa/179 aa]), and ORF13 showed homology to an ATPase (30% local identity [64 aa/213 aa]) on the amino acid level. The latter contained a P-loop NTPase (P-loop containing nucleoside triphosphate hydrolase) domain and an HerA domain, as predicted using NCBI's conserved domains (20). These ATPases are known to be involved in viral genome packing and segregation (21). For ORF6, a ribbon-helix-helix domain, which is often part of DNA binding proteins like transcriptional regulators, was predicted using Phyre2 (22). ORF22 contains an Env-gp36 domain, which is part of an envelope-forming protein in the lentivirus feline immunodeficiency virus (FIV) (23), and this finding potentially suggests that ORF22 represents the structural coating protein. Three ORFs showed similarities to hypothetical proteins (ORF3, ORF9, and ORF16). Furthermore, secondary-structure predictions were performed using the Phyre2 software. Thereby seven ORFs with larger amounts of beta sheets were detected; however, no jelly roll structures were found, which are characteristic of major capsid proteins of icosahedral viruses (Table 1).
FIG 2.

Genome map of MetSV. All predicted ORFs with a size of >90 aa are depicted. Dark gray arrows indicate ORFs with a predicted function. All predicted ORFs are located on the same strand and thus have the same direction of transcription. The nucleotide sequences of the inverted terminal repeats (ITRs) on both ends are depicted.
TABLE 1.
Predicted ORFs of MetSV
| Predicted ORF | Genome position (nt) | BRE and TATA box sequences (starting positions)a | No. of amino acids (predicted protein size [Da]) | Secondary structure predictionb | DELTA-BLAST result |
Predicted domain (predicted function)c | Proteome analysisd | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Similar protein(s) (organism) | Query coverage (%) | E value | % Identity | |||||||
| ORF1 | 295–465 | GAATTTTAAAT (−28, −25) | 56 (6,160) | 47% Disordered, 78% α-helix, 7% β-strand | Pyruvate formate-lyase (Desulfuromonas sp. strain TF) | 89 | 6.7 | 42 | +/− | |
| ORF2 | 608–733 | GAA-N2-TATAGTTATA (−38, −33) | 41 (4,510) | 27% Disordered, 80% α-helix, 0% β-strand | None | + | ||||
| ORF3 | 733–876 | AAA-N2-TAATGAAAAA (−35, −30) | 47 (5,170) | 23% Disordered, 49% α-helix, 17% β-strand | Predicted protein piccolo, partial (Myotis brandtii) | 81 | 0.078 | 41 | DUF4451, domain with unknown function | |
| ORF4 | 903–1025 | Not detected | 41 (4,510) | 29% Disordered, 29% α-helix, 32% β-strand | None | + | ||||
| ORF5 | 1167–1445 | Not detected | 92 (10,120) | 14% Disordered, 14% α-helix, 57% β-strand | None | + | ||||
| ORF6 | 1417–2151 | CAA-N-AATATA (−25, −21) | 244 (26,840) | 25% Disordered, 84% α-helix, 0% β-strand | Uncharacterized protein Dsimw501_GD16851 (Drosophila simulans) | 55 | 5.8 | 18 | Ribbon-helix-helix-domain (potential DNA-binding protein) | +/− |
| ORF7 | 2313–3392 | GTA-N4-AAAAGTAAA (−50, −43) | 359 (39,490) | 9% Disordered, 32% α-helix, 22% β-strand | Type B DNA polymerase (Bacillus cereus) | 72 | 2e−16 | 30 | Conserved protein domain family, DNA_pol_B_2 (DNA polymerase) | +/− |
| ORF8 | 3728–3967 | GAA-N3-TAAAATAT (−45, −39) | 79 (8,690) | 35% Disordered, 38% α-helix, 16% β-strand | Hypothetical protein AHOG_08260 (Actinoalloteichus hoggarensis) | 75 | 2.9 | 35 | +/− | |
| ORF9 | 3999–4187 | Not detected | 62 (6,820) | 10% Disordered, 90% α-helix, 0% β-strand | Hypothetical protein (Bacillus sp. strain FJAT-22058) | 66 | 0.068 | 44 | Conserved protein domain family, urocanase | + |
| ORF10 | 4344–4697 | GAC-N5-TTTAAAGTATATA (−35, −27) | 117 (12,870) | 35% Disordered, 68% α-helix, 2% β-strand | Hypothetical protein A2020_10980 (Lentisphaerae bacterium GWF2_45_14) | 32 | 0.34 | 45 | Conserved protein domain family HTH/conserved protein domain family, Zn-ribbon_8 | + |
| ORF11 | 4679–4822 | Not detected | 47 (5,170) | 15% Disordered, 0% α-helix, 89% β-strand | Hypothetical protein SBOR_1092 (Sclerotinia borealis F-4128) | 85 | 1.6 | 38 | +/− | |
| ORF12 | 4819–4944 | Not detected | 41 (4,510) | 32% Disordered, 7% α-helix, 51% β-strand | None | |||||
| ORF13 | 5263–5871 | GAG-N-TTTTAAAAATTAT (−31, −27) | 202 (22,220) | 8% Disordered, 51% α-helix, 18% β-strand | ATPase (Halodesulfurarchaeum formicicum) | 84 | 1e−10 | 30 | (ATPase) | +/− |
| ORF14 | 6007–6108 | Not detected | 33 (3,630) | 67% Disordered, 0% α-helix, 27% β-strand | None | |||||
| ORF15 | 6020–6328 | GAC-N4-TTTTTTTAATT (−52, −45) | 102 (11,220) | 24 % Disordered, 36 % α-helix, 27 % β-strand | MCE family protein (Mycobacterium obuense) | 98 | 1.3 | 32 | + | |
| ORF16 | 6403–6843 | CAACACATTAAA (−24, −18) | 146 (16,060) | 25% Disordered, 24% α-helix, 45% β-strand | Hypothetical protein APR63_09490 (Desulfuromonas sp. strain SDB) | 88 | 3e−06 | 34 | + | |
| ORF17 | 7024–7191 | GAC-N3-AATAAT (−24, −18) | 55 (6,050) | 11% Disordered, 91% α-helix, 0% β-strand | Hypothetical protein SAMN05444173_2079 (Opitutus sp. strain GAS368) | 73 | 0.89 | 36 | +/− | |
| ORF18 | 7202–7522 | GAC-N-ATTTAAA (−31, −27) | 106 (11,660) | 50% Disordered, 52% α-helix, 14% β-strand | Hypothetical protein (Pseudoalteromonas sp. strain S3431) | 61 | 2.3 | 35 | + | |
| ORF19 | 7519–7791 | Not detected | 90 (9,900) | 16% Disordered, 16% α-helix, 49% β-strand | None | + | ||||
| ORF20 | 7804–8547 | GAGTATTA (−29, −26) | 247 (27,170) | 11% Disordered, 13% α-helix, 54% β-strand | Hypothetical protein (Bacillus sp. strain JCM 19047) | 22 | 3.5 | 40 | + | |
| ORF21 | 8560–9393 | GACTATTT (−42, −39) | 277 (30,470) | 46% Disordered, 34% α-helix, 11% β-strand | None | + | ||||
| ORF22 | 9409–10413 | GAAAAATTAA (−25, −22) | 334 (36,740) | 9% Disordered, 16% α-helix, 48% β-strand | Beta-galactosidase (uncultured Ruminococcus sp.) | 32 | 3.9 | 27 | An Env-gp36 domain (putative envelope protein) | + |
The sequence of the BRE box is in boldface, and that of the TATA box is underlined. Starting nucleotide positions are given for the BRE and TATA boxes in respective order.
Predictions made using Phyre2.
For all ORFs except ORF6, functions were predicted based on an NCBI search. Phyre2 was used for ORF6.
Sequence coverage is represented as follows: +, about 50 %; +/−, between 30 and 49%.
Screening for MetSV-derived spacers in methanosarcinal CRISPR arrays.
M. mazei type strain DSM 3647 contains two CRISPR/Cas systems, which belong to the subtypes IB and IIIC according to the nomenclature from Makarova et al. and Vestergaard et al. (24–26). Both CRISPR arrays were screened for MetSV-derived spacers. No matches were detected in the type strain (M. mazei DSM 3647). Draft genomes of additional Methanosarcina isolates recently reported by Youngblut et al. (27) and available genomes from the NCBI database were also screened for specific MetSV-derived spacers. No spacers without a mismatch were identified. Only spacers with one to six mismatches were identified in Methanosarcina genomes (see Table S1 in the supplemental material). Responsible for the selection of certain protospacers on the viral genome is the protospacer-adjacent motif (PAM), which is defined by the respective CRISPR/Cas system. By analyzing the protospacers in MetSV, the potential PAM motif TTA/T was predicted.
Expression of viral genes during infection: RNA and protein patterns.
