

Abstract
Periodontitis is an inflammatory disease of the supporting structures of the dentition that is initiated by bacterial that form a biofilm on the surface of the teeth. The pathogenesis of the disease is a result of complex interactions between the biofilm and the host response that results in dysbiosis of the microbiome and dysregulation of the inflammatory response. Current data suggest that the excess inflammation associated with periodontitis is due to a failure of resolution of inflammation pathways. In this review, the relationship between inflammation and microbial dysbiosis is examined in the context of pro-inflammation and pro-resolution mediators and their ability to modify the course of disease. The impact of local oral inflammation on systemic inflammation and the relationship of periodontitis to other inflammatory diseases, including type 2 diabetes and cardiovascular disease is reviewed. Active resolvers of inflammation, including the lipoxins and resolvins, show great promise as therapeutics for the treatment of periodontitis and other inflammatory diseases.
Introduction
Periodontitis is an inflammatory disease of the supporting structures of the teeth. The periodontium comprises the alveolar bone, the periodontal ligament (the connective tissue fibers connecting the tooth to the alveolar bone) and the cementum on the surface of the tooth root into which the connective tissue fibers insert (Figure 1A). In the oral cavity, the crown of the tooth is exposed to and colonized by the commensal microflora in the mouth in the form of organized bacterial biofilms, commonly known as dental plaque. There are sequential stages of periodontal disease development beginning with gingivitis. Gingivitis is, as the name implies, an inflammation of the gingiva that is induced by chronic exposure to dental plaque (Figure 1B). The pathological stages of gingivitis range from the initial neutrophil dominated lesion, which with time develops into a mature immune lesion dominated by B and plasma cells. The advanced lesion of gingivitis is characterized by loss of gingival collagen, but importantly, it is reversible with the removal of the plaque. The shift to periodontitis is characterized by an irreversible loss of attachment apparatus of the tooth with concomitant alveolar bone loss Figure 1B). The composition of the biofilm associated with periodontitis changes and becomes more complex and more dominated by gram negative bacteria. Removal of the bacterial insult does not result in return to tissue homeostasis with regeneration of lost tissues. The trigger for conversion of a gingivitis lesion to periodontitis remains unknown.
Figure 1. Characteristics of Periodontitis.
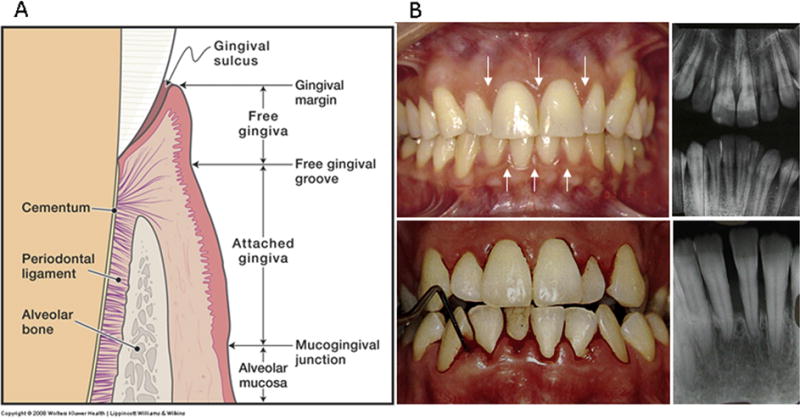
1A: Anatomy of the normal periodontium illustrates the components of the normal periodontal organ. Dental plaque accumulation in the gingival sulcus initiates inflammation of the gingiva that can lead to loss of connective tissue collagen, destruction of the periodontal ligament fibers that traverse from the bone and insert into root surface cementum with active loss of alveolar bone. 1B: The clinical and radiographic picture of mild gingivitis vs. advanced periodontitis. The upper panel illustrates mild swelling and redness of the gingiva in mild gingivitis (arrows); radiographs illustrate normal alveolar bone height with no apparent bone loss. The lower panel shows significant redness and swelling of the gingiva that easily bleeds with gentle probing in advanced periodontitis. The radiographs show loss of 60–70% of the alveolar bone height.
The relationship between the oral microbiome and the development of periodontitis is complex. The initial assumption in the literature that specific pathogens cause the disease is no longer considered valid, since it now realized that the bacteria associated with disease are actually commensals and the “pathogens” associated with disease result from an overgrowth of minor components of the biolfilm creating a “dysbiotic” microbiome. The driver for the shift to a dysbiotic microflora appears to be inflammation induced changes in the growth environment. This concept was first recognized in the early 1990’s and was called the ecological plaque hypothesis (1). In this hypothesis, it was proposed that the subgingival environment dictates or selects the specific microbial composition and this in turn drives the change from health to disease. Thus, the nonspecific accumulation of plaque leads to inflammation within the gingival tissues and gingivitis. Inflammation then drives the environmental changes within the gingival sulcus favoring the growth of gram negative and proteolytic species of bacteria. The gram negative proteolytic bacteria amplify the inflammatory response further enriching the environment with tissue breakdown products that enhance their growth. These and other recent findings have led to a paradigm shift with respect to the etiology and pathogenesis of periodontitis from an infectious disease to an inflammatory disease. This distinction changes the treatment paradigm from trying to control the composition of the commensal flora to control of inflammation (2, 3).
In general, the gingival microbiome associated with periodontal health remains stable over time in a state of dynamic equilibrium with the host. However, the host inflammatory and immune response can be overwhelmed by excessive plaque accumulation, systemic perturbations (immune disorders, changes in hormonal balance or systemic diseases such as diabetes) or environmental factors (e.g. smoking, diet, and stress) leading to a chronic inflammatory lesion. With time, the inflammatory lesion becomes a mature immune lesion as seen in most chronic inflammatory diseases. Disease-associated bacteria are relatively minor components of the subgingival flora in health and increase significantly with the development of periodontal pockets and periodontitis (4–7). In health, these organisms seem to be regulated by the interspecies competition creating microbial homeostasis. With the onset of chronic inflammation, the nutrient environment locally in the gingival sulcus becomes enriched with collagen peptides, increases in plasma proteins and haemoglobin from bleeding that select for assaccharolytic bacteria that use essential amino acids and hemin as their energy source. Several bacteria associated with periodontitis have heme proteins as growth requirements, which make bleeding and vascular permeability resulting from inflammation important variables. Therefore, periodontitis is associated with an overgrowth of specific subsets of microbes within the subgingival dental plaque as a result of changes in the microenvironment (1, 8). Dysbiosis of the periodontal microbiome (9, 10) is clearly associated with periodontitis; whether dysbiosis causes disease or results from disease has not been definitively demonstrated. However, it is likely that this is a dynamic two-way interaction.
Bacteria are undoubtedly the principal cause of gingivitis; it is the host response that dictates whether disease progresses (11, 12). Overwhelming evidence has accrued to demonstrate that it is uncontrolled inflammatory and immune responses that largely drive the tissue destruction (13, 14).
Pro-resolving Mediators in Periodontitis
Pro-inflammatory mediators, including cytokines, chemokines and metalloproteinases are known to increase dramatically in periodontal tissues and gingival crevicular fluid (GCF) in periodontitis (15–17). In particular, early response lipid mediators, including leukotriene B4 (LTB4) and prostaglandin E2 (PGE2) are markedly increased (18). In fact, much of the pathophysiology of periodontitis can be explained by the actions of these lipid mediators (19–21). PGE2 is significantly elevated in GCF of periodontitis patients, the increases correlate well with disease severity and ongoing activity, and can be used to predict future disease episodes (18, 22). Moreover, COX activity, particularly COX-2 activity, is increased in periodontal tissue in disease and in response to periodontal pathogens in vitro (23–27).
Early investigations of pro-resolving mediators in periodontitis revealed that neutrophils from periodontal patients produce increased LXA4 compared to subjects without periodontal disease (27), especially patients with Localized Aggressive Periodontitis. This study was the first reported LC-MS/MS- based analysis of eicosanoids produced by neutrophils from periodontitis patients. LTB4 and PGE2 were detected as previously reported (18), and this study revealed LXA4 production by neutrophils and in the GCF of periodontal lesions on LAP patients (27). The finding of LXA4 upregulation and in vivo neutrophil priming was also observed in asthma patients (28) and can be experimentally induced with cytokine in vitro (29). The data suggest that that the chronic periodontal lesion is characterized by hyperactivated neutrophils with resultant production of activation of lipoxin pathways. Importantly, the lipoxins were either in insufficient quantity or receptor binding was dysfunctional as resolution of inflammation was not achieved with the naturally produced lipoxins.
The question of whether sufficient lipoxin could be produced to prevent periodontal disease was addressed in a rabbit model of ligature induced periodontitis that compared wild type rabbits to 15-lipoxygenase (LO) overexpressing transgenic rabbits that have elevated circulating levels of LXA4 (30). The 15-LO product, 15-hydroxyeicosatetraenoic acid (HETE) is a known substrate for myeloid 5-LO to produce LXA4. 15-LO overexpressing transgenic rabbits exhibited reduced neutrophil recruitment and release of granule associated enzymes that play a major role in periodontal tissue destruction. In acute periodontitis, the 15-LO transgenic rabbits were protected from periodontal tissue damage and bone loss (Figure 2). These data show that sufficient quantities of lipoxin were capable of preventing tissue destruction in bacterially induced periodontal disease.
Figure 2. Resistance of 15-LO TG rabbits to periodontal destruction.
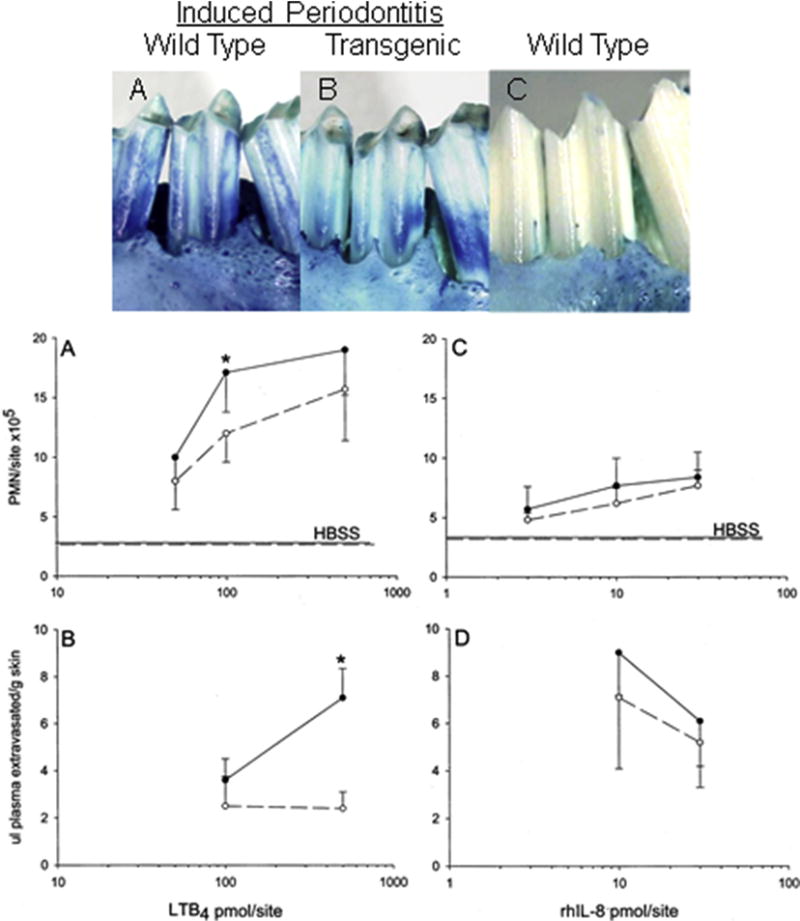
Periodontitis was induced in transgenic and wild type rabbits with ligature and P. gingivalis as described in the text. Ligature alone served as control. Upper photos: A- Clinical outcome 6 weeks after induction of periodontitis. B- Clinical picture of a TG animal that received P. gingivalis following the same regimen as the animal in A. C- Wild type animal receiving ligature alone; no P. gingivalis, no appreciable bone loss. Lower panel: 15-LO-overexpressing transgenic rabbits exhibit reduced leukocyte recruitment to skin and reduced plasma extravasation (quantified using Evans blue from skin biopsies) with LTB4. MPO activity was monitored in rabbit skin as an index of neutrophil infiltration. Wild type rabbits (solid line) exhibited significantly higher (p < 0.05) levels of leukocyte recruitment (A) and leukocyte-dependent plasma leakage (B) in response to LTB4 compared with 15-LO transgenic rabbits (dashed line). Statistical significance was not observed with human rIL-8 addition to the skin (C and D).
