

Abstract
Maternal obesity increases placental transport of macronutrients, resulting in fetal overgrowth and obesity later in life. Choline participates in fatty acid metabolism, serves as a methyl donor, and influences growth signaling, which may modify placental macronutrient homeostasis and affect fetal growth. Using a mouse model of maternal obesity, we assessed the effect of maternal choline supplementation on preventing fetal overgrowth and restoring placental macronutrient homeostasis. C57BL/6J mice were fed either a high-fat (HF, 60% kcal from fat) diet or a normal (NF, 10% kcal from fat) diet with a drinking supply of either 25 mM choline chloride or control purified water, respectively, beginning 4 weeks prior to mating until gestational day 12.5. Fetal and placental weight, metabolites, and gene expression were measured. HF feeding significantly (P < 0.05) increased placental and fetal weight in the HF-control (HFCO) versus NF-control (NFCO) animals, whereas the HF choline-supplemented (HFCS) group effectively normalized placental and fetal weight to the levels of the NFCO group. Compared to HFCO, the HFCS group had lower (P < 0.05) glucose transporter 1 (GLUT1) and fatty acid transport protein 1 (FATP1) expression as well as lower accumulation of glycogen in the placenta. The HFCS group also had lower (P < 0.05) placental 4E-binding protein 1 and ribosomal protein s6 phosphorylation, which are indicators of mechanistic target of rapamycin complex 1 (mTORC1) activation favoring macronutrient anabolism. In summary, our results suggest that maternal choline supplementation prevented fetal overgrowth in obese mice at mid-gestation and improved biomarkers of placental macronutrient homeostasis.
Keywords: choline, obesity, gestational diabetes, fetal overgrowth, placenta, nutrient transport
1. Introduction
Maternal obesity has emerged as a significant health problem that increases the risk of various pregnancy complications [1], including gestational diabetes mellitus (GDM), which is characterized by hyperglycemia and glucose intolerance during pregnancy [2]. Babies born to obese mothers have a higher risk of macrosomia (birth weight greater than 4kg or 9 lbs) [3, 4] and are more likely to develop childhood obesity, which predisposes them to type 2 diabetes and cardiovascular disease as they age [1, 3]. Development of GDM in obese women further exacerbates the risk of macrosomia [5].
The mechanism by which maternal obesity leads to fetal overgrowth is not well understood, but the placenta is increasingly recognized as an important mediator dictating the rate of maternal nutrients transported to the fetus [6]. The insulin/insulin-like growth factor (IGF) pathway, composed of insulin, IGFs, IGF receptors and binding proteins, is proposed to enhance placental nutrient transport, thereby promoting fetal growth [7]. Another critical regulator of placental nutrient transport is the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mechanistic target of rapamycin (mTOR) pathway. Maternal obesity activated mTOR complex 1 (mTORC1) in the placenta, which enhanced macronutrient synthesis whilst upregulating glucose transporters and System A and System L amino acid transporters in mice fed a high-fat/high-sugar diet [8]. Specifically, maternal obesity upregulated glucose transporters (GLUT) 1 and 3, which are two major glucose transporters expressed in cytotrophoblasts that promote facilitated diffusion of glucose across the placenta, and thus may increase glucose accretion by the fetus [9]. Maternal obesity also results in excess fetal adiposity in both rodents and humans [10, 11]. The fetus acquires lipids mostly from maternal circulation through placental fatty acid transport due to limited de novo lipogenesis [12]. Fatty acid transport in the placenta is initiated with the action of lipases which release fatty acids from triglycerides, phospholipids, and lipoproteins. Thereafter the free fatty acids are transported by fatty acid binding proteins (FABPs), fatty acid transporters (FATPs) 1–4, and fatty acid translocase (CD36) to the fetus [10]. Maternal obesity is associated with upregulation of the fatty acid transport related proteins and marked increases in triglyceride content in the placenta [10, 13].
Dietary approaches or the use of bioactive compounds to normalize placental nutrient metabolism and transport in cases of maternal obesity and GDM are largely unexplored. Prior studies demonstrate that choline, an essential micronutrient and the precursor of phosphatidylcholine, acetylcholine, and betaine [14], interacts with macronutrient metabolism [15, 16]. Serving as a structural component of lipoproteins, phosphatidylcholine is needed for very low density lipoprotein (VLDL) synthesis and hepatic lipid output [17–19]. Choline deficiency results in non-alcoholic fatty liver disease [15]. A species of phosphatidylcholine (1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine) serves as an endogenous ligand for peroxisome proliferator-activated receptor alpha (PPAR-α) and activates this transcription factor to suppress de novo fatty acid synthesis and increase fatty acid catabolism [20]. These observations suggest that choline plays an important role in preventing ectopic fat accumulation. Since maternal obesity increases placental lipid accumulation which is associated with fetal adiposity [21], increasing the availability of choline may attenuate placental lipid overload and reduce lipid transport to the fetus. Furthermore, phosphatidylcholine deficiency activates the mitogenic pathway involving protein kinase Cs (PKCs) and promotes cell proliferation [22, 23]. Maintaining choline status may then prevent excessive placental and fetal growth. Via oxidation to betaine, choline provides methyl groups for methylation reactions including DNA methylation. Several critical growth factors of the placenta, such as IGF2, are controlled by DNA methylation and thus may be responsive to varied choline availability [24, 25]. Previous studies also support the importance of choline for normal placental functioning such as improving angiogenesis and reducing stress biomarkers and inflammation [26, 27]. Placental angiogenesis and inflammation also affect nutrient metabolism. In sum, choline is a promising modifier of placental macronutrient metabolism under the condition of maternal obesity.
Liang et al. reported that high-fat (HF) feeding induced hyperglycemia and glucose intolerance in female C57BL/6J mice [28]. HF feeding also led to fetal overgrowth in a similar mouse model [29]. These phenotypes correlate well with obesity and GDM arising from excessive energy consumption in humans. In this study, we examined whether maternal choline supplementation prevents HF feeding-induced fetal overgrowth in mice. We also examined the effect of choline on placental macronutrient metabolism.
2. Methods
2.1. Animals and diets
Six-week-old C57BL/6J mice were obtained from Jackson Laboratory. The mice were housed at 22°C, humidity 40–60%, and 12-hour light/dark cycle with regular bedding and enrichment. After 2 weeks of acclimation with ad libitum access to regular lab diet (Laboratory Rodent Diet 5001, LabDiet, St. Louis, MO), the female mice were divided into 4 groups: the normal-fat control (NFCO) group received a diet (D12450J, Research Diets, New Brunswick, NJ) containing 10% kcal from fat which is a normal dietary fat content for mice and purified drinking water; the NF choline supplemented (NFCS) group received the NF diet and purified drinking water supplemented with 25mM choline chloride; the high-fat control (HFCO) group received a HF diet (D12492, Research Diets) containing 60% kcal from fat and purified drinking water; and the HF choline supplemented (HFCS) group received the HF diet and purified drinking water supplemented with 25mM choline chloride. Male mice for mating received the NFCO diet and purified drinking water. All animals had free access to food and water. Composition of experimental diets was previously described by others [30, 31]. Choline bitartrate was added to both diets on a 2 g/4057 kcal basis. Because the HF diet had a higher calorie density than the NF diet (5.24 versus 3.85 kcal/g), choline bitartrate content/g diet was higher in the HF than the NF diet. We also quantified choline moieties in non-chemically defined ingredients in the diets (e.g. lard) using liquid chromatography-mass spectrometry (LC-MS)/MS (Supplemental Table 1). We estimated that the HF diet had 10.3 mmol/kg and the NF diet had 7.6 mmol/kg total choline.
After 4 weeks of feeding on assigned diets, two female mice were housed with one male mouse for timed-mating in each cage. If a vaginal plug was detected in the morning, the female mouse was transferred to a separate cage and time was recorded as embryonic day (E) 0.5. Female mice continued to receive their assigned diets during gestation. Animals were weighed every week to assess weight gain throughout the study. There were 8–13 female mice in each group that were fed experimental diets initially. However, only 6–8 animals (70%) in each group conceived successfully from which samples and data were collected.
2.2 Food intake and water consumption
Food intake and water consumption of the animals were measured and calculated weekly by subtracting the weight of food left on each cage and the volume of water left in each bottle from the amount/volume initially given to the animals. Total choline intake from food and water was calculated as the concentration of choline in food multiplied by the amount of food consumed + the concentration of choline in water multiplied by the volume of water consumed.
