

ABSTRACT
CM2 is the second membrane protein of the influenza C virus and has been demonstrated to play a role in the uncoating and genome packaging processes in influenza C virus replication. Although the effects of N-linked glycosylation, disulfide-linked oligomerization, and palmitoylation of CM2 on virus replication have been analyzed, the effect of the phosphorylation of CM2 on virus replication remains to be determined. In this study, a phosphorylation site(s) at residue 78 and/or 103 of CM2 was replaced with an alanine residue(s), and the effects of the loss of phosphorylation on influenza C virus replication were analyzed. No significant differences were observed in the packaging of the reporter gene between influenza C virus-like particles (VLPs) produced from 293T cells expressing wild-type CM2 and those from the cells expressing the CM2 mutants lacking the phosphorylation site(s). Reporter gene expression in HMV-II cells infected with VLPs containing the CM2 mutants was inhibited in comparison with that in cells infected with wild-type VLPs. The virus production of the recombinant influenza C virus possessing CM2 mutants containing a serine-to-alanine change at residue 78 was significantly lower than that of wild-type recombinant influenza C virus. Furthermore, the virus growth of the recombinant viruses possessing CM2 with a serine-to-aspartic acid change at position 78, to mimic constitutive phosphorylation, was virtually identical to that of the wild-type virus. These results suggest that phosphorylation of CM2 plays a role in efficient virus replication, probably through the addition of a negative charge to the Ser78 phosphorylation site.
IMPORTANCE It is well-known that many host and viral proteins are posttranslationally modified by phosphorylation, which plays a role in the functions of these proteins. In influenza A and B viruses, phosphorylation of viral proteins NP, M1, NS1, and the nuclear export protein (NEP), which are not integrated into the membranes, affects the functions of these proteins, thereby affecting virus replication. However, it was reported that phosphorylation of the influenza A virus M2 ion channel protein, which is integrated into the membrane, has no effect on virus replication in vitro or in vivo. We previously demonstrated that the influenza C virus CM2 ion channel protein is modified by N-glycosylation, oligomerization, palmitoylation, and phosphorylation and have analyzed the effects of these modifications, except phosphorylation, on virus replication. This is the first report demonstrating that phosphorylation of the influenza C virus CM2 ion channel protein, unlike that of the influenza A virus M2 protein, plays a role in virus replication.
KEYWORDS: CM2 protein, influenza C virus, ion channel protein, phosphorylation, virus replication
INTRODUCTION
The genome of the influenza C virus consists of seven single-stranded RNA segments of negative polarity (1). RNA segment 6 (M gene) of influenza C/Yamagata/1/88 is 1,181 nucleotides in length and encodes the matrix (M1) and CM2 proteins. The predominant mRNA of this RNA segment lacks the region from nucleotide 755 to 982 and encodes the 242-amino-acid (aa) M1 protein (2). Unspliced mRNA from RNA segment 6, synthesized in small amounts, encodes a 374-aa protein, designated P42, which contains an additional 132 amino acids from the C terminus of M1 (3, 4). P42 is cleaved by signal peptidase at the C-terminal side of alanine residue 259 to generate CM2, composed of the C-terminal 115 amino acids, in addition to the M1′ protein, composed of the N-terminal 259 amino acids (5, 6).
CM2 is an integral membrane protein that is oriented in membranes with a 23-aa N-terminal extracellular domain, a 23-aa transmembrane domain, and a 69-aa C-terminal cytoplasmic domain (7–9). CM2 is modified posttranslationally by the addition of an N-linked oligosaccharide chain at asparagine residue 11, which converts CM20, the nonglycosylated form of CM2 (Mr of ∼16,000), into CM2a (Mr of ∼18,000) (7, 8). Conversion of the carbohydrate chain from a high-mannose type to a complex type converts CM2a into CM2b with heterogeneous electrophoretic mobility (Mr of ∼22,000 to 30,000) (7). CM2 is also modified by palmitoylation at cysteine residue 65 (7, 10). CM2 is further modified by phosphorylation, which occurs exclusively at the serine residues (11). Alanine replacement of the candidate phosphorylation sites, the three serine residues at 78, 103, and 108, demonstrated that serine 78 and serine 103 are the major and minor sites for phosphorylation, respectively, with no phosphorylation of serine 108 detected (10). Furthermore, CM2 is modified by the formation of intermolecular disulfide bonds at cysteine residues 1, 6, and 20, generating disulfide-linked dimers and tetramers (7, 8, 10). CM2 is also known to be transported to the cell surface (7, 8, 10) and is incorporated into virions (7).
We demonstrated that CM2 forms a voltage-activated ion channel permeable to chloride ions (12). Another group indicated that CM2 is capable of elevating the trans-Golgi complex pH via proton permeability although to a lesser extent than that observed in the M2 proton channel of influenza A virus (13).
The M2 protein of influenza A virus has been shown to possess ion channel activity selective for H+ ions and to play important roles in the virus life cycle by facilitating uncoating, genome segment packaging into virions, and virion morphology (1). As the biochemical properties of CM2 are very similar to those of M2 (7, 8) and as CM2 appears to possess proton permeability similar to that of M2 (13), it is conceivable that CM2 also has a role in influenza C virus replication. We therefore examined the effect of CM2 on virus replication using an influenza C virus-like particle (VLP)-generating system (14) and revealed that CM2 plays a role in the packaging of the reporter gene into VLPs as well as in the uncoating of the VLPs, suggesting that CM2 is essential for virus replication (15).
The effect of the posttranslational modification of CM2 on influenza C virus replication has been investigated using a reverse genetics approach with recombinant viruses lacking CM2 modification sites (16). The loss of sites for N-glycosylation and disulfide bond formation reduced virus growth, suggesting that these posttranslational modifications in the extracellular domain of CM2 are required for efficient virus replication (17, 18). In contrast, no inhibition in growth was observed for a recombinant virus lacking a palmitoylation site in the cytoplasmic domain of CM2, suggesting that the palmitoylation of CM2 does not affect virus replication (19). However, the effect of the phosphorylation of CM2 on influenza C virus replication has not yet been analyzed. To this end, we generated VLPs and recombinant influenza C virus possessing CM2 lacking a phosphorylation site(s) and analyzed growth. The resultant data suggested that the phosphorylation of CM2 plays a role in influenza C virus replication.
RESULTS
Expression of CM2 phosphorylation-deficient mutants.
To investigate the effects of CM2 protein phosphorylation on influenza C virus replication, we constructed transient expression vectors that encoded CM2 mutants lacking a phosphorylation site(s) in the cytoplasmic domain, CM2-S78A, CM2-S103A, and CM2-S78/103A, in which either or both of the residues S78 and S103 were changed to alanines (Fig. 1A). The transfected cells expressing wild-type CM2 or the above CM2 mutants were lysed at 48 h posttransfection, and the expressed wild-type CM2 or CM2 mutants were separated by electrophoresis under reducing conditions and detected by Western blotting with an anti-glutathione S-transferase (GST)/CM2 serum. No significant differences were observed in the expression levels of the wild-type CM2 and the CM2 mutants (data not shown). Furthermore, we examined the expression of the wild-type CM2 and CM2 mutants as well as the degree of phosphorylation of the CM2 mutants by metabolic labeling. The 293T cells transfected with plasmids expressing wild-type CM2 or the CM2 mutants lacking a phosphorylation site(s) were pulse-labeled with [35S]methionine (lanes S) or [32P]orthophosphate (lanes P) for 4 h at 48 h posttransfection and immunoprecipitated with anti-GST/CM2 serum (Fig. 1B). No significant differences were observed in the expression levels of the wild-type CM2 and the CM2 mutants (Fig. 1B, lanes S). Furthermore, the maturation of CM2 from CM2a to CM2b observed in the cells expressing the CM2 mutants was virtually identical to that in the cells expressing the wild-type CM2, indicating that phosphorylation of CM2 did not affect the N-glycosylation processing of CM2 from a high-mannose type to a complex type. As shown in lanes P of Fig. 1B, a small amount of the phosphorylated CM2b was detected in cells expressing wild-type CM2, and a smaller amount of phosphorylated CM2b was detected in cells expressing CM2-S78A and CM2-S103A. To confirm the phosphorylation levels, the metabolically labeled cells were immunoprecipitated with anti-GST/CM2 serum, and the resulting immunoprecipitates were treated with N-glycanase and analyzed by SDS-PAGE (Fig. 1C). After N-glycanase treatment, the glycosylated CM2a and CM2b proteins shown in Fig. 1B were converted into nonglycosylated CM20 and electrophoretically migrated as almost a single band. The total amounts of the CM2 proteins migrating as CM20 did not differ significantly between the wild-type CM2 and the CM2 mutants (Fig. 1C, upper panel). The amount of phosphorylated CM2-S103A was similar to that of phosphorylated wild-type CM2, while the amount of phosphorylated CM2-S78A was much less than that of phosphorylated wild-type CM2 (Fig. 1C, lower panel). No phosphorylation of CM2-S78/103A was detected. These data indicate that phosphorylation predominantly occurred at Ser78, with a lesser degree of phosphorylation occurring at Ser103. Furthermore, the CM20 of CM2-S78A and that of CM2-S103A were electrophoretically migrated slightly more and less quickly, respectively, and both were detected in wild-type CM2 (Fig. 1C, lower panel). This result indicates that the faster- and slower-migrating CM20 proteins were phosphorylated at S103 and S78, respectively.