RNA was isolated at different time points after infection of M. mazei cultures with MetSV. Transcription of predicted MetSV genes ORF6, ORF7, ORF13, ORF15, ORF19, and ORF22 was evaluated by reverse transcription-PCR (RT-PCR) using specific primer pairs (see Table S2). Generally, 30 min after MetSV infection, transcripts of all tested MetSV ORFs were detected. Transcripts were still detectable after 90 min and 210 min postinfection (p.i.), as shown in Fig. 3.
FIG 3.
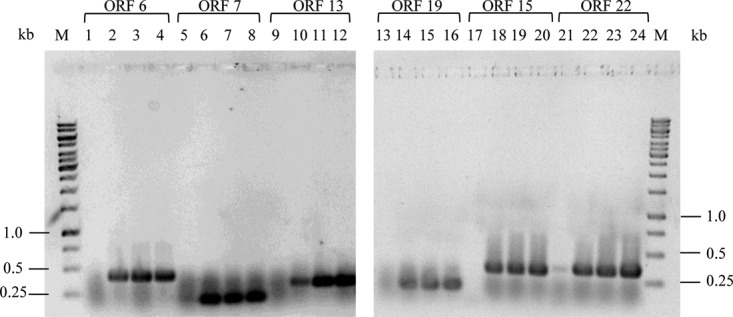
Detection of MetSV transcripts after M. mazei infection. At 0, 30, 90, and 210 min postinfection with MetSV, transcripts of the predicted ORFs 6, 7, 13, 19, and 22 were analyzed by RT-PCR using specific primer sets (see Table S2 in the supplemental material). The respective RT-PCR products were analyzed by agarose gel electrophoresis. ORFs are identified above the lane numbers. Time points are represented as follows: 0 min, lanes 1, 5, 9, 13, 17, and 21; 30 min, lanes 2, 6, 10, 14, 18, and 22; 90 min, lanes 3, 7, 11, 15, 19, and 23; 210 min, lanes 4, 8, 12, 16, 20, and 24. M, 1-kb marker (ThermoFisher Scientific, Darmstadt, Germany).
Furthermore, we performed stacking gel separation coupled to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis (28) of the purified MetSV proteome. Out of two samples, we identified 12 predicted ORF gene products with, on average, sequence coverage greater than 50% (see Tables S3 and S4). In addition to the 12 confidently identified protein species, single proteolytic peptides from three further virus proteins were found (data not shown). Additionally, two prominent protein bands with a size of approximately 11 kDa and 16 kDa were separately analyzed showing that the major components were gene products of ORF16 (16-kDa band) and ORF 15 (11-kDa band) (Fig. 4; see also Tables S5 and S6).
FIG 4.
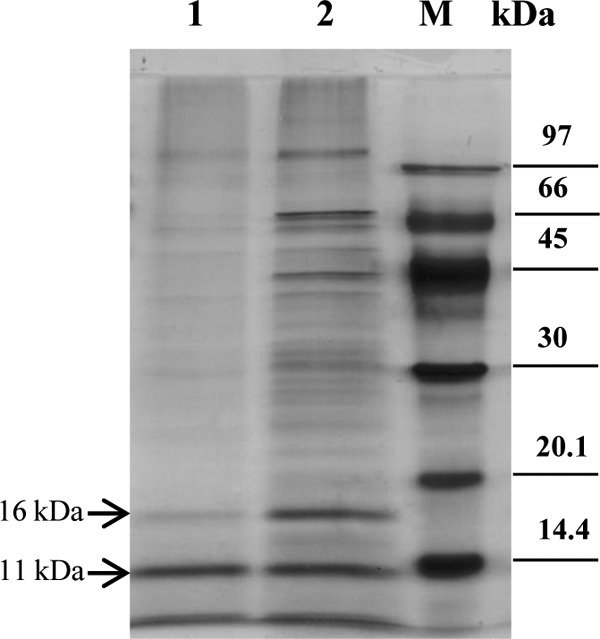
Protein pattern of MetSV. Total protein extracts of MetSV were separated by SDS-PAGE, followed by silver staining. Lane 1, MetSV protein extract after a second washing step of the virus particles; lane 2, MetSV protein extract without a second washing step; M, low-molecular-weight marker (GE Healthcare Europe GmbH, Freiburg, Germany).
Core lipid analysis.
M. mazei DSM 3647 and the purified virus fraction were analyzed by atmospheric pressure chemical ionization (APCI) mass spectrometry for their di- and tetraether content. We found that both samples contained the same core lipids in similar distributions but in significantly different quantities. Archaeol was the dominant membrane lipid that constituted 1318.85 and 40.81 ng/g dry cell material in the archaeal host and MetSV, respectively (Table 2 and Fig. 5). In addition, traces of glycerol dialkyl glycerol tetraether without a cyclopentane moiety (GDGT-0) were detected in M. mazei DSM 3647 (1.76 ng/g dry cell material) and MetSV (0.07 ng/g dry cell material). These results indicate that MetSV contains an internal membrane and incorporates lipids obtained from the host. To exclude the possibility of contamination of the virus fraction by archaeal lipids during sample processing, a control experiment with the filtered supernatant of a mature M. mazei culture was performed in which no core lipids were detected.
TABLE 2.
Core lipids of M. mazei DSM 3647 and MetSV measured by APCI mass spectrometry
| Sample | Amt of lipid (ng/g dry biomass [%]) |
|
|---|---|---|
| Archaeol | GDGT-0 | |
| M. mazei DSM 3647 | 1,318.85 (99.87) | 1.76 (0.13) |
| MetSV | 40.06 (99.83) | 0.07 (0.17) |
FIG 5.

HPLC-APCI-MS chromatogram. Distribution of archaeol and glycerol dialkyl glycerol tetraether without a cyclopentane moiety (GDGT-0) in M. mazei DSM 3647 (A), MetSV (B), and the control sample (C).
Host range of MetSV.
Different Methanosarcina strains available from the DSMZ were tested for sensitivity to MetSV. Actively growing cultures were incubated with MetSV, and the optical turbidity was monitored at 600 nm. Most of the tested strains growing as single cells were lysed by MetSV within 4 h (M. mazei strains DSM 3647/DSM 2244/DSM 4556/DSM 6300/DSM 7222 and M. barkeri strain DSM 1311). The only exception was Methanosarcina acetivorans DSM 2834TC2A growing as single cells, which, however, was not lysed by MetSV (summarized in Table 3). In contrast, all strains typically growing as sarcina-like aggregates were not affected by MetSV (strains M. mazei DSM 9195, M. barkeri DSM 800, Methanosarcina soligelidi DSM 26065, Methanosarcina sp. strain DSM 11855, and Methanosarcina siciliae DSM 3028). Control strains that included other methanogenic archaea such as Methanosphaera stadtmanae, Methanosaeta thermophila, or the Gram-negative bacterium Escherichia coli were not sensitive to MetSV (data not shown), strongly indicating a narrow host range of MetSV.
TABLE 3.
Host specificity and morphological phenotypes of different Methanosarcina strains
| Strain | Culture morphology | Isolate source | Effect of MetSV challenge |
|---|---|---|---|
| M. mazei Gö1* (3A) | Single cells | Laboratory derived strain | Lysis |
| M. mazei DSM 3647 | Single cells | Anaerobic sewage digester | Lysis |
| M. mazei DSM 2244 | Single cells | Cow dung | Lysis |
| M. mazei DSM 4556 | Single cells | Alkaline mud, oil exploration drilling site | Lysis |
| M. mazei DSM 6300 | Single cells | Biomethanation granules | Lysis |
| M. mazei DSM 7222 | Single cells | Anaerobic sewage digester | Lysis |
| M. mazei DSM 9195 | Sarcina-like aggregates | Paddy field soil | No lysis |
| M. barkeri DSM 800 | Sarcina-like aggregates | Anaerobic sewage digester | No lysis |
| M. barkeri DSM 1311 | Single cells | Anaerobic sediment | Lysis |
| M. acetivorans DSM 2834TC2A | Single cells | Marine mud | No lysis |
| M. soligelidi DSM 26065 | Sarcina-like aggregates | Permafrost-affected soil | No lysis |
| Methanosarcina sp. DSM 11855 | Sarcina-like aggregates | Ovine rumen | No lysis |
| M. siciliae DSM 3028 | Sarcina-like aggregates | Lake sediment | No lysis |
Challenging M. mazei with MetSV and establishment of viral resistance.