2.3. Intraperitoneal glucose tolerance (IGT) tests
At E11.5, the animals were fasted for 6 hours starting from the beginning of the light cycle. To conduct the IGT tests, the mice were injected with 20% D-glucose (2g glucose/kg body weight) intraperitoneally. A drop of blood was obtained by tail nicking to measure blood glucose using a glucometer at baseline (0 minute), 15, 30, 60, 90, and 120 minutes after glucose injection. The Area Under the Curve (AUC) was calculated based on the method by Tai et al. [32].
2.4. Sample collection
At E12.5, the pregnant mice were euthanized after a 6-hour fast by carbon dioxide inhalation. Previous research suggested that both maternal blood glucose and weight gain exhibited greatest differences between HF and NF animals at this time point [28]. Maternal blood was retrieved by cardiac puncture immediately after euthanasia and collected in a serum separator tube (BD, Franklin Lakes, NJ). The blood was centrifuged at 12,000 × g for 10 minutes to obtain serum. Maternal liver, abdominal fat, placentas, and fetuses were retrieved, rinsed in phosphate buffered saline, and dried on absorbent paper. The samples were weighed on an analytical balance. Thereafter, they were flash frozen in liquid nitrogen and stored at −80 C° after dissection, or immersed in RNAlater® (Thermo Scientific, Grand Island, NY) overnight before being stored at −80 C° until analysis, or immersed in formalin and stored at room temperature. The study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at Brooklyn College.
2.5. Analytical measurements
For analytical measurement of placental samples, we excluded dams with a litter size lower than 5 or higher than 10 to prevent the potential confounding effect of litter size on metabolic parameters. Two samples were randomly selected from each litter unless specified otherwise.
2.5.1. Embryo sexing
Sex of all fetuses was determined by PCR of a sequence specific to the mouse sex-determining region Y (Sry) gene on the Y chromosome according to the method by McClive et al. [33]. A sequence specific to the myogenin (Myog) gene on chromosome 1 was amplified to serve as a control.
2.5.2. Non-esterified fatty acid (NEFA) measurements
Maternal NEFAs were measured with the HR Series NEFA-HR(2) colorimetric reagents (Wako Diagnostics, Richmond, VA) according to the manufacturer’s instructions.
2.5.3. Insulin, leptin, and IGF-1 measurements
Maternal serum insulin, leptin, and IGF-1 were measured with enzyme-linked immunosorbent assay (ELISA) kits (ALPCO, Salem, NH) according to the manufacturer’s instructions.
2.5.4. Choline measurements
Whole livers and placentas were pulverized in liquid nitrogen. A weighed portion of the samples (about 50 mg) was used for choline quantification. Measurements of choline and its derivatives were conducted using the LC-MS/MS methodology [34]. One placenta from each dam was randomly chosen for measurement. Six livers and placentas from each group were randomly chosen for choline quantification.
2.5.5 Placental morphology
Formalin fixed placental samples from the NFCO, HFCO, and HFCS groups were sectioned at 5 μm thickness, stained with hematoxylin and eosin, and scanned with the ImageScope software (Leica Biosystems Inc., Buffalo Grove, IL) by Histowicz, Inc (Brooklyn, NY). Placental slides (2 per dam) from 3 dams of each group were analyzed using the ImageJ software (National Institutes of Health, Bethesda, MD). Thickness of the 3 placental layers - decidua, junctional zone, and labyrinth - was measured at the thickest point of the placenta [35]. The relative thickness of the layers was calculated as the thickness of each layer/total thickness of all three layers.
2.5.6. RNA extraction and quantitative real-time PCR
RNA was extracted using the TRIzol® reagent (Thermo Fisher). Reverse transcription was conducted using the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher) following the manufacturer’s instructions. Gene transcript abundance was analyzed by quantitative real-time PCR with SYBR green detection using the CFX384 Touch™ Real-Time PCR Detection System (Bio-Rad, Hercules, California). The reaction conditions were as follows: 95°C for 3 min, followed by 39 cycles with 20 s at 95°C, 30 s at 58°C, and 20 s at 72°C. Data were expressed as the fold difference of the gene of interest relative to the housekeeping gene, beta-actin (Actb), using the ΔΔCt method [36]. All primers were designed using GeneRunner Version 3.01 (http://www.softpedia.com) (Supplemental Table 2). Expression of the following genes was analyzed: peroxisomal acyl-coenzyme A oxidase 1 (Acox1), fatty acid translocase (CD36 antigen, Cd36), carnitine palmitoyltransferase 1b (Cpt1b), fatty acid synthase (Fas), fatty acid transporter 1 (Fatp1), Fatp4, glucose transporter 1 (Glut1), Glut3, insulin-like growth factor I receptor (Igf1r), Igf2, interleukin 1 beta (Il1b), interleukin 6 (Il6), lipoprotein lipase (Lpl), and tumor necrosis factor alpha (Tnfa).
2.5.7. Western blot
The abundance of protein was analyzed by western blot. Total proteins were extracted from two placentas of each dam with the radioimmunoprecipitation assay (RIPA) lysis buffer containing the Halt™ protease and phosphatase inhibitor cocktail (Thermo Fisher). The proteins were separated by SDS-PAGE electrophoresis and transferred onto nitrocellulose membranes. After transfer, membranes were blocked with a commercial blocking buffer (Superblock™, Thermo Fisher) for 2 hours and incubated with primary antibodies overnight. The primary antibodies include: GLUT1, CD36, β-actin (Abcam, Cambridge, MA); FATP1, ribosomal protein S6 (Santa Cruz, Dallas, Texas); eukaryotic translation initiation factor 4E-binding protein 1(4E-BP1), Phospho-4E-BP1(Thr 37/46), Phospho-S6 (Ser 235/236) (Cell Signaling, Danvers, MA); and phospho-AKT-(Thr308) and AKT (Signalway Antibody, College Park, MD). After incubation, the membranes were incubated with HRP-conjugated antibodies (GE Healthcare, Chicago, IL) for 1 hour. Target protein bands were visualized using chemiluminescence detection reagents (GE Healthcare) and quantified by the ImageJ software. Relative protein abundance was expressed as the fold difference of intensity in the target proteins relative to the reference protein (β-actin).
2.5.8. Glycogen and triglyceride measurements
Placental glycogen and triglyceride concentrations were quantified with the Glycogen Fluorometric Assay Kit (Cayman, Ann Arbor, MI) and Triglyceride Colorimetric Assay Kit (Cayman) according to the manufacturer’s instructions.
2.6. Statistical analyses
General linear models (GLMs) followed by post-hoc Fisher’s least significant difference (LSD) tests were constructed to assess the difference in the dependent variables (e.g., embryonic weight, placenta weight, and gene expression) among the four groups of mice. For the assays in which multiple embryos or placentas from the same dam were analyzed, dam was adjusted in the model as a random factor. Fetal sex and litter size were also included in the model as independent variables if they significantly modified the dependent variable. However, we were not able to analyze results in the two sexes separately due to the relatively small sample size. The difference in maternal weight gain was analyzed with repeated measures Analysis of Variance (rANOVA). Dependent variables deviating from the normal distribution (fetal Cpt1b mRNA; placental Acox1, Glut3, and Lpl mRNA) were logarithmically transformed before analysis. A P value < 0.05 was considered as significant. Values are presented as means ± standard error of mean (SEM).
3. Results
3.1. Food and choline intake
The HFCO group had lower (P = 0.02) average weekly food intake than the other 3 groups (Table 1). The other 3 groups had similar food intake. However, since the HF diet had a higher calorie density than the NF diet, the HFCO group still tended to have higher (P = 0.06) caloric intake per week than the NFCO group, whereas the HFCS group had the highest caloric intake versus the other 3 groups (P < 0.01). Water consumption was not significantly different among the groups (P = 0.24). Total choline intake from food and water was on average 4.5 fold higher in the choline supplemented groups versus the control groups regardless of diet. Choline intake was not significantly different between the HFCS and NFCS groups. However, the HFCO group had 17% higher (P = 0.04) weekly choline consumption than the NFCO group.
Table 1.