FIG 1.
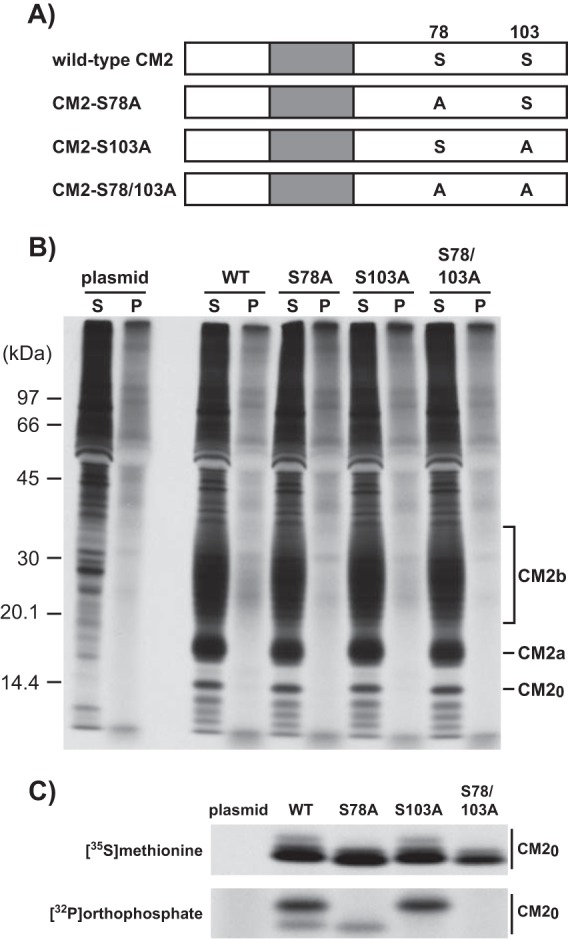
Construction of CM2 mutants lacking a phosphorylation site(s) and metabolic labeling of the CM2 mutants. (A) We constructed transient expression vectors that encoded CM2 mutants lacking a phosphorylation site(s) in the cytoplasmic domain, CM2-S78A, CM2-S103A, and CM2-S78/103A, by the substitution of alanines for serines at residue 78 (S78), S103, and S78 plus S103, respectively. (B and C) 293T cells transfected with the plasmids expressing wild-type CM2 or CM2 mutants lacking a phosphorylation site(s) (Fig. 1A) were pulse-labeled with [35S]methionine or [32P]orthophosphate for 4 h at 48 h posttransfection. The cells were disrupted in RIPA buffer and then immunoprecipitated with anti-GST/CM2 serum. The resulting immunoprecipitates were untreated (B) or treated with N-glycanase at 37°C for 16 h (C) and analyzed by SDS-PAGE on 17.5% gels containing 4 M urea under reducing conditions and processed for analysis by fluorography.
CM2 phosphorylation does not affect the generation of VLPs.
We next investigated the effect of CM2 phosphorylation on the generation of influenza C virus-like particles (VLPs). Wild-type VLPs (WT-VLPs), VLPs possessing CM2-S78A, CM2-S103A, or CM2-S78/103A, and CM2-deficient VLPs (ΔCM2-VLPs) as a negative control were generated by the transfection of the 293T cells with plasmids, as described in Materials and Methods. To compare the number of WT-VLPs and VLPs possessing CM2 mutants, the supernatant from transfected 293T cells in three 100-mm petri dishes was clarified through 30% sucrose in NTE (100 mM NaCl, 10 mM Tris-HCl, 1 mM EDTA) buffer, and the pellet containing the VLPs was subjected to hemagglutination and determination of the protein concentration. Four independent experiments showed that the hemagglutination titers and protein amounts for the clarified WT-VLPs, VLPs possessing CM2-S78A, CM2-S103A, or CM2-S78/103A, and ΔCM2-VLPs were virtually identical at 128 to 256 hemagglutinating units (HAU)/ml and 7.07 to 8.06 μg, respectively (Table 1). This indicates that there were no significant differences in the numbers of VLPs generated. Thus, CM2 phosphorylation appears not to affect VLP generation. We further examined whether the phosphorylation of CM2 affects the composition of VLPs. Equal amounts of clarified WT-VLPs, VLPs possessing CM2 mutants lacking a phosphorylation site(s), and ΔCM2-VLPs, based on protein amounts, were analyzed by Western blotting using anti-GST/CM2 serum or monoclonal antibodies against the major structural proteins, hemagglutinin-esterase (HE) and M1 (Fig. 2). We were unable to detect any differences in the amounts or the electrophoretic patterns of CM2 between the WT-VLPs and the VLPs possessing CM2 mutants lacking a phosphorylation site(s), suggesting that CM2 phosphorylation does not affect the incorporation of CM2 proteins into VLPs (Fig. 2A). Further, the amounts of the major structural proteins, HE and M1, were almost identical between the WT-VLPs and the VLPs possessing CM2 mutants lacking a phosphorylation site(s), suggesting that CM2 phosphorylation does not affect the incorporation of major structural proteins into VLPs (Fig. 2B).
TABLE 1.
Quantification of VLPs
| VLPa | Amt of protein (μg)b |
|---|---|
| WT | 7.83 ± 1.14 |
| S78A | 7.64 ± 0.87 |
| S103A | 7.87 ± 1.70 |
| S78/103A | 8.06 ± 0.69 |
| ΔCM2 | 7.07 ± 1.11 |
WT, S78A, S103A, S78/103A, and ΔCM2 indicate wild-type VLPs, VLPs possessing CM2-S78A, CM2-S103A, or CM2-S78/103A, and CM2-deficient VLPs, respectively.
Data from four independent experiments are presented as means ± standard deviations. The HA titer was 128 to 256 HAU/ml for all viruses.
FIG 2.