M. mazei DSM 3647 was grown to a turbidity of approximately 0.2 to 0.3 at 600 nm, and then MetSV-containing lysate was added. Growth immediately stopped, and cell lysis was observed after approximately 4 h of challenge; approximately 12 h later the majority of the cells were lysed (Fig. 6A). However, after several weeks of incubation at 37°C, the infected cultures started to grow again, and cells tended to form large aggregates. These cells grew as typical sarcina-like aggregates and were no longer affected by the addition of MetSV at various growth stages. Representative growth analyses are shown in Fig. 6B. Chromosomal DNA was isolated from four independent aggregated cultures and confirmed to represent M. mazei chromosomal DNA. To exclude an involvement of the CRISPR/Cas system during resistance formation, the CRISPR arrays were examined for newly acquired spacers against MetSV. Therefore, the first 29 spacers/repeats of the CRISPR IB array and the first 9 spacers/repeats of the CRISPR IIIC array were PCR amplified and analyzed by sequencing, demonstrating that a MetSV-derived spacer was not integrated in either array IB or IIIC. However, in several cases a rearrangement of spacers was observed, and spacers from the 3′ end of the array were moved to the front or directly after the leader, as depicted in Fig. 6C.
FIG 6.

Challenging M. mazei with MetSV and establishment of virus resistance. (A) Representative growth curve of early-exponential-phase growing M. mazei cultures (50 ml) (turbidity at 600 nm, ∼0.2) challenged with 0.5 ml of filtered MetSV-lysed culture supernatant (indicated by the arrow). After approximately 4 h most of the cells were lysed by MetSV. (B) At approximately 200 h p.i. with MetSV, the M. mazei (DSM 3467) culture started to form significant aggregates which were not susceptible to the virus. (C) The CRISPR IB arrays of four independent aggregated cultures (1 to 4) were cloned and sequenced, but no integration of an MetSV-specific spacer could be detected. However, rearrangements of the existing spacers (original numbering) were obtained. wt, wild type.
Effects on global transcription of the resistant M. mazei strain in the presence of MetSV.
Transcriptome analysis was performed using whole genomic DNA microarrays and cDNAs generated from total RNAs, which were isolated from uninfected cells and cells growing as aggregates at 200 h postinfection (p.i.) with MetSV. In three independent biological analyses, transcripts of 2,334 ORFs were detectable; of these 219 ORFs and 107 ORFs were identified with, respectively, 3-fold-higher and 3-fold-lower transcript abundances in the aggregated cells at 200 h p.i. with MetSV compared to levels in the noninfected cultures (Table 4 and Table S7). Several ORFs which have homologies to proteins involved in envelope formation and 35 ORFs annotated as transporters had significantly elevated transcript levels (>3-fold), for example, the potential operon MM_3095-MM_3098 encoding two S-layer proteins and two ABC-transporter genes (>10- to 20-fold). A total of 107 ORFs had lower transcription levels in the aggregated cells at 200 h p.i. compared to levels in uninfected cells. Thirty-three of these ORFs code for ribosomal proteins, indicating a general slowdown of growth. Transcription levels of several ORFs were verified by quantitative RT-PCR (qRT-PCR) as listed in Table 5. For ORF MM_0490 with the highest fold change in the transcript amounts (annotated as phycocyanin alpha-subunit phycocyanobilin lyase) and MM_3096 (encoding a putative S-layer protein), the significantly increased transcript levels were verified by qRT-PCR (442.74 ± 43.46 and 10.18 ± 1.14).
TABLE 4.
Microarray analyses of aggregated cells after MetSV infection versus transcript levels in uninfected cells
| Gene transcript level in infected vs uninfected cells and function | No. of genes |
|---|---|
| Higher transcript levels | |
| Transporter | 35 |
| Transcription regulation | 11 |
| Metabolism | 58 |
| Stress | 13 |
| RNA/DNA modification | 2 |
| Conserved/hypothetical proteins | 95 |
| Amino acid biosynthesis/RNA processing | 5 |
| Lower transcript levels | |
| Ribosomal proteins | 33 |
| Envelope/transporter | 9 |
| Conserved/hypothetical proteins | 17 |
| Metabolism | 21 |
| DNA/RNA synthesis | 5 |
| Transcription regulation | 3 |
| Amino acid biosynthesis | 19 |
TABLE 5.
Transcription levels of MetSV-infected aggregates versus uninfected cells determined by qRT-PCR and microarray analyses
| Methanosarcina mazei ORF | Predicted function | Transcription levela |
|
|---|---|---|---|
| qRT-PCR | Microarray | ||
| MM_0002 | Dipeptide ABC transporter, binding protein | 1.68 ± 0.26 | 0.13 ± 0.08 |
| MM_0276 | Superfamily II DNA and RNA helicase | 2.72 ± 0.61 | 9.11 ± 12.02 |
| MM_0490 | Phycocyanin alpha-subunit phycocyanobilin lyase | 442.74 ± 43.46 | 113.01 96.7 |
| MM_0807 | Protein translation initiation factor 6 (IF-6) | 0.48 ± 0.06 | 0.24 ± 0.25 |
| MM_1009 | Protein translocase, subunit SecE | 0.47 ± 0.02 | 0.06 ± 0.04 |
| MM_1601 | Cobalamin biosynthesis protein CobN | 265.87 ± 62.27 | 49.82 ± 57.2 |
| MM_2021 | Conserved protein | 2.89 ± 1.33 | 12.07 ± 15.59 |
| MM_2833 | ABC transporter, ATP-binding protein | 5.15 ± 0.47 | 74.89 ± 61.67 |
| MM_3096 | Hypothetical protein (S-layer domain protein) | 10.18 ± 1.14 | 17.35 ± 21.08 |
Fold change in infected versus uninfected cells.
DISCUSSION
Since the first discovery of methanoarchaeon-infecting viruses in 1989, Methanosarcina spherical virus (MetSV) is the only virus which has been isolated and characterized for methanoarchaea for some time. In addition to the spherical virus morphology, the internal membrane, and the presence of a potential ATPase, which might be involved in genome packaging into procapsids, we did not observe a protein which was similar to the known major capsid proteins; therefore, we could not classify MetSV into one of the known virus families. In this respect it is tempting to speculate that MetSV belongs to a new virus family. In contrast, all other isolated methanoarchaeon-infecting viruses possess heads and tails and belong to either the Myoviridae or Siphoviridae family (reviewed in reference 3).
Genome organization and viral replication.
The genome of MetSV is double stranded and linear and has a size of 10,567 bp. This is in line with all other known archaeal viruses, all containing DNA (reviewed in reference 3) and showing genome sizes from 5 to 144 kbp (reviewed in reference 29). There are inverted terminal repeats (ITRs) with lengths of 59 nt on both ends of the MetSV genome. Quite a number of linear archaeal viruses contain ITRs, including the Haloarcula hispanica-infecting viruses SH1 and HHIV-2, Sulfolobus virus SIRV1, the haloviruses His1 and His2, and the Pyrobaculum-infecting virus PFV1 (2, 30–34). The presence of ITRs strongly suggests that MetSV, similar to the bacteriophages PRD1 and Ø29 or the archaeoviruses His1, His2, and SH1, also replicates via protein-primed DNA replication (30, 33, 35, 36). A further indication of protein-primed DNA replication is the predicted type B polymerase, encoded at one end of the MetSV genome. Type B polymerases are able to use proteins attached to the 5′ ends of linear dsDNA to prime DNA replication (reviewed in reference 29). Functions were assigned for two other ORFs. First, ORF13 encodes an ATPase which contains an HerA (HAS-barrel/ATPase domain) and a P-loop NTPase domain, indicating that this ATPase is involved in DNA packing into the capsid, as has been shown for phage PDR1 ATPase (reviewed in reference 3). Second, the ORF22 gene product contains an Env-gp36 domain, which is part of a lentivirus surface glycoprotein (feline immunodeficiency retrovirus); thus, we speculate that the ORF22 gene product might be part of the MetSV envelope. In addition, we predicted for several ORFs both archaeal promoter elements, the TATA and BRE boxes, which show high similarity to the M. mazei promoter. This indicates that MetSV is using the transcription machinery of the host, as has been reported for, e.g., SH1 virus (37). In the experimental setup, however, it was not ascertainable that MetSV ORFs show different transcription patterns at various time points after infection. Thus, we could not distinguish between early and late viral genes, as reported for other viruses (38–42). In addition to the 11 large ORFs, 11 small ORFs (sORFs) (≤90 aa) were predicted (Table 1). These sORFs encode small proteins ranging from 3.6 to 10 kDa. Some of them (i.e., ORF11 and ORF17) contain up to 43% hydrophobic amino acids. Small proteins are described for viruses infecting eukaryotic cells, like influenza virus, human immunodeficiency virus (HIV), fibropapillomavirus, poxvirus, or paramyxovirus. Based on the high ratio of hydrophobic amino acids, most of the small proteins have transmembrane domains and are able to form so-called viroporins, or they function as regulators for expression or activity of larger cellular transmembrane proteins by binding to their membrane-spanning domains (reviewed in reference 43). The identified MetSV sORFs might also be involved in envelope formation or modulating other large protein functions. The shortest ribosomally produced peptides from a phage genome have been recently reported to be involved in the decision between the lysogenic and lytic cycles of temperate phages by functioning as a viral quorum sensing system (44).
Host range.
