

Abstract
Interactions between polymer molecules and inorganic nanoparticles can play a dominant role in nanocomposite material mechanics, yet control of such interfacial interaction dynamics remains a significant challenge particularly in water. This study presents insights on how to engineer hydrogel material mechanics via nanoparticle interface-controlled cross-link dynamics. Inspired by the adhesive chemistry in mussel threads, we have incorporated iron oxide nanoparticles (Fe3O4 NPs) into a catechol-modified polymer network to obtain hydrogels cross-linked via reversible metal-coordination bonds at Fe3O4 NP surfaces. Unique material mechanics result from the supra-molecular cross-link structure dynamics in the gels; in contrast to the previously reported fluid-like dynamics of transient catechol–Fe3+ cross-links, the catechol–Fe3O4 NP structures provide solid-like yet reversible hydrogel mechanics. The structurally controlled hierarchical mechanics presented here suggest how to develop hydrogels with remote-controlled self-healing dynamics.
Keywords: nanocomposite hydrogels, organic–inorganic interface, supra-molecular assembly, bio-inspired metal-coordinate polymers, polymer physics, rheology
Graphical Abstract
Interest in engineering the mechanics of hydrogels continues to grow at a rapid pace fueled in large part by their promising performance as soft tissue implants.1–3 Recent advances in hydrogel design have removed past material limitations dictated by traditionally low strength and high fragility (low toughness), which has opened the door for more widespread tissue-implant applications. For example, incorporation of inorganic nanoparticles (NPs) has been demonstrated to reinforce the hydrogel matrix, resulting in stronger and tougher nanocomposite gels.4,5 Through specific interactions with the hydrogel polymer chains, nanoparticles can effectively contribute to the polymer network elasticity and thereby increase the mechanical strength of the gels.6 Furthermore, if the polymer–particle interfacial cross-links are mechanically reversible (achieved for example via electrostatic interactions or hydrogen bonds), dynamic hydrogels with intrinsic self-healing capabilities can result.7,8 Finally, nanoparticles can introduce a variety of inorganic materials’ functionalities, such as electronic conductivity9 and magnetic response,10 which may lead to stimuli-responsive hydrogel applications in the future.11,12 Nevertheless, while successful incorporation of reversibly cross-linked nanoparticles has been achieved in a few select hydrogel systems, control over polymer–nanoparticle interaction dynamics is still limited in aqueous media.4,7,12,13 An expanded repertoire of controllable polymer–particle interfacial bond dynamics could help to broadly advance the functional engineering of nanocomposite hydrogel mechanics.
Metal–organic interfacial bonding inspired from marine biological materials such as the mussel holdfast may provide solutions to this problem. Mussels adhere their byssal threads onto a variety of solid surfaces under water, and investigations have revealed that the catecholic side-group “DOPA” plays a central role in this water-resistive adhesion14–16 by forming coordination bonds with transition metal ions in the bulk thread,17,18 and with metal atoms at the adhesive interface.19–21 Inspired by the mussel, catechol-containing adhesives have now been developed for multiple surfaces,22,23 and catechol-modified polymer side groups are extensively applied in nanomaterials surface engineering.24–26 In addition, since the metal coordination bonds are mechanically reversible, they have been demonstrated to function as dynamic cohesive cross-links in bulk self-healing of polymer materials.27–30
Here, we report a novel approach to assemble nano-composite hydrogels via mussel-inspired metal-coordination chemistry at the polymer–particle interface using iron oxide nanoparticles (Fe3O4 NP) and catechol–polymer as building blocks. As a result of the catechol–Fe(III) interfacial bond dynamics and the unique network structure, the assembled polymer–nanoparticle composite demonstrates strikingly different relaxation mechanics compared to conventional covalent-cross-linked gels or metallogels: the gel network is intrinsically dynamic, while its bulk shape can be maintained for extended periods of time. We further found that the bond kinetics dictating the relaxation mechanics can be finely tuned through design of the cross-link geometry at the polymer–nanoparticle interface. By demonstrating direct control over composite mechanics via polymer–particle metal–organic bond dynamics, this work presents a clear strategy for how to advance the design of nanocomposite hydrogels toward stimuli-responsive self-healing materials.
RESULTS AND DISCUSSION
Preparation and Structure of Self-Assembled NP Gels
Fe3O4 NPs and catechol–polymers are assembled into a composite hydrogel network in a simple two-step procedure (see Figure 1a,b). First, we predecorate Fe3O4 NPs with stabilizing hydrophilic ligands (linear monofunctionalized polyethylene glycol carboxylic acid, mPEG-COOH), and second, we mix the stabilized Fe3O4 NPs with 4-arm catechol-terminated polyethylene glycol (4cPEG) in unbuffered water (pH ≈ 4–5) at 50 °C. As discussed further below, if the NP concentration is 1 vol % or above, a solid Fe3O4 NP-cross-linked hydrogel (NP gel) will form due to –COOH ligand substitution with catechol (see Figure S1 for gelation kinetics of a 1.9 vol % NP gel). Unlike the previously reported slow-flowing viscoelastic fluid-like properties of Fe3+ metal-ion cross-linked networks,27 the metal–NP cross-linked networks are self-standing gels that maintain their shape at room temperature. Due to the magnetic properties of the Fe3O4 NPs, the NP–polymer gels respond to magnetic fields as they can be attracted by a magnet (see Figure 1b).