Localized Aggressive Periodontitis, as the name implies, is a rapidly progressing form of periodontitis that seemed refractory to endogenous levels of lipoxins (27). Further investigations of dysregulation of resolution in LAP were reported by Fredman et al. (31). In this study, aberrant LO activity was demonstrated in whole blood of LAP patients. This finding was associated with increased surface P-selectin expression on LAP platelets and increased CD18 expression on circulating neutrophils and monocytes, which translated into significantly greater platelet-neutrophil and platelet-monocytes aggregates in circulating whole blood of LAP patients. Defective macrophage phagocytosis of opsonized particles in LAP was also noted. Taken together, these observations suggest a failure to resolve local inflammatory insults induced by bacteria in the periodontium leading to periodontal disease progression.
Interestingly, Fredman reported that the in vitro abnormalities in LAP were all reversed with exogenous addition of RvE1, a resolvin of the E series derived from EPA. Further investigation of the neutrophil response to proresolution mediators by Hasturk and co-workers (32) revealed that LAP neutrophils respond to resolvins, but not lipoxins. These findings bring up the interesting prospect of selective abnormalities of the response to individual pro-resolving mediators and raise the question of the potential of these mediators as therapeutics in periodontitis.
Pro-resolving Mediators as Therapeutics
Based upon a growing body of literature that shows omega-3 fatty acids containing EPA (C20:5) are protective in a variety of human inflammatory diseases (33–36), the potential of metabolites of EPA, such as RvE1 for the prevention and treatment of periodontitis was investigated. The arachidonic acid derived proinflammatory eicosanoids play a key role in the onset and pathogenesis of inflammatory lesions in periodontitis (18, 37, 38). The etiologic stimulus is bacteria and the putative pathogens Porphyromas gingivalis, Tannerella Forsythia and Treponema denticola are strongly associated with clinical disease (39). At the time, it was still unclear how large a role inflammation played in the pathogenesis of periodontitis.
In order to evaluate the role of inflammation in periodontitis pathogenesis and to determine the therapeutic potential of the EPA derived resolution agonist RvE1, experiments were undertaken to evaluate the actions of RvE1 in rabbit periodontitis initiated by the human pathogen Porphyromonas gingivalis (P.g.). The rabbit model was established showing that ligature of specific teeth (in this case lower second premolars) with 5-0 silk sutures with topical application of 108 P.g. to the ligature 3 times per week induced severe periodontitis within 6 weeks. In the experiment, RvE1 was applied topically to the ligature in a 1 μg/ml PBS solution containing 5% ethanol (4 μl per application) from baseline for 6 weeks; controls received the PBS vehicle alone. The treatment was applied topically as a monotherapy three times per week at the same time as the P.g. Other controls in the experiment included a group that received ligature, but no microbial challenge; and another group neither ligature nor microbial challenge. To directly demonstrate the etiologic role of P.g., a final group received systemic metronidazole to kill the P.g. and also to provide a direct means of assessing antimicrobial therapy vs. control of inflammation through the actions of the endogenous proresolution agonist, RvE1.
The results revealed that application of P.g. to the ligature caused severe periodontal disease easily visible to the naked eye. Direct bone measurements after sacrifice revealed destruction of about 50% of the alveolar bone around the affected teeth. 50 mg IM administration of metronidazole inhibited onset of disease; the no ligature, no P.g. group did not develop disease. Topical treatment with 4μg of RvE1 three times per week at the ligature site inhibited bone and tissue destruction by >95% (Figure 3). The intrinsic antibacterial activity of RvE1 was assessed and none was found. Importantly, the rabbits tolerated the procedure well and showed no evidence of bacterial overgrowth of pathogens with the application of RvE1, which was resolving the inflammatory response. Histological evaluations showed a robust inflammatory infiltrate in the animals not receiving RvE1, whereas animals that received RvE1 exhibited essentially no neutrophils in the periodontal tissues. Bone histology revealed active osteoclastic bone resorption in the non-RvE1 animals and no osteoclast-like cells in the bone of the RvE1 treated animals. These first proof-of-principle studies demonstrated that RvE1 was effective in the prevention of periodontitis and confirmed the inflammatory nature of periodontitis demonstrating that control of inflammation with resolvins in an infectious disease leads to clearance of the infection and does not lead to disseminating infection (32).
Figure 3. Prevention of Porphyromonas gingivalis Associated Periodontitis.
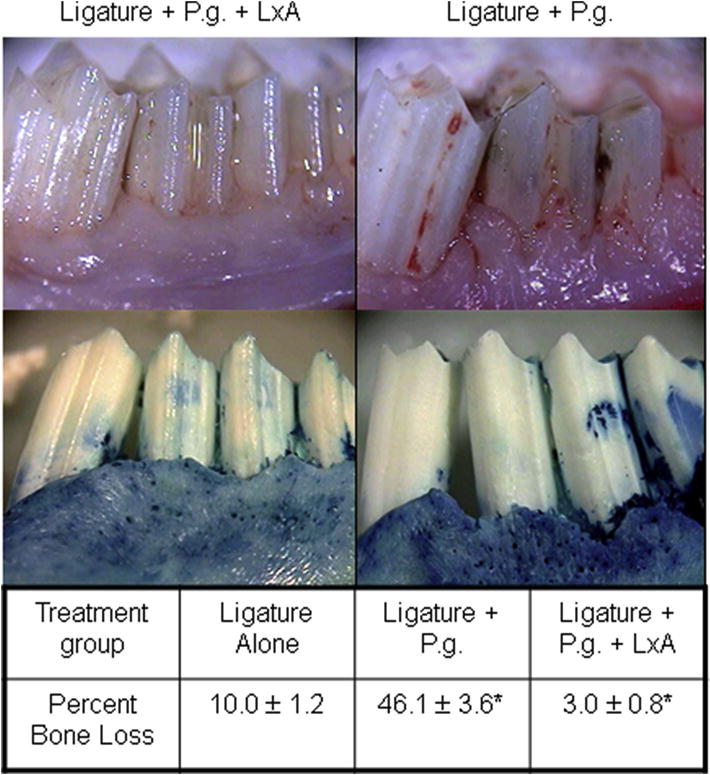
The impact of local application of an aspirin-triggered lipoxin analog (LxA) on experimental periodontitis in rabbits induced by topical application of the human pathogen Porphyromonas gingivalis was quantified by direct measurements of alveolar bone destruction. The photographs in the upper panels illustrate the marked reduction of soft tissue inflammation and alveolar bone loss that result from topical application of 4 μl of a 1 μg/ml LXA solution in PBS three times per week. The quantitative data below illustrate that topical application of LxA prevented 95% of the alveolar bone loss compared to PBS alone. Ligature alone did not cause significant bone loss indicating the significant pro-inflammatory properties of the human pathogen.
The potential of resolvins and lipoxins for treating existing, established periodontitis was first established in the same rabbit model of ligature and P.g. induced periodontitis; however, in this case, the disease was allowed to progress for 6 weeks in all experimental animals. After establishment of disease at 6 weeks, P.g. application was stopped, one group of animals was sacrificed to assess the level of disease, and the remaining animals were treated with RvE1 or vehicle as above for an additional six weeks. In these experiments, the composition of the biofilm associated with the affected teeth was monitored before disease, before therapy and after therapy. As additional controls, two groups of animals were treated with topical application of LTB4 and PGE2 instead of RvE1 (8).
The results of the treatment experiment revealed that topical treatment with RvE1 stopped disease progression and resulted in regeneration of periodontal tissues lost to prior disease, including bone and connective tissue attachment of the tooth to the bone (8). The important control in this experiment was the vehicle treated animals in which the disease progressed in spite of ceasing administration of P.g. As would be expected, topical application of PGE2 and LTB4 enhanced disease profession (8) (Figure 4).
Figure 4. Treatment of Periodontitis with Resolvin E1.
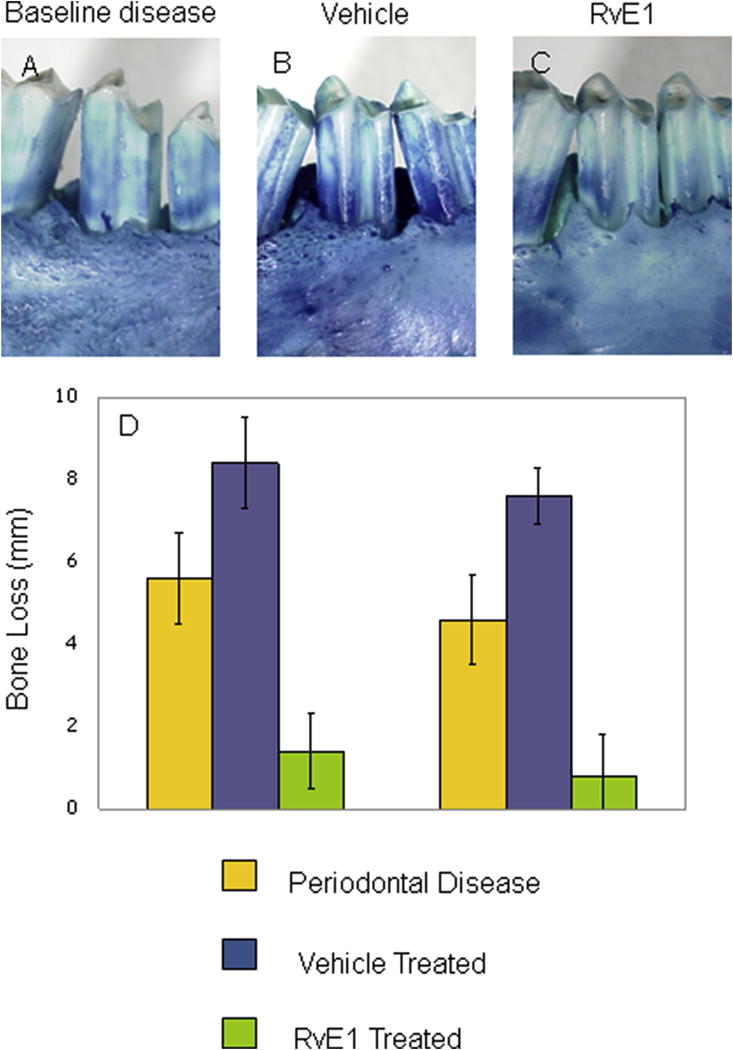
Periodontitis was established in the rabbit with ligature and Porphyromonas gingivalis for 6 weeks that resulted in significant alveolar bone loss at 6 weeks (Panel A). Beginning at 6 weeks, Resolvin E1 was applied topically three times per week (4 μl of a 1 μg/ml solution in PBS) for an additional 6 weeks. The vehicle treated group exhibited further bone loss (Panel B and D), while the RvE1 treated group exhibited regrowth on bone to near pre-disease levels (Panels C and D).