Generally, the host range of methanogenic viruses can vary from rather narrow (e.g., ΦF3, which infects only M. thermoautotrophicum) to broad (e.g., virus ΦF1, which infects several different M. thermoautotrophicum strains) (8). MetSV was able to infect a variety of different Methanosarcina strains (Table 3) but not strains of either Methanosphaera stadtmanae or Methanosaeta thermophile. Consequently, MetSV represents the first virus specifically infecting the genus Methanosarcina. This is in line with the identification of different MetSV-derived spacers with one to six mismatches in their sequences exclusively in Methanosarcina (draft) genomes. Functional spacers can include mismatches unless the seed region is not mutated (45). Challenge assays clearly showed that the lytic virus MetSV can infect only Methanosarcina strains growing as single cells and not as sarcina-like aggregates. Interestingly, M. acetivorans, which also grows as single cells under laboratory conditions, was not infected by MetSV. The reason for this phenomenon might be that MetSV is sensitive to high salt concentrations since M. acetivorans was grown in high-salt medium. Additional evidence for this is provided by the observation that MetSV was also not infectious for M. mazei DSM 3647 when the strain was grown in a medium supplemented with 500 mM sodium chloride (data not shown). Further spacer analyses in different Methanosarcina draft genomes showed that MetSV is a ubiquitously distributed virus and that strains with MetSV-derived spacers might have an active CRISPR/Cas system, in contrast to M. mazei DSM 3647 (46).
Resistance formation.
M. mazei resistance against MetSV infection was detected after a change of morphology phenotype from single cells to aggregates. It is known that in some cases bacteria change their surface layer proteins in a reversible manner by different expression patterns, resulting in a phenotypically heterogenic population. This phase variation increases overall fitness and reduces sensitivity to phage infection (reviewed in reference 47). One example is the different expression of the phases Bvg+ and Bvg− of BvgAS, a two-component system from Bordetella which is involved in the regulation of the expression of surface proteins. In Bvg+-phase cells (BvgAS is active), pertactin is expressed, which is the recognition site for phage BPP-1. Consequently in Bvg−-phase cells (BvgAS is inactive), pertactin expression is suppressed, resulting in increased protection against BPP-1 infection (48–50). Transcriptome analysis of MetSV-induced aggregated M. mazei cells versus that of nonchallenged cells revealed several differently transcribed genes that are potentially involved in altering the cell surface structure (Table 4). These ORFs encode S-layer proteins or ABC transporters, which are known to be involved in segregation of S-layer proteins (reviewed in reference 51). The ORF with highest transcript levels in the aggregated cells, MM_0490, annotated as phycocyanin alpha-subunit phycocyanobilin lyase, is almost conserved in the Methanosarcinales. Using Phyre2 prediction, a possible function in cell adhesion was predicted. Furthermore, Saunders et al. (52) reported a PBS lyase HEAT repeat domain-containing protein from M. barkeri that had homologies to secreted proteins from Methanococcoides burtonii involved in envelope formation. Considering these findings and the highly elevated transcript levels of MM_0490 in the aggregated cells after MetSV infection, it is tempting to speculate that this protein is crucially involved in envelope formation and the formation of sarcina-like aggregations. Consequently, we propose that resistance against MetSV is mediated by phase variation of the morphology phenotype from single cells to aggregates caused by differences in envelope structures through different expression levels of surface proteins and not by CRISPR-derived immunity.
MATERIALS AND METHODS
Strains and plasmids.
Strains and plasmids used in this study are listed in Table 6.
TABLE 6.
Strains and the virus and plasmid used in this study
| Name | Description | Reference or source |
|---|---|---|
| Strains | ||
| M. mazei DSM 3647 | Type strain | DSMZ |
| M. mazei DSM 2244 | DSMZ | |
| M. mazei DSM 4556 | DSMZ | |
| M. mazei DSM 6300 | DSMZ | |
| M. mazei DSM 7222 | DSMZ | |
| M. mazei DSM 9195 | DSMZ | |
| M. barkeri DSM 1311 | DSMZ | |
| M. barkeri DSM 800 | Type strain | DSMZ |
| M. acetivorans DSM 2834 | Type strain | DSMZ |
| M. soligelidi DSM 26065 | Type strain | DSMZ |
| Methanosarcina sp. DSM 11855 | DSMZ | |
| M. siciliae DSM 3028 | Type strain | DSMZ |
| E. coli DH5a | General cloning strain | 75 |
| Virus | ||
| Methanosarcina spherical virus (MetSV) | This study | |
| Plasmid | ||
| pCR2.1-TOPO | Plac lacZα-CCB, Kanr Ampr, pUC ori; cloning vector | Life Technologies, (Darmstadt Germany) |
Growth conditions.
All archaeal strains were grown under strictly anaerobic conditions. The growth media were generally supplemented with 100 μg/ml ampicillin to prevent bacterial contamination. M. mazei isolates (DSM 3647, 2244, 4556, 6300, and 9195), M. soligelidi (DSM 26065), and M. barkeri (DSM 800 and 1311) strains were grown in minimal medium under a N2/CO2 (80:20) atmosphere at 37°C without shaking, as described previously (53, 54). The medium was supplemented with 150 mM methanol (MeOH), 40 mM acetate, 2 mM cysteine, and 1 mM sodium sulfide. In the case of Methanosarcina sp. DSM 11855, minimal medium was additionally supplemented with 5% (vol/vol) rumen fluid (clarified), and the strain was incubated at 55°C. M. acetivorans (DSM 2834) was grown in high-salt medium as described by Sowers and Schreier (55). M. siciliae (DSM 3028) was grown in DSMZ medium number 324 (Methanolobus II medium). Growth was monitored by determining the turbidity (optical density [OD]) of the cultures at 600 nm (OD600).
Virus enrichment and isolation.
Digester sludge was filtered (0.2-μm-pore-size filter, polyethersulfone membrane; Sarstedt, Nümbrecht, Germany) under anaerobic conditions, and the filtrate was added to 5 ml of an exponentially growing M. mazei DSM 3647 culture. After cell lysis of the culture, the virus-containing supernatant was again anaerobically filtered, and 10% virus filtrate (final concentration) was added to a fresh M. mazei DSM 3647 culture. After 10 passages only one type of virus was detectable by transmission electron microscopy.
Transmission electron microscopy.
One liter of M. mazei culture was grown to a turbidity of ∼0.2 at 600 nm and supplemented with MetSV-containing filtrate to a 1% final concentration and further incubated at 37°C. After cell lysis (24 h), viruses were harvested using a modified protocol as described by Colombet et al. (56). Briefly, after the addition of 10% polyethylene glycol (PEG) 8000 and 0.6% NaCl (Roth, Karlsruhe, Germany), the cultures were incubated for approximately 24 h in the dark at 4°C. The white layer, which contains the viruses, was transferred to a sterile centrifugation tube and harvested by centrifugation at 8,000 × g for 20 min at 4°C. The pellet was resuspended in 300 μl of SM buffer (0.1 M NaCl, 8 mM MgSO4·7H2O, 50 mM Tris-HCl, 0.005% glycerol [wt/vol], pH 7.0). Then, 1 M KCl (final concentration) was added, and the mixture was incubated for 20 min on ice. The mixture was centrifuged at 12,000 × g for 10 min at 4°C. The virus-containing supernatant was transferred to a new tube and stored at −80°C. After negative staining with 2% (wt/vol) uranyl acetate (Serva, Heidelberg, Germany), grid samples were analyzed with a transmission electron microscope (Tecnai10; FEI Thermo Fisher Scientific, Eindhoven, the Netherlands) equipped with a Megaview G2 charge-coupled-device (CCD) camera (Emsis, Münster, Germany) as described in detail previously (57).
MetSV infection.
M. mazei cultures (50 ml each) were grown in closed bottles to a turbidity of ∼0.2 at 600 nm; then 0.5 ml of filtered (0.2-μm pore size) supernatant of an MetSV-lysed culture was added subsequently. The cultures were further incubated at 37°C until the cells were completely lysed (8 to 12 h) or aggregate formation was obtained (within 2 weeks).
Isolation of viral DNA.
Lysed M. mazei cultures were filtered (0.2-μm pore size), and the filtrate was centrifuged at 30,000 × g for 6 h at 4°C. The pellet was resuspended overnight in 200 μl of nuclease-free water (Roth, Karlsruhe, Germany) at 4°C. DNase I (Life Technologies, Darmstadt, Germany) treatment was performed for 30 min at 37°C to destroy contaminating M. mazei DNA. After DNase I treatment and separation of DNase I, DNA was isolated using a QiaAmp Mini Elute virus spin kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The obtained elution fraction was treated with RNase A (Life Technologies, Darmstadt, Germany) for 30 min at 37°C, followed by a proteinase K (Life Technologies, Darmstadt, Germany) treatment for 30 min at 37°C. To inactivate and remove proteins, ethanol precipitation was performed.