Figure 1.

(a) Structures of polymers 4cPEG and mPEG-COOH. (b) Schematic illustration of the preparation procedures of Fe3O4 nanoparticle-cross-linked hydrogel (NP gel), as well as representative photos of samples at each stage. From left to right: Fe3O4 NP dry powder, stabilized Fe3O4 NPs in aqueous dispersion before gel assembly, the self-standing solid hydrogel obtained after assembly with 4cPEG, and magnetic attraction of the resulting gel.
As shown in SEM and TEM images, NPs are evenly dispersed within the gel networks with an average interparticle distance (dp-p) of a few nanometers (see Figure 2a,b) in good agreement with the hydrodynamic diameter of 4cPEG (dpol) ~6 nm (see Figures S2 and S3 for size distribution of Fe3O4 NPs and 4cPEG, respectively). Long-range nanoscale assemblies are furthermore visible in the NP–4cPEG gels, whereas no long-range features are observed in covalently cross-linked 4cPEG without NPs (see Figure S4). To confirm the elastically active role of 4cPEG-NP cross-link structures in the gel network, we performed oscillatory rheology of Fe3O4 NP–4cPEG samples with various NP concentrations. The elastic modulus G′ and loss modulus G″ measured at 1% strain, 1 rad s−1 angular frequency, and room temperature are plotted as a function of NP volume fraction in Figure 2c. At low NP concentrations, the samples behave as fluids (G″ > G′), but at ~1.0 vol % NP (52 mg/mL), a viscoelastic gel-like material starts to form (G′ ≈ G″), and above 1.0 vol %, solid gels are obtained (G′ > G″). The G′ of the elastic polymer–particle network that form beyond this gel point increase by more than 3 orders of magnitude as the NP concentration increases from 1.0 to 1.5 vol % (see Figure 2c). Our data suggests this gel point is percolation controlled (see insets in Figure 2c). That is, in the fluid regime below 1 vol % NP, dp-p > dpol and 4cPEG chains cannot bridge adjacent particles. However, with increasing NP concentration, the interparticle distance reaches the percolation threshold at ~1 vol % NP where dp-p ≈ dpol and 4cPEG is able to connect NPs into a network. In the regime above 1 vol % NP, dp-p < dpol and solid gel networks are formed with a network stiffness (G′) controlled by the NP concentration demonstrating the elastically active particle–polymer interfacial coupling (see Figure S5).
Figure 2.

(a) Cross-sectional SEM (scale bar: 200 nm) of freeze-dried NP gel. (b) TEM image (scale bar: 50 nm) of ultramicro-cut of a NP gel embedded in resin. (c) G′ and G″ of the Fe3O4 NP–4cPEG solutions as a function of NP volume fraction (strain = 1%, angular frequency = 1 rad s−1, temperature = 20 °C). Insets: photos of Fe3O4 NP–4cPEG solutions and schematics of proposed material assembly at different NP concentrations. For gels in (a) and (b), the NP volume fraction = 1.9 vol %).
Coupling Polymer–NP Interfacial Chemistry to Gel Mechanics
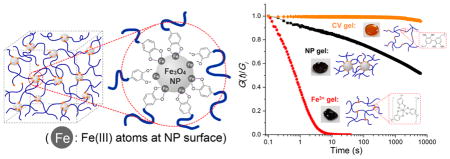
To investigate the coupling between bulk gel mechanics and coordinate cross-link dynamics at the NP–polymer interface, two additional hydrogels with the same 4cPEG backbone but different types of network cross-links were prepared for comparison following previously reported methods:27 tris-catechol-Fe3+ coordination cross-linking at pH ≈ 12 (Fe3+ gel) and intercatechol covalent cross-linking (CV gel) (see Figure 3a). 4cPEG concentrations were kept constant (100 mg/mL) in all prepared gel samples for comparison. Raman spectroscopy confirmed the presence of phenyl ring vibration peaks from ~1250 to ~1500 cm−1 in NP gel, Fe3+ gel, CV gel, and 4cPEG18,31 (see Figure 3b). However, vibrations indicative of catechol–Fe coordinate interactions (from ~550 to ~650 cm−1) were only present in the Fe3+ gel and NP gel spectra. As previously reported,27 while Raman shift positions of different coordination states (mono-, bis-, and tris- complex) are almost constant, the distinct signature peaks associated with strong bidentate Fe–catechol coordination is only observed in the high pH Fe3+ gel. In contrast, the NP gel displays a broad Fe–catechol vibrational spectrum indicative of weak coordination observed upon low pH induced catechol protonation. Previous studies have reported that high pH-induced catechol oxidation into a quinone can lead to intercatechol coupling and thereby covalent cross-linking.32,33 However, with a gel pH ≈ 4.1, oxidation-induced intercatechol covalent cross-linking is unlikely to occur in NP gels,34,35 which is additionally confirmed by UV–vis spectral analysis (see Figure S6). Additionally, a NP gel will disassemble and dissolve after we induce excess catechol oxidation, which supports a load-bearing role of catechol–NP coordinate interactions in the NP gel network (see Figure S7a). Similarly, NP gels can be completely dissolved in excess water upon mild heating or sonication, in support of fully reversible catechol–NP coordinate interfacial cross-links. In contrast, CV gels with permanent intercatechol cross-links cannot be redissolved even after heating in H2O for 5 days (Figure S7b,c). All of the above data support that dynamic polymer–particle interfacial catechol–Fe coordination bonds serve as the dominant cross-links of the 4cPEG–Fe3O4 NP gel networks.