Average weekly food and choline intake of dams1
| NFCO (n = 8) |
NFCS (n = 6) |
HFCO (n = 8) |
HFCS (n = 7) |
|
|---|---|---|---|---|
| Food intake (g/week) | 17.6 ± 0.7b | 18.1 ± 0.8b | 15.0 + 0.8a | 18.3 ± 0.7b |
| Caloric intake (kcal/week) | 68 ± 3a | 69 ± 4a | 79 ± 4(b) | 96 ± 4c |
| Water intake (mL/week) | 20.4 ± 0.6a | 20.7 ± 0.8a | 19.5 ± 0.6a | 19.0 ± 0.7a |
| Total choline intake (μmol/week) | 133 ± 10a | 663 ± 12c | 155 ± 13b | 664 ± 12c |
Different diets were fed to dams from 4 weeks before timed-mating to gestational day 12.5. Data were analyzed using the general linear model. Different letters indicate P < 0.05;
: P = 0.06. Values represent means ± standard error of mean (SEM). NF: normal-fat diet; HF: high-fat diet; CO: control; CS: choline supplemented
3.2. Maternal weight, glucose tolerance, and biomarkers
3.2.1. Choline supplementation did not improve maternal weight gain or glucose tolerance
The HF groups had significantly higher (P < 0.01) weight gain before and during gestation than the NF groups, confirming the development of maternal obesity in these mice (Fig. 1A). Fasting blood glucose levels were higher (P < 0.01) in the HF (HFCO: 159 ± 5 mg/dL, HFCS: 158 ± 5 mg/dL) than the NF (NFCO: 125 ± 5 mg/dL, NFCS: 119 ± 7 mg/dL) groups at E11.5. Furthermore, HF feeding led to impaired glucose tolerance at E11.5 (HF versus NF groups, P < 0.01) (Fig. 1B and C), suggesting disturbances in blood glucose control typically observed in GDM. However, choline supplementation did not affect weight gain or blood glucose control. The groups did not differ in timed-mating success rate or litter size either (data not shown).
Fig. 1.
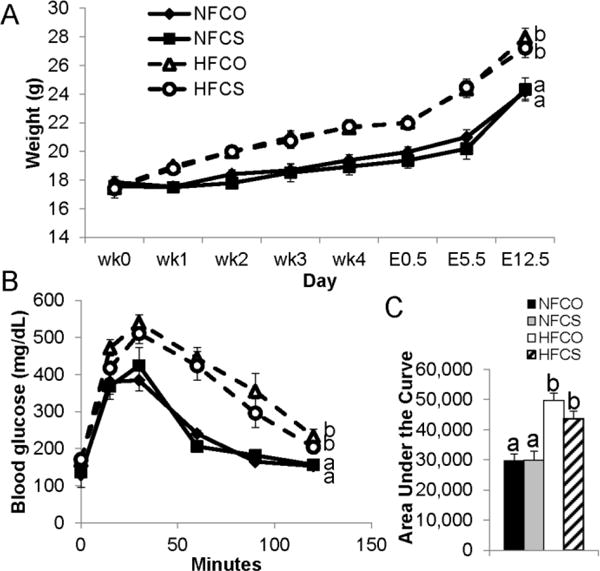
Weight gain and intraperitoneal glucose tolerance (IGT) of dams fed different diets. (A) Weight gain was measured both before timed-mating and during gestation (E0.5–E12.5). (B) IGT tests were conducted at E11.5 with 2g/kg D-glucose injected. (C) The area under the curve of the IGT tests. NFCO: n = 8; NFCS: n = 6; HFCO: n = 8; HFCS: n = 7. Values are mean ± standard error of mean (SEM); different letters indicate P < 0.05. NF: normal-fat diet; HF: high-fat diet; CO: control; CS: choline supplemented.
3.2.2. Maternal choline supplementation mildly altered hepatic choline metabolite contents
Since the liver is the main organ where choline is metabolized, we quantified hepatic choline derivative contents of these animals. Choline supplementation led to approximately 80% higher (P < 0.01) hepatic betaine concentrations in both the NFCS and HFCS groups than the NFCO and HFCO groups (Table 2). Sphingomyelin concentrations were increased by HF feeding regardless of choline intake (HF versus NF groups, P < 0.01). Other choline derivatives were neither altered by HF feeding nor choline supplementation (P > 0.05). These results suggest that maternal HF feeding and choline supplementation had only modest influence on hepatic choline metabolite status of the dams.
Table 2.
Choline-derived metabolite concentrations in the maternal liver at gestational day 12.51
| (nmol metabolite/g tissue) | NFCO (n = 6) |
NFCS (n = 6) |
HFCO (n = 6) |
HFCS (n = 6) |
|---|---|---|---|---|
| Choline | 171 ± 29a | 192 ± 31a | 164 +29a | 247 ± 31a |
| Betaine | 291 ± 56a | 529 ± 61b | 231 ± 56a | 418 ± 61b |
| Methionine | 97 ± 11a | 90 ± 12a | 99 ± 11a | 105 ± 12a |
| Glycerophosphocholine | 328 ± 25a | 330 ± 28a | 305 ± 25a | 335 ± 28a |
| Phosphocholine | 280 ± 61a | 242 ± 66a | 105 ± 61a | 180 ± 66a |
| Phosphatidylcholine | 15,209 ± 1,542a | 17,663 ± 1,689a | 17,997 ± 1,542a | 19,956 ± 1,689a |
| Sphingomyelin | 2,075 ± 83a | 1,820 ± 91a | 2,452 ± 83b | 2,348 ± 91b |
Different diets were fed to dams from 4 weeks before timed-mating to gestational day 12.5. Livers from 6 animals were randomly chosen from each group for quantification. Data were analyzed using the general linear model. Different letters indicate P < 0.05; Values represent means ± standard error of mean (SEM). NF: normal-fat diet; HF: high-fat diet; CO: control; CS: choline supplemented
3.2.3. Biomarkers related to maternal obesity were largely unchanged by choline supplementation
To delineate the metabolic changes underlying maternal obesity, we examined several markers of fat metabolism. We observed a two-fold increase (P < 0.01) in abdominal fat weight in both HF groups versus the NF groups (Table 3). However, liver weight (P = 0.7) and liver triglyceride contents were unaltered by HF feeding (HF versus NF, P = 0.9). Interestingly, serum NEFAs and triglycerides did not increase with HF feeding either (HF versus NF, P > 0.05). Choline intake did not modify maternal fat measurements (CO versus CS, P > 0.05).
Table 3.
Maternal metabolic markers related to visceral obesity at gestational day 12.51
| NFCO (n = 8) |
NFCS (n = 6) |
HFCO (n = 8) |
HFCS (n = 7) |
|
|---|---|---|---|---|
| Liver weight (g) | 1.24 ± 0.05a | 1.25 ± 0.06a | 1.33 ± 0.05a | 1.29 ± 0.06a |
| Liver triglycerides (mg/g tissue) | 23.6 ± 2.3a | 21.8 ± 2.9a | 23.3 ± 2.5a | 22.9 ± 2.7a |
| Abdominal fat (g) | 0.42 ± 0.15a | 0.49 ± 0.18a | 1.04 ± 0.16b | 0.91 ± 0.17b |
| Serum insulin (ng/mL) | 0.55 ± 0.08a | 0.76 ± 0.11a | 0.70 ± 0.10a | 0.74 ± 0.11a |
| Serum triglycerides (mg/dL) | 55.7 ± 4.0a | 47.9 ± 5.1a | 48.5 ± 4.0a | 55.1 ± 4.3a |
| Serum IGF-1 (mg/mL) | 0.82 ± 0.22a | 1.09 ± 0.26a | 1.55 ± 0.23b | 1.23 ± 0.24a,b |
| Serum NEFAs (mmol/L) | 0.77 ± 0.04a | 0.66 ± 0.05a | 0.50 ± 0.05a | 0.66 ± 0.05a |
| Serum leptin (ng/mL) | 1.93 ± 0.65a | 1.93 ± 0.94a | 6.93 ± 0.76b | 5.38 ± 0.83b |
Different diets were fed to dams from 4 weeks before timed-mating to gestational day 12.5. Data were analyzed using the general linear model. Different letters indicate P < 0.05; Values represent means ± standard error of mean (SEM). IGF-1, insulin-like growth factor 1; NEFA, non-esterified fatty acid; NF: normal-fat diet; HF: high-fat diet; CO: control; CS: choline supplemented
We next examined the changes in maternal hormone secretion associated with maternal obesity. Regardless of choline intake, HF feeding led to a 3-fold increase in serum leptin concentrations (HF versus NF, P < 0.01), but did not significantly alter insulin levels (HF versus NF, P = 0.1). We also measured serum concentrations of IGF-1, which promotes placental and fetal growth. Interestingly, there was an increase (88%, P = 0.03) in IGF-1 concentrations in the HFCO group but not in the HFCS group (P = 0.2), when compared to the NFCO group.