Virus genome: sequencing, assembly, and annotation.
Viral DNA was isolated. Afterwards, the DNA was processed according to the Rapid Library Preparation Method Manual (Roche, Mannheim, Germany) (58). Multiplex identifier (MID) adaptors for the Rapid Library (Roche, Mannheim, Germany) were ligated to the DNA fragments. The DNA fragments were cleaned and quantified using an Agilent 2100 Bioanalyzer High Sensitivity DNA analysis kit (Agilent Technologies, Waldbronn, Germany). The individual samples were combined in a library pool. The sample of the final library was run on an Agilent 2100 Bioanalyzer (Agilent Technologies, Waldbronn, Germany) prior to emulsion PCR and sequencing as recommended by Roche. The library was subsequently sequenced on a 454 GS-FLX using Titanium sequencing chemistry. Contigs were reconstructed using Mira, version 4.0 (59), with the job options “genome, de novo, accurate.” In addition, a shotgun cloning library was generated for closing the gaps. The viral DNA was restricted with HindIII (NEB, Frankfurt, Germany) and cloned into a pCR-TOPO 2.1 vector (Life Technologies, Darmstadt, Germany). The contigs were aligned to the shotgun sequences using the CodonCode aligner (CodonCode Corporation, Centerville, MA, USA). ORFs were predicted using ORF Finder (National Center for Biotechnology Information [NCBI; http://www.ncbi.nlm.nih.gov/]). Phyre2 was used to predict protein domains by homology detections (22).
Sequencing CRISPR arrays of M. mazei DSM 3647.
CRISPR array IB (spacers 1 to 29) was amplified using the primers C1for/C1rev2. For CRISPR array IIIC (spacers 1 to 9) the primers CRISPR_IIIC_for/CRISPR_IIICsp_r were used (listed in Table S2 in the supplemental material). The obtained PCR products were TOPO-TA cloned into a pCR-TOPO 2.1 vector (Life Technologies, Darmstadt, Germany) according to the manufacturer's instructions. Plasmid DNA was generally transformed into E. coli DH5α according to the method of Inoue et al. (60). The insert of the resulting plasmids was sequenced by the method of Sanger (at the Institute of Clinical Molecular Biology [IKMB], Kiel, Germany).
Bioinformatic analysis of Methanosarcina draft genomes.
All Methanosarcina mazei draft genomes used in this study were downloaded from Youngblut et al. (27) and the NCBI in 2015 (for accession numbers, see Table S1). CRISPR loci were predicted using an in-house tool on all draft genomes. CRISPR locus orientations were predicted using the CRISPRstrand tool (61). The consensus direct repeat for each CRISPR locus is the most frequent direct repeat in this CRISPR array. All CRISPR spacers were compiled into a single database and compared against the MetSV sequence using the FASTA tool, version 36.3.6 (62), optimizing the number of allowed hits for MetSV. To estimate the significance of a hit between the MetSV sequence and the CRISPR spacer with a certain number of mismatches, we used a shuffling approach, which allowed us to distinguish between a random match (false positive) and a true match between spacer sequences and MetSV. Thus, for each spacer sequence in these data sets, a di- or trinucleotide shuffle was generated. According to the literature, the number of mismatches can be very high in some instances (63). Therefore, we used a modified version of IntaRNA (64) to predict RNA-DNA duplexes. A protospacer-adjacent motif (PAM) plays a significant role in CRISPR-mediated immunity. In our study, we determined the consensus PAM by extracting five nucleotides upstream of the putative protospacers from the virus genome and then aligning them.
Protein analysis by SDS-PAGE and mass spectrometry.
Filtered MetSV-lysed M. mazei DSM 2647 cultures were centrifuged at 30,000 × g for 6 h at 4°C. The resulting pellet was resuspended in deionized H2O overnight at 4°C. Protein extracts were generated by adding SDS-PAGE loading buffer (125 mM Tris-HCl [pH 6.8], 140 mM SDS, 0.3 mM bromophenol blue, 10% β-mercaptoethanol [vol/vol]) and incubating the resuspended pellet at 100°C for 10 min. Samples were analyzed by 12.5% SDS-PAGE according to Laemmli (65). Prior to silver staining, the gels were washed with solution 1 (20 ml of ethanol, 4 ml of glacial acetic acid, 16 ml of H2O) overnight and two times for 20 min with solution 2 (39 ml of ethanol, 91 ml of H2O) at room temperature. Silver staining was performed as described by Rabilloud et al. (66).
Additionally, stacking gel-based SDS-PAGE followed by LC-MS analysis (28) was employed for the analysis of the proteome of two MetSV samples. Briefly, 10-μg aliquots of the protein extract, either without or with washing of the filtered extract (samples 1 and 2, respectively) were suspended in SDS-PAGE loading buffer, heated at 95°C for 5 min, and loaded onto 12% SDS-PAGE gels. The samples were migrated into the gels via constant voltage (40 V) for 15 min, followed by 80 V for a further 5 min to allow the proteins to enter the resolving gel; then Coomassie brilliant blue staining was performed.
In-gel digestion.
For in-gel digestion, protein bands and protein-containing regions (ca. 25 mm2 of the stacking gel-cleaned SDS-PAGE gel) were excised and destained with 30% acetonitrile (ACN)–70% ammonium bicarbonate (50 mM, pH 7.4). Destained gel slices were dehydrated by addition of 100% ACN. Seventy microliters of 10 mM 1,4-dl-dithiothreitol was added for disulfide reduction (60 min at 56°C). The supernatant was removed, and 70 μl of 55 mM iodoacetamide was added for cysteine alkylation incubation in the dark for 60 min at room temperature. Gel slices were dehydrated with 100% ACN. For enzymatic digestion of the protein bands, 1.5 μg of the enzyme (trypsin or GluC, sequencing grade; Promega, Madison, WI, USA) or 0.3 μg for the stacking gel protein-containing regions was added in 10 μl of 50 mM ammonium bicarbonate, pH 7.4. After a 10-min incubation, an additional 90 μl of ammonium bicarbonate buffer was added, and digestion was continued for 16 h at 37°C. Supernatants were transferred to new tubes, and the peptides were extracted from the gel pieces with increasing concentrations of acetonitrile (60% ACN–70% ammonium bicarbonate and 100% ACN), pooled with the supernatants, and dried by applying a vacuum in a SpeedVac (Concentrator plus; Eppendorf, Wesseling-Berzdorf, Germany). For LC-MS/MS analysis, samples were resuspended in 20 μl of 3% acetonitrile–0.05% formic acid.
LC-MS/MS analysis of peptides.
For LC-MS/MS analysis, 5 μl of each peptide sample was used. Peptide separation was performed on a Dionex Ultimate 3000 ultrahigh-performance liquid chromatography (UHPLC) system (Acclaim PepMap 100 analytical column; 2-μm particle size, 100-Å pore size, 75 μm by 500 mm), coupled online to a Thermo Q Exactive Plus Orbitrap mass spectrometer. For chromatographic separation of peptides from the protein bands, 0.05% formic acid (FA) as buffer A and 80% ACN–0.04% FA as buffer B were used for the following gradient: 0 min, 5% B; 5 min, 5% B; 95 min, 70% B; 100 min, 95% B; 110 min, 95% B; 110.1 min, 5% B; 120 min, 5% B. For chromatographic separation of peptides from the stacking gel approach, the same eluents were used across the following gradient: 0 min, 4% B; 2 min, 4% B; 82 min, 20% B; 122 min, 40% B; 130 min, 90% B; 140 min, 90% B; 141 min, 4% B; 150 min, 4% B. The flow rate was set to 300 nl/min with a column oven temperature of 30°C for protein band peptides and 45°C for the peptides from the stacking gel approach. For MS/MS of the protein band peptides, the 10 most intensive ions were fragmented by using high-energy collision dissociation (HCD) at a normalized collision energy of 25 (full-scan MS, 300 to 2,000 m/z; resolution, 70,000). For MS/MS of the stacking gel peptides, the 15 most intensive ions were fragmented by HCD at a normalized collision energy of 27.5 (full-scan MS, 350 to 1,300 m/z; resolution, 70,000). For automatic data interpretation, a SequestHT database search was performed on a Proteome Discoverer (version 2.2.0.388) against FASTA databases containing the predicted MetSV ORFs, the proteome of Methanosarcina mazei Gö1 (UniProt; accessed 26 June 2017), and common impurities (common Repository of Adventitious Proteins [cRAP]). The resulting filter settings were as follows: peptide confidence, high; peptide rank, 1; minimum, 1% of base peak; precursor mass tolerance, 7 ppm; fragment mass tolerance, 0.02 Da; enzyme specificity, trypsin or GluC; maximum missed cleavage sites, 2; length, 6 to 144 amino acids; static modifications, carbamidomethyl (Cys); dynamic modifications, oxidation (Met) and acetylation (protein N terminus). To be classified as identified, proteins required at least two high-confidence peptides and a minimum of one unique (proteotypic) peptide.