Figure 3.

Correlating gel mechanics with different types of cross-link dynamics. (a) Schematic of 4cPEG gel network formed by metal-ion coordination (Fe3+ gel) and covalent (CV gel) cross-linking, and photo images of the respective gel samples. (b) Resonance Raman spectra of NP gel, Fe3+ gel, CV gel, and comparison with 4cPEG alone. (c) Frequency sweeps (strain = 1%) and (d) step strain (10%) relaxation curves of NP gel, Fe3+ gel, and CV gel at 20 °C. G(t) is normalized by the recorded initial relaxation modulus Gi. (e) Failure experiment switching between 1% and 1000% oscillatory shear strain in NP gels showing repeatable cycles of full G′ recovery (frequency = 1 rad s−1). (f) Schematic of strain energy dissipation in NP gel by reversible association–dissociation of catechol groups at NP surfaces.
To characterize the chemo-mechanical coupling between the 4cPEG–NP interface cross-link dynamics and composite gel mechanics, we studied the rheological behaviors of NP gels in comparison with Fe3+ gel and CV gel. Frequency sweeps (see Figure 3c) show that the elastic plateau moduli of the three different gels are of similar order of magnitude (104 Pa). However, as previously reported,27 the Fe3+ gel displays a clear cross over between G″ and G′ in accordance with its transient metal-ion coordinate cross-links, whereas the CV gel displays a dominant elastic solid behavior with G′ ≫ G″ over the entire frequency range in agreement with its permanent covalent network cross-links. In contrast, the NP gel displays a viscoelastic solid-like behavior with no cross over between G″ and G′ and a damping ratio G″/G′ between the CV and the Fe3+ gel (Figure S8). This energy-dissipative yet solid-like behavior of NP gel networks is in agreement with the individually transient yet combined stable cross-link behavior of the polymer–particle interface (to be discussed in greater detail below). As a result, NP gels both hold their shape over extended periods of time (days) and rapidly recover elastic modulus after repeated large-amplitude oscillatory shear-induced failure (see Figure 3e and Figure S9). We stress this important difference in properties between the NP gels reported here and Fe3+ and CV gels reported previously;27 NP gels are solid-like (similar to CV gels), yet mechanically reversible at room temperature (similar to the fluid-like Fe3+ gels). These significant differences in bulk material mechanics are a direct result of the distinct stress-relaxation dynamics dictated by the different catechol cross-link structures in the three gels, which will be further discussed below.
Network Relaxation Controlled by Cross-Link Structure
To further investigate the coupling between gel mechanics and catechol cross-link bond dynamics, we performed step-strain relaxation experiments, where a step strain (γ = 10%) is applied to the gels and the relaxation modulus G(t) = σ(t)/γ is monitored with time (σ, stress; t, time). The results offer quantitative insights on how the dynamics of the three types of catechol cross-links dictate dramatically different network mechanics. CV gel networks are cross-linked by permanent covalent intercatechol bonds that cannot rearrange to dissipate strain energy and hence the CV gel does not relax (see Figure 3d). The NP and Fe3+ gels both relax the induced stress since their reversible coordination cross-links can dissociate and reconnect into a new network, whereby stress is relaxed (see Figure 3f). Yet, despite the fundamental similarities in the underlying coordinate cross-link bond dynamics, the bulk stress relaxation mechanics operate over significantly different time periods in NP and Fe3+ gels (see Figure 3d). To better understand how fundamentally similar bond dynamics can result in such different bulk mechanics, we investigated the characteristic relaxation time and activation energy of the network cross-linking dissociation process in NP and Fe3+ gels. Many viscoelastic polymer materials obey Kohlrausch’s stretched-exponential relaxation model:
where G0 is the plateau modulus, τ is the characteristic relaxation time, and a is a fitting parameter dictated by the physical constraints of the individual material system.36,37 If the cross-link dissociation mechanism is a thermally activated process, its associated activation energy barrier Ea can be estimated since the relaxation time τ will follow Arrhenius’s behavior:36
Step-strain relaxation curves from NP and Fe3+ gels at different temperatures are well fitted by the above stretched-exponential model (see Figure 4) wherefrom the activation energies of the catechol–Fe3O4 NP and tris-catechol-Fe3+ cross-link dissociation mechanism can be estimated to be Ea,NP ~ 58 kJ mol−1 and Ea,ion ~ 86 kJ mol−1, respectively (see Figure 4b,d). The significantly lower pH in NP gels (pH ≈ 4 vs pH ≈ 12 in Fe3+ gels) is likely the main reason for this difference in activation energy barrier, since, as supported by our Raman data described above, the protonation on catechol oxygen atoms weakens the Fe–catechol coordination interaction which substantially lowers the activation barrier to dissociation.38
Figure 4.