3.3. Placental and embryonic outcomes
3.3.1. Maternal choline supplementation prevented HF feeding-induced fetal overgrowth
We further examined whether maternal obesity led to placental and fetal overgrowth as was observed in humans [4], and whether choline supplementation provided a modifying effect on these outcomes. At E12.5, the HFCO group had higher (P < 0.01) placental (0.081 ± 0.002 vs. 0.074 ± 0.002 g) and fetal (0.103 ± 0.003 vs. 0.09 ± 0.003 g) weight than the NFCO group, suggestive of fetal overgrowth. However, maternal choline supplementation in the HFCS group completely normalized placental (HFCS versus NFCO, P = 0.6) and fetal (HFCS versus NFCO, P = 0.3) weight (Fig. 2). These data indicate that maternal choline supplementation may prevent fetal overgrowth at mid-gestation in these mice despite continued maternal obesity.
Fig. 2.
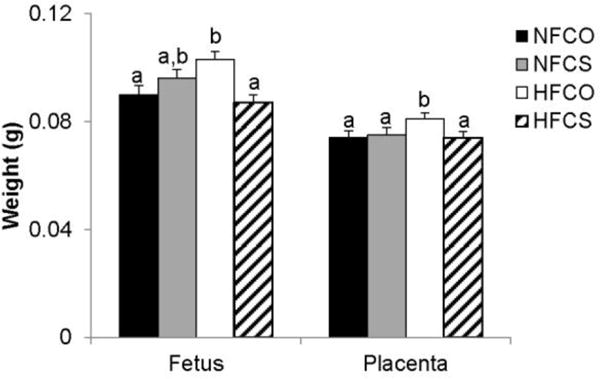
Fetal and placental weight at E12.5. Different diets were fed to dams from 4 weeks before timed-mating to gestational day 12.5. Solid bars, NFCO, n = 8; shaded bars, NFCS, n = 6; open bars, HFCO, n = 8; hatched bars, HFCS, n = 7. n is the number of dams. All placentas and fetuses in each dam were included in the analysis. Values are mean ± standard error of mean (SEM); different letters indicate P < 0.05. NF: normal-fat diet; HF: high-fat diet; CO: control; CS: choline supplemented.
3.3.2 Thickness of placental layers and mRNA abundance of inflammatory markers did not differ among the groups
We examined the relative thickness of placental layers as an assessment of placental morphology that may influence nutrient transport among the NFCO, HFCO, and HFCS groups. However, there was no significant difference (P > 0.05) in overall thickness (data not shown) or relative thickness of the decidua, junctional zone, or labyrinth among the groups (Fig. 3). Inflammation of the placenta may also influence placental growth, but we did not observe significance differences in the placental mRNA abundance of pro-inflammatory markers Il6, Il1b, or Tnfa among the groups either (data not shown).
Fig. 3.
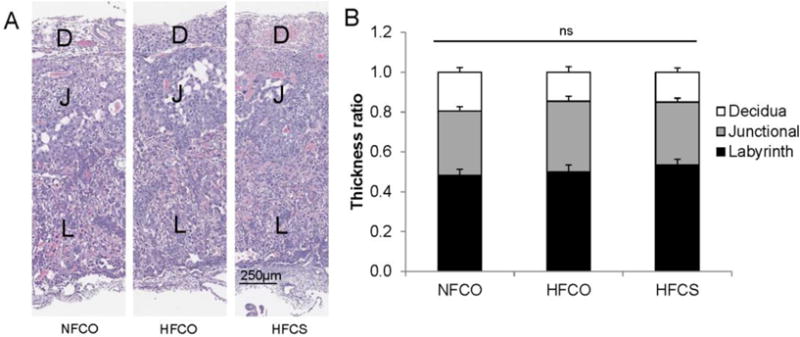
Thickness of placental layers at E12.5. (A) Histological appearance of representative placentas. (B) Thickness ratios of placental layers in the NFCO (n = 3), HFCO (n = 3), and HFCS (n = 3) groups. Different diets were fed to dams from 4 weeks before timed-mating to gestational day 12.5. n is the number of dams. Two placentas in each dam were included in the analysis. Values are mean ± standard error of mean (SEM); different letters indicate P < 0.05; ns: not significant. D, decidua; J, junctional zone; L, labyrinth; NF: normal-fat diet; HF: high-fat diet; CO: control; CS: choline supplemented.
3.3.3. Maternal choline supplementation during HF feeding downregulated macronutrient transporters in the placenta
Next we examined whether molecular indicators of placental nutrient transport were different among the groups. We observed higher mRNA abundance of glucose and fatty acid transporters Glut1 (P = 0.05) and Cd36 (P = 0.03) in the HFCO group than in the NFCO group (Fig. 4A). mRNA abundance of Cpt1b, which is a target of PPAR-α, was also upregulated (P = 0.03) in the HFCO versus the NFCO group, yet the mRNA abundance of Acox1, another target of PPAR-α, was unaltered (P = 0.2). Choline supplementation in the HFCS group normalized the mRNA abundance of Glut1, Cd36, and Cpt1b to the levels of the NFCO group (P > 0.05). The difference in transporter abundance was also confirmed at the protein level for GLUT1 (P = 0.01) but not for CD36 (P = 0.9) (Fig. 4B). In addition, the HFCO group had higher abundance of FATP1 (P = 0.03), another fatty acid transporter, at the protein level when compared to the NFCO group, although there was no difference in mRNA abundance between the two groups (P = 0.7). The HFCS group had consistently lower (P < 0.05) abundance of FATP1 at both mRNA and protein levels versus the HFCO group. Interestingly, mRNA abundance of Fatp4 which preferentially transports polyunsaturated fatty acids (PUFAs) through the placenta [37] was upregulated in the HFCS group than the other 3 groups (P = 0.04). Expression of other placental genes, including Glut3, Lpl, and Fas was not altered by maternal HF diet or choline supplementation.
Fig. 4.
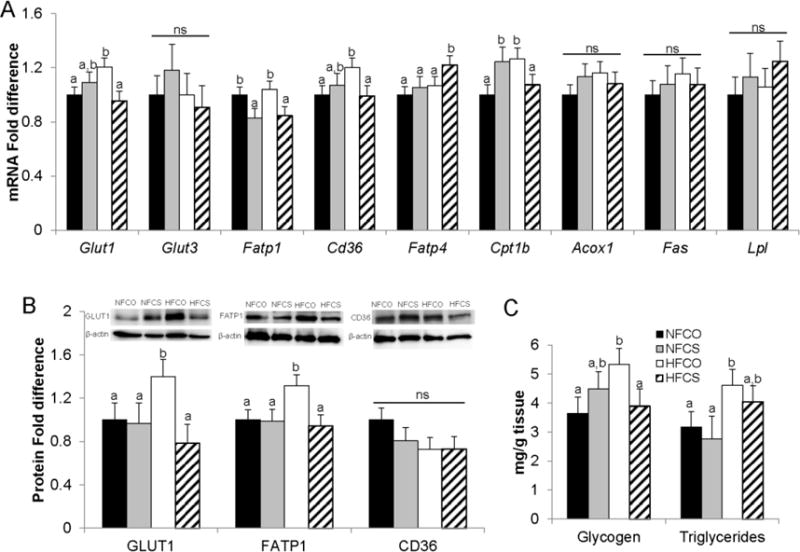
Placental glucose and fatty acid metabolism at E12.5. Different diets were fed to dams from 4 weeks before timed-mating to gestational day 12.5. (A) mRNA abundance was measured by real-time PCR. (B) Macronutrient transport proteins were assessed by western blot. (C) Glycogen and triglyceride accumulation in the placenta were measured using assay kits. Solid bars, NFCO, n = 8; shaded bars, NFCS, n = 6; open bars, HFCO, n = 7; hatched bars, HFCS, n = 6. n is the number of dams. Two placentas in each dam were included in the analysis. Values are mean ± standard error of mean (SEM); different letters indicate P < 0.05; ns: not significant. Acox1: Peroxisomal acyl-coenzyme A oxidase 1; Cd36: fatty acid translocase; Cpt1b: Carnitine palmitoyltransferase 1b; Fas: fatty acid synthase; Fatp: fatty acid transport protein; Glut: glucose transporter; Lpl: lipoprotein lipase. NF: normal-fat diet; HF: high-fat diet; CO: control; CS: choline supplemented.