An important aspect of this study was the monitoring of the lesion associated biofilm. Before initiation of disease, the rabbit periodontal microbiome was found to be simple with a number of non-pathogenic commensals dominating the flora. Initiation of disease with ligature and P.g. resulted in the expected large increase in P.g., but also a marked increase in the complexity of the biofilm and the overall bacterial load. Among the wide array of inflammatory diseases that afflict man, periodontitis is in some ways unique, because the etiology is known; it is bacteria. However, the causative organisms have been a matter of debate for some time. These studies directly support the earlier work of Marsh (1), that demonstrated in chemostat biofilm systems that the addition of gram negative pathogens, such as P.g. have a big impact on the stability and dynamics of the biofilm. It was demonstrated in this experiment that introduction of P.g. resulted in overgrowth of other gram negative pathogens. Perhaps the more significant finding was that control of inflammation with RvE1 reversed the biofilm changes with elimination of P.g. and resolution of the microbial dysbiosis (8).
Regeneration of soft and hard tissues lost to disease is a prominent problem in many inflammatory diseases, including periodontitis and arthritis. Currently, there are few, if any, effective treatments that predictably lead to regeneration of lost tissue, likely due to the inhibition to control inflammation. Inhibitors of inflammation through blocking of enzymes or receptors do not control inflammation as much as it controls the symptoms of inflammation. It is noteworthy that >95% of lost tissues were regenerated with RvE1 treatment, albeit in a rabbit. However, it is also noteworthy that tin the control animals, disease progressed and tissue damage got worse. This was not a spontaneously healing model. Thus, these studies emphasize the role of natural control of inflammation in tissue regeneration also noting that the principles of tissue engineering, providing exogenous scaffold, cells and mediators, were not necessary.
Periodontitis is characterized by the destruction of soft and hard tissue (connective tissue and bone) supporting the teeth mediated by the host response (40). Lipid mediators play an important role in both the onset and natural resolution of these lesions. PGE2 and LTB4 are markedly elevated in progressive, active disease (41, 42) and appear to be the main determinants of maintaining chronic inflammation. Conversely, the drivers of resolution of inflammation and return to homeostasis are the resolution pathways and their products the lipoxins and resolvins. The question of production and response of the lipoxins and resolvins in disease and why they fail to resolve active lesions is an open question, but periodontitis provides a convenient disease model to unravel these complexities. It is now clear that reversal of chronic inflammation and return to homeostasis requires the active removal of immune and inflammatory cells from the lesion (43).
Pro-resolving Mediators and the Microbiome
With the current availability of molecular tools that directly extract and sequence cloned DNA from communities of microorganisms, our understanding of the relationship between human cells and the microbes that colonize the human body has completely changed (44).
Literally thousands of previously uncultured and unknown microbes have been identified in and on human tissue. The systematic study of the human microbiome has revealed bacterial associations with inflammatory diseases and conditions previously thought to be sterile events or in which the composition of the microflora was regarded as commensal (45–50). To that end, the relationship of inflammation to the microbiome clearly is of major impact in inflammatory disease and the relationship is bi-directional. Pathogens that can persist, particularly intracellular pathogens, can cause significant inflammatory dysregulation (51–53) and upregulation of systemic inflammation, as in obesity and type 2 diabetes, can cause dysbiosis of the gut microbiome (49).
These findings raise the question of the temporal relationship of the periodontal microbiome to inflammation in periodontal disease. It has been demonstrated that there is dysbiosis of the oral microbiome associated with periodontitis (9), but the temporal sequence of onset of each and the interplay between bacteria and inflammation is just beginning to be described (54, 55).
The dogma in periodontal therapy for the past 60+ years has been that removal of plaque on the teeth (the dental biofilm) is sufficient to reduce inflammation, but the effect is transient and inflammation rapidly returns (56). With the more recent realization that susceptibility to periodontitis and the destructive pathogenesis is mediated by the host response to bacteria (57–59), severe periodontitis has been associated with excess inflammation that includes, oxidative stress (60, 61) and cytokine production (62). Longitudinal studies demonstrate that inflammation predicts disease progression (63, 64) and the associated dysbiosis in periodontitis observed in human cross-sectional studies occurs after onset of disease (63).
As noted above, in studies of periodontitis progression and treatment with RvE1 in rabbits, resolution of inflammation prevented and reversed disease and promoted bone remodeling (8, 32, 65, 66). A relevant observation in these studies was that treatment with RvE1 caused spontaneous disappearance of the periodontal pathogen, P.g., without mechanical or antimicrobial therapy. In a subsequent study by Lee and co-workers, the temporal dynamics of inflammation-induced dysbiosis of the periodontal microbiota and the impact of RvE1 were examined in rat periodontitis (54). Periodontitis was induced in rats using ligature placement alone to observe naturally occurring dysbiosis of the rat microbiome associated with progressive periodontitis, rather than adding a human pathogen to induce disease. To obtain an unbiased assessment of inflammatory changes, global differential gene expression in periodontal tissue was determined in health, periodontitis and periodontitis treated with topical RvE1. To determine the impact of inflammatory status on the composition of the microbiome, 16S rDNA sequencing was performed in parallel.
As demonstrated in rabbits and mice before, topical application of RvE1 in rat periodontitis both prevented and successfully treated ligature-induced periodontitis in the rat with significant regeneration of lost periodontal soft tissues and bone. Concomitantly, shifts in the local microbiota induced by inflammation were markedly changed by the control of inflammation with RvE1, and treatment of established periodontitis with RvE1 reversed to a significant degree the shifts associated with disease progression (Figure 5).
Figure 5. RvE1 and the Dysbiotic Microbiome.
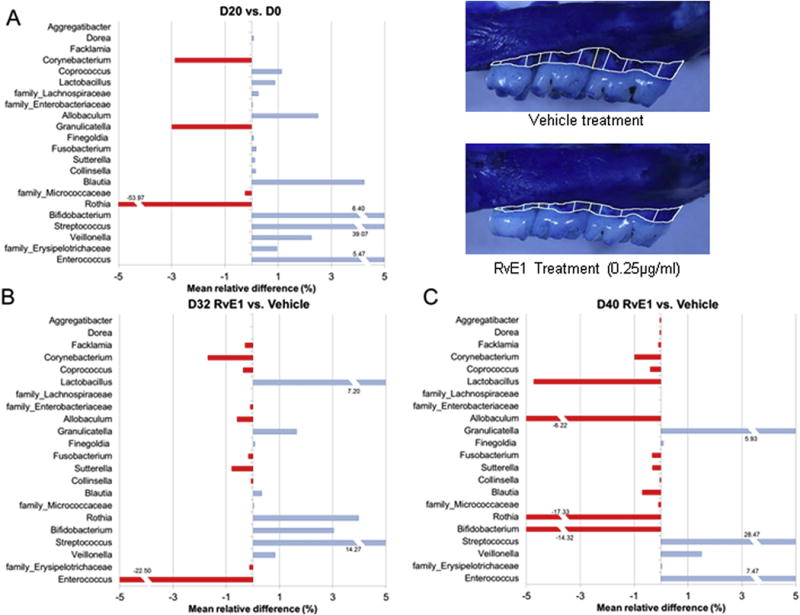
Periodontitis was induced in the rat model with ligature alone to monitor natural dysbiotic shifts of the rat microbiome in response to inflammation (Panel A). The established periodontitis was treated with topical RvE1, which reserved alveolar bone loss (inset). The impact of RvE1 treatment on the microbiome at 32 and 40 days was compared to vehicle treatment and expressed as percent mean relative difference (Panels B and C, respectively). The data suggest that control of excess inflammation reverses the pathogenic shift of the microbiome. The complexity of the microbiome (alpha diversity) and the bacterial biomass was also reduced (data not shown). (Adapted from Lee et al. 2016 (54)).
These findings reveal important aspects of the role of inflammation in host/microbiome interactions in health and disease that are generalizable and not limited to periodontitis. In active disease, species that are associated with disease clinically overgrow due to inflammation-induced changes in the local ecology obtaining required nutrients. The disease associated microbial community becomes more diverse in diseased sites (α diversity), while the microbial composition of diseased site within the individual become more similar (β diversity) (67–71). These results emphasize two important biological principles: 1. there is a direct link between local environmental conditions and the activity and composition of the microbiota, and 2. the inflammation induced impact on the microbiome is modifiable by active resolution of inflammation. These changes are not observed with inhibition of inflammation with drugs, including NSAIDs (72).
The shifts in the microbiome induced by inflammation are certainly not benign. Growth conditions that favor overgrowth of certain commensals also provide a growth environment that changes their physiology, pathogenicity and expression of virulence factors. Global differential gene expression analyses have also been performed on the microflora associated with progressive disease. The analysis of the microbiome transcriptome revealed that virulence factors are upregulated by both presumed pathogens and also health associated commensals (73, 74). Taken together, a picture is emerging in which the inflammatory response and infection are linked in a bi-directional balance in homeostasis and a bi-directional imbalance in inflammatory diseases that have an infectious component. The inflammatory response can contribute to bacterial proportional changes and expression of bacterial virulence and control of excess inflammation can benefit management of infection and clearance. These principles are not limited to oral diseases such as periodontitis, but have been demonstrated in sepsis, IBD and other diseases as well (75, 76).
Diseases resulting from the apparent disruption of the equilibrium between the host and resident biofilm have been suggested to be related to gene-by-environment interactions (55). The basic assumption is that the disease is manifest by polygenic causes modified by the environment. In a recent report, Sima and co-workers (55) investigated these interactions using inbred mice as genetic reference populations to identify quantitative trait loci (QTLs). QTLs are identified by associating the observable disease variations in inbred mouse strains with the known genetic variation. Earlier studies linked the correlation between phenotype and genotype in other complex disease, including diabetes and cancer (77). Using genetic marker regression analysis, loci that contain genes potentially contributing to the expression of a disease trait in response to the periodontal microbiome were identified. The analysis confirmed that inflammatory alveolar bone loss is a polygenic trait and that, as expected, there are shifts in the microflora associated with periodontal disease progression. Interestingly, the disease changes in the microbiome were related to a marked increase in Firmicutes in disease with a concomitant decrease in Bacteriodes. Spirochetes were significantly increased on the tongue in periodontitis suggesting that the tongue serves as a reservoir for periodontal pocket colonization. The QTL identified in the analysis combined with the microbial shifts suggest that loci associated with macrophage and osteoclast functions are influenced associated with shifts in the oral microbiome. These data emphasize the dynamic interplay required for maintenance of homeostasis and periodontal health and the central role of inflammation is creation of dysbiosis and disease.
Pro-resolving Mediators in Periodontal Regeneration
In chronic osteolytic inflammatory diseases such as periodontitis, sustained microbial challenge and a failure of endogenous resolution pathways result in tissue destruction (57). The ideal outcome of acute inflammation is its complete resolution (78, 79). Regeneration of hard and soft tissues lost to periodontal disease is limited and not predictable (80–82). It is now widely appreciated that uncontrolled acute inflammation can lead to tissue injury, chronic inflammation, tissue scarring and fibrosis (83–85). In the face of uncontrolled host defense mechanisms, tissue engineering, regeneration and reconstruction of both diseased and injured oral and craniofacial tissues are significantly hampered (86, 87). In periodontal disease (and in any contaminated surgical or traumatic wound), an uncontrolled inflammatory response causes neutrophil-mediated tissue injury that in turn can lead to irreversible bone loss. Periodontal infections and non-healing or scarring facial wounds are a major public health concern and financial burden (87–89). New data reveal that endogenous control of inflammation directly enhances bone healing and regeneration. Mediators of resolution of inflammation have actions beyond control of neutrophil function; they exhibit receptor mediated control of osteoclasts (65, 66), as well as stem cells (90–92) that differentiate into fibroblasts, and osteoblasts that function in wound healing and bone regeneration.
Importantly, the actions of lipoxins and resolvins are not limited to inflammatory cells. Mesenchymal stem cells and downstream stromal cells including fibroblasts and osteoblasts express receptors for and positively respond to resolution agonists. Lipoxin and resolvin treated mesenchymal stem cells are resistant to apoptosis in vivo and in vitro and enhanced viability has been shown to be mediated through PI3K-Akt signaling (90). In addition, treated mesenchymal stem cells up-regulate secretion of growth factors (93). In regenerating tissues in mice, treatment with resolvin resulted in differentiation and activation of osteoblasts (65).