Step strain relaxation plots of NP gel (a) and Fe3+ gel (c) at different temperatures with stretched-exponential model fits (solid black lines), and Arrhenius plot of stress relaxation time (ln τ) versus inverse temperature (1/T) of NP gel (b) and Fe3+ gel (d). Thermal activation energy is calculated from the slope.
With the energy barrier for cross-link dissociation in NP gels lower than in Fe3+ gels (i.e., Ea,NP < Ea,ion), this cannot account for the orders of magnitude slower relaxation of NP gels shown in Figure 3d. We propose instead that the different gel relaxation mechanics are a result of different hierarchically controlled cross-link structure dynamics. In Fe3+ gels, the stress relaxation mechanism of each cross-link structure (an octahedral tris-catechol-Fe3+ coordinate complex, see Figure 3a) involves only 3 elastically active chains. In contrast, on the basis of calculations from the NP’s average surface area (~280 nm2) and the reported PEG chain density (~2 chains/nm2) in a similar system,24 we estimate an upper bound of ~102 chains constitute each cross-linking structure in NP gels (see Figure 1b), whereby stress relaxation requires simultaneous dissociation of multiple chains in a slower and more complicated process (see Figure 3f). To test this hypothesis, we designed two separate experiments to reduce the number of elastically active polymer chains on each NP: (1) adding free catechol-terminated ligands to compete with 4cPEG; (2) decreasing the NP size (see Figure 5a). Specifically, in experiment 1 different amounts of free mPEG-dopa ligand were added during NP gel preparation, and in experiment 2, NP gels with smaller Fe3O4 NPs (d = 6.6 ± 1.2 nm) were prepared (Figure S10). As predicted, the NP gels relax faster with both increasing amount of mPEG-dopa and smaller NPs (Figure 5b,c) and the comparison of relaxation times calculated from stretched-exponential fitting demonstrates a decrease in relaxation time by orders of magnitude (see Figure 5d). These results strongly support that the stress-relaxation mechanism of NP gels is indeed controlled by the number of actively stress-bearing polymer chains bound to each NP. With this knowledge in hand, we will be able to better control bulk mechanics of NP gels by direct engineering of polymer–particle interfacial dynamics at the nanoscale.
Figure 5.

(a) Schematic of two strategies for reducing the number of elastically active polymer chains cross-linked at each NP in order to accelerate gel network relaxation; decreasing NP size or introducing ligand competition at the polymer–particle interface. Step strain relaxation plots and fitted relaxation curves (solid lines) by stretched-exponential model of NP gels with (b) increasing competing ligand mPEG-dopa concentration (inset: molecular formula of mPEG-dopa) and (c) decreasing NP diameter d are shown. (d) Comparison of relaxation times of original NP gel (NP diameter = 9.4 nm, black column), NP gel with smaller NPs (diameter = 6.6 nm, red column), and NP gels with different concentrations of mPEG-dopa ligands (blue columns).
CONCLUSIONS
By mimicking the interfacial adhesion chemistry of mussel threads, we have developed a facile approach to assemble Fe3O4 nanoparticles into hydrogel networks by cross-linking polymer chains at the NP surfaces via catechol–Fe coordination bonds. The resulting hydrogel mechanics are directly coupled to the reversible coordinate cross-link dynamics at the polymer–nanoparticle interface in the gel network. This work highlights the relationship between macroscale mechanics of nano-composite materials and nanoscale dynamics at the polymer–particle interface, which helps shed light on how dynamic mechanics in polymer–nanoparticle systems can be controlled. In addition, this work demonstrates how to easily incorporate functional (e.g., magnetic) nanoparticles as cross-link structures in hydrogel networks, which lays the foundation for future design of stimuli-responsive smart hydrogels with remote-controlled dynamic mechanical properties.
MATERIALS AND METHODS
Materials
A 4-arm PEG (MW = 10 kDa) was purchased from JenKem Technology USA, Inc. (Allen, TX). mPEG-COOH (MW = 2.6 kDa) was purchased from Laysan Bio, Inc. Sodium periodate (NaIO4), sodium sulfate (Na2SO4), sodium acetate buffer solution (NaAc/HAc buffer, 3 M, pH = 5.2 ± 0.1), iron(III) acetylacetonate (Fe(acac)3), iron(II) acetate (Fe(ac)2), hydrochloric acid (HCl), dopamine hydrochloride, N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU), 1-hydroxybenzotriazole hydrate (HOBt), triethylamine (TEA), methanol (MeOH), ethanol (MeOH), dichloromethane (DCM), N,N-dimethylformamide (DMF), diethyl ether (Et2O), and chloroform (CHCl3) were purchased from Sigma-Aldrich. 4-Nitrophenyl chloroformate (4NPC) was purchased from Fisher Scientific. All chemicals were used without further purification.