Consistent with the alterations in transport proteins, placental triglyceride and glycogen concentrations were elevated by about 50% in the HFCO group (P = 0.04 for both markers) versus the NFCO group. Choline supplementation in the HFCS group partially or completely normalized placental triglyceride and glycogen concentrations to the levels of the NFCO group (Fig. 4C).
3.3.4. Maternal choline supplementation during HF feeding downregulated components of growth-promoting pathways in the placenta
To further delineate the effect of HF feeding and choline supplementation on placental growth and macronutrient transport, we examined the expression or activity of IGFs and mTORC1, both growth-promoting proteins and upstream regulators of placental nutrient transporters. We showed in section 3.2.3 that maternal blood IGF-1 concentrations increased in the HFCO group but not in the HFCS group when compared to NFCO. Placental Igf2 mRNA expression was lower (P = 0.03) in both CS groups than the CO groups (Fig. 5A). However, mRNA abundance of Igf1r, a receptor of IGF1, was not altered by HF feeding or choline supplementation.
Fig. 5.
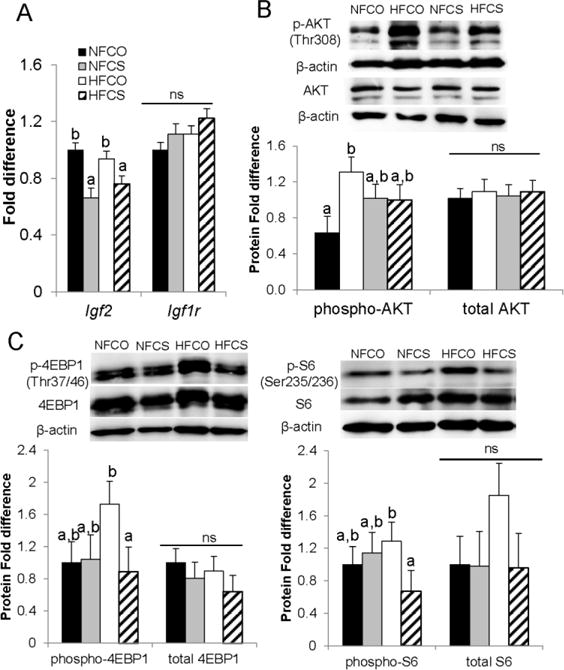
mRNA abundance of the IGF system and activation of the AKT/mTOR pathway in the placenta at E12.5. Different diets were fed to dams from 4 weeks before timed-mating to gestational day 12.5. (A) mRNA abundance was measured by real-time PCR. (B) AKT phosphorylation and (C) mTOR-regulated proteins were assessed by western blot. Solid bars, NFCO, n = 8; shaded bars, NFCS, n = 6; open bars, HFCO, n = 7; hatched bars, HFCS, n = 6. n is the number of dams. Two placentas in each dam were included in the analysis. Values are mean ± standard error of mean (SEM); different letters indicate P < 0.05; ns: not significant. AKT: protein kinase B; IGF, insulin-like growth factor; Igf1r, insulin-like growth factor 1 receptor; mTOR, mechanistic target of rapamycin; S6, ribosomal protein S6; 4E-BP1, 4E-binding protein 1. NF: normal-fat diet; HF: high-fat diet; CO: control; CS: choline supplemented.
Phosphorylation of AKT at Thr308 [p-AKT(Thr308)] is needed for its activation, which further activates mTOR. p-AKT(Thr308) was significantly higher in the HFCO (P < 0.01) but not the HFCS (P = 0.1) group when compared to the NFCO group (Fig. 5B). Phosphorylation of 4E-BP1 at Thr 37/46 and S6 at Ser 235/236 are two well-known indicators of mTORC1 activation [8]. Although we observed numerical increases in both 4E-BP1 and S6 phosphorylation in the HFCO group versus the other 3 groups, statistical significance was only observed between the HFCO and HFCS group, i.e. phosphorylation of these two mTORC1 targets were both decreased (P < 0.05) in the placentas of the HFCS group versus the HFCO group (Fig. 5C), suggesting a reduction in mTORC1 activity.
3.3.5. Maternal choline supplementation prevented the decline in placental phosphatidylcholine concentrations during HF feeding
Lastly we examined changes in placental choline metabolite concentrations, which indicate alterations in the metabolic fates of choline that may influence placental and fetal growth. HF feeding decreased (P = 0.045) phosphatidylcholine concentrations in the HFCO versus the NFCO placentas. However, the HFCS group completely restored phosphatidylcholine concentrations (HFCS versus NFCO, P = 0.7) (Table 4). Choline supplementation in the NFCS group led to higher (P < 0.01) acetylcholine concentrations versus the NFCO group, consistent with a previous study in the human placenta [34]. However, acetylcholine content was not altered in the HFCS group compared to the NFCO or HFCO groups. Other choline-derived metabolites, including betaine, glycerophosphocholine, phosphocholine, sphingomyelin, and methionine were not altered by HF feeding or choline supplementation (P > 0.05).
Table 4.
Placental choline-derived metabolites at E12.51
| (nmol metabolite/g tissue) | NFCO (n = 6) |
NFCS (n = 6) |
HFCO (n = 6) |
HFCS (n = 6) |
|---|---|---|---|---|
| Choline | 392 ± 39a | 432 ± 39a | 317 +39a | 359 ± 39a |
| Betaine | 3,854 ± 1,831a | 5,801 ± 1,831a | 7,826 ± 1,831a | 5,060 ± 1,831a |
| Acetylcholine | 0.035 ± 0.010a | 0.075 ± 0.010b | 0.024 ± 0.010a | 0.030 ± 0.010a |
| Methionine | 196 ± 16a | 156 ± 16a | 193 ± 16a | 182 ± 16a |
| Glycerophosphocholine | 317 ± 30a | 357 ± 30a | 292 ± 30a | 309 ± 30a |
| Phosphocholine | 781 ± 80a | 617 ± 80a | 640 ± 80a | 712 ± 80a |
| Phosphatidylcholine | 9,069 ± 517b | 8,789 ± 562b | 7,230 ± 562a | 9,288 ± 562b |
| Sphingomyelin | 2,253 ± 311a | 1,952 ± 364a | 2,287 ± 412a | 2,477 ± 412a |
Different diets were fed to dams from 4 weeks before timed-mating to gestational day 12.5. Data were analyzed using the general linear model. Different letters indicate P < 0.05; n is the number of dams. One placenta was used for analysis for each dam. Values represent means ± standard error of mean (SEM). NF: normal-fat diet; HF: high-fat diet; CO: control; CS: choline supplemented
4. Discussion
Many studies have demonstrated that maternal obesity results in fetal overgrowth [1, 5, 38–40] and increases the risk for obesity later in life [4]. Dietary interventions that focused on calorie and glycemic control during pregnancy improved maternal metabolism but often failed to translate into improved fetal growth outcomes, e.g. lowering the risk of macrosomia [41–43]. We demonstrated for the first time that maternal choline supplementation prevented fetal overgrowth at mid-gestation in HF feeding-induced obese mice. Choline supplementation also attenuated the expression or activity of proteins involved with placental macronutrient transport and growth signaling, suggesting a role of choline in maintaining placental metabolic homeostasis.
We first confirmed that C57BL/6J mice fed a HF diet entered gestation in the obese state and remained to be obese during gestation. The HFCO dams had impaired glucose tolerance at E11.5 and higher placental and fetal weight compared to the NFCO dams. Therefore, these HF mice served as a relevant model of maternal obesity induced by excessive energy intake [29] leading to GDM-like phenotypes and fetal overgrowth [5, 28]. Interestingly, the HFCS dams had higher caloric intake than the HFCO dams but did not gain more weight. Whether it is related to higher energy expenditure or lower nutrient absorption in the HFCS group remains to be discerned.
We demonstrated that maternal choline supplementation in the HF-fed dams completely prevented placental and fetal overgrowth. Moreover, the influence of choline on fetal weight appeared to be specific to the HFCS group, since the NFCS group did not show signs of restricted growth compared to NFCO. This result suggests that choline itself does not hinder energy accretion of the fetus. The influence of choline on growth is likely to be mediated through its interaction with macronutrient metabolism when fat and glucose are in excess. This crosstalk between choline and macronutrient metabolism has been reviewed by others [15, 16].