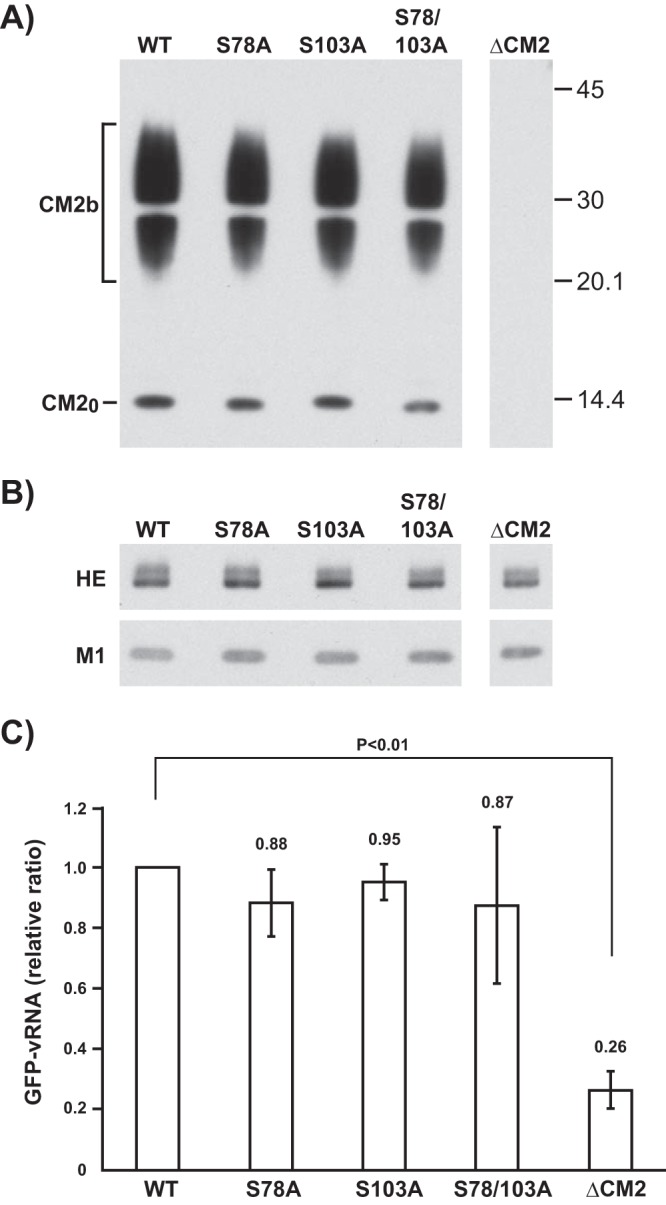
Western blotting of WT-VLPs and VLPs possessing CM2 mutants and quantification of reporter GFP-vRNA in VLPs. (A and B) The purified WT-VLPs as a positive control, VLPs containing CM2-S78A, CM2-S103A, or CM2-S78/103A, and CM2-deficient VLPs (ΔCM2-VLPs) as a negative control were analyzed by Western blotting using either anti-GST/CM2 serum (A) or monoclonal antibodies against the major structural proteins, HE and M1 (B). (C) RNA was extracted from an identical amount of WT-VLPs, VLPs containing CM2-S78A, CM2-S103A, or CM2-S78/103A, and ΔCM2-VLPs and subjected to real-time RT-PCR using a pair of primers specific for the GFP sequence. The copy number of GFP-vRNA in the WT-VLPs was used for normalization, and the statistical analysis of four independent experiments is shown. All data are presented as the ratio of means ± SEM unless otherwise indicated. For comparisons between all possible pairs in each experiment group, analysis of variance (ANOVA) with Tukey's post hoc test was used, with a P of <0.05 considered to be statistically significant.
Phosphorylation of CM2 might not affect reporter gene packaging into VLPs.
We next examined the amount of green fluorescent protein (GFP) gene in viral RNA (vRNA) sense (GFP-vRNA) in the VLPs. RNA was extracted from identical amounts of WT-VLPs, VLPs containing CM2 mutants lacking a phosphorylation site(s), and ΔCM2-VLPs, based on protein amounts, and then subjected to real-time reverse transcription-PCR (RT-PCR) using a pair of primers specific for the GFP sequence. Four independent experiments revealed that the amount of GFP-vRNA in the ΔCM2-VLPs was significantly lower than that in the WT-VLPs (Fig. 2C), as described previously (15). However, there were no significant differences in the amounts of GFP-vRNA between the WT-VLPs and the VLPs containing CM2 mutants lacking a phosphorylation site(s). Furthermore, Western blotting revealed that there were no significant differences in the expression levels of the major viral proteins in the VLP-producing 293T cells (data not shown), and no differences were observed in the amounts of GFP-vRNA between cells producing the WT-VLPs and those producing the VLPs containing the CM2 mutants, CM2-S78A, CM2-S103A, or CM2-S78/103A (data not shown). These data indicated that the phosphorylation of CM2 may not affect the packaging efficiency of the reporter gene into VLPs.
Phosphorylation of CM2 is necessary for efficient gene transfer by VLPs.
We previously reported that WT-VLPs are capable of transferring GFP-vRNA to HMV-II cells (gene transfer) (14), indicating the transcription and translation of GFP-vRNA in the HMV-II cells infected with WT-VLPs. In the present study, in order to investigate the gene transfer ability of VLPs containing CM2 mutants lacking a phosphorylation site(s), equal amounts of WT-VLPs as a positive control, VLPs containing CM2-S78A, CM2-S103A, or CM2-S78/103A, and ΔCM2-VLPs as a negative control, based on the amounts of GFP-vRNA, were inoculated onto HMV-II cell monolayers, followed by superinfection with the helper virus. Representative microscopic data for four independent experiments are shown in Fig. 3A. Considerable GFP expression was detected in the HMV-II cells infected with the WT-VLPs, whereas only a few HMV-II cells infected with ΔCM2-VLPs expressed GFP. The number of GFP-positive cells infected with the VLPs containing CM2-S78A or CM2-S78/103A was considerably smaller than the number infected with WT-VLPs. However, the number of GFP-positive cells infected with VLPs containing CM2-S103A was only a little smaller than the number infected with WT-VLPs. To quantify the results, the GFP proteins expressed in VLPs-infected cells were further analyzed by Western blotting, followed by densitometric analysis. Representative data for three independent Western blotting experiments are shown in Fig. 3B. The amount of GFP protein expressed in the HMV-II cells infected with VLPs containing CM2-S78A or CM2-S78/103A was much lower than that in the cells infected with WT-VLPs, while the amount of GFP expressed in the cells infected with VLPs containing CM2-S103A was only slightly lower than that in the cells infected with WT-VLPs. The densitometric analysis of these three independent experiments revealed that the amount of GFP expressed in the HMV-II cells infected with VLPs containing CM2-S78A, CM2-S103A, or CM2-S78/103A was approximately 40%, 80%, or 30%, respectively, of that in the cells infected with WT-VLPs (Fig. 3C). There was a statistically significant difference in GFP expression levels between the cells infected with WT-VLPs and those infected with VLPs containing the CM2-S78A or CM2-S78/103A but not between the cells infected with WT-VLPs and those infected with VLPs containing CM2-S103A. These data indicated that the phosphorylation of the VLP CM2 protein, particularly that at S78, is involved in efficient gene transfer.
FIG 3.