Synthesis of Fe3O4 NP
Bare Fe3O4 NPs was synthesized following previously reported methods with minor modifications.39 Briefly, 687 mg of Fe(acac)3 was dissolved in 9 mL of benzyl alcohol. The mixture was heated to 170 °C with reflux and stirring at 700 rpm for 24 h. The reaction mixture was cooled down and mixed with 35 mL of EtOH, then centrifuged at 10 000 rpm for 10 min. The supernatant was discarded, and the precipitate was washed with 40 mL of EtOH for twice. The NP precipitate was redispersed in 80 mL of 1:1 (v/v) solution of CHCl3 and DMF, and 100 mg of mPEG-COOH was added. The mixture was homogenized and equilibrated by pulsed sonication (pulse, 10 s on + 4 s off; power, 125 W) for 1 h. Then, the mixture was centrifuged at 10 000 rpm for 10 min to remove any aggregates, and rotary evaporated at 50 °C, 30 mbar for >8 h to remove CHCl3. After CHCl3 was confirmed to be completely removed, the NP dispersion in DMF was dialyzed with water (MWCO = 3500 Da) for 1 day (water exchanged for >3 times) to exchange solvent. Finally, the NP aqueous solution was lyophilized, and hydrophilic Fe3O4 NPs decorated by mPEG-COOH were stored as powder. From thermal gravimetric analysis (TGA) results, the Fe3O4 weight percent in the NP powder was determined to be 45–60 wt % (Figure S11).
Synthesis of 4cPEG
4cPEG is synthesized following previously reported procedures.40 Briefly, 10 g of 4-arm PEG was added to a two-neck round-bottom flask, which was purged with Ar. Anhydrous DCM (80 mL) was added to dissolve the PEG. TEA (2.5× mol equiv relative to –OH) was added, and the solution was stirred for 15 min. 4NPC (2.5× mol equiv relative to –OH) was dissolved separately in anhydrous DCM (20 mL) and slowly added to the 4-arm PEG solution. The reaction was allowed to proceed for ~18 h at room temperature in an inert environment of Ar. The volume was reduced by rotary evaporation, and the activated 4-arm PEG-NPC was collected by precipitation in cold Et2O and −20 °C MeOH (2×). The product was then dried under vacuum overnight.
Purified 4-arm PEG-NPC was combined with 90 mL of a 2:1 (v/v) solution of DMF and DCM. Once dissolved, dopamine hydrochloride (2.5× mol equiv relative to –NPC) was added. When the solution was homogeneous, the reaction was activated by the addition of TEA (2.5× mol equiv relative to –NPC) and stirred for ~18 h. The volume was reduced by rotary evaporation, and the PEO was collected by precipitation in acidified cold ether and acidified MeOH at −20 °C. The product was then dried under vacuum overnight and dissolved in ~100 mL of 12.1 mM HCl. The solution was filtered, dialyzed (3500 MWCO) against water at pH 4 for 24 h, dialyzed against Milli-Q water for 4 h, frozen at −80 °C, and lyophilized. The final purified 4cPEG was obtained as a white solid and stored under Ar at −20 °C until needed. 1H NMR (D2O) δ (ppm): 6.6–6.8 (m, 3H, aromatic), 3.4–4.1 (m, –O–CH2–CH2–), 3.3 (t, 2H, CH2 adjacent to aromatic ring), 2.7 (t, 2H, –CH2–NH–CO–). The conjugation efficiency is 94% calculated from the absorbance at 280 nm (Figure S12b).
Synthesis of mPEG-Dopa
A total of 500 mg (0.19 mmol) of mPEG-COOH was dissolved in 6 mL of a 2:1 (v/v) solution of DCM and DMF. Then, 72 mg (0.38 mmol) of dopamine hydrochloride and 90 mg (0.67 mmol) of HOBt were added, and the mixture was bubbled with N2 for 15 min. After that, 153 mg (0.38 mmol) of HBTU and 100 μL (0.66 mmol) of TEA were added, and the mixture was stirred in N2 atmosphere at room temperature for 2 h. The solution volume was reduced by rotary evaporation; then, the solution was acidified by mixing with 10 mL of 1 M HCl. The aqueous solution was extracted with 10 mL of CHCl3 thrice; then, the organic phase was collected and dried with Na2SO4. After the solution was concentrated to <2 mL by rotary evaporation, it was precipitated in 30 mL of cold ether at −20 °C. The crude product (white precipitate) was purified by redissolving in ~2 mL of DCM and precipitation in 30 mL of cold ether at −20 °C for two more times. Then, the precipitate was redissolved in H2O and lyophilized. The final purified mPEG-dopa was obtained as a white solid powder and stored at −20 °C until needed. 1H NMR (D2O) δ (ppm): 6.7–6.8 (m, 3H, aromatic), 3.3–4.0 (m, –O–CH2–CH2–), 3.4 (t, 2H, CH2 adjacent to aromatic ring), 2.7 (t, 2H, –CH2–NH–CO–). The conjugation efficiency is 99% calculated from the absorbance at 280 nm (Figure S12b).