Lipid extraction.
Acid hydrolysis was carried out on freeze-dried biomass of M. mazei DSM 3647 and purified cell material of MetSV to release di- and tetraether lipids. In addition, a virus-free culture of M. mazei was harvested during its late stationary growth phase and purified as described above to test whether archaeal lipids were successfully retained during the cleanup procedure. All organic tissue (between 3.1 and 20.1 mg) was reacted with 7.5 ml of 5% hydrochloric acid (HCl) in MeOH for 4 h at 80°C and then allowed to cool to room temperature. The supernatant was separated from the biomass by centrifugation (4,696 × g; 10 min) and then transferred to an 80-ml centrifuge tube. Subsequently, the cell material was extracted three times with 5 ml of a solvent mixture of dichloromethane (DCM)-MeOH (3:1, vol/vol). High-performance liquid chromatography (HPLC)-grade water (∼10 ml) was added to the combined supernatants, resulting in a phase separation. The lipid-containing organic bottom layer was transferred to a round-bottom flask, and the remaining water layer was washed twice each with 11 ml of DCM. The combined lipid extract was then reduced under vacuum, transferred to a preweighed vial, and dried under a gentle stream of nitrogen. All lipid extracts were stored at −20°C and redissolved in DCM-MeOH (9.1, vol/vol) at a concentration of 1 mg/ml prior to analysis.
Lipid analysis.
Archaeal core lipids were analyzed according to the analytical procedure originally described by Hopmans et al. (67) and afterwards modified by Liu et al. (68). Target compounds were eluted using an Alliance 2695 (Waters, United Kingdom) HPLC system interfaced to a ZQ (Micromass, United Kingdom) single quadruple mass spectrometer. The HPLC was fitted with a Grace Prevail Cyano HPLC column (3-μm particle size; 150 mm by 2.1-mm inner diameter [i.d.]) and a guard column of the same material. Separation was achieved at a constant temperature of 30°C with a flow rate of 0.2 ml/min and the following gradient: 90% A (n-hexane)–10% B (n-hexane–propan-2-ol, 90:10, vol/vol) held isocratically for 5 min, followed by a linear gradient to 81% A–19% B in 20 min and then a linear gradient to 100% B in 35 min, which was held for 5 min. The column was reequilibrated with 90% A–10% B at 0.2 ml/min for 15 min before the next injection.
The mass spectrometer was equipped with an atmospheric pressure chemical ionization (APCI) interface source operated in positive ion mode. MS settings were as follows: source, 150°C; vaporizer, 500°C; corona, 2 μA; cone voltage, 40 V; extractor, 3 V; radio frequency (RF) lens, 0.1 V; desolvation gas, N2 at 8 liters/min. Detection of archaeal core lipids was achieved by single ion recording of their protonated molecular ions [M+H]+ (dwell time, 234 ms) as reported by Hopmans et al. (67) and Liu et al. (68). Absolute concentrations of the target compounds were determined after peak integration in a QuanLynx application manager and comparison of area counts with those of an external standard curve obtained by serial dilutions of a synthetic tetraether diol with 77 carbon atoms (69). Similar response factors were assumed for archaeal lipids and the external standard.
RNA isolation.
M. mazei total RNA from different growth phases was isolated by Isol-RNA lysis reagent extraction as reported by Nickel et al. (46) (5′ Prime GmbH, Hilden, Germany), followed by DNase I treatment (Life Technologies, Darmstadt, Germany). Quantity and purity of RNA were determined as described previously (70).
RT-PCR.
A One-Step RT-PCR kit (Qiagen, Hilden, Germany) was used for reverse transcription-PCR (RT-PCR) according to the manufacturer's instructions. Samples of 0.5 μg total RNA from M. mazei DSM 3647 (wild type) isolated at different time points after infection with MetSV (0, 60, 90, and 210 min) and the primers listed in Table S2 were used. The respective PCR fragments were evaluated on a 1% agarose gel.
qRT-PCR.
Quantitative RT-PCR (qRT-PCR) was performed as described by Veit et al. (71) using a QuantiTect SYBR green RT-PCR kit (Qiagen, Hilden, Germany) and a ViiA 7 cycler (Applied Biosystems by Life Technologies, Darmstadt, Germany). Primer pairs used for quantitative RT-PCRs are listed in Table S2. Fold change in transcript abundance for each ORF was determined by comparison with the threshold cycle (CT) of transcripts of three control M. mazei genes (i.e., MM1621, MM2181, and MM1215) (71, 72). The fold change in the abundance of a transcript was calculated using the following formula: fold change = 2−ΔΔCT (73).
Microarray chip generation.
Oligonucleotides (length, 70 nt) for the 3,371 predicted ORFs and additional 42 new ORFs from M. mazei identified during whole-transcriptome shotgun sequencing (RNA-seq) analyses (74) were generated (Eurofins, Ebersberg, Germany). All oligonucleotides were bioinformatically analyzed before synthesis to exclude unspecific hybridization by MWG Eurofins Genomics (Ebersberg, Germany). The oligonucleotides were resolved in dimethyl sulfoxide to a final concentration of 0.3 ng/nl and then spotted onto aminosilane-coated Type-7* microarray slides (GE Healthcare Europe GmbH, Freiburg, Germany) with a Lucidea Array Spotter Generation V (GE Healthcare Europe GmbH, Freiburg, Germany). Each oligonucleotide was spotted in duplicate on the slides and fixed by UV light. The microarray chips were stored in a vacuum desiccator at 20°C.
RNA labeling, hybridization, and microarray analysis.
The cDNA generation and simultaneous labeling with fluorescent Cy3 and Cy5 dyes were performed by using a SuperScript III Reverse Transcriptase kit (Life Technologies, Darmstadt, Germany). A Lucidea SlidePro hybridization chamber was used for hybridization to microarray slides. Hybridization, scanning, data normalization, and evaluation were performed using GenePix Pro software, version 6.0 (Axon Instruments, Union City, CA, USA) as described previously (70, 72). Three independent biological replicates, including a dye swap experiment, were performed. Differences in transcript levels of selected ORFs were additionally verified by qRT-PCR as detailed above.
Accession number(s).
The MetSV sequence determined in this study was deposited in the NCBI database under accession number MF186604.
Supplementary Material
ACKNOWLEDGMENTS
We thank Daniela Hallack, Christopher Noack, and Heinke Buhtz for technical support as well as Alena Wiegandt for MS analysis of the 11- and 16-kDa protein bands. Armin Ehrenreich (Department of Microbiology, Technical University of Munich, Germany) is acknowledged for spotting microarrays and Thierry Benvegnu for providing an aliquot of the tetraether diol standard.
This study was supported by the DFG (FOR1680 and SCHM1052/12-1 and 2).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00955-17.