Reporter gene expression in HMV-II cells infected with VLPs. (A) HMV-II cells were infected with equal amounts of WT-VLPs, VLPs containing CM2-S78A, CM2-S103A, or CM2-S78/103A, and ΔCM2-VLPs, based on the amounts of GFP-vRNA, superinfected with the helper virus, and then incubated for 48 h. The representative microscopic data from four independent experiments are shown. (B) The cells in the experiment shown in panel A were lysed and analyzed by Western blotting using anti-GFP polyclonal antibodies. The representative data for three independent experiments are shown. α-Tubulin, as an internal control, is shown in the lower panel. (C) The three independent Western blotting experiments shown in Fig. 3B were next analyzed by densitometry, followed by statistical analysis. The amount of GFP proteins expressed in the cells infected with WT-VLPs was used for normalization. All data are presented as the ratio of means ± SEM unless otherwise indicated. For comparisons between all possible pairs in each experiment group, analysis of variance (ANOVA) with Tukey's post hoc test was used, with a P value of <0.05 considered to be statistically significant (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (D) HMV-II cells were mock infected (control) or infected with equal amounts of WT-VLPs, VLPs containing CM2-S78A, CM2-S103A, or CM2-S78/103A, and ΔCM2-VLPs, superinfected with the helper virus, and then incubated for 48 h, followed by flow cytometry. Representative data for three independent experiments are shown. Vertical and horizontal lines indicate GFP intensities and forward scattered light (FSC) corresponding to the size of cells, respectively. Each dot indicates an individual cell. Upper right and lower right squares represent GFP-positive and GFP-negative cells, respectively. Numbers shown at the upper right of each panel indicate the rate (percentage) of the GFP-positive cells per total number of cells. (E) The three independent flow cytometry experiments were analyzed statistically as described in for panel C. Analysis of variance (ANOVA) with Tukey's post hoc test was used, with a P value of <0.05 considered to be statistically significant (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
To further quantify the number of GFP-positive cells and the intensity of GFP expression in each cell, HMV-II cells infected with equal amounts of WT-VLPs (as a positive control), VLPs containing CM2-S78A, CM2-S103A, or CM2-S78/103A, and ΔCM2-VLPs, based on the amounts of GFP-vRNA, were superinfected with the helper virus and analyzed by flow cytometry. Representative data for three independent experiments are shown in Fig. 3D. The rate (percentage) of the GFP-positive cells per total number of cells infected with WT-VLPs, VLPs containing CM2-S78A, CM2-S103A, or CM2-S78/103A, ΔCM2-VLPs, or the helper virus alone (control) was 5.20, 2.66, 4.78, 2.63, 0.43, or 0.46, respectively. The statistical analysis of the three independent experiments is shown in Fig. 3E. The rate (percentage) of the GFP-positive cells per total number of cells infected with VLPs containing CM2-S78A or CM2-S78/103A (2.67 ± 0.34 or 3.09 ± 0.63) was significantly smaller than that with WT-VLPs (5.17 ± 0.52). However, there was no significant difference in the GFP-positive rate between the cells infected with WT-VLPs and those infected with VLPs containing CM2-S103A (4.49 ± 0.64). The GFP-positive rate observed for infection with ΔCM2-VLPs (0.66 ± 0.23) was the same as that for infection with the helper virus alone (0.66 ± 0.17) as a background. These data also indicated that the phosphorylation of the VLP CM2 protein, particularly that at S78, is involved in reporter gene expression. We previously demonstrated that CM2 is involved in the uncoating process during virus replication, thereby affecting reporter gene expression (15). Therefore, the reduced gene expression shown in the cells infected with VLPs containing CM2-S78A or CM2-S78/103A was speculated to result from impaired uncoating due to the loss of phosphorylation. To examine this possibility, HMV-II cells infected with WT-VLPs or VLPs containing CM2 mutants were divided into nuclear and cytoplasmic fractions at 60 min postinfection (p.i.), and the GFP-vRNA of the respective fractions was quantified by real-time PCR (15) in three independent experiments. However, we could not find any significant differences in the uncoating process between WT-VLP and VLPs containing CM2-S78A or CM2-S78/103A (data not shown).
Phosphorylation of CM2 affects influenza C virus replication.
To determine whether the phosphorylation of CM2 affects influenza C virus replication, recombinant viruses possessing wild-type CM2, CM2-S78A, CM2-S103A, or CM2-S78/103A (designated rWT, rS78A, rS103A, or rS78/103A, respectively) were generated by reverse genetics. MDCK cells were infected with these recombinant viruses at a multiplicity of infection (MOI) of 0.001 in the presence of 20 μg/ml tosylamide phenylmethyl chloromethyl ketone (TPCK)-trypsin. The culture medium supernatants at 0.5, 1, 2, 3, 4, 5, 6, and 7 days p.i. were processed for plaque assays to quantify virus yields (Fig. 4A). The virus yields of rS78A and rS78/103A were significantly lower than the yield of rWT virus from 3 days p.i. to 7 days p.i. However, there was no significant difference in virus yields between the rS103A and rWT viruses. Furthermore, the plaque sizes of both rS78A and rS78/103A were markedly smaller than the plaque size of rWT. However, the plaque size of rS103A was almost identical to that of rWT (Fig. 4B). To quantify the plaque size of the recombinant viruses, the sizes of all plaques observed in six representative plates in the plaque assays for rWT, rS78A, rS103A, and rS78/103A (Fig. 4B) were measured, and the numbers of plaques larger and smaller than 0.5 mm in diameter were counted. The ratio of the number of plaques larger than 0.5 mm per total plaque number for each plate of rWT, rS78A, rS103A, and rS78/103A and the mean of the ratios are shown in Fig. 4C. The ratios of large plaques for both rS78A and rS78/103A were significantly smaller than the ratio for the rWT virus; however, that for rS103A was virtually identical to that for rWT. These data indicated that phosphorylation at S78 is required for efficient virus replication.
FIG 4.
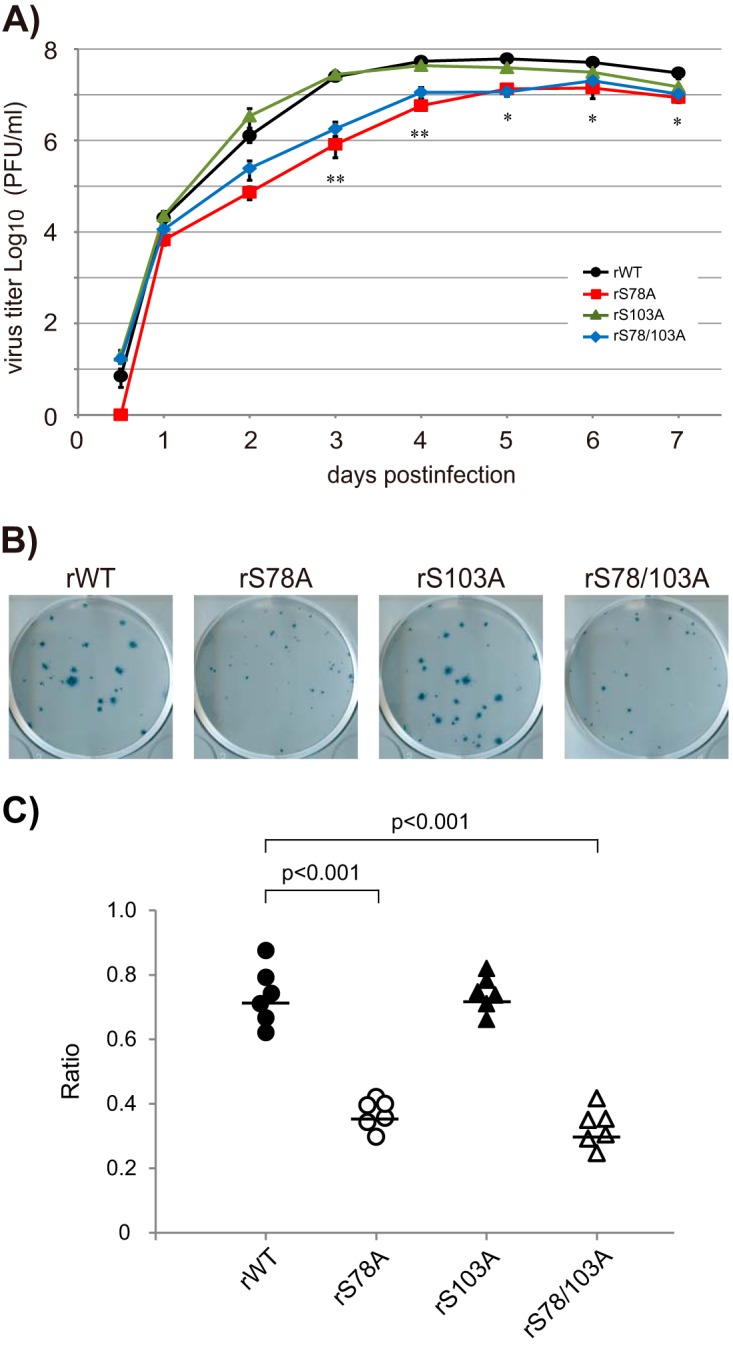
Growth kinetics of recombinant virus possessing CM2 mutants lacking a phosphorylation site(s). (A) The recombinant viruses possessing wild-type CM2, CM2-S78A, CM2-S103A, or CM2-S78/103A were generated by reverse genetics as described in Materials and Methods and designated rWT, rS78A, rS103A, or rS78/103A, respectively. MDCK cells were infected with these recombinant viruses at an MOI of 0.001 in the presence of 20 μg/ml TPCK-trypsin. The culture medium supernatants at 0.5, 1, 2, 3, 4, 5, 6, and 7 days p.i. were processed for plaque assays to quantify infectious virus titers. The results for three independent experiments were statistically analyzed. All data are presented as the ratio of means ± SEM unless otherwise indicated. For comparisons between all possible pairs in each experiment group, analysis of variance (ANOVA) with Tukey's post hoc test was used, with a P value of <0.05 considered to be statistically significant (*, P < 0.05; **, P < 0.01). (B) The representative plaque morphology at 4 days p.i. for three independent plaque assays of rWT, rS78A, rS103A, and rS78/103A (panel A) are shown. (C) The sizes of all plaques observed in six representative plates from the plaque assays for rWT, rS78A, rS103A, and rS78/103A (Fig. 4B) were measured, and the numbers of plaques larger or smaller than 0.5 mm were counted. The ratios of the number of plaques larger than 0.5 mm per total plaque number for each plate of rWT, rS78A, rS103A, and rS78/103A virus were plotted as closed circles, open circles, closed triangles, and open triangles, respectively, and the means of the ratios are shown as bars.