Preparation of Gel Samples
NP Gel
For each gel sample, Fe3O4 NPs and 20 mg of 4cPEG were dissolved in 200 μL of H2O. The Fe3O4 concentration in the mixture can be determined by controlling the amount of Fe3O4 NP powders added (note: concentration is calculated in Fe3O4 weight; actual NP powder weight added was calculated based on Fe3O4 concentration and Fe3O4 weight ratio in NP powders determined by TGA). Unless specified, Fe3O4 concentration in gel samples is 100 mg/mL. The solution mixture was transferred into a mold and sealed, and a solid gel was obtained after curing in a 50 °C oven for 24 h. The pH of gels was measured using a pH-meter with a flat-surface electrode designed for solids, semisolids and liquids.
Fe3+ Gel
Preparation procedure is similar to reported protocol.27 For each sample, 100 μL of 200 mg/mL 4cPEG aqueous solution was mixed with 33 μL of 80 mM FeCl3 solution (catechol/Fe3+ molar ratio of 3:1), resulting in a dark green solution. Then, it was mixed with 67 μL of 1 M NaOH solution and a dark red gel formed.
CV Gel
Preparation procedure is similar to reported protocol.27 For each sample, 100 μL of 200 mg/mL 4cPEG aqueous solution was mixed with 100 μL of 40 mM NaIO4 solution (catechol/IO4− molar ratio of 2:1). The solution mixture was transferred into a mold and sealed. An orange, elastic solid gel was obtained after curing at room temperature overnight.
Resonance Raman Spectroscopy
For Raman spectroscopic studies, a continuous laser beam was focused on the samples through a HoloLab Series 5000 Raman microscope (Kaiser Optical Systems, Inc.). A 785 nm near-infrared laser excitation (Invictus, Kaiser Optical Systems, Inc.) was used in combination with a 50× microscope objective. Samples were loaded on a glass substrate coated with gold. A laser power of 20 mW combined with a short integration time of 0.5 s was used for each measurement to prevent the sample from burning by the laser beam. Each collected spectra consisted of 120 accumulations of a 0.5 s integration time. The obtained spectra were smoothed with a Savitzky-Golay smoothing filter, and the baseline was flattened.
Transmission Electron Microscopy (TEM)
For TEM and HRTEM analysis of Fe3O4 nanoparticles, a JEOL 2010 TEM operating at 200 kV was used. The sample was prepared from drying aqueous solution of mPEG-COOH stabilized Fe3O4 NPs on Cu grids. The size distribution of NPs was counted by Pebbles v2.0 software.41
For TEM imaging of NP hydrogels, a FEI Technai Spirit TEM operating at 80 kV was used. The gel samples were first dehydrated by ethanol and propylene oxide, and then embedded in Embed 812 resin (Electron Microscopy Sciences). The samples were cut with a Reichert Ultracut E ultramicrotome.
Scanning Electron Microscopy (SEM)
For SEM analysis, a Zeiss Merlin operating in high vacuum was used. An acceleration voltage of kV was chosen for recording the images. The gel samples were first freeze-dried, cut for cross-section by a razor, and then coated with a thin layer of gold in order to avoid charging effects.
Thermal Gravimetric Analysis (TGA)
Freeze-dried stabilized Fe3O4 NP powder was analyzed by a TGA Q500 (TA Instruments). Samples were equilibrated at 35 °C for 30 min, and then heated from 35 to 600 °C at 10 °C/min in air atmosphere with a flow rate of 60.0 mL/min. The mass profile with increasing temperature was recorded.
Nuclear Magnetic Resonance (NMR)
The solvents for preparing 4cPEG and mPEG-dopa samples were deuterium oxide (D2O, 99.9% atom %, Sigma-Aldrich). The spectra were obtained on a Varian Mercury 300 MHz NMR spectrometer.
Dynamic Light Scattering (DLS)
DLS measurements were performed on a DynaPro NanoStar Light Scatterer. The sample was prepared by filtering 0.50 mg/mL 4cPEG aqueous solution through a 0.2 μm cutoff syringe filter prior to measurements. To check reproducibility, each measurement was repeated three times on the same sample, and 10 acquisitions were collected for each measurement.
UV–Vis Spectroscopy
UV–vis absorption spectra ranging from 800 to 220 nm were collected on a Cary 500i UV–vis-NIR Dual-Beam Spectrophotometer. The samples were measured in a quartz cuvette with a path length of 1 cm, and the background absorption was subtracted before measurement.
To quantitatively determine the amount and conjugation efficiency of catecholic groups in 4cPEG and mPEG-dopa samples, all samples were dissolved in 0.1 M NaAc/HAc buffer (pH = 5.2) to eliminate the influence of pH. Spectra of dopamine samples with different concentrations ranging from 0.10 to 0.51 mmol L−1 were collected, and a standard curve was obtained by linear fitting from their absorbance at 280 nm. The amounts of catecholic groups in polymers can then be calculated by applying the absorbance at 280 nm into the standard curve (see Figure S12).
To study the oxidation of 4cPEG after treating with Fe3O4 NPs, excess amount of 4cPEG (50 mg) was added into 3 mL of 3.3 mg/mL Fe3O4 NP aqueous dispersion, and the mixture was stirred at 50 °C for 24 h (same as gelation condition). Then, the solution was filtered through a centrifugal filter with a cutoff size of 100 kDa so only the free polymers penetrate through the filter. After filtration, the colorless solution at the bottom was diluted by 5 times and UV–vis spectra were collected on this sample. For comparison, samples of a pristine 4cPEG solution as well as an oxidized 4cPEG solution (by mixing 4cPEG and NaIO4 in 1:1 mol ratio) were also prepared for UV–vis measurement (see Figure S2).