Unlike the significant influence that choline had on fetal growth, choline supplementation did not prevent maternal obesity or glucose intolerance of the HF mice. This result was consistent with previous studies in humans indicating that choline intake had only moderate influence on maternal biomarkers [26, 44]. The lack of changes in maternal metabolism raises the possibility that choline modulates fetal growth by altering the maternal to fetal nutrient transport through the placenta.
Placental nutrient transport is determined by several factors, including placental structure and vasculature, maternal nutrient supply, hormonal influence, and placental nutrient sensing and growth signals. Maternal obesity is often characterized by elevated levels of blood triglycerides which are transported across the placenta, thereby increasing fetal fat deposition [6, 45, 46]. While maternal circulating NEFA and triglyceride levels were not affected by HF feeding in the current study, we did observe increased accumulation of triglycerides in placentas from the HFCO versus NFCO dams. These triglycerides in the placenta may be released into the growing fetus and promote fetal adiposity at late gestation [21, 47]. Concentrations of glycogen in the placenta were also higher in the HFCO versus NFCO animals, corroborating previous reports on diabetes-complicated pregnancies [48]. Although its function is not entirely clear, placental glycogen may serve as a reservoir of glucose which provides carbohydrate energy not only for local endothelial cells [49], but also for the fetus [50]. Interestingly, choline supplementation alleviated placental triglyceride and glycogen accumulation of the HF animals, and thus may reduce the placental pool of macronutrients that can be mobilized for fetal uptake.
Previous studies suggested that in response to maternal obesity, the macronutrient overloaded placenta also upregulated its expression of nutrient transporters, thereby enhancing transplacental nutrient transport [9]. We observed consistent increases in GLUT1 at both mRNA and protein levels in the HFCO versus NFCO placentas. Although the placenta may sequester some GLUTs from the plasma membrane to prevent a substantial increase in glucose transport [51], this compensatory mechanism is often overridden in pregnancies present with macrosomia in which GLUT1 overexpression is associated with accelerated fetal growth [52]. We also observed upregulation of the placental FATP1 protein by HF feeding, despite unchanged mRNA abundance compared to the NFCO animals, suggesting the possibility of post-transcriptional regulation [53]. FATP1 was reported to determine tissue distribution of dietary fat [54]. Its upregulation may promote fat deposition, which corroborates the positive associations observed between abundance of this transporter and placental and fetal adiposity in human and animal models of maternal obesity [13, 55]. Importantly, choline supplementation seemed to consistently lower both the mRNA and protein levels of GLUT1 and FATP1 in the HF animals, which gives rise to a potential pathway to preventing excess nutrient transport and eventually aiding in normalizing fetal growth. Nevertheless, a direct measurement of the placental to fetal nutrition flow is necessary to verify the impact of maternal HF feeding and choline supplementation on placental nutrient transport. It should also be noted that not every transporter or enzyme that we tested was responsive to HF feeding or choline supplementation. CD36 showed an increase in mRNA but not protein abundance when comparing the HFCO with the NFCO placentas. Placental LPL and GLUT3 levels were reported to increase during HF feeding at late gestation [9, 10] but were not altered at E12.5 in this study. Interestingly, Fatp4 mRNA levels were elevated in the HFCS group versus the other 3 groups. However, since FATP4 is the major placental transporter of long-chain PUFAs, its increase may actually have a positive impact on docosahexaenoic acid (DHA) transport that is often downregulated in GDM pregnancies [37].
Several pathways promote placental growth and nutrient transport. IGFs are well-known for their growth-promoting effect during the prenatal period [7, 56], favoring the proliferation and survival of cytotrophoblasts [49]. Maternal IGF-1 levels increased in GDM complicated pregnancies [49] as well as in the HFCO versus NFCO mice in the current study. However, IGF-2 mRNA abundance in the placenta was not altered by HF feeding. In contrast, maternal choline supplementation moderately decreased maternal IGF-1 in the HF-fed mice, while significantly decreased placental IGF2 mRNA abundance in both HF and NF conditions. These results suggest that HF feeding and choline supplementation may have opposite effects on IGF signaling, although they may target different components in the pathway.
Another key promoter of placental growth and nutrient transport is the PI3K/AKT/mTOR signaling pathway, which is activated in placentas from both obese and GDM pregnancies [8, 57]. This pathway also responds to IGF signals via the IGF receptor and subsequent activation of the insulin receptor substrate [57]. We detected an increase in p-AKT (Thr308) in the HFCO versus NFCO placentas, consistent with the activation of this pathway by maternal HF feeding which provides a possible mechanism to enhance placental growth. Choline supplementation seemed to attenuate the increase but did not completely normalize AKT phosphorylation. mTOR is considered as the central nutrient sensor of the placenta which promotes macronutrient synthesis and transport [6]. Mice fed the Western diet had higher 4E-BP1 and S6 phosphorylation, indicating activation of mTOR [8]. However, these two markers were not significantly altered by maternal HF feeding in our study despite numerical increases. Therefore, it is unclear whether mTOR is a major downstream effector of AKT that mediates metabolic disturbances in the current HF model. Interestingly, maternal choline supplementation during HF feeding seemed to decrease the phosphorylation of 4E-BP1 and S6, suggesting attenuated mTOR activity. Taken together, it is possible that HFCS dampens IGF, AKT, and mTOR signaling, thereby preventing nutrient accumulation and transport in the placenta, even if the negative effect of maternal HF feeding on placental transport is not completely mediated by the same mechanism. Other AKT-regulated proteins such as forkhead box O3 (FOXO3) or maternal hormones such as leptin may serve as alternative mechanisms that mediate the impact of HF feeding, which requires further investigation. Additionally, although we did not observe differences in the thickness of placental layers, further examination on placental vasculature or cell type may reveal structural differences of the placenta among the groups that can affect placental growth and transport.
It is unclear by what mechanisms that choline interacts with the aforementioned growth signaling pathways. Data on placental metabolite concentrations suggest that maternal HF feeding decreased placental phosphatidylcholine content which was reversed by choline supplementation. Phosphatidylcholine deficiency has been shown to activate uncontrolled cell growth by increasing the accumulation of the second messenger diacylglycerol which activates the PKC-mediated mitogenic pathways [22, 23]. Further studies are needed to validate whether maintaining the level of phosphatidylcholine is indeed important for the attenuation of placental growth signals and improvement of fetal growth outcomes.
There are several limitations of the current study. The NFCO and HFCO groups did not have identical choline consumption which introduced an additional variable besides HF feeding. However, it was very unlikely that the additional choline in the HFCO group versus the NFCO group resulted in maternal obesity since the much higher choline intake difference in the CS groups versus the CO groups did not induce or exacerbate obesity. The method that we used to measure intake could not account for food and water loss in cage bedding which may limit the accurate calculation of choline intake. Nevertheless, the approximate 4-fold difference in choline intake was consistent with what was used in previous studies to demonstrate beneficial effects of maternal choline supplementation on cognitive outcomes of offspring [58, 59] and seemed to be sufficient to overcome fetal overgrowth in the current study. It should be noted that this study only investigated the effect of choline at mid-gestation because a previous study showed that hyperglycemia was the most prominent at this time point [28]. Longer-term studies are needed to examine the impact of prenatal choline availability on late gestation and postnatal outcomes.
In summary, HF feeding induced GDM-like phenotypes and fetal overgrowth at mid-gestation in C57BL/6J mice, whereas maternal choline supplementation prevented fetal overgrowth. The influence of choline on placental macronutrient transport and accumulation may play a role in normalizing fetal growth, yet more mechanistic studies are needed to confirm this association. Overall, this study provides promising evidence that choline may counteract some of the negative influence of maternal obesity on placental metabolism and fetal growth in mice.
Supplementary Material
Acknowledgments
We thank Dr. Martha Stipanuk, Cornell University for helpful discussion. J.N. conducted research, analyzed data, and wrote the paper; E.G., C.J.R., T.T.A., O.V.M, E.S., and K.N. conducted research; M.A.C., A.S., and K.A. interpreted the data; X.J. designed the study, analyzed data, and has primary responsibility for final content. All authors read and approved the final manuscript. This work was supported by the National Institutes of Health (1SC2DK108292) (X.J.).