REFERENCES
- 1.Snyder JC, Bolduc B, Young MJ. 2015. 40 Years of archaeal virology: expanding viral diversity. Virology 479–480:369–378. doi: 10.1016/j.virol.2015.03.031. [DOI] [PubMed] [Google Scholar]
- 2.Pina M, Bize A, Forterre P, Prangishvili D. 2011. The archeoviruses. FEMS Microbiol Rev 35:1035–1054. doi: 10.1111/j.1574-6976.2011.00280.x. [DOI] [PubMed] [Google Scholar]
- 3.Pietila MK, Demina TA, Atanasova NS, Oksanen HM, Bamford DH. 2014. Archaeal viruses and bacteriophages: comparisons and contrasts. Trends Microbiol 22:334–344. doi: 10.1016/j.tim.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Meile L, Jenal U, Studer D, Jordan M, Leisinger T. 1989. Characterization of OM1, a virulent phage of Methanobacterium thermoautotrophicum Marburg. Arch Mikrobiol 152:105–110. doi: 10.1007/BF00456085. [DOI] [Google Scholar]
- 5.Jordan M, Meile L, Leisinger T. 1989. Organization of Methanobacterium thermoautotrophicum bacteriophage psi M1 DNA. Mol Gen Genet 220:161–164. doi: 10.1007/BF00260872. [DOI] [PubMed] [Google Scholar]
- 6.Pfister P, Wasserfallen A, Stettler R, Leisinger T. 1998. Molecular analysis of Methanobacterium phage psiM2. Mol Microbiol 30:233–244. doi: 10.1046/j.1365-2958.1998.01073.x. [DOI] [PubMed] [Google Scholar]
- 7.Luo Y, Pfister P, Leisinger T, Wasserfallen A. 2001. The genome of archaeal prophage psiM100 encodes the lytic enzyme responsible for autolysis of Methanothermobacter wolfeii. J Bacteriol 183:5788–5792. doi: 10.1128/JB.183.19.5788-5792.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nölling J, Groffen A, De Vos WM. 1993. ΦFl and ΦF3, two novel virulent, archaeal phages infecting different thermophilic strains of the genus Methanobacterium. J Gen Microbiol 139:2511–2516. doi: 10.1099/00221287-139-10-2511. [DOI] [Google Scholar]
- 9.Wood AG, Whitman WB, Konisky J. 1989. Isolation and characterization of an archaebacterial viruslike particle from Methanococcus voltae A3. J Bacteriol 171:93–98. doi: 10.1128/jb.171.1.93-98.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krupovic M, Bamford DH. 2008. Archaeal proviruses TKV4 and MVV extend the PRD1-adenovirus lineage to the phylum Euryarchaeota. Virology 375:292–300. doi: 10.1016/j.virol.2008.01.043. [DOI] [PubMed] [Google Scholar]
- 11.Lai MC, Chen SC. 2001. Methanofollis aquaemaris sp. nov., a methanogen isolated from an aquaculture fish pond. Int J Syst Evol Microbiol 51:1873–1880. doi: 10.1099/00207713-51-5-1873. [DOI] [PubMed] [Google Scholar]
- 12.Borrel G, Colombet J, Robin A, Lehours AC, Prangishvili D, Sime-Ngando T. 2012. Unexpected and novel putative viruses in the sediments of a deep-dark permanently anoxic freshwater habitat. ISME J 6:2119–2127. doi: 10.1038/ismej.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chien IC, Meschke JS, Gough HL, Ferguson JF. 2013. Characterization of persistent virus-like particles in two acetate-fed methanogenic reactors. PLoS One 8:e81040. doi: 10.1371/journal.pone.0081040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leahy SC, Kelly WJ, Altermann E, Ronimus RS, Yeoman CJ, Pacheco DM, Li D, Kong Z, McTavish S, Sang C, Lambie SC, Janssen PH, Dey D, Attwood GT. 2010. The genome sequence of the rumen methanogen Methanobrevibacter ruminantium reveals new possibilities for controlling ruminant methane emissions. PLoS One 5:e8926. doi: 10.1371/journal.pone.0008926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samuel BS, Hansen EE, Manchester JK, Coutinho PM, Henrissat B, Fulton R, Latreille P, Kim K, Wilson RK, Gordon JI. 2007. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc Natl Acad Sci U S A 104:10643–10648. doi: 10.1073/pnas.0704189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen EE, Lozupone CA, Rey FE, Wu M, Guruge JL, Narra A, Goodfellow J, Zaneveld JR, McDonald DT, Goodrich JA, Heath AC, Knight R, Gordon JI. 2011. Pan-genome of the dominant human gut-associated archaeon, Methanobrevibacter smithii, studied in twins. Proc Natl Acad Sci U S A 108(Suppl 1):S4599–S4606. doi: 10.1073/pnas.1000071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Makino S, A N, Koike H, Suzuki M. 1999. Prophages inserted in archaebacterial genomes. Proc Japan Acad 75:166–171. doi: 10.3792/pjaa.75.166. [DOI] [Google Scholar]
- 18.Haring M, Peng X, Brugger K, Rachel R, Stetter KO, Garrett RA, Prangishvili D. 2004. Morphology and genome organization of the virus PSV of the hyperthermophilic archaeal genera Pyrobaculum and Thermoproteus: a novel virus family, the Globuloviridae. Virology 323:233–242. doi: 10.1016/j.virol.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Deppenmeier U, Johann A, Hartsch T, Merkl R, Schmitz RA, Martinez-Arias R, Henne A, Wiezer A, Baumer S, Jacobi C, Bruggemann H, Lienard T, Christmann A, Bomeke M, Steckel S, Bhattacharyya A, Lykidis A, Overbeek R, Klenk HP, Gunsalus RP, Fritz HJ, Gottschalk G. 2002. The genome of Methanosarcina mazei: evidence for lateral gene transfer between bacteria and archaea. J Mol Microbiol Biotechnol 4:453–461. [PubMed] [Google Scholar]
- 20.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH. 2015. CDD: NCBI's conserved domain database. Nucleic Acids Res 43:D222–D226. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iyer LM, Makarova KS, Koonin EV, Aravind L. 2004. Comparative genomics of the FtsK-HerA superfamily of pumping ATPases: implications for the origins of chromosome segregation, cell division and viral capsid packaging. Nucleic Acids Res 32:5260–5279. doi: 10.1093/nar/gkh828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olmstedt RA, Hirsch VM, Purcello RH, Johnson PR. 1989. Nucleotide sequence analysis of feline immunodeficiency virus: genome organization and relationship to other lentiviruses. Proc Natl Acad Sci U S A 86:8088–8092. doi: 10.1073/pnas.86.20.8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. 2011. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJ, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, van der Oost J, Backofen R, Koonin EV. 2015. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vestergaard G, Garrett RA, Shah SA. 2014. CRISPR adaptive immune systems of Archaea. RNA Biol 11:156–167. doi: 10.4161/rna.27990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Youngblut ND, Wirth JS, Henriksen JR, Smith M, Simon H, Metcalf WW, Whitaker RJ. 2015. Genomic and phenotypic differentiation among Methanosarcina mazei populations from Columbia River sediment. ISME J 9:2191–2205. doi: 10.1038/ismej.2015.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hung CW, Klein T, Cassidy L, Linke D, Lange S, Anders U, Bureik M, Heinzle E, Schneider K, Tholey A. 2016. Comparative proteome analysis in Schizosaccharomyces pombe identifies metabolic targets to improve protein production and secretion. Mol Cell Proteomics 15:3090–3106. doi: 10.1074/mcp.M115.051474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Peng N, Shah SA, Huang L, She Q. 2015. Archaeal extrachromosomal genetic elements. Microbiol Mol Biol Rev 79:117–152. doi: 10.1128/MMBR.00042-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bamford DH, Ravantti JJ, Ronnholm G, Laurinavicius S, Kukkaro P, Dyall-Smith M, Somerharju P, Kalkkinen N, Bamford JK. 2005. Constituents of SH1, a novel lipid-containing virus infecting the halophilic euryarchaeon Haloarcula hispanica. J Virol 79:9097–9107. doi: 10.1128/JVI.79.14.9097-9107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaakkola ST, Penttinen RK, Vilen ST, Jalasvuori M, Ronnholm G, Bamford JK, Bamford DH, Oksanen HM. 2012. Closely related archaeal Haloarcula hispanica icosahedral viruses HHIV-2 and SH1 have nonhomologous genes encoding host recognition functions. J Virol 86:4734–4742. doi: 10.1128/JVI.06666-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blum H, Zillig W, Mallok S, Domdey H, Prangishvili D. 2001. The genome of the archaeal virus SIRV1 has features in common with genomes of eukaryal viruses. Virology 281:6–9. doi: 10.1006/viro.2000.0776. [DOI] [PubMed] [Google Scholar]
- 33.Bath C, Cukalac T, Porter K, Dyall-Smith ML. 2006. His1 and His2 are distantly related, spindle-shaped haloviruses belonging to the novel virus group, Salterprovirus. Virology 350:228–239. doi: 10.1016/j.virol.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 34.Rensen EI, Mochizuki T, Quemin E, Schouten S, Krupovic M, Prangishvili D. 2016. A virus of hyperthermophilic archaea with a unique architecture among DNA viruses. Proc Natl Acad Sci U S A 113:2478–2483. doi: 10.1073/pnas.1518929113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Savilahti H, Bamford DH. 1993. Protein-primed DNA replication: role of inverted terminal repeats in the Escherichia coli bacteriophage PRD1 life cycle. J Virol 67:4696–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez-Huici V, Salas M, Hermoso JM. 2000. Sequence requirements for protein-primed initiation and elongation of phage O29 DNA replication. J Biol Chem 275:40547–40553. doi: 10.1074/jbc.M007170200. [DOI] [PubMed] [Google Scholar]
- 37.Porter K, Russ BE, Yang J, Dyall-Smith ML. 2008. The transcription programme of the protein-primed halovirus SH1. Microbiology 154:3599–3608. doi: 10.1099/mic.0.2008/019422-0. [DOI] [PubMed] [Google Scholar]