Rheology Measurements
The dynamic mechanical properties of the hydrogels were tested on an Anton Paar rheometer with parallel plate geometry (PP-10 probe, 10 mm diameter, flat). All tests except for gelation test were done immediately after transferring the gel sample onto the sample stage. A Peltier hood with a water cell was used for all experiments to control measuring temperature and prevent sample dehydration. Unless specified, all experiments were conducted at 20 °C. Oscillatory frequency sweep testing of gels was performed at constant 1% strain and angular frequency ranging from 100 to 0.1 rad s−1 on a logarithmic scale while monitoring the storage modulus (G′) and loss modulus (G″). Relaxation tests were performed by applying a 10% step strain γ, and then the stress σ and relaxation modulus G(t) = σ/γ were monitored over time. Each experiment was performed on 3 parallel samples to ensure repeatability and determine the deviation in relaxation time. Healing tests were performed by straining the gel sample from γ = 0.1% to γ = 1000% (nonlinear regime which induces gel network failure) at oscillatory angular frequency ω = 1 rad s−1, then immediately followed by linear condition (γ = 1%, ω = 1 rad s−1) while monitoring the recovery of the storage modulus (G′). Gelation tests were performed by placing a drop (50 μL) of Fe3O4 NP + 4cPEG solution before assembly between the stage and the probe plate. Then, G′ and G″ were monitored over time by oscillatory measurements (γ = 1%, ω = 1 rad s−1) at 50 °C, and the sample was sealed by a layer of mineral oil to prevent dehydration in long time tests.
Supplementary Material
Acknowledgments
Q. Li and N. Holten-Andersen acknowledge support from MIT Research Committee and MIT Sea Grant via the Doherty Professorship in Ocean Utilization. D. G. Barrett and P. B. Messersmith were supported by the National Institutes of Health under award number R37DE014193. This work made use of the Shared Experimental Facilities supported in part by the MRSEC Program of the National Science Foundation under award number DMR – 1419807. We thank E. Amstad and M. J. Harrington for helpful feedback, and J. P. McGann and J. C. Grossman for assistance in thermal gravimetric analysis.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.5b06692.
Structural characterizations (TEM images of Fe3O4 NPs, SEM of NP gel and CV gel, and DLS size distribution of 4cPEG), supplementary mechanical characterizations (NP gel’s gelation curve, frequency sweeps with increasing NP concentrations, comparison of tan δ curves and healing curves), and characterization of materials’ chemical compositions (determination of 4cPEG’s oxidation by UV–vis spectra, Fe3O4NP’s thermal gravimetric analysis, and measurement of catechol coupling efficiency on PEG by UV–vis) (PDF)
References
- 1.Kopecek J. Hydrogels: From Soft Contact Lenses and Implants to Self-Assembled Nanomaterials. J Polym Sci, Part A: Polym Chem. 2009;47:5929–5946. doi: 10.1002/pola.23607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glassman MJ, Chan J, Olsen BD. Reinforcement of Shear Thinning Protein Hydrogels by Responsive Block Copolymer Self-Assembly. Adv Funct Mater. 2013;23:1182–1193. doi: 10.1002/adfm.201202034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang C, Javadi A, Ghaffari M, Gong S. A pH-Sensitive Molecularly Imprinted Nanospheres/Hydrogel Composite as a Coating for Implantable Biosensors. Biomaterials. 2010;31:4944–4951. doi: 10.1016/j.biomaterials.2010.02.073. [DOI] [PubMed] [Google Scholar]
- 4.Haraguchi K, Takehisa T. Nanocomposite Hydrogels: A Unique Organic-Inorganic Network Structure with Extraordinary Mechanical, Optical, and Swelling/De-swelling Properties. Adv Mater. 2002;14:1120–1124. [Google Scholar]
- 5.Schexnailder P, Schmidt G. Nanocomposite Polymer Hydrogels. Colloid Polym Sci. 2009;287:1–11. [Google Scholar]
- 6.Haraguchi K, Li H, Matsuda K, Takehisa T, Elliott E. Mechanism of Forming Organic/Inorganic Network Structures during In-situ Free-Radical Polymerization in PNIPA-Clay Nanocomposite Hydrogels. Macromolecules. 2005;38:3482–3490. [Google Scholar]