Animals models of periodontal disease have been used to demonstrate regeneration of periodontal tissues mediated by lipoxins and resolvins, endogenous regulators if inflammation (8, 54, 94). The capacity for regeneration in small animals is greater than in large animals and man. However, models were developed that were able to demonstrate progression of disease in control animals and complete healing with >95% regeneration in animals topically treated with resolvin or lipoxin alone.
Regeneration of tissues lost to disease is a common problem in inflammatory diseases, including arthritis and periodontitis. The therapeutic strategies used try to follow the principles of tissue engineering to attempt to recapitulate development of the organ in question. This includes providing scaffolds, adding back appropriate stem or progenitor cells and soluble mediators. In just about all instances, failed tissue engineering attempts are due to uncontrolled inflammation. Inhibitors of inflammation, e.g. COX inhibitors or TNFα receptors antagonists, do not alleviate the problem. Feed forward, receptor mediated active resolution of inflammation does.
In the rabbit periodontitis model, active periodontitis was established with ligature of teeth and exogenous addition of the human pathogen P.g. Treatment of established periodontitis with RvE1 (8) or a lipoxin A4 analog resulted in complete resolution of the inflammatory lesion and >95% regeneration of bone and connective tissue attachment (periodontal ligament regeneration) to the tooth (Figure 4). In animals treated with vehicle alone (no lipoxin), the disease progressed with further loss of attachment and bone. When the proinflammatory mediators PGE2 or LTB4 were topically applied, disease progression was exacerbated (8). The important principles demonstrated by these studies are that resolvins and lipoxins act to modulate the inflammatory response essentially aborting and reversing the chronic phase, promoting more rapid resolution, and that elimination of inflammation in a healing lesion promotes tissue regeneration. In small animals, these findings are not limited to the rabbit, but have been reproduced in mice and rats as well (54, 95).
Harnessing the biomimetic properties of resolvins and lipoxins in large animals, including humans, presents a new set of problems. Large animals spontaneously develop periodontitis, like in humans, and do not spontaneously regenerate periodontal tissue under any circumstances, again, like humans. Thus, to more replicate the treatment of periodontal disease in humans, a lipoxin analog was used as an adjunct to periodontal surgical therapy to evaluate the impact on periodontal regeneration (94). Using the Hanford Miniature Swine with chronic periodontitis, surgical debridement of the periodontal lesion was followed by placement of lipoxin analog in a neutrophil microparticle carrier in the lesion before suturing. The active particle carrier with lipoxin was termed nano-proresolving medicine (NPRM). The NPRM were prepared from membrane-shed vesicles from neutrophils (96) that have been shown to target specific tissues without dilution or inactivation of the proresolving mediators (97). The controls included the components alone (lipoxin or unloaded particles) and surgery alone.
The results revealed that the components alone had small benefits, each producing significantly more bone that the surgery alone; however, the NPRM treated lesions produced significantly greater bone and periodontal ligament regeneration than the components alone (Figure 6). The NPRM served as a mimetic of endogenous resolving mechanisms and exhibited potent bioactions in tissue engineering in the treatment of an osteolytic inflammatory disease.
Figure 6. Alveolar Bone Regeneration in a Large Animal Model.
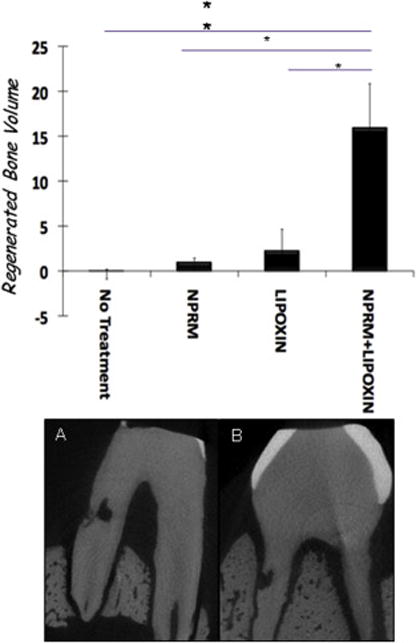
To assess the impact of therapeutic resolution of inflammation in a clinical periodontology relevant setting, the impact of locally delivered lipoxin analog in a targeted slow release nanoparticle was evaluated after periodontal surgery. Periodontitis treatment comprising surgical debridement was performed to treat infrabony pockets in a minipig. Benzo-lipoxin A4 was formulated in de-humanized neutrophil microparticles designated nano-proresolving medicines (NPRM). The NPRM was compared to surgery alone, lipoxin analog alone and microparticles alone. Quantification of regenerated bone volume (top panel) revealed that surgical debridement alone had not regeneration of bone. The benzo-lipoxin analog alone and the microparticles alone exhibited some limited activity. The NPRM generated significantly greater volume of alveolar bone in the model. The lower radiographs show that the notch marking the bone height at time of surgery was covered by new bone after NPRM treatment with no activity in the surgery alone control.
The actions of lipoxins and resolvins in tissue regeneration bring up the question of the biologic targets for this class of molecules. Are the observed actions solely in response to modification of inflammatory cell phenotype? This question was investigated in a series of experiments that determined receptor expression and function for lipoxins and resolvins on bone cells in periodontal regeneration. The direct actions of RvE1 were first determined on osteoclasts examining osteoclast development and bone resorption (98). Osteoclasts express receptors for RvE1, both ERV1 and BLT-1 (99), but signal through BLT-1. RvE1 inhibited osteoclast differentiation reducing the number multinucleated cells and attenuated RANKL-induced nuclear translocation of the p50 subunit of NFκB without altering apoptosis or osteoclast survival. Thus, the bone sparing actions of RvE1 are mediated in part through inhibition of osteoclast growth and differentiation in addition to inflammation resolution. Further, examining bone formation, it was demonstrated that osteoblasts also express receptors for RvE1, but the functional receptor is ERV1. RvE1 promotes osteoblast differentiation and bone formation (65).
Taken together, there is substantial evidence for a new mechanism for tissue regeneration in inflammatory diseases like periodontitis. Lipoxins and resolvins are functionally transferred to bone cell membrane receptors in addition to leukocytes modulating both responses to promote bone formation. The complex remodeling of bone is directly impacted by inflammation. The cellular events regulated by lipoxins and resolvins efficiently control the bacterial biofilm and its environment, including suppression or elimination of pathogens, resulting in significant regeneration of bone and connective tissues for the reestablishment of the periodontal organ.
Pro-resolving Mediators, Periodontitis and Systemic Disease
Excessive inflammation is now recognized as a central component of the most prevalent diseases in developed societies, including periodontitis, Type 2 diabetes (T2D), and cardiovascular disease. The trigger for excess inflammation remains elusive with associations to obesity and other risk factors, but the role of the microbiome is just beginning to be explored. The complications of diabetes mellitus, particularly Type 2, include periodontitis and cardiovascular disease (CVD). Fifty percent of the US population has at least some periodontal disease; T2D doubles risk for periodontitis (100). CVD risk is increased four times (Framingham Heart Study) and accounts for the majority of premature deaths in Type 2 diabetics (101). A major link between T2D and its complications is inflammation (101, 102). In periodontitis, after acute infection, the shift to chronicity and persistence of pathogens (dysbiosis) may in fact result from increased inflammation (8, 103–106) and lead to leukocyte-mediated tissue destruction. New data clearly demonstrate that Type 2 diabetics are refractory to standard periodontal therapy, which further emphasizes this interrelationship (107, 108).
The mechanism of the interactions between obesity, T2D and its complications of periodontitis and CVD is unclear. Phagocyte interactions with platelets are dysregulated in people with periodontitis or with T2D (109, 110). Thus, mechanisms related to inflammation in periodontitis may be linked with the primary cause of mortality in diabetics, CVD. It is also well-established that sufficient proresolution agonist concentrations are necessary to prevent tissue damage in inflammation (8, 32) and that these pathways are deficient in obesity and T2D (111).
Periodontitis, T2D, and cardiovascular disease are complex and involve inflammation-mediated microvascular and macrovascular damage, disruption of lipid metabolism, glycosylation of proteins, and other abnormalities that induce or are the result of dysregulation of inflammation. In periodontitis and in T2D, uncontrolled local and systemic inflammation induces dysbiosis (overgrowth of commensal pathogens) in the mouth and in the gut. The dysbiosis in the gut is associated with changes in gut permeability leading to increased systemic inflammation and insulin resistance. The resultant dysregulation of inflammation is central to phagocyte-mediated tissue injury in T2D and its periodontal complications. The return of inflamed tissues to health and homeostasis through active resolution of inflammation appears to be compromised in obesity and T2D (112). Omega 3 fatty acids and an increased omega-3/omega-6 ratio are associated with lower prevalence of coronary artery disease in Greenland Inuits and reduced incidence of diabetes in different populations (113). Similarly, omega-3 active metabolites, particularly resolvins, are protective against obesity associated inflammation and insulin resistance (114, 115).
Type 2 Diabetes and Periodontitis
The increase in inflammation induced by T2D directly contributes to the increased prevalence and severity of periodontitis in T2D (116); however, the mechanism of the interactions between T2D and its complications is unclear. The relationship between diabetes and periodontal disease is reciprocal. Infections, including periodontal infections, have a significant impact on diabetic control, and diabetes is a significant risk factor for the development and severity of periodontal disease (117). In 2011, 8.3% of the children and adults in the United States (25.8 million people) were reported to have diagnosed (18M) and undiagnosed (7M) diabetes; another 79 million have pre-diabetes (American Diabetes Association 2011 Diabetes Fact Sheet). The resulting health care and lost productivity costs in 2013 were $246 billion. The substantial increase in obesity, which is the main risk factor for T2D, and the high rates of impaired fasting glucose and impaired glucose tolerance found in NHANES III, indicates that diabetes will grow as a major health problem in the U.S. (118).
Emerging evidence suggests that nonresolving inflammation is a critical underlying factor of both periodontitis and T2D that is sustained by a deficiency of lipoxins and resolvins (31, 119–121). To further investigate the comorbidity of periodontitis and T2D, Herrera and coworkers investigated neutrophil function in an ERV1 (RvE1 receptor) overexpressing diabetic transgenic mouse.
Several important observations stemmed from these studies. First, the ERV1 transgenic mouse was demonstrated to be an excellent gain-of-function model for the actions of resolvins (65). ERV1 transgenic mice exhibit a reduced inflammatory phenotype and respond to RvE1 better than wild type. The ERV1 transgenic mouse is resistant to periodontitis. Diabetic mice are significantly more susceptible to periodontitis than wild type, and ERV1 transgenic mice with diabetes are protected from periodontitis (Figure 7). Additional of topical exogenous RvE1 enhances the response. Looking at the mechanism behind increased susceptibility to periodontitis, it was noted that diabetic mice (db/db leptin receptor deficient strain) exhibit impaired neutrophil phagocytosis of the periodontal pathogen P.g. in vitro and clearance of the same pathogen in the murine dorsal airpouch model (122) was deficient and more severe tissue damage was seen. RvE1 added to the phagocytosis assay increased P.g. phagocytosis and killing by neutrophils in the transgenic diabetic animals. In the airpouch, RvE1 addition decreased inflammatory cell influx and increased P.g. clearance suggesting that excess inflammation impedes normal defense mechanisms. The activation of neutrophil signaling pathways in phagocytosis supported this observation. Incubation of neutrophils with P.g. induced phosphorylation of Akt and MAPK that was dampened by RvE1. Taken together, the data suggest that RvE1 increases efficiency of phagocytosis and reduces the potential for collateral tissue damage.
Figure 7. Failure of Resolution Pathways in Type 2 Diabetes-associated Periodontitis.