We next investigated whether the CM2 phosphorylation-mediated effect on virus replication is dependent on the negative charge of phosphoserine. To examine this possibility, recombinant viruses possessing CM2 with a serine-to-aspartic acid (D) change at position 78 (CM2-S78D) or 103 (CM2-S103D) (designated rS78D or rS103D, respectively) were generated to mimic constitutive phosphorylation (Fig. 5A). MDCK cells were infected with either rWT, rS78D, or rS103D at an MOI of 0.001 in the presence of 20 μg/ml TPCK-trypsin, and virus titers were determined by plaque assays as described above. The virus yields of rS78D and rS103D were virtually identical to the yield of rWT from 0.5 days p.i. to 7 days p.i. (Fig. 5B). These data suggest that phosphorylation of CM2 plays a role in efficient virus replication, probably by the addition of a negative charge at the phosphorylation site(s).
FIG 5.
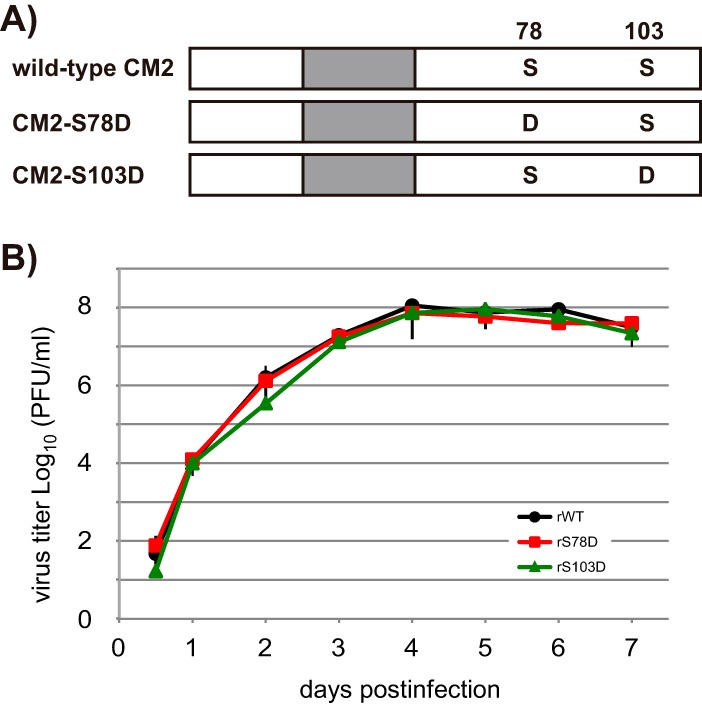
Growth kinetics of recombinant virus containing CM2 mutants possessing aspartic acid substitutions at S78 or S103. (A) To mimic constitutive phosphorylation, CM2 mutants possessing an aspartic acid (D) substitution at S78 or S103 were constructed and designated CM2-S78D or CM2-S103D, respectively. (B) Recombinant viruses containing CM2-S78D or CM2-S103D (designated rS78D or rS103D, respectively) were generated by reverse genetics. MDCK cells were infected with these recombinant viruses at an MOI of 0.001 in the presence of 20 μg/ml TPCK-trypsin. Virus growth was quantified by plaque assays as described in the legend of Fig. 4A. Three independent experiments were statistically analyzed, and all data are presented as the ratio of means ± SEM.
DISCUSSION
Phosphorylation of the viral proteins of influenza A and B viruses has been demonstrated, and phosphorylation sites have also been identified and found to be conserved (20). Phosphorylation of influenza viral proteins plays a number of important roles in virus replication as follows. Phosphorylation of NP contributes to its nuclear-cytoplasmic shuttling, self-oligomerization, and vRNP activity, thereby affecting viral growth (20–24). Phosphorylation of NS1 regulates its dimerization and nuclear localization (20), and phosphorylation of threonine at residues 49 and 80 of NS1 suppresses its interferon-antagonistic activity and binding affinity with RIG-I, respectively, leading to attenuation of virus replication (25, 26). Phosphorylation of the nuclear export protein (NEP) regulates its nuclear export activity or polymerase activity-enhancing function (27). The matrix protein M1 is phosphorylated at multiple sites, which regulates the nuclear import of M1, thereby controlling virus replication (20, 28). However, few studies have investigated whether phosphorylation of hemagglutinin (HA) and neuraminidase (NA), the glycosylated integral membrane proteins of influenza A and B viruses, affects virus replication (20). In contrast, one report revealed that the phosphorylation of the ion channel protein M2, which is a nonglycosylated integral membrane protein of the influenza A virus, is not required for the in vitro or in vivo replication of influenza A virus (29).
With regard to influenza C virus, we previously demonstrated that the HE, NP, and M1 proteins are phosphorylated (30). We further demonstrated that the CM2 protein is also modified by phosphorylation and identified its phosphorylation sites (10, 11). The phosphorylated CM2 molecules are incorporated into virions (11), and the CM2 phosphorylation sites, at S78 and S103, are highly conserved (31, 32), which raised the possibility that the phosphorylation of CM2 may affect its function during infection. To examine this possibility, we investigated the effect of CM2 phosphorylation on influenza C virus replication in this study using VLP generation systems. There was no difference in VLP formation levels between the cells expressing wild-type CM2 and those expressing CM2-S78A, CM2-S103A, or CM2-S78/103A (Table 1). These results demonstrate that phosphorylation of CM2 is not required for influenza C VLP generation. We also demonstrated that CM2 is involved in the packaging of the reporter gene into VLPs (15). Recently, it has been demonstrated that the loss of either the N-glycosylation site or cysteine residues involved in disulfide linkage of CM2 reduces the incorporation of reporter GFP-vRNA into VLPs, suggesting that these posttranslational modifications are involved in genome packaging into virions (17, 18). In this study, we could not find any significant difference in the packaging of reporter GFP-vRNA between WT-VLPs and VLPs containing CM2-S78A, CM2-S103A, or CM2-S78/103A (Fig. 2C), suggesting that phosphorylation of CM2 is not necessary for genome packaging.
The amounts of GFP expressed in the HMV-II cells infected with VLPs containing CM2-S78A, CM2-S103A, and CM2-S78/103A were approximately 40%, 80%, and 30%, respectively, of the amount in the cells infected with WT-VLPs (Fig. 3C), suggesting that phosphorylation of CM2 plays a role in reporter gene expression. The phosphorylation of CM2-S78/103A was negligible (Fig. 1C); however, the GFP expression detected in the cells infected with VLPs containing CM2-S78/103A was approximately 30% of that in the cells infected with WT-VLPs (Fig. 3C). These data suggested that the efficiency of gene expression is improved, in part, by CM2 phosphorylation. We previously demonstrated that CM2 is involved in the uncoating process during virus replication, thereby affecting reporter gene expression (15). Therefore, the reduced gene expression shown in the cells infected with VLPs containing CM2-S78A or CM2-S78/103A (Fig. 3) might result from impaired uncoating due to the loss of phosphorylation.