Abbreviations
- Acox1
peroxisomal acyl-coenzyme A oxidase 1
- AKT
protein kinase B
- CD36
fatty acid translocase
- CPT1B
carnitine palmitoyltransferase 1b
- E
embryonic day
- FATP
fatty acid transport protein
- GDM
gestational diabetes mellitus
- GLUT
glucose transporter
- HF
high-fat
- IGF
insulin-like growth factor
- NF
normal-fat
- LC-PUFA
long-chain polyunsaturated fatty acid
- LPL
lipoprotein lipase
- mTOR
mechanistic target of rapamycin
- NEFA
non-esterified fatty acids
- PI3K
Phosphoinositide 3-kinase
- PPAR-α
peroxisome proliferator-activated receptor alpha
- S6
ribosomal protein s6
- 4E-BP1
4E-binding protein 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors declare no conflicts of interest.
References
- 1.Leddy MA, Power ML, Schulkin J. The impact of maternal obesity on maternal and fetal health. Rev Obstet Gynecol. 2008;1(4):170–8. [PMC free article] [PubMed] [Google Scholar]
- 2.Chu SY, Callaghan WM, Kim SY, Schmid CH, Lau J, England LJ, et al. Maternal obesity and risk of gestational diabetes mellitus. Diabetes Care. 2007;30(8):2070–6. doi: 10.2337/dc06-2559a. [DOI] [PubMed] [Google Scholar]
- 3.Gillman MW, Rifas-Shiman S, Berkey CS, Field AE, Colditz GA. Maternal gestational diabetes, birth weight, and adolescent obesity. Pediatrics. 2003;111(3):e221–6. doi: 10.1542/peds.111.3.e221. [DOI] [PubMed] [Google Scholar]
- 4.Vinturache AE, McDonald S, Slater D, Tough S. Perinatal outcomes of maternal overweight and obesity in term infants: a population-based cohort study in Canada. Sci Rep. 2015;5:9334. doi: 10.1038/srep09334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kc K, Shakya S, Zhang H. Gestational diabetes mellitus and macrosomia: a literature review. Ann Nutr Metab. 2015;66(Suppl 2):14–20. doi: 10.1159/000371628. [DOI] [PubMed] [Google Scholar]
- 6.Brett KE, Ferraro ZM, Yockell-Lelievre J, Gruslin A, Adamo KB. Maternal-fetal nutrient transport in pregnancy pathologies: the role of the placenta. Int J Mol Sci. 2014;15(9):16153–85. doi: 10.3390/ijms150916153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sferruzzi-Perri AN, Owens JA, Pringle KG, Roberts CT. The neglected role of insulin-like growth factors in the maternal circulation regulating fetal growth. J Physiol. 2011;589(Pt 1):7–20. doi: 10.1113/jphysiol.2010.198622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosario FJ, Powell TL, Jansson T. Activation of placental insulin and mTOR signaling in a mouse model of maternal obesity associated with fetal overgrowth. Am J Physiol Regul Integr Comp Physiol. 2016;310(1):R87–93. doi: 10.1152/ajpregu.00356.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosario FJ, Kanai Y, Powell TL, Jansson T. Increased placental nutrient transport in a novel mouse model of maternal obesity with fetal overgrowth. Obesity (Silver Spring) 2015;23(8):1663–70. doi: 10.1002/oby.21165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiao L, Guo Z, Bosco C, Guidotti S, Wang Y, Wang M, et al. Maternal high-fat feeding increases placental lipoprotein lipase activity by reducing SIRT1 expression in mice. Diabetes. 2015;64(9):3111–20. doi: 10.2337/db14-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaar JL, Crume T, Brinton JT, Bischoff KJ, McDuffie R, Dabelea D. Maternal obesity, gestational weight gain, and offspring adiposity: the exploring perinatal outcomes among children study. J Pediatr. 2014;165(3):509–15. doi: 10.1016/j.jpeds.2014.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cetin I, Alvino G, Cardellicchio M. Long chain fatty acids and dietary fats in fetal nutrition. J Physiol. 2009;587(Pt 14):3441–51. doi: 10.1113/jphysiol.2009.173062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu MJ, Ma Y, Long NM, Du M, Ford SP. Maternal obesity markedly increases placental fatty acid transporter expression and fetal blood triglycerides at midgestation in the ewe. Am J Physiol Regul Integr Comp Physiol. 2010;299(5):R1224–31. doi: 10.1152/ajpregu.00309.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang X, West AA, Caudill MA. Maternal choline supplementation: a nutritional approach for improving offspring health? Trends Endocrinol Metab. 2014;25(5):263–73. doi: 10.1016/j.tem.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Corbin KD, Zeisel SH. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr Opin Gastroenterol. 2012;28(2):159–65. doi: 10.1097/MOG.0b013e32834e7b4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeisel SH. Metabolic crosstalk between choline/1-carbon metabolism and energy homeostasis. Clin Chem Lab Med. 2013;51(3):467–75. doi: 10.1515/cclm-2012-0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao ZM, Vance DE. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J Biol Chem. 1988;263(6):2998–3004. [PubMed] [Google Scholar]
- 18.Li Z, Vance DE. Phosphatidylcholine and choline homeostasis. J Lipid Res. 2008;49(6):1187–94. doi: 10.1194/jlr.R700019-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Vance JE, Vance DE. The role of phosphatidylcholine biosynthesis in the secretion of lipoproteins from hepatocytes. Can J Biochem Cell Biol. 1985;63(8):870–81. doi: 10.1139/o85-108. [DOI] [PubMed] [Google Scholar]
- 20.Chakravarthy MV, Lodhi IJ, Yin L, Malapaka RR, Xu HE, Turk J, et al. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell. 2009;138(3):476–88. doi: 10.1016/j.cell.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radaelli T, Lepercq J, Varastehpour A, Basu S, Catalano PM, Hauguel-De Mouzon S. Differential regulation of genes for fetoplacental lipid pathways in pregnancy with gestational and type 1 diabetes mellitus. Am J Obstet Gynecol. 2009;201(2):209.e1–e10. doi: 10.1016/j.ajog.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.da Costa KA, Garner SC, Chang J, Zeisel SH. Effects of prolonged (1 year) choline deficiency and subsequent re-feeding of choline on 1,2-sn-diradylglycerol, fatty acids and protein kinase C in rat liver. Carcinogenesis. 1995;16(2):327–34. doi: 10.1093/carcin/16.2.327. [DOI] [PubMed] [Google Scholar]
- 23.Blusztajn JK, Zeisel SH. 1,2-sn-diacylglycerol accumulates in choline-deficient liver. A possible mechanism of hepatic carcinogenesis via alteration in protein kinase C activity? FEBS Lett. 1989;243(2):267–70. doi: 10.1016/0014-5793(89)80142-5. [DOI] [PubMed] [Google Scholar]
- 24.Gong L, Pan YX, Chen H. Gestational low protein diet in the rat mediates Igf2 gene expression in male offspring via altered hepatic DNA methylation. Epigenetics. 2010;5(7):619–26. doi: 10.4161/epi.5.7.12882. [DOI] [PubMed] [Google Scholar]
- 25.Kovacheva VP, Mellott TJ, Davison JM, Wagner N, Lopez-Coviella I, Schnitzler AC, et al. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J Biol Chem. 2007;282(43):31777–88. doi: 10.1074/jbc.M705539200. [DOI] [PubMed] [Google Scholar]
- 26.Jiang X, Yan J, West AA, Perry CA, Malysheva OV, Devapatla S, et al. Maternal choline intake alters the epigenetic state of fetal cortisol-regulating genes in humans. FASEB J. 2012;26(8):3563–74. doi: 10.1096/fj.12-207894. [DOI] [PubMed] [Google Scholar]
- 27.Jiang X, Bar HY, Yan J, Jones S, Brannon PM, West AA, et al. A higher maternal choline intake among third-trimester pregnant women lowers placental and circulating concentrations of the antiangiogenic factor fms-like tyrosine kinase-1 (sFLT1) FASEB J. 2013;27(3):1245–53. doi: 10.1096/fj.12-221648. [DOI] [PubMed] [Google Scholar]