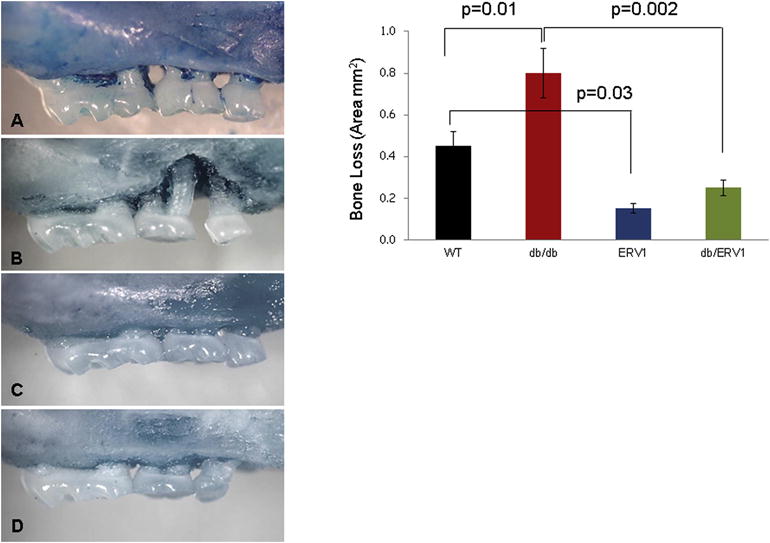
To assess the role of resolution of inflammation in T2D-associated periodontitis, the ERV1 transgenic mouse overexpressing the receptor for RvE1 was employed in gain-of-function studies (95). Periodontitis was induced in wild type (Panel A), ERV1 transgenic (panel B), T2D (db/db leptin receptor deficient, Panel C) and T2D (db/db) mice overexpressing ERV1 (Panel D) by P. gingivalis gavage). Bone loss area is quantified in the right panel. ERV1 transgenic mice are protected from periodontitis (Panel A vs C). T2D-associated periodontitis is more severe that in Wild type (Panel A vs. B). Overexpression of ERV1 in T2D mice affords protection from T2D-associated periodontitis implicating a failure of resolution pathways in T2D pathology and suggesting that failure of resolution can be therapeutically corrected with addition of RvE1 in T2D as it is in periodontitis.
A direct impact of RvE1 on the pathogenesis of diabetes was also noted. ERV1 transgenic diabetic mice exhibited significantly reduced fat accumulation. Concomitantly, the macrophage accumulation in the fat was significantly reduced suggesting reduced production of inflammatory mediators by fat tissue in the transgenic animals that is likely contributing to the non-inflammatory phenotype and protection from disease (95).
Cardiovascular Disease and Periodontitis
Cardiovascular diseases (CVD) and subsequent ischemic complications, including myocardial infarction and stroke, are the most common causes of morbidity and mortality in the United States (123) with 70% of the aging population suffering from CVD (124). The underlying cause is atherosclerosis, which can progress for years without symptoms. A catastrophic acute ischemic event (thrombosis) occurs when an atheromatous plaque disrupts (125, 126). Inflammation is a major determinant of plaque formation and rupture (127, 128). Elevated systemic inflammation predicts CVD events and local inflammatory foci both coincide with, and induce systemic inflammation (129–131). Periodontitis is such a chronic inflammatory focus. Patients with periodontal disease share many of the same risk characteristics as patients with CVD; they are older, predominantly male, and exhibit similar stress and smoking behaviors (132). Periodontal disease and CVD co-segregate in the population (133, 134). Periodontitis clearly imparts excess risk for CVD (135) and enhances CVD in animal models (136, 137).
Periodontitis increases large vessel atherogenesis in the high fat diet rabbit model (136, 138). The New Zealand White (NZW) rabbit serves as a model for periodontitis with bacterial challenge with similar characteristics to human. High fat diet-induced atherogenesis can be induced simultaneously and P.g. elicited inflammatory changes locally and in the vessel wall can be tracked.
The direct impact of treatment of periodontitis with RvE1 on atherogenesis was demonstrated in this model (138). The investigators found that oral-topical administration of low dose RvE1 to the gingiva (4.0 μg and 0.4 μg) provided dose dependent protection against the onset of periodontitis and atherogenesis (Figure 8). Interestingly, in the absence of periodontal inflammation, oral-topical administration of RvE1 provided significantly more protection against atherogenesis, with reduced intima/media ratio and decreased inflammatory cell infiltrate, demonstrating the impact of local inflammatory foci on the initiation of CVD. RvE1 significantly reduced serum CRP.
Figure 8. Atherogenesis that is accelerated by Periodontitis is slowed by RvE1.
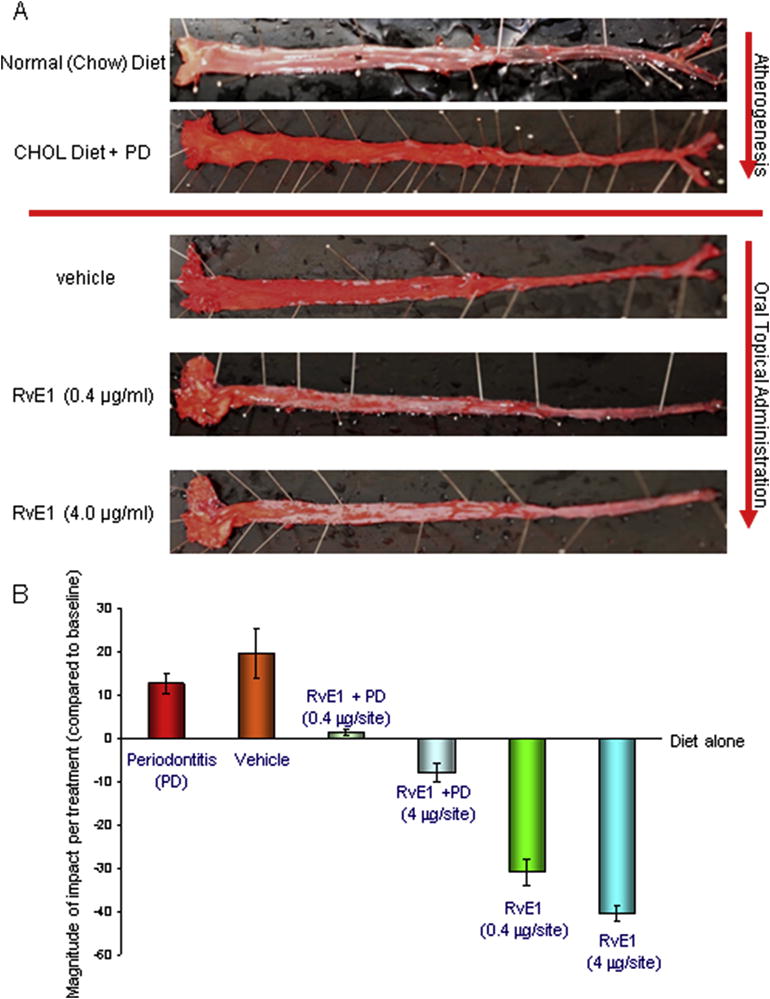
0.5% cholesterol in the New Zealand White Rabbit diet induces atherogenic changes in the aorta characterized by fatty streaks that is worsened by concomitant periodontitis (136) (Panel A). The ability of RvE1 treatment of periodontitis to reverse fatty streak development is seen with increased doses of RvE1. Interestingly, in the absence of periodontitis, local application of RvE1 to the gingiva (4 μg) offered significantly greater protection from atheromatous changes (Panel B). The data confirm that periodontitis has a significant impact on the progression of atheromas in this model and suggest a potential therapeutic benefit for regulation of inflammation with resolvins in atherogenesis. [Adapted from (138)]
These data suggest that RvE1 is rapidly absorbed through the mucosa to provide systemic actions. Oral-topical RvE1 blocks periodontal disease and its impact on systemic inflammation. RvE1 is also extremely potent exhibiting marked reductions in systemic inflammation blocking atherogenesis at extremely low doses. The targets and mechanism of action are also beginning to be unraveled.
In atherogenesis, macrophage activation and class switching are critical for foam cell formation (139). Recent data suggest that this activity is associated with CD36 activation, which works as a scavenging receptor and is a critical molecule in internalization of oxo-LDL (140). CD36 can be activated by a high cholesterol diet and results in macrophage mediated foam cell formation. Porphyromonas gingivalis stimulates CD36 and oxo-LDL activation through ERK signaling and leads to the activation of NF-κB and generation of IL-1β (141). CD36/oxo-LDL activity and macrophage-foam cell formation lead to increased IL-8, IL-6, IL-18, TNF-α, MCP-1, MMP-2 and MMP-9 and decreased IL-10 production (142, 143). P. gingivalis-mediated stimulation of this mechanism can be mediated through TLR2 and TLR4 recognition and TLR2 binding to LPS results in pro-atherogenic events (144). Docosahexaenoic acid (DHA), which is an omega-3 fatty acid precursor of D-class resolvins, has been found to decrease IL-6 and IL-8 production through CD36-mediated activity (145).
Conclusion
With a greater understanding of regulation pathways of inflammation has come an increased understanding of the etiology and pathogenesis of periodontal diseases. Today, we think of periodontitis as an inflammatory disease initiated by commensal bacteria. This is a very different conclusion than previously adhered to infectious disease principles that suggested that one “caught” periodontitis by acquiring exogenous pathogens. Current evidence points to inflammation driving changes in the commensal microbiome that further amplify the inflammatory response. This realization changes the treatment target.
The active, ligand driven, resolution of inflammation provides new targets for natural control of inflammation. In periodontitis, the evidence points to the disease being directly related to a failure of resolution pathways, albeit, the level at which the failure occurs is unknown, and may vary from patient to patient. The two most obvious reasons are a failure to produce sufficient pro-resolving mediator and a failure of the receptor to be expressed or to respond.
Importantly, in periodontitis, it has been demonstrated that adding sufficient exogenous pro-resolving mediator reverses the inflammation and the disease. Characterizing this response has led to several important observations:
Pro-resolving mediators can control inflammatory disease
Pro-resolving mediators can reverse pathologic changes in the human microbiome.
Control of inflammation with pro-resolution mediators, but not anti-inflammatory inhibitors, promotes tissue regeneration and return to homeostasis
Local inflammatory foci, such as periodontitis, have a significant impact on systemic inflammation and systemic inflammatory disease
Pro-resolution mediators have a significant positive impact on resolving systemic inflammatory diseases with oral topical administration.
The actions of SPMs in periodontitis are summarized in Figure 9.
Figure 9. Actions of SPMs in periodontitis.
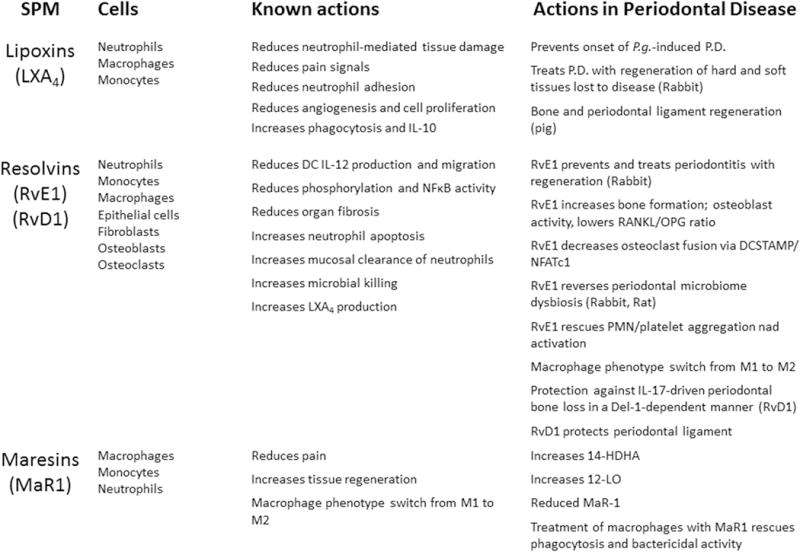
Several of the known agonists of resolution of inflammation have been investigated in periodontal diseases, including lipoxins, E and D series resolvins, and maresins. The cell type and known actions of the SPM are compared to the reported actions in periodontal diseases.