Furthermore, the data indicating that the phosphorylation of CM2 was involved in gene expression (Fig. 3) also suggested that the phosphorylation of CM2 may affect virus replication. To examine this, the growth of the recombinant virus possessing CM2 phosphorylation-deficient mutants was compared with that of wild-type recombinant virus (Fig. 4). The results indicated that CM2 phosphorylation at S78 is necessary for efficient virus replication. The recombinant virus rS78/103A, in which both the major and minor CM2 phosphorylation sites (at S78 and S103, respectively) were replaced with alanine and no phosphorylation was detected (Fig. 1), replicated to a significantly lower level than did rWT although the virus yield of rS78/103A reached 107 PFU/ml (Fig. 4A). These data indicated that phosphorylation of CM2 affects virus replication at least in part.
The growth of the recombinant rS78D virus, which possessed CM2 with a serine-to-aspartic acid (D) change at position 78 to mimic constitutive phosphorylation, was virtually identical to that of rWT (Fig. 5), suggesting that phosphorylation of CM2 plays a role in efficient virus replication, probably by the addition of a negative charge to phosphorylation site S78. Therefore, the reduced growth of rS78A (Fig. 4) might be explained as follows. As the mutation of phosphorylation site S78 to an alanine residue may result in the loss of the negative charge, the appropriate conformation of CM2 and protein-protein interactions might be impaired, thereby attenuating the protein functions during virus replication, most likely those involved in the uncoating process. A similar phenomenon was previously reported as follows. When the phosphorylation sites, i.e., the serine residues of influenza A virus NEP, were changed to alanines, nuclear export activity was impaired, but this activity was rescued by the phosphomimetic change of Ser to aspartic acid (27).
The data obtained in this study indicated that CM2 phosphorylation at S78 was important for efficient virus replication although the underlying mechanism was not fully elucidated. It remains to be tested whether the phosphorylation level of CM2 has a direct effect on the replication/transcription of viral RNAs. Since CM2 is a glycosylated integral membrane protein and is not localized in the nucleus, it is thought unlikely that the phosphorylation level of CM2 has a direct effect on the replication/transcription of viral RNAs.
In conclusion, phosphorylation of CM2, the ion channel protein of influenza C virus, was found to affect virus replication. This finding is strikingly different from the effect of the phosphorylation of M2, the influenza A virus ion channel protein, on virus replication as the phosphorylation of M2 does not affect in vitro or in vivo influenza A virus replication (29).
MATERIALS AND METHODS
Ethics statement.
The present study was approved by the Safety Committee for Gene Recombination Experiments of Yamagata University (no. 26-41).
Cells, viruses, and antibodies.
We used 293T cells, HMV-II cells (a human malignant melanoma cell line), and MDCK cells maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum (FBS) (14–19), RPMI 1640 medium containing 10% newborn calf serum (11), and Eagle's minimum essential medium (MEM) containing 10% fetal bovine serum, respectively. Rabbit antiserum against the GST fusion protein containing the CM2 protein (anti-GST/CM2 serum) was prepared as described previously (3). Monoclonal antibodies (MAbs) against the HE, NP, and M1 proteins of influenza C/Ann Arbor/1/50 were prepared as reported previously (33, 34).
Construction of CM2 mutants lacking phosphorylation sites.
We constructed mutants pME18S/Met-CM2-S78A, pME18S/Met-CM2-S103A, and pME18S/Met-CM2-S78/103A expressing CM2 mutants lacking a phosphorylation site(s) by the substitution of alanines for serines at residues 78 (S78), S103, and S78 plus S103, respectively (Fig. 1A). Briefly, pME18S/Met-CM2-S78A was constructed by inverse PCR using a KOD-Plus mutagenesis kit (Toyobo) with CM2-S78A sense(5′-ATTGACGCGATGGAAAAAGATATTGC-3′) as a sense primer, CM2-Ala(76–78) R (5′-TTCAGGCATAATTGTGGTCT-3′) as an antisense primer, and pME18S/Met-CM2-YA (14) as a template. The plasmids expressing CM2 mutants lacking a phosphorylation site(s) were purified for transient expression, and the sequences of all constructs described above were verified by sequencing. The primer sequences and PCR protocols are available from the authors upon request.
Metabolic labeling and immunoprecipitation.
The 293T cells transfected with plasmids expressing wild-type CM2 or CM2 mutants lacking a phosphorylation site(s) were pulse-labeled with 1.2 MBq/ml of [35S]methionine (ARC) or 3 MBq/ml of [32P]orthophosphate (PerkinElmer) for 4 h at 48 h posttransfection (10, 11). The cells were disrupted in radioimmunoprecipitation assay (RIPA) buffer and then immunoprecipitated with anti-GST/CM2 serum as described previously (3, 7). The immunoprecipitates obtained were either left untreated or treated with N-glycanase (N-glycosidase F, recombinant; Roche) for 16 h at 37°C (7, 10) and subsequently analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) on 17.5% gels containing 4 M urea and processed for analysis by fluorography (35).
Generation and purification of VLPs.
For the generation of wild-type VLPs (WT-VLPs), 293T cells in a 35-mm petri dish were transfected with the following 10 plasmids as described previously (14, 15): 0.5 μg of pPolI/NP-AA.GFP(−), 0.125 μg of pcDNA/PB2-AA, 0.25 μg of pcDNA/PB1-AA, 0.25 μg of pcDNA/P3-AA, 0.25 μg of pCAGGS.MCS/NP-AA, 1.25 μg of pME18S/HE-AA, 0.3 μg of pCAGGS.MCS/M1-AA, 0.03125 μg of pME18S/Met-CM2-YA, 0.7 μg of pME18S/NS1-YA, and 1.0 μg of pME18S/NS2-YA. For the generation of VLPs possessing CM2 mutants lacking a phosphorylation site(s), 293T cells were transfected with a mixture containing the same amounts of the above 10 plasmids, except that 0.03125 μg each of pME18S/Met-CM2-S78A, pME18S/Met-CM2-S103A, and pME18S/Met-CM2-S78/103A was transfected instead of pME18S/Met-CM2-YA. As a negative control, CM2-deficient VLPs (ΔCM2-VLPs) were generated by transfection with the above nine plasmids, excluding pME18S/Met-CM2-YA, in which 0.03125 μg of pME18S was included instead of pME18S/Met-CM2-YA to adjust the total amount of plasmid DNA (15). To prepare a large quantity of VLPs, 293T cells in a 100-mm petri dish were transfected with proportional amounts of the plasmids described above, and VLPs in the culture medium were collected, purified, and suspended in 10% glycerol in phosphate-buffered saline (PBS) as described previously (15). The protein concentration of the purified VLPs was determined using a Pierce bicinchoninic acid (BCA) protein assay kit (Thermo SCIENTIFIC) according to the manufacturer's instructions.
Immunoblotting of 293T cells and VLPs.
Transfected 293T cells and purified VLPs were resolved by SDS-PAGE on 17.5% gels containing 4 M urea under reducing conditions (35). After SDS-PAGE, immunoblotting using the MAbs and antisera was carried out as described previously (14, 36). The proteins were detected by Amersham ECL Prime Western blotting detection reagent (GE Healthcare) according to the manufacturer's instructions. Band intensities were measured by Image J, version 1.48, software (Wayne Rasband, National Institutes of Health [http://rsb.info.nih.gov/ij/]).
RNA extraction and real-time RT-PCR.
The RNAs were extracted from the plasmid-transfected 293T cells and purified VLPs using an RNeasy minikit (Qiagen) according to the manufacturer's instructions. The RNA preparations were treated with Turbo DNA-free DNase (Ambion), purified, reverse transcribed using a viral GFP (vGFP)-reverse transcriptase (RT) primer complementary to GFP-vRNA, and then subjected to real-time PCR to quantify GFP-vRNA as described previously (15, 37). As a loading control for RNAs extracted from the cells, β-actin mRNAs were quantified by real-time PCR using β-actin forward and reverse primers (38). The primer sequences and real-time PCR protocols are available upon request.
Infection of HMV-II cells with VLPs.