- 28.Liang C, DeCourcy K, Prater MR. High-saturated-fat diet induces gestational diabetes and placental vasculopathy in C57BL/6 mice. Metabolism. 2010;59(7):943–50. doi: 10.1016/j.metabol.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 29.Jones HN, Woollett LA, Barbour N, Prasad PD, Powell TL, Jansson T. High-fat diet before and during pregnancy causes marked up-regulation of placental nutrient transport and fetal overgrowth in C57/BL6 mice. FASEB J. 2009;23(1):271–8. doi: 10.1096/fj.08-116889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dhar MS, Sommardahl CS, Kirkland T, Nelson S, Donnell R, Johnson DK, et al. Mice heterozygous for Atp10c, a putative amphipath, represent a novel model of obesity and type 2 diabetes. J Nutr. 2004;134(4):799–805. doi: 10.1093/jn/134.4.799. [DOI] [PubMed] [Google Scholar]
- 31.Pruznak AM, Kazi AA, Frost RA, Vary TC, Lang CH. Activation of AMP-activated protein kinase by 5-aminoimidazole-4-carboxamide-1-beta-D-ribonucleoside prevents leucine-stimulated protein synthesis in rat skeletal muscle. J Nutr. 2008;138(10):1887–94. doi: 10.1093/jn/138.10.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tai MM. A mathematical model for the determination of total area under glucose tolerance and other metabolic curves. Diabetes Care. 1994;17(2):152–4. doi: 10.2337/diacare.17.2.152. [DOI] [PubMed] [Google Scholar]
- 33.McClive PJ, Sinclair AH. Rapid DNA extraction and PCR-sexing of mouse embryos. Mol Reprod Dev. 2001;60(2):225–6. doi: 10.1002/mrd.1081. [DOI] [PubMed] [Google Scholar]
- 34.Yan J, Jiang X, West AA, Perry CA, Malysheva OV, Devapatla S, et al. Maternal choline intake modulates maternal and fetal biomarkers of choline metabolism in humans. Am J Clin Nutr. 2012;95(5):1060–71. doi: 10.3945/ajcn.111.022772. [DOI] [PubMed] [Google Scholar]
- 35.Kim DW, Young SL, Grattan DR, Jasoni CL. Obesity during pregnancy disrupts placental morphology, cell proliferation, and inflammation in a sex-specific manner across gestation in the mouse. Biol Reprod. 2014;90(6):130. doi: 10.1095/biolreprod.113.117259. [DOI] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Larqué E, Demmelmair H, Gil-Sánchez A, Prieto-Sánchez MT, Blanco JE, Pagán A, et al. Placental transfer of fatty acids and fetal implications. Am J Clin Nutr. 2011;94(6 Suppl):1908S–13S. doi: 10.3945/ajcn.110.001230. [DOI] [PubMed] [Google Scholar]
- 38.Gaudet L, Ferraro ZM, Wen SW, Walker M. Maternal obesity and occurrence of fetal macrosomia: a systematic review and meta-analysis. Biomed Res Int. 2014;2014:640291. doi: 10.1155/2014/640291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ornoy A. Prenatal origin of obesity and their complications: Gestational diabetes, maternal overweight and the paradoxical effects of fetal growth restriction and macrosomia. Reprod Toxicol. 2011;32(2):205–12. doi: 10.1016/j.reprotox.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 40.van Hoorn J, Dekker G, Jeffries B. Gestational diabetes versus obesity as risk factors for pregnancy-induced hypertensive disorders and fetal macrosomia. Aust N Z J Obstet Gynaecol. 2002;42(1):29–34. doi: 10.1111/j.0004-8666.2002.00035.x. [DOI] [PubMed] [Google Scholar]
- 41.Han S, Crowther CA, Middleton P, Heatley E. Different types of dietary advice for women with gestational diabetes mellitus. Cochrane Database Syst Rev. 2013;3:CD009275. doi: 10.1002/14651858.CD009275.pub2. [DOI] [PubMed] [Google Scholar]
- 42.Bain E, Crane M, Tieu J, Han S, Crowther CA, Middleton P. Diet and exercise interventions for preventing gestational diabetes mellitus. Cochrane Database Syst Rev. 2015;4:CD010443. doi: 10.1002/14651858.CD010443.pub2. [DOI] [PubMed] [Google Scholar]
- 43.Poston L, Bell R, Croker H, Flynn AC, Godfrey KM, Goff L, et al. Effect of a behavioural intervention in obese pregnant women (the UPBEAT study): a multicentre, randomised controlled trial. Lancet Diabetes Endocrinol. 2015 doi: 10.1016/S2213-8587(15)00227-2. [DOI] [PubMed] [Google Scholar]
- 44.Jiang X, Bar HY, Yan J, West AA, Perry CA, Malysheva OV, et al. Pregnancy induces transcriptional activation of the peripheral innate immune system and increases oxidative DNA damage among healthy third trimester pregnant women. PLoS One. 2012;7(11):e46736. doi: 10.1371/journal.pone.0046736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hellmuth C, Lindsay KL, Uhl O, Buss C, Wadhwa PD, Koletzko B, et al. Association of maternal prepregnancy BMI with metabolomic profile across gestation. Int J Obes (Lond) 2016 doi: 10.1038/ijo.2016.153. [DOI] [PubMed] [Google Scholar]
- 46.Scifres CM, Catov JM, Simhan HN. The impact of maternal obesity and gestational weight gain on early and mid-pregnancy lipid profiles. Obesity (Silver Spring) 2014;22(3):932–8. doi: 10.1002/oby.20576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindegaard ML, Damm P, Mathiesen ER, Nielsen LB. Placental triglyceride accumulation in maternal type 1 diabetes is associated with increased lipase gene expression. J Lipid Res. 2006;47(11):2581–8. doi: 10.1194/jlr.M600236-JLR200. [DOI] [PubMed] [Google Scholar]
- 48.Desoye G, Hauguel-de Mouzon S. The human placenta in gestational diabetes mellitus. The insulin and cytokine network. Diabetes Care. 2007;30(Suppl 2):S120–6. doi: 10.2337/dc07-s203. [DOI] [PubMed] [Google Scholar]
- 49.Hiden U, Glitzner E, Hartmann M, Desoye G. Insulin and the IGF system in the human placenta of normal and diabetic pregnancies. J Anat. 2009;215(1):60–8. doi: 10.1111/j.1469-7580.2008.01035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Desoye G, Gauster M, Wadsack C. Placental transport in pregnancy pathologies. Am J Clin Nutr. 2011;94(6 Suppl):1896S–902S. doi: 10.3945/ajcn.110.000851. [DOI] [PubMed] [Google Scholar]
- 51.Desoye G, Korgun ET, Ghaffari-Tabrizi N, Hahn T. Is fetal macrosomia in adequately controlled diabetic women the result of a placental defect?–a hypothesis. J Matern Fetal Neonatal Med. 2002;11(4):258–61. doi: 10.1080/jmf.11.4.258.261. [DOI] [PubMed] [Google Scholar]
- 52.Gaither K, Quraishi AN, Illsley NP. Diabetes alters the expression and activity of the human placental GLUT1 glucose transporter. J Clin Endocrinol Metab. 1999;84(2):695–701. doi: 10.1210/jcem.84.2.5438. [DOI] [PubMed] [Google Scholar]
- 53.Glatz JF, Luiken JJ, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol Rev. 2010;90(1):367–417. doi: 10.1152/physrev.00003.2009. [DOI] [PubMed] [Google Scholar]
- 54.Wu Q, Ortegon AM, Tsang B, Doege H, Feingold KR, Stahl A. FATP1 is an insulin-sensitive fatty acid transporter involved in diet-induced obesity. Mol Cell Biol. 2006;26(9):3455–67. doi: 10.1128/MCB.26.9.3455-3467.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hirschmugl B, Desoye G, Catalano P, Klymiuk I, Scharnagl H, Payr S, et al. Maternal obesity modulates intracellular lipid turnover in the human term placenta. Int J Obes (Lond) 2017;41(2):317–23. doi: 10.1038/ijo.2016.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dimasuay KG, Boeuf P, Powell TL, Jansson T. Placental Responses to Changes in the Maternal Environment Determine Fetal Growth. Front Physiol. 2016;7:12. doi: 10.3389/fphys.2016.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, et al. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab. 2013;98(1):105–13. doi: 10.1210/jc.2012-2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCann JC, Hudes M, Ames BN. An overview of evidence for a causal relationship between dietary availability of choline during development and cognitive function in offspring. Neurosci Biobehav Rev. 2006;30(5):696–712. doi: 10.1016/j.neubiorev.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 59.Zhu CH, Wu T, Jin Y, Huang BX, Zhou RF, Wang YQ, et al. Prenatal choline supplementation attenuates spatial learning deficits of offspring rats exposed to low-protein diet during fetal period. J Nutr Biochem. 2016;32:163–70. doi: 10.1016/j.jnutbio.2015.09.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.