As our knowledge of the actions of pro-resolution mediators and their use as therapeutics evolves, the potential for the production of new mimetic drugs for the control of inflammatory diseases, without the side-effect profile that plagues the current anti-inflammatory pharmacopoeia, seems boundless.
Acknowledgments
Supported in part by USPHS Grants DE025020 and 025383 from the National Institutes of Dental and Craniofacial Research.
References
- 1.Marsh PD. Microbial ecology of dental plaque and its significance in health and disease. Advances in dental research. 1994;8:263–271. doi: 10.1177/08959374940080022001. [DOI] [PubMed] [Google Scholar]
- 2.Van Dyke TE. Inflammation and periodontal diseases: a reappraisal. Journal of periodontology. 2008;79:1501–1502. doi: 10.1902/jop.2008.080279. [DOI] [PubMed] [Google Scholar]
- 3.Van Dyke TE. The management of inflammation in periodontal disease. Journal of periodontology. 2008;79:1601–1608. doi: 10.1902/jop.2008.080173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffen AL, Becker MR, Lyons SR, Moeschberger ML, Leys EJ. Prevalence of Porphyromonas gingivalis and periodontal health status. J Clin Microbiol. 1998;36:3239–3242. doi: 10.1128/jcm.36.11.3239-3242.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McNabb H, Mombelli A, Gmur R, Mathey-Dinc S, Lang NP. Periodontal pathogens in the shallow pockets of immigrants from developing countries. Oral microbiology and immunology. 1992;7:267–272. doi: 10.1111/j.1399-302x.1992.tb00586.x. [DOI] [PubMed] [Google Scholar]
- 6.Papapanou PN, Baelum V, Luan WM, Madianos PN, Chen X, Fejerskov O, et al. Subgingival microbiota in adult Chinese: prevalence and relation to periodontal disease progression. Journal of periodontology. 1997;68:651–666. doi: 10.1902/jop.1997.68.7.651. [DOI] [PubMed] [Google Scholar]
- 7.Riep B, Edesi-Neuss L, Claessen F, Skarabis H, Ehmke B, Flemmig TF, et al. Are putative periodontal pathogens reliable diagnostic markers? J Clin Microbiol. 2009;47:1705–1711. doi: 10.1128/JCM.01387-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. Journal of immunology. 2007;179:7021–7029. doi: 10.4049/jimmunol.179.10.7021. [DOI] [PubMed] [Google Scholar]
- 9.Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nature reviews Microbiology. 2012;10:717–725. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darveau RP, Hajishengallis G, Curtis MA. Porphyromonas gingivalis as a potential community activist for disease. Journal of dental research. 2012;91:816–820. doi: 10.1177/0022034512453589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kornman KS, Page RC, Tonetti MS. The host response to the microbial challenge in periodontitis: assembling the players. Periodontology 2000. 1997;14:33–53. doi: 10.1111/j.1600-0757.1997.tb00191.x. [DOI] [PubMed] [Google Scholar]
- 12.Page RC, Kornman KS. The pathogenesis of human periodontitis: an introduction. Periodontology 2000. 1997;14:9–11. doi: 10.1111/j.1600-0757.1997.tb00189.x. [DOI] [PubMed] [Google Scholar]
- 13.Page RC, Offenbacher S, Schroeder HE, Seymour GJ, Kornman KS. Advances in the pathogenesis of periodontitis: summary of developments, clinical implications and future directions. Periodontology 2000. 1997;14:216–248. doi: 10.1111/j.1600-0757.1997.tb00199.x. [DOI] [PubMed] [Google Scholar]
- 14.Vandenberg JI, Conigrave A, King GF, Kirk K. From kinetics to imaging: an NMR odyssey–a festschrift symposium in honour of Philip William Kuchel. European biophysics journal: EBJ. 2013;42:1–2. doi: 10.1007/s00249-012-0883-8. [DOI] [PubMed] [Google Scholar]
- 15.Romanelli R, Mancini S, Laschinger C, Overall CM, Sodek J, McCulloch CA. Activation of neutrophil collagenase in periodontitis. Infection and immunity. 1999;67:2319–2326. doi: 10.1128/iai.67.5.2319-2326.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gainet J, Chollet-Martin S, Brion M, Hakim J, Gougerot-Pocidalo MA, Elbim C. Interleukin-8 production by polymorphonuclear neutrophils in patients with rapidly progressive periodontitis: an amplifying loop of polymorphonuclear neutrophil activation. Lab Invest. 1998;78:755–762. [PubMed] [Google Scholar]
- 17.Assuma R, Oates T, Cochran D, Amar S, Graves DT. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. Journal of immunology. 1998;160:403–409. [PubMed] [Google Scholar]
- 18.Offenbacher S, Odle BM, Van Dyke TE. The use of crevicular fluid prostaglandin E2 levels as a predictor of periodontal attachment loss. Journal of periodontal research. 1986;21:101–112. doi: 10.1111/j.1600-0765.1986.tb01443.x. [DOI] [PubMed] [Google Scholar]
- 19.Solomon LM, Juhlin L, Kirschenbaum MB. Prostaglandin on cutaneous vasculature. The Journal of investigative dermatology. 1968;51:280–282. [PubMed] [Google Scholar]
- 20.Raisz LG, Koolemans-Beynen AR. Inhibition of bone collagen synthesis by prostaglandin E2 in organ culture. Prostaglandins. 1974;8:377–385. doi: 10.1016/0090-6980(74)90113-0. [DOI] [PubMed] [Google Scholar]
- 21.Klein DC, Raisz LG. Prostaglandins: stimulation of bone resorption in tissue culture. Endocrinology. 1970;86:1436–1440. doi: 10.1210/endo-86-6-1436. [DOI] [PubMed] [Google Scholar]
- 22.Offenbacher S, Odle BM, Gray RC, Van Dyke TE. Crevicular fluid prostaglandin E levels as a measure of the periodontal disease status of adult and juvenile periodontitis patients. Journal of periodontal research. 1984;19:1–13. doi: 10.1111/j.1600-0765.1984.tb01190.x. [DOI] [PubMed] [Google Scholar]
- 23.ElAttar TM. Prostaglandin E2 in human gingiva in health and disease and its stimulation by female sex steroids. Prostaglandins. 1976;11:331–341. doi: 10.1016/0090-6980(76)90155-6. [DOI] [PubMed] [Google Scholar]
- 24.Albers HK, Loning T, Lisboa BP. Biochemical and morphologic studies on prostaglandins E and F in the normal and inflamed gingiva. Deutsche zahnarztliche Zeitschrift. 1979;34:440–443. [PubMed] [Google Scholar]
- 25.ElAttar TM, Lin HS, Tira DE. The relationship between the concentration of female sex steroids and prostaglandins production by human gingiva in vitro. Prostaglandins Leukot Med. 1982;8:447–458. [PubMed] [Google Scholar]
- 26.ElAttar TM, Lin HS, Tira DE. Arachidonic acid metabolism in inflamed gingiva and its inhibition by anti-inflammatory drugs. Journal of periodontology. 1984;55:536–539. doi: 10.1902/jop.1984.55.9.536. [DOI] [PubMed] [Google Scholar]
- 27.Pouliot M, Clish CB, Petasis NA, Van Dyke TE, Serhan CN. Lipoxin A(4) analogues inhibit leukocyte recruitment to Porphyromonas gingivalis: a role for cyclooxygenase-2 and lipoxins in periodontal disease. Biochemistry. 2000;39:4761–4768. doi: 10.1021/bi992551b. [DOI] [PubMed] [Google Scholar]
- 28.Chavis C, Vachier I, Chanez P, Bousquet J, Godard P. 5(S),15(S)-dihydroxyeicosatetraenoic acid and lipoxin generation in human polymorphonuclear cells: dual specificity of 5-lipoxygenase towards endogenous and exogenous precursors. The Journal of experimental medicine. 1996;183:1633–1643. doi: 10.1084/jem.183.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fiore S, Serhan CN. Formation of lipoxins and leukotrienes during receptor-mediated interactions of human platelets and recombinant human granulocyte/macrophage colony-stimulating factor-primed neutrophils. The Journal of experimental medicine. 1990;172:1451–1457. doi: 10.1084/jem.172.5.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serhan CN, Jain A, Marleau S, Clish C, Kantarci A, Behbehani B, et al. Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. Journal of immunology. 2003;171:6856–6865. doi: 10.4049/jimmunol.171.12.6856. [DOI] [PubMed] [Google Scholar]
- 31.Fredman G, Oh SF, Ayilavarapu S, Hasturk H, Serhan CN, Van Dyke TE. Impaired phagocytosis in localized aggressive periodontitis: rescue by Resolvin E1. PloS one. 2011;6:e24422. doi: 10.1371/journal.pone.0024422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hasturk H, Kantarci A, Ohira T, Arita M, Ebrahimi N, Chiang N, et al. RvE1 protects from local inflammation and osteoclast- mediated bone destruction in periodontitis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2006;20:401–403. doi: 10.1096/fj.05-4724fje. [DOI] [PubMed] [Google Scholar]
- 33.Albert CM, Campos H, Stampfer MJ, Ridker PM, Manson JE, Willett WC, et al. Blood levels of long-chain n-3 fatty acids and the risk of sudden death. The New England journal of medicine. 2002;346:1113–1118. doi: 10.1056/NEJMoa012918. [DOI] [PubMed] [Google Scholar]
- 34.Hibbeln JR. Fish consumption and major depression. Lancet. 1998;351:1213. doi: 10.1016/S0140-6736(05)79168-6. [DOI] [PubMed] [Google Scholar]
- 35.Olfson M, Marcus SC, Druss B, Elinson L, Tanielian T, Pincus HA. National trends in the outpatient treatment of depression. JAMA: the journal of the American Medical Association. 2002;287:203–209. doi: 10.1001/jama.287.2.203. [DOI] [PubMed] [Google Scholar]
- 36.Shinmura K, Tang XL, Wang Y, Xuan YT, Liu SQ, Takano H, et al. Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:10197–10202. doi: 10.1073/pnas.97.18.10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Birkedal-Hansen H. Role of cytokines and inflammatory mediators in tissue destruction. Journal of periodontal research. 1993;28:500–510. doi: 10.1111/j.1600-0765.1993.tb02113.x. [DOI] [PubMed] [Google Scholar]
- 38.Birkedal-Hansen H, Moore WG, Bodden MK, Windsor LJ, Birkedal-Hansen B, DeCarlo A, et al. Matrix metalloproteinases: a review. Critical reviews in oral biology and medicine: an official publication of the American Association of Oral Biologists. 1993;4:197–250. doi: 10.1177/10454411930040020401. [DOI] [PubMed] [Google Scholar]
- 39.Van Dyke TE, Serhan CN. Resolution of inflammation: a new paradigm for the pathogenesis of periodontal diseases. Journal of dental research. 2003;82:82–90. doi: 10.1177/154405910308200202. [DOI] [PubMed] [Google Scholar]
- 40.Genco RJ. Host responses in periodontal diseases: current concepts. Journal of periodontology. 1992;63:338–355. doi: 10.1902/jop.1992.63.4s.338. [DOI] [PubMed] [Google Scholar]
- 41.Offenbacher S, Heasman PA, Collins JG. Modulation of host PGE2 secretion as a determinant of periodontal disease expression. Journal of periodontology. 1993;64:432–444. doi: 10.1902/jop.1993.64.5s.432. [DOI] [PubMed] [Google Scholar]
- 42.Offenbacher S, Collins JG, Heasman PA. Diagnostic potential of host response mediators. Advances in dental research. 1993;7:175–181. doi: 10.1177/08959374930070020801. [DOI] [PubMed] [Google Scholar]
- 43.Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annual review of immunology. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- 44.Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, et al. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic acids research. 2012;40:D571–579. doi: 10.1093/nar/gkr1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DiGiulio DB, Romero R, Amogan HP, Kusanovic JP, Bik EM, Gotsch F, et al. Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: a molecular and culture-based investigation. PloS one. 2008;3:e3056. doi: 10.1371/journal.pone.0003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome biology. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franceschi F, Niccoli G, Ferrante G, Gasbarrini A, Baldi A, Candelli M, et al. CagA antigen of Helicobacter pylori and coronary instability: insight from a clinico-pathological study and a meta-analysis of 4241 cases. Atherosclerosis. 2009;202:535–542. doi: 10.1016/j.atherosclerosis.2008.04.051. [DOI] [PubMed] [Google Scholar]