The purified VLPs suspended in PBS were treated with tosylamide phenylmethyl chloromethyl ketone (TPCK)-treated trypsin (20 μg/ml) at 37°C for 10 min, followed by the addition of soybean trypsin inhibitor (15). The HMV-II cell monolayers were infected with an identical amount of the above VLPs containing wild-type CM2 or CM2 mutants lacking a phosphorylation site(s), based on the amounts of GFP-vRNAs quantified by real-time RT-PCR of VLPs, and incubated at 33°C for 60 min. They were subsequently infected with the helper virus (C/Ann Arbor/1/50) at a multiplicity of infection (MOI) of 5 and incubated for up to 48 h. GFP-positive HMV-II cells were observed under a DMI 3000B fluorescence microscope (Leica Microsystems) and photographed. Furthermore, the amounts of GFP proteins expressed in the VLP-infected HMV-II cells were measured by Western blotting with anti-GFP polyclonal antibody (pAb) (MBL) as a primary antibody and horseradish peroxidase (HRP)-conjugated goat anti-rabbit Ig (BioSource International) as a secondary antibody, and quantified using Image J, version 1.48, software. The α-tubulin in cells was measured by Western blotting using monoclonal anti-α-tubulin antibody produced in mouse (Sigma-Aldrich) as an internal control.
Flow cytometry.
VLP-infected HMV-II cells were harvested at 48 h postinfection by treatment with Accutase (Innovative Cell Technologies) and fixed with 1% paraformaldehyde for 10 min at room temperature. Fixed cells were suspended in 3% FBS in PBS and analyzed using a flow cytometer (BD FACSCant II; BD Biosciences). The data were analyzed with FlowJo software (FlowJo, LLC).
Generation of recombinant influenza C viruses by reverse genetics and multistep growth analysis.
The recombinant influenza C virus possessing wild-type CM2 (designated rWT) was generated by reverse genetics as described previously (16). The recombinant viruses possessing CM2 mutants lacking a phosphorylation site(s), in which the phosphorylation sites S78, S103, and S78 and S103 were replaced with alanine residues, were generated by reverse genetics and designated rS78A, rS103A, and rS78/103A, respectively. For the generation of rS78A, rS103A, and rS78/103A, pPolI/CM2-S78A, pPolI/CM2-S103A, and pPolI/CM2-S78/103A, respectively, were transfected into 293T cells instead of pPolI/M, together with the other six pPolI plasmids and nine protein-expressing plasmids (16). The culture medium of the transfected cells was collected at 72 h posttransfection and inoculated into the amniotic cavity of 9-day-old chicken eggs to prepare stock viruses. The M gene sequences of the rS78A, rS103A, and rS78/103A viruses were determined to ensure that no unwanted mutations were present. MDCK cells were infected with these recombinant viruses at an MOI of 0.001 in the presence of 20 μg/ml TPCK-trypsin. The culture medium supernatants were processed for determination of infectious virus titers by plaque assays.
Plaque assays.
The infectious titers of the recombinant viruses were determined by plaque assays according to the procedures described previously (19, 39). Briefly, viruses were serially 10-fold diluted and inoculated onto MDCK cell monolayers. After incubation at 34°C for 60 min, the cells were overlaid with MEM containing 1% Avicel and 5 μg/ml TPCK-trypsin and incubated at 34°C. At 4 days postinfection (p.i.), the cells were fixed, incubated with anti-NP monoclonal antibody, H27 (40), as a primary antibody and goat anti-mouse IgG(H+L), horseradish peroxidase conjugate as a secondary antibody, and visualized with True Blue (KPL) (39).
Statistical analysis.
All data are presented as the ratio of means ± standard errors of the means (SEM) unless otherwise indicated. For comparisons between all possible pairs in each experimental group, analysis of variance (ANOVA) with Tukey's post hoc test was used to test differences, with a P value of <0.05 considered to be statistically significant. Statistical analysis was carried out using IBM SPSS Statistics software, version 19 (IBM SPSS).
ACKNOWLEDGMENTS
This work was supported by JSPS KAKENHI grant numbers JP23590536, JP24590560, and JP16K19136 and The Association for Research on Lactic Acid Bacteria (Yakult).
We thank Takako Okuwa (Kanazawa Medical University) for her technical advice. We also thank Hironobu Asao, Yuji Takeda, and Akemi Araki (Yamagata University) for their technical advice.
We have no potential conflicts of interest to declare related to this article.
REFERENCES
- 1.Shaw ML, Palese P. 2013. Orthomyxoviridae, p 1151–1185. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Rancaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Yamashita M, Krystal M, Palese P. 1988. Evidence that the matrix protein of influenza C virus is coded for by a spliced mRNA. J Virol 62:3348–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hongo S, Sugawara K, Nishimura H, Muraki Y, Kitame F, Nakamura K. 1994. Identification of a second protein encoded by influenza C virus RNA segment 6. J Gen Virol 75:3503–3510. doi: 10.1099/0022-1317-75-12-3503. [DOI] [PubMed] [Google Scholar]
- 4.Hongo S, Gao P, Sugawara K, Muraki Y, Matsuzaki Y, Tada Y, Kitame F, Nakamura K. 1998. Identification of a 374-amino-acid protein encoded by RNA segment 6 of influenza C virus. J Gen Virol 79:2207–2213. doi: 10.1099/0022-1317-79-9-2207. [DOI] [PubMed] [Google Scholar]
- 5.Pekosz A, Lamb RA. 1998. Influenza C virus CM2 integral membrane glycoprotein is produced from a polypeptide precursor by cleavage of an internal signal sequence. Proc Natl Acad Sci U S A 95:13233–13238. doi: 10.1073/pnas.95.22.13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hongo S, Sugawara K, Muraki Y, Matsuzaki Y, Takashita E, Kitame F, Nakamura K. 1999. Influenza C virus CM2 protein is produced from a 374-amino-acid protein (P42) by signal peptidase cleavage. J Virol 73:46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hongo S, Sugawara K, Muraki Y, Kitame F, Nakamura K. 1997. Characterization of a second protein (CM2) encoded by RNA segment 6 of influenza C virus. J Virol 71:2786–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pekosz A, Lamb RA. 1997. The CM2 protein of influenza C virus is an oligomeric integral membrane glycoprotein structurally analogous to influenza A virus M2 and influenza B virus NB proteins. Virology 237:439–451. doi: 10.1006/viro.1997.8788. [DOI] [PubMed] [Google Scholar]
- 9.Hay AJ. 1998. Functional properties of the virus ion channels, p 78–81. In Nicholson K, Webster RG, Hay AJ (ed), Textbook of influenza. Blackwell Science, Oxford, United Kingdom. [Google Scholar]
- 10.Li ZN, Hongo S, Sugawara K, Sugahara K, Tsuchiya E, Matsuzaki Y, Nakamura K. 2001. The sites for fatty acylation, phosphorylation and intermolecular disulphide bond formation of influenza C virus CM2 protein. J Gen Virol 82:1085–1093. doi: 10.1099/0022-1317-82-5-1085. [DOI] [PubMed] [Google Scholar]
- 11.Tada Y, Hongo S, Muraki Y, Matsuzaki Y, Sugawara K, Kitame F, Nakamura K. 1998. Phosphorylation of influenza C virus CM2 protein. Virus Res 58:65–72. doi: 10.1016/S0168-1702(98)00103-8. [DOI] [PubMed] [Google Scholar]
- 12.Hongo S, Ishii K, Mori K, Takashita E, Muraki Y, Matsuzaki Y, Sugawara K. 2004. Detection of ion channel activity in Xenopus laevis oocytes expressing influenza C virus CM2 protein. Arch Virol 149:35–50. doi: 10.1007/s00705-003-0209-3. [DOI] [PubMed] [Google Scholar]