- 38.Kessler A, Brinkman AB, van der Oost J, Prangishvili D. 2004. Transcription of the rod-shaped viruses SIRV1 and SIRV2 of the hyperthermophilic archaeon sulfolobus. J Bacteriol 186:7745–7753. doi: 10.1128/JB.186.22.7745-7753.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maaty WS, Steffens JD, Heinemann J, Ortmann AC, Reeves BD, Biswas SK, Dratz EA, Grieco PA, Young MJ, Bothner B. 2012. Global analysis of viral infection in an archaeal model system. Front Microbiol 3:411. doi: 10.3389/fmicb.2012.00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frols S, Dyall-Smith M, Pfeifer F. 2012. Biofilm formation by haloarchaea. Environ Microbiol 14:3159–3174. doi: 10.1111/j.1462-2920.2012.02895.x. [DOI] [PubMed] [Google Scholar]
- 41.Okutan E, Deng L, Mirlashari S, Uldahl K, Halim M, Liu C, Garrett RA, She Q, Peng X. 2013. Novel insights into gene regulation of the rudivirus SIRV2 infecting Sulfolobus cells. RNA Biol 10:875–885. doi: 10.4161/rna.24537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ortmann AC, Brumfield SK, Walther J, McInnerney K, Brouns SJ, van de Werken HJ, Bothner B, Douglas T, van de Oost J, Young MJ. 2008. Transcriptome analysis of infection of the archaeon Sulfolobus solfataricus with Sulfolobus turreted icosahedral virus. J Virol 82:4874–4883. doi: 10.1128/JVI.02583-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DiMaio D. 2014. Viral miniproteins. Annu Rev Microbiol 68:21–43. doi: 10.1146/annurev-micro-091313-103727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erez Z, Steinberger-Levy I, Shamir M, Doron S, Stokar-Avihail A, Peleg Y, Melamed S, Leavitt A, Savidor A, Albeck S, Amitai G, Sorek R. 2017. Communication between viruses guides lysis-lysogeny decisions. Nature 541:488–493. doi: 10.1038/nature21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, van der Oost J, Brouns SJ, Severinov K. 2011. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci U S A 108:10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nickel L, Weidenbach K, Jager D, Backofen R, Lange SJ, Heidrich N, Schmitz RA. 2013. Two CRISPR-Cas systems in Methanosarcina mazei strain Go1 display common processing features despite belonging to different types I and III. RNA Biol 10:779–791. doi: 10.4161/rna.23928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dy RL, Richter C, Salmond GP, Fineran PC. 2014. Remarkable mechanisms in microbes to resist phage infections. Annu Rev Virol 1:307–331. doi: 10.1146/annurev-virology-031413-085500. [DOI] [PubMed] [Google Scholar]
- 48.Decker KB, James TD, Stibitz S, Hinton DM. 2012. The Bordetella pertussis model of exquisite gene control by the global transcription factor BvgA. Microbiology 158:1665–1676. doi: 10.1099/mic.0.058941-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stibitz S, Aaronson W, Monack D, Falkow S. 1989. Phase variation in Bordetella pertussis by frameshift mutation in a gene for a novel two-component system. Nature 338:266–269. doi: 10.1038/338266a0. [DOI] [PubMed] [Google Scholar]
- 50.Liu M, Deora R, Doulatov SR, Gingery M, Eiserling FA, Preston A, Maskell DJ, Simons RW, Cotter PA, Parkhill J, Miller JF. 2002. Reverse transcriptase-mediated tropism switching in Bordetella bacteriophage. Science 295:2091–2094. doi: 10.1126/science.1067467. [DOI] [PubMed] [Google Scholar]
- 51.Davidson AL, Dassa E, Orelle C, Chen J. 2008. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol Mol Biol Rev 72:317–364. doi: 10.1128/MMBR.00031-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saunders NF, Ng C, Raftery M, Guilhaus M, Goodchild A, Cavicchioli R. 2006. Proteomic and computational analysis of secreted proteins with type I signal peptides from the Antarctic archaeon Methanococcoides burtonii. J Proteome Res 5:2457–2464. doi: 10.1021/pr060220x. [DOI] [PubMed] [Google Scholar]
- 53.Deppenmeier U, Blaut M, Mahlmann A, Gottschalk G. 1990. Reduced coenzyme F420: heterodisulfide oxidoreductase, a proton-translocating redox system in methanogenic bacteria. Proc Natl Acad Sci U S A 87:9449–9453. doi: 10.1073/pnas.87.23.9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ehlers C, Veit K, Gottschalk G, Schmitz RA. 2002. Functional organization of a single nif cluster in the mesophilic archaeon Methanosarcina mazei strain Gö1. Archaea 1:143–150. doi: 10.1155/2002/362813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sowers KR, Schreier HJ (ed). 1995. Archaea: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 56.Colombet J, Robin A, Lavie L, Bettarel Y, Cauchie HM, Sime-Ngando T. 2007. Virioplankton “pegylation”: use of PEG (polyethylene glycol) to concentrate and purify viruses in pelagic ecosystems. J Microbiol Methods 71:212–219. doi: 10.1016/j.mimet.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 57.Casey E, Mahony J, Neve H, Noben JP, Dal Bello F, van Sinderen D. 2015. Novel phage group infecting Lactobacillus delbrueckii subsp. lactis, as revealed by genomic and proteomic analysis of bacteriophage Ldl1. Appl Environ Microbiol 81:1319–1326. doi: 10.1128/AEM.03413-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roche. 2010. Rapid library preparation method manual. Roche, Mannheim, Germany. [Google Scholar]
- 59.Chevreux B, Wetter T, Suhai S. 1999. Genome sequence assembly using trace signals and additional sequence information, p 45–56. In Proceedings of the German conference on bioinformatics GCB ′99, Hannover, Germany, 4 to 6 October 1999. [Google Scholar]
- 60.Inoue H, Nojima H, Okayama H. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 96:23–28. doi: 10.1016/0378-1119(90)90336-P. [DOI] [PubMed] [Google Scholar]
- 61.Alkhnbashi OS, Costa F, Shah SA, Garrett RA, Saunders SJ, Backofen R. 2014. CRISPRstrand: predicting repeat orientations to determine the crRNA-encoding strand at CRISPR loci. Bioinformatics 30:i489–i496. doi: 10.1093/bioinformatics/btu459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pearson WR, Lipman DJ. 1988. Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A 85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manica A, Zebec Z, Steinkellner J, Schleper C. 2013. Unexpectedly broad target recognition of the CRISPR-mediated virus defence system in the archaeon Sulfolobus solfataricus. Nucleic Acids Res 41:10509–10517. doi: 10.1093/nar/gkt767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Busch A, Richter AS, Backofen R. 2008. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 24:2849–2856. doi: 10.1093/bioinformatics/btn544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 66.Rabilloud T, Carpentier G, Tarroux P. 1988. Improvement and simplification of low-background silver staining of proteins by using sodium dithionite. Electrophoresis 9:288–291. doi: 10.1002/elps.1150090608. [DOI] [PubMed] [Google Scholar]
- 67.Hopmans EC, Schouten S, Pancost RD, van der Meer MT, Sinninghe Damste JS. 2000. Analysis of intact tetraether lipids in archaeal cell material and sediments by high performance liquid chromatography/atmospheric pressure chemical ionization mass spectrometry. Rapid Commun Mass Spectrom 14:585–589. doi:. [DOI] [PubMed] [Google Scholar]
- 68.Liu XL, Summons RE, Hinrichs KU. 2012. Extending the known range of glycerol ether lipids in the environment: structural assignments based on tandem mass spectral fragmentation patterns. Rapid Commun Mass Spectrom 26:2295–2302. doi: 10.1002/rcm.6355. [DOI] [PubMed] [Google Scholar]
- 69.Brard M, Laine C, Rethore G, Laurent I, Neveu C, Lemiegre L, Benvegnu T. 2007. Synthesis of archaeal bipolar lipid analogues: a way to versatile drug/gene delivery systems. J Org Chem 72:8267–8279. doi: 10.1021/jo071181r. [DOI] [PubMed] [Google Scholar]
- 70.Weidenbach K, Gloer J, Ehlers C, Sandman K, Reeve JN, Schmitz RA. 2008. Deletion of the archaeal histone in Methanosarcina mazei Go1 results in reduced growth and genomic transcription. Mol Microbiol 67:662–671. doi: 10.1111/j.1365-2958.2007.06076.x. [DOI] [PubMed] [Google Scholar]
- 71.Veit K, Ehlers C, Schmitz RA. 2005. Effects of nitrogen and carbon sources on transcription of soluble methyltransferases in Methanosarcina mazei strain Go1. J Bacteriol 187:6147–6154. doi: 10.1128/JB.187.17.6147-6154.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Veit K, Ehlers C, Ehrenreich A, Salmon K, Hovey R, Gunsalus RP, Deppenmeier U, Schmitz RA. 2006. Global transcriptional analysis of Methanosarcina mazei strain Go1 under different nitrogen availabilities. Mol Genet Genomics 276:41–55. doi: 10.1007/s00438-006-0117-9. [DOI] [PubMed] [Google Scholar]
- 73.Talaat AM, Lyons R, Howard ST, Johnston SA. 2004. The temporal expression profile of Mycobacterium tuberculosis infection in mice. Proc Natl Acad Sci U S A 101:4602–4607. doi: 10.1073/pnas.0306023101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jager D, Sharma CM, Thomsen J, Ehlers C, Vogel J, Schmitz RA. 2009. Deep sequencing analysis of the Methanosarcina mazei Go1 transcriptome in response to nitrogen availability. Proc Natl Acad Sci U S A 106:21878–21882. doi: 10.1073/pnas.0909051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.