- 48.Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, et al. Toward defining the autoimmune microbiome for type 1 diabetes. The ISME journal. 2011;5:82–91. doi: 10.1038/ismej.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PloS one. 2010;5:e9085. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rajendhran J, Shankar M, Dinakaran V, Rathinavel A, Gunasekaran P. Contrasting circulating microbiome in cardiovascular disease patients and healthy individuals. International journal of cardiology. 2013;168:5118–5120. doi: 10.1016/j.ijcard.2013.07.232. [DOI] [PubMed] [Google Scholar]
- 51.Helaine S, Cheverton AM, Watson KG, Faure LM, Matthews SA, Holden DW. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science. 2014;343:204–208. doi: 10.1126/science.1244705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miskinyte M, Sousa A, Ramiro RS, de Sousa JA, Kotlinowski J, Caramalho I, et al. The genetic basis of Escherichia coli pathoadaptation to macrophages. PLoS pathogens. 2013;9:e1003802. doi: 10.1371/journal.ppat.1003802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu Y, Xie J, Li Y, Yue J, Chen J, Chunyu L, et al. Using a cDNA microarray to study cellular gene expression altered by Mycobacterium tuberculosis. Chinese medical journal. 2003;116:1070–1073. [PubMed] [Google Scholar]
- 54.Lee CT, Teles R, Kantarci A, Chen T, McCafferty J, Starr JR, et al. Resolvin E1 Reverses Experimental Periodontitis and Dysbiosis. Journal of immunology. 2016;197:2796–2806. doi: 10.4049/jimmunol.1600859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sima C, Cheng Q, Rautava J, Levesque C, Sherman P, Glogauer M. Identification of quantitative trait loci influencing inflammation-mediated alveolar bone loss: insights into polygenic inheritance of host-biofilm disequilibria in periodontitis. Journal of periodontal research. 2016;51:237–249. doi: 10.1111/jre.12303. [DOI] [PubMed] [Google Scholar]
- 56.Haffajee AD, Socransky SS. Introduction to microbial aspects of periodontal biofilm communities, development and treatment. Periodontology 2000. 2006;42:7–12. doi: 10.1111/j.1600-0757.2006.00190.x. [DOI] [PubMed] [Google Scholar]
- 57.Van Dyke TE. Proresolving lipid mediators: potential for prevention and treatment of periodontitis. Journal of clinical periodontology. 2011;38(Suppl 11):119–125. doi: 10.1111/j.1600-051X.2010.01662.x. [DOI] [PubMed] [Google Scholar]
- 58.Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med. 2014;6:229ra240. doi: 10.1126/scitranslmed.3007696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moutsopoulos NM, Chalmers NI, Barb JJ, Abusleme L, Greenwell-Wild T, Dutzan N, et al. Subgingival microbial communities in Leukocyte Adhesion Deficiency and their relationship with local immunopathology. PLoS pathogens. 2015;11:e1004698. doi: 10.1371/journal.ppat.1004698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kantarci A, Oyaizu K, Van Dyke TE. Neutrophil-mediated tissue injury in periodontal disease pathogenesis: findings from localized aggressive periodontitis. Journal of periodontology. 2003;74:66–75. doi: 10.1902/jop.2003.74.1.66. [DOI] [PubMed] [Google Scholar]
- 61.Chapple IL, Matthews JB. The role of reactive oxygen and antioxidant species in periodontal tissue destruction. Periodontology 2000. 2007;43:160–232. doi: 10.1111/j.1600-0757.2006.00178.x. [DOI] [PubMed] [Google Scholar]
- 62.Graves D. Cytokines that promote periodontal tissue destruction. Journal of periodontology. 2008;79:1585–1591. doi: 10.1902/jop.2008.080183. [DOI] [PubMed] [Google Scholar]
- 63.Tanner AC, Kent R, Jr, Kanasi E, Lu SC, Paster BJ, Sonis ST, et al. Clinical characteristics and microbiota of progressing slight chronic periodontitis in adults. Journal of clinical periodontology. 2007;34:917–930. doi: 10.1111/j.1600-051X.2007.01126.x. [DOI] [PubMed] [Google Scholar]
- 64.Kinney JS, Morelli T, Oh M, Braun TM, Ramseier CA, Sugai JV, et al. Crevicular fluid biomarkers and periodontal disease progression. Journal of clinical periodontology. 2014;41:113–120. doi: 10.1111/jcpe.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao L, Faibish D, Fredman G, Herrera BS, Chiang N, Serhan CN, et al. Resolvin E1 and chemokine-like receptor 1 mediate bone preservation. Journal of immunology. 2013;190:689–694. doi: 10.4049/jimmunol.1103688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhu M, Van Dyke TE, Gyurko R. Resolvin E1 regulates osteoclast fusion via DC-STAMP and NFATc1. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2013;27:3344–3353. doi: 10.1096/fj.12-220228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, et al. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. The ISME journal. 2012;6:1176–1185. doi: 10.1038/ismej.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu B, Faller LL, Klitgord N, Mazumdar V, Ghodsi M, Sommer DD, et al. Deep sequencing of the oral microbiome reveals signatures of periodontal disease. PloS one. 2012;7:e37919. doi: 10.1371/journal.pone.0037919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, et al. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. The ISME journal. 2013;7:1016–1025. doi: 10.1038/ismej.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li Y, He J, He Z, Zhou Y, Yuan M, Xu X, et al. Phylogenetic and functional gene structure shifts of the oral microbiomes in periodontitis patients. The ISME journal. 2014;8:1879–1891. doi: 10.1038/ismej.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shi B, Chang M, Martin J, Mitreva M, Lux R, Klokkevold P, et al. Dynamic changes in the subgingival microbiome and their potential for diagnosis and prognosis of periodontitis. MBio. 2015;6:e01926–01914. doi: 10.1128/mBio.01926-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kornman KS, Blodgett RF, Brunsvold M, Holt SC. Effects of topical applications of meclofenamic acid and ibuprofen on bone loss, subgingival microbiota and gingival PMN response in the primate Macaca fascicularis. Journal of periodontal research. 1990;25:300–307. doi: 10.1111/j.1600-0765.1990.tb00919.x. [DOI] [PubMed] [Google Scholar]
- 73.Duran-Pinedo AE, Chen T, Teles R, Starr JR, Wang X, Krishnan K, et al. Community-wide transcriptome of the oral microbiome in subjects with and without periodontitis. The ISME journal. 2014;8:1659–1672. doi: 10.1038/ismej.2014.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yost S, Duran-Pinedo AE, Teles R, Krishnan K, Frias-Lopez J. Functional signatures of oral dysbiosis during periodontitis progression revealed by microbial metatranscriptome analysis. Genome Med. 2015;7:27. doi: 10.1186/s13073-015-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, et al. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461:1287–1291. doi: 10.1038/nature08541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:7671–7676. doi: 10.1073/pnas.0409271102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cotran RS, K V, Collins T, editors. Robbins Pathologic Basis of Disease. 6th. Philadelphia: W.B. Saunders Co.; 1999. [Google Scholar]
- 79.Rock KL, Kono H. The inflammatory response to cell death. Annual review of pathology. 2008;3:99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sculean A, Nikolidakis D, Schwarz F. Regeneration of periodontal tissues: combinations of barrier membranes and grafting materials - biological foundation and preclinical evidence: a systematic review. Journal of clinical periodontology. 2008;35:106–116. doi: 10.1111/j.1600-051X.2008.01263.x. [DOI] [PubMed] [Google Scholar]
- 81.Reynolds MA, Aichelmann-Reidy ME, Branch-Mays GL, Gunsolley JC. The efficacy of bone replacement grafts in the treatment of periodontal osseous defects. A systematic review. Annals of periodontology / the American Academy of Periodontology. 2003;8:227–265. doi: 10.1902/annals.2003.8.1.227. [DOI] [PubMed] [Google Scholar]
- 82.Murphy KG, Gunsolley JC. Guided tissue regeneration for the treatment of periodontal intrabony and furcation defects. A systematic review Annals of periodontology / the American Academy of Periodontology. 2003;8:266–302. doi: 10.1902/annals.2003.8.1.266. [DOI] [PubMed] [Google Scholar]
- 83.Serhan CN, Yacoubian S, Yang R. Anti-inflammatory and proresolving lipid mediators. Annual review of pathology. 2008;3:279–312. doi: 10.1146/annurev.pathmechdis.3.121806.151409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 1987;237:1171–1176. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- 85.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 86.Dee KC, P D, Bizios R. An Introduction to Tissue-Biomaterial Interactions. Hoboken, NJ: John Wiley & Sons; 2002. [Google Scholar]
- 87.Lumelsky NL. Commentary: engineering of tissue healing and regeneration. Tissue engineering. 2007;13:1393–1398. doi: 10.1089/ten.2007.0100. [DOI] [PubMed] [Google Scholar]
- 88.Offenbacher S, Barros SP, Beck JD. Rethinking periodontal inflammation. Journal of periodontology. 2008;79:1577–1584. doi: 10.1902/jop.2008.080220. [DOI] [PubMed] [Google Scholar]
- 89.Williams RC. Understanding and managing periodontal diseases: a notable past, a promising future. Journal of periodontology. 2008;79:1552–1559. doi: 10.1902/jop.2008.080182. [DOI] [PubMed] [Google Scholar]
- 90.Hong S, Lu Y, Yang R, Gotlinger KH, Petasis NA, Serhan CN. Resolvin D1, protectin D1, and related docosahexaenoic acid-derived products: Analysis via electrospray/low energy tandem mass spectrometry based on spectra and fragmentation mechanisms. Journal of the American Society for Mass Spectrometry. 2007;18:128–144. doi: 10.1016/j.jasms.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Circulation Editors’ Picks: Most Read Articles on the Topic of Biomarkers. Circulation. 2012;125:e299–e314. [Google Scholar]
- 92.Wada K, Arita M, Nakajima A, Katayama K, Kudo C, Kamisaki Y, et al. Leukotriene B4 and lipoxin A4 are regulatory signals for neural stem cell proliferation and differentiation. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2006;20:1785–1792. doi: 10.1096/fj.06-5809com. [DOI] [PubMed] [Google Scholar]
- 93.Abela GS, Aziz K. Cholesterol crystals cause mechanical damage to biological membranes: a proposed mechanism of plaque rupture and erosion leading to arterial thrombosis. Clin Cardiol. 2005;28:413–420. doi: 10.1002/clc.4960280906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Van Dyke TE, Hasturk H, Kantarci A, Freire MO, Nguyen D, Dalli J, et al. Proresolving nanomedicines activate bone regeneration in periodontitis. Journal of dental research. 2015;94:148–156. doi: 10.1177/0022034514557331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Herrera BS, Hasturk H, Kantarci A, Freire MO, Nguyen O, Kansal S, et al. Impact of resolvin E1 on murine neutrophil phagocytosis in type 2 diabetes. Infection and immunity. 2015;83:792–801. doi: 10.1128/IAI.02444-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Distler JH, Distler O. Inflammation: Microparticles and their roles in inflammatory arthritides. Nature reviews Rheumatology. 2010;6:385–386. doi: 10.1038/nrrheum.2010.87. [DOI] [PubMed] [Google Scholar]
- 97.Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. 2012;120:e60–72. doi: 10.1182/blood-2012-04-423525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Herrera BS, Ohira T, Gao L, Omori K, Yang R, Zhu M, et al. An endogenous regulator of inflammation, resolvin E1, modulates osteoclast differentiation and bone resorption. British journal of pharmacology. 2008;155:1214–1223. doi: 10.1038/bjp.2008.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arita M, Ohira T, Sun YP, Elangovan S, Chiang N, Serhan CN. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. Journal of immunology. 2007;178:3912–3917. doi: 10.4049/jimmunol.178.6.3912. [DOI] [PubMed] [Google Scholar]