- 13.Betakova T, Hay AJ. 2007. Evidence that the CM2 protein of influenza C virus can modify the pH of the exocytic pathway of transfected cells. J Gen Virol 88:2291–2296. doi: 10.1099/vir.0.82785-0. [DOI] [PubMed] [Google Scholar]
- 14.Muraki Y, Washioka H, Sugawara K, Matsuzaki Y, Takashita E, Hongo S. 2004. Identification of an amino acid residue on influenza C virus M1 protein responsible for formation of the cord-like structures of the virus. J Gen Virol 85:1885–1893. doi: 10.1099/vir.0.79937-0. [DOI] [PubMed] [Google Scholar]
- 15.Furukawa T, Muraki Y, Noda T, Takashita E, Sho R, Sugawara K, Matsuzaki Y, Shimotai Y, Hongo S. 2011. Role of the CM2 protein in the influenza C virus replication cycle. J Virol 85:1322–1329. doi: 10.1128/JVI.01367-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muraki Y, Murata T, Takashita E, Matsuzaki Y, Sugawara K, Hongo S. 2007. A mutation on influenza C virus M1 protein affects virion morphology by altering the membrane affinity of the protein. J Virol 81:8766–8773. doi: 10.1128/JVI.00075-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okuwa T, Muraki Y, Himeda T, Ohara Y. 2012. Glycosylation of CM2 is important for efficient replication of influenza C virus. Virology 433:167–175. doi: 10.1016/j.virol.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 18.Muraki Y, Okuwa T, Himeda T, Hongo S, Ohara Y. 2013. Effect of cysteine mutations in the extracellular domain of CM2 on the influenza C virus replication. PLoS One 8:e60510. doi: 10.1371/journal.pone.0060510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muraki Y, Okuwa T, Furukawa T, Matsuzaki Y, Sugawara K, Himeda T, Hongo S, Ohara Y. 2011. Palmitoylation of CM2 is dispensable to influenza C virus replication. Virus Res 157:99–105. doi: 10.1016/j.virusres.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 20.Hutchinson EC, Denham EM, Thomas B, Trudgian DC, Hester SS, Ridlova G, York A, Turrell L, Fodor E. 2012. Mapping the phosphoproteome of influenza A and B viruses by mass spectrometry. PLoS Pathog 8:e1002993. doi: 10.1371/journal.ppat.1002993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bui M, Myers JE, Whittaker GR. 2002. Nucleo-cytoplasmic localization of influenza virus nucleoprotein depends on cell density and phosphorylation. Virus Res 84:37–44. doi: 10.1016/S0168-1702(01)00413-0. [DOI] [PubMed] [Google Scholar]
- 22.Turrell L, Hutchinson EC, Vreede FT, Fodor E. 2015. Regulation of influenza A virus nucleoprotein oligomerization by phosphorylation. J Virol 89:1452–1455. doi: 10.1128/JVI.02332-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mondal A, Potts GK, Dawson AR, Coon JJ, Mehle A. 2015. Phosphorylation at the homotypic interface regulates nucleoprotein oligomerization and assembly of the influenza virus replication machinery. PLoS Pathog 11:e1004826. doi: 10.1371/journal.ppat.1004826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng W, Li J, Wang S, Cao S, Jiang J, Chen C, Ding C, Qin C, Ye X, Gao GF, Liu W. 2015. Phosphorylation controls the nuclear-cytoplasmic shuttling of influenza A virus nucleoprotein. J Virol 89:5822–5834. doi: 10.1128/JVI.00015-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kathum OA, Schräder T, Anhlan D, Nordhoff C, Liedmann S, Pande A, Mellmann A, Ehrhardt C, Wixler V, Ludwig S. 2016. Phosphorylation of influenza A virus NS1 protein at threonine 49 suppresses its interferon antagonistic activity. Cell Microbiol 18:784–791. doi: 10.1111/cmi.12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng W, Cao S, Chen C, Li J, Zhang S, Jiang J, Niu Y, Fan W, Li Y, Bi Y, Gao GF, Sun L, Liu W. 2016. Threonine 80 phosphorylation of non-structural protein 1 regulates the replication of influenza A virus by reducing the binding affinity with RIG-I. Cell Microbiol 19:e12643. doi: 10.1111/cmi.12643. [DOI] [PubMed] [Google Scholar]
- 27.Reuther P, Giese S, Götz V, Riegger D, Schwemmle M. 2014. Phosphorylation of highly conserved serine residues in the influenza A virus nuclear export protein NEP plays a minor role in viral growth in human cells and mice. J Virol 88:7668–7673. doi: 10.1128/JVI.00854-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang S, Zhao Z, Bi Y, Sun L, Liu X, Liu W. 2013. Tyrosine 132 phosphorylation of influenza A virus M1 protein is crucial for virus replication by controlling the nuclear import of M1. J Virol 87:6182–6191. doi: 10.1128/JVI.03024-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas JM, Stevens MP, Percy N, Barclay WS. 1998. Phosphorylation of the M2 protein of influenza A virus is not essential for virus viability. Virology 252:54–64. doi: 10.1006/viro.1998.9384. [DOI] [PubMed] [Google Scholar]
- 30.Nishimura H, Sugawara K, Gao P, Muraki Y, Hongo S, Kitame F, Nakamura K. 1995. Identification of influenza C virus phosphoproteins. Microbiol Immunol 39:737–740. doi: 10.1111/j.1348-0421.1995.tb03251.x. [DOI] [PubMed] [Google Scholar]
- 31.Tada Y, Hongo S, Muraki Y, Sugawara K, Kitame F, Nakamura K. 1997. Evolutionary analysis of influenza C virus M genes. Virus Genes 15:53–59. doi: 10.1023/A:1007915215958. [DOI] [PubMed] [Google Scholar]
- 32.Matsuzaki Y, Sugawara K, Furuse Y, Shimotai Y, Hongo S, Oshitani H, Mizuta K, Nishimura H. 2016. Genetic lineage and reassortment of influenza C viruses circulating between 1947 and 2014. J Virol 90:8251–8265. doi: 10.1128/JVI.00969-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugawara K, Nishimura H, Hongo S, Kitame F, Nakamura K. 1991. Antigenic characterization of the nucleoprotein and matrix protein of influenza C virus with monoclonal antibodies. J Gen Virol 72:103–109. doi: 10.1099/0022-1317-72-1-103. [DOI] [PubMed] [Google Scholar]
- 34.Sugawara K, Nishimura H, Hongo S, Muraki Y, Kitame F, Nakamura K. 1993. Construction of an antigenic map of the haemagglutinin-esterase protein of influenza C virus. J Gen Virol 74:1661–1666. doi: 10.1099/0022-1317-74-8-1661. [DOI] [PubMed] [Google Scholar]
- 35.Yokota M, Nakamura K, Sugawara K, Homma M. 1983. The synthesis of polypeptides in influenza C virus-infected cells. Virology 130:105–117. doi: 10.1016/0042-6822(83)90121-6. [DOI] [PubMed] [Google Scholar]
- 36.Grambas S, Hay AJ. 1992. Maturation of influenza A virus hemagglutinin-estimates of the pH encountered during transport and its regulation by the M2 protein. Virology 190:11–18. doi: 10.1016/0042-6822(92)91187-Y. [DOI] [PubMed] [Google Scholar]
- 37.Moeller F, Nielsen FC, Nielsen LB. 2003. New tools for quantifying and visualizing adoptively transferred cells in recipient mice. J Immunol Methods 282:73–82. doi: 10.1016/j.jim.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 38.Abrahamsen HN, Steiniche T, Nexo E, Hamilton-Dutoit SJ, Sorensen BS. 2003. Towards quantitative mRNA analysis in paraffin-embedded tissues using real-time reverse transcriptase-polymerase chain reaction: a methodological study on lymph nodes from melanoma patients. J Mol Diagn 5:34–41. doi: 10.1016/S1525-1578(10)60449-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matrosovich M, Matrosovich T, Garten W, Klenk HD. 2006. New low-viscosity overlay medium for viral plaque assays. Virol J 3:63. doi: 10.1186/1743-422X-3-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugawara K, Muraki Y, Takashita E, Matsuzaki Y, Hongo S. 2006. Conformational maturation of the nucleoprotein synthesized in influenza C virus-infected cells. Virus Res 122:45–52. doi: 10.1016/j.virusres.2006.06.009. [DOI] [PubMed] [Google Scholar]