- 100.Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ, Cdc Periodontal Disease Surveillance workgroup: James Beck GDRP Prevalence of periodontitis in adults in the United States: 2009 and 2010. Journal of dental research. 2012;91:914–920. doi: 10.1177/0022034512457373. [DOI] [PubMed] [Google Scholar]
- 101.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 102.Dandona P, Aljada A, Chaudhuri A, Mohanty P, Garg R. Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation. 2005;111:1448–1454. doi: 10.1161/01.CIR.0000158483.13093.9D. [DOI] [PubMed] [Google Scholar]
- 103.Andoh A, Tsujikawa T, Sasaki M, Mitsuyama K, Suzuki Y, Matsui T, et al. Faecal microbiota profile of Crohn’s disease determined by terminal restriction fragment length polymorphism analysis. Aliment Pharmacol Ther. 2009;29:75–82. doi: 10.1111/j.1365-2036.2008.03860.x. [DOI] [PubMed] [Google Scholar]
- 104.Champion OL, Valdez Y, Thorson L, Guttman JA, Menendez A, Gaynor EC, et al. A murine intraperitoneal infection model reveals that host resistance to Campylobacter jejuni is Nramp1 dependent. Microbes Infect. 2008;10:922–927. doi: 10.1016/j.micinf.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 105.Braun J, Wei B. Body traffic: ecology, genetics, and immunity in inflammatory bowel disease. Annual review of pathology. 2007;2:401–429. doi: 10.1146/annurev.pathol.1.110304.100128. [DOI] [PubMed] [Google Scholar]
- 106.Shih DQ, Targan SR, McGovern D. Recent advances in IBD pathogenesis: genetics and immunobiology. Curr Gastroenterol Rep. 2008;10:568–575. doi: 10.1007/s11894-008-0104-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Borgnakke WS, C I, Genco RJ, Armitage GC, Bartold PM, D’Aiuto F, Eke PI, Giannobile WV, Kocher T, Kornman KS, Lang NP, Madianos PN, Murakami S, Nishimura F, Offenbacher S, Preshaw PM, Rahman Au, Sanz M, Slots J, Tonetti MS, Van Dyke TE. The randomized controlled trial (RCT) published by the Journal of the American Medical Association (JAMA) on the impact of periodontal therapy on glycated hemoglobin (HbA1c) has fundamental problems. J Evid Base Dent Pract. 2014;14:127–132. doi: 10.1016/j.jebdp.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Engebretson SP, Hyman LG, Michalowicz BS, Schoenfeld ER, Gelato MC, Hou W, et al. The effect of nonsurgical periodontal therapy on hemoglobin A1c levels in persons with type 2 diabetes and chronic periodontitis: a randomized clinical trial. JAMA: the journal of the American Medical Association. 2013;310:2523–2532. doi: 10.1001/jama.2013.282431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fredman G, Van Dyke TE, Serhan CN. Resolvin E1 regulates adenosine diphosphate activation of human platelets. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:2005–2013. doi: 10.1161/ATVBAHA.110.209908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tuttle HA, Davis-Gorman G, Goldman S, Copeland JG, McDonagh PF. Platelet-neutrophil conjugate formation is increased in diabetic women with cardiovascular disease. Cardiovascular diabetology. 2003;2:12. doi: 10.1186/1475-2840-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PloS one. 2010;5:e9539. doi: 10.1371/journal.pone.0009539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Freire MO, Dalli J, Serhan CN, Van Dyke TE. Neutrophil Resolvin E1 Receptor Expression and Function in Type 2 Diabetes. Journal of immunology. 2017;198:718–728. doi: 10.4049/jimmunol.1601543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bang HO, Dyerberg J, Sinclair HM. The composition of the Eskimo food in north western Greenland. Am J Clin Nutr. 1980;33:2657–2661. doi: 10.1093/ajcn/33.12.2657. [DOI] [PubMed] [Google Scholar]
- 114.Claria J, Dalli J, Yacoubian S, Gao F, Serhan CN. Resolvin D1 and resolvin D2 govern local inflammatory tone in obese fat. Journal of immunology. 2012;189:2597–2605. doi: 10.4049/jimmunol.1201272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gonzalez-Periz A, Horrillo R, Ferre N, Gronert K, Dong B, Moran-Salvador E, et al. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: a role for resolvins and protectins. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2009;23:1946–1957. doi: 10.1096/fj.08-125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Iacopino AM. Periodontitis and diabetes interrelationships: role of inflammation. Annals of periodontology / the American Academy of Periodontology. 2001;6:125–137. doi: 10.1902/annals.2001.6.1.125. [DOI] [PubMed] [Google Scholar]
- 117.Loe H. Periodontal disease. The sixth complication of diabetes mellitus. Diabetes care. 1993;16:329–334. [PubMed] [Google Scholar]
- 118.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 119.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 120.Oh SF, Dona M, Fredman G, Krishnamoorthy S, Irimia D, Serhan CN. Resolvin E2 formation and impact in inflammation resolution. Journal of immunology. 2012;188:4527–4534. doi: 10.4049/jimmunol.1103652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Merched AJ, Ko K, Gotlinger KH, Serhan CN, Chan L. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2008;22:3595–3606. doi: 10.1096/fj.08-112201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sin YM, Sedgwick AD, Chea EP, Willoughby DA. Mast cells in newly formed lining tissue during acute inflammation: a six day air pouch model in the mouse. Annals of the rheumatic diseases. 1986;45:873–877. doi: 10.1136/ard.45.10.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, et al. Heart disease and stroke statistics–2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480–486. doi: 10.1161/CIRCULATIONAHA.108.191259. [DOI] [PubMed] [Google Scholar]
- 124.Papadopoulos G, Shaik-Dasthagirisaheb YB, Huang N, Viglianti GA, Henderson AJ, Kantarci A, et al. Immunologic environment influences macrophage response to Porphyromonas gingivalis. Molecular oral microbiology. 2016 doi: 10.1111/omi.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Verbrugghe E, Boyen F, Gaastra W, Bekhuis L, Leyman B, Van Parys A, et al. The complex interplay between stress and bacterial infections in animals. Vet Microbiol. 2012;155:115–127. doi: 10.1016/j.vetmic.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 126.Feltes TF, Bacha E, Beekman RH, 3rd, Cheatham JP, Feinstein JA, Gomes AS, et al. Indications for cardiac catheterization and intervention in pediatric cardiac disease: a scientific statement from the American Heart Association. Circulation. 2011;123:2607–2652. doi: 10.1161/CIR.0b013e31821b1f10. [DOI] [PubMed] [Google Scholar]
- 127.Lindy O, Suomalainen K, Makela M, Lindy S. Statin use is associated with fewer periodontal lesions: A retrospective study. BMC Oral Health. 2008;8:16. doi: 10.1186/1472-6831-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Campbell LA, Rosenfeld ME. Infection and Atherosclerosis Development. Archives of medical research. 2015;46:339–350. doi: 10.1016/j.arcmed.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chung HY, Lee EK, Choi YJ, Kim JM, Kim DH, Zou Y, et al. Molecular inflammation as an underlying mechanism of the aging process and age-related diseases. Journal of dental research. 2011;90:830–840. doi: 10.1177/0022034510387794. [DOI] [PubMed] [Google Scholar]
- 130.Subramanian S, Emami H, Vucic E, Singh P, Vijayakumar J, Fifer KM, et al. High-dose atorvastatin reduces periodontal inflammation: a novel pleiotropic effect of statins. J Am Coll Cardiol. 2013;62:2382–2391. doi: 10.1016/j.jacc.2013.08.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kantarci A, Yen S, Will LA. Conclusion and Future Directions. Frontiers of oral biology. 2016;18:130. doi: 10.1159/000351910. [DOI] [PubMed] [Google Scholar]
- 132.Bertoni A, Rastoldo A, Sarasso C, Di Vito C, Sampietro S, Nalin M, et al. Dehydroepiandrosterone-sulfate inhibits thrombin-induced platelet aggregation. Steroids. 2012;77:260–268. doi: 10.1016/j.steroids.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 133.Berr SS, Brookeman JR. On MR imaging of atheromatous lipids in human arteries. J Magn Reson Imaging. 1995;5:373–374. doi: 10.1002/jmri.1880050325. [DOI] [PubMed] [Google Scholar]
- 134.Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:2761–2796. doi: 10.1161/CIR.0b013e318223e230. [DOI] [PubMed] [Google Scholar]
- 135.Dietrich T, Sharma P, Walter C, Weston P, Beck J. The epidemiological evidence behind the association between periodontitis and incident atherosclerotic cardiovascular disease. Journal of clinical periodontology. 2013;40(Suppl 14):S70–84. doi: 10.1111/jcpe.12062. [DOI] [PubMed] [Google Scholar]
- 136.Jain A, Batista EL, Jr, Serhan C, Stahl GL, Van Dyke TE. Role for periodontitis in the progression of lipid deposition in an animal model. Infection and immunity. 2003;71:6012–6018. doi: 10.1128/IAI.71.10.6012-6018.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Giugliano RP, Braunwald E. The year in non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol. 2009;54:1544–1555. doi: 10.1016/j.jacc.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 138.Hasturk H, Abdallah R, Kantarci A, Nguyen D, Giordano N, Hamilton J, et al. Resolvin E1 (RvE1) Attenuates Atherosclerotic Plaque Formation in Diet and Inflammation-Induced Atherogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2015;35:1123–1133. doi: 10.1161/ATVBAHA.115.305324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Di Pietro N, Formoso G, Pandolfi A. Physiology and pathophysiology of oxLDL uptake by vascular wall cells in atherosclerosis. Vascular pharmacology. 2016;84:1–7. doi: 10.1016/j.vph.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 140.Jay AG, Chen AN, Paz MA, Hung JP, Hamilton JA. CD36 binds oxidized low density lipoprotein (LDL) in a mechanism dependent upon fatty acid binding. The Journal of biological chemistry. 2015;290:4590–4603. doi: 10.1074/jbc.M114.627026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Brown PM, Kennedy DJ, Morton RE, Febbraio M. CD36/SR-B2-TLR2 Dependent Pathways Enhance Porphyromonas gingivalis Mediated Atherosclerosis in the Ldlr KO Mouse Model. PloS one. 2015;10:e0125126. doi: 10.1371/journal.pone.0125126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bzowska M, Nogiec A, Skrzeczynska-Moncznik J, Mickowska B, Guzik K, Pryjma J. Oxidized LDLs inhibit TLR-induced IL-10 production by monocytes: a new aspect of pathogen-accelerated atherosclerosis. Inflammation. 2012;35:1567–1584. doi: 10.1007/s10753-012-9472-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:1731–1738. doi: 10.1161/ATVBAHA.114.303887. [DOI] [PubMed] [Google Scholar]
- 144.Chavez-Sanchez L, Garza-Reyes MG, Espinosa-Luna JE, Chavez-Rueda K, Legorreta-Haquet MV, Blanco-Favela F. The role of TLR2, TLR4 and CD36 in macrophage activation and foam cell formation in response to oxLDL in humans. Human immunology. 2014;75:322–329. doi: 10.1016/j.humimm.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 145.Shikama Y, Kudo Y, Ishimaru N, Funaki M. Possible Involvement of Palmitate in Pathogenesis of Periodontitis. Journal of cellular physiology. 2015;230:2981–2989. doi: 10.1002/jcp.25029. [DOI] [PubMed] [Google Scholar]