- 7.Wang Q, Mynar JL, Yoshida M, Lee E, Lee M, Okuro K, Kinbara K, Aida T. High-Water-Content Mouldable Hydrogels by Mixing Clay and a Dendritic Molecular Binder. Nature. 2010;463:339–343. doi: 10.1038/nature08693. [DOI] [PubMed] [Google Scholar]
- 8.Haraguchi K, Uyama K, Tanimoto H. Self-healing in Nanocomposite Hydrogels. Macromol Rapid Commun. 2011;32:1253–1258. doi: 10.1002/marc.201100248. [DOI] [PubMed] [Google Scholar]
- 9.Pardo-Yissar V, Gabai R, Shipway AN, Bourenko T, Willner I. Gold Nanoparticle/Hydrogel Composites with Solvent-Switchable Electronic Properties. Adv Mater. 2001;13:1320–1323. [Google Scholar]
- 10.Zhang Y, Yang B, Zhang X, Xu L, Tao L, Li S, Wei Y. A Magnetic Self-healing Hydrogel. Chem Commun. 2012;48:9305–9307. doi: 10.1039/c2cc34745h. [DOI] [PubMed] [Google Scholar]
- 11.Balazs AC, Emrick T, Russell TP. Nanoparticle Polymer Composites: Where Two Small Worlds Meet. Science. 2006;314:1107–1110. doi: 10.1126/science.1130557. [DOI] [PubMed] [Google Scholar]
- 12.Wu CJ, Gaharwar AK, Schexnailder PJ, Schmidt G. Development of Biomedical Polymer-Silicate Nanocomposites: A Materials Science Perspective. Materials. 2010;3:2986–3005. [Google Scholar]
- 13.Dawson JI, Oreffo RO. Clay: New Opportunities for Tissue Regeneration and Biomaterial Design. Adv Mater. 2013;25:4069–4086. doi: 10.1002/adma.201301034. [DOI] [PubMed] [Google Scholar]
- 14.Waite JH, Tanzer ML. Polyphenolic Substance of Mytilus edulis: Novel Adhesive Containing L-dopa and Hydroxyproline. Science. 1981;212:1038–1040. doi: 10.1126/science.212.4498.1038. [DOI] [PubMed] [Google Scholar]
- 15.Papov VV, Diamond TV, Biemann K, Waite JH. Hydroxyarginine-Containing Polyphenolic Proteins in the Adhesive Plaques of the Marine Mussel Mytilus edulis. J Biol Chem. 1995;270:20183–20192. doi: 10.1074/jbc.270.34.20183. [DOI] [PubMed] [Google Scholar]
- 16.Waite JH, Qin X. Polyphosphoprotein from the Adhesive Pads of Mytilus edulis. Biochemistry. 2001;40:2887–2893. doi: 10.1021/bi002718x. [DOI] [PubMed] [Google Scholar]
- 17.Holten-Andersen N, Jaishankar A, Harrington MJ, Fullenkamp DE, DiMarco G, He L, McKinley GH, Messersmith PB, Lee KYC. Metal-Coordination: Using One of Nature’s Tricks to Control Soft Material Mechanics. J Mater Chem B. 2014;2:2467–2472. doi: 10.1039/C3TB21374A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harrington MJ, Masic A, Holten-Andersen N, Waite JH, Fratzl P. Iron-Clad Fibers: A Metal-Based Biological Strategy for Hard Flexible Coatings. Science. 2010;328:216–220. doi: 10.1126/science.1181044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee H, Scherer NF, Messersmith PB. Single-Molecule Mechanics of Mussel Adhesion. Proc Natl Acad Sci U S A. 2006;103:12999–13003. doi: 10.1073/pnas.0605552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang DS, Harrington MJ, Lu Q, Masic A, Zeng H, Waite JH. Mussel Foot Protein-1 (mcfp-1) Interaction with Titania Surfaces. J Mater Chem. 2012;22:15530–15533. doi: 10.1039/C2JM32439C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu J, Wei W, Menyo MS, Masic A, Waite JH, Israelachvili JN. Adhesion of Mussel Foot Protein-3 to TiO2 Surfaces: The Effect of pH. Biomacromolecules. 2013;14:1072–1077. doi: 10.1021/bm301908y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee H, Lee BP, Messersmith PB. A Reversible Wet/Dry Adhesive Inspired by Mussels and Geckos. Nature. 2007;448:338–341. doi: 10.1038/nature05968. [DOI] [PubMed] [Google Scholar]
- 23.Stepuk A, Halter JG, Schaetz A, Grass RN, Stark WJ. Mussel-Inspired Load Bearing Metal-Polymer Glues. Chem Commun. 2012;48:6238–6240. doi: 10.1039/c2cc31996a. [DOI] [PubMed] [Google Scholar]
- 24.Amstad E, Gillich T, Bilecka I, Textor M, Reimhult E. Ultrastable Iron Oxide Nanoparticle Colloidal Suspensions Using Dispersants with Catechol-Derived Anchor Groups. Nano Lett. 2009;9:4042–4048. doi: 10.1021/nl902212q. [DOI] [PubMed] [Google Scholar]
- 25.Na HB, Palui G, Rosenberg JT, Ji X, Grant SC, Mattoussi H. Multidentate Catechol-Based Polyethylene Glycol Oligomers Provide Enhanced Stability and Biocompatibility to Iron Oxide Nanoparticles. ACS Nano. 2012;6:389–399. doi: 10.1021/nn203735b. [DOI] [PubMed] [Google Scholar]
- 26.Studart AR, Amstad E, Gauckler LJ. Colloidal Stabilization of Nanoparticles in Concentrated Suspensions. Langmuir. 2007;23:1081–1090. doi: 10.1021/la062042s. [DOI] [PubMed] [Google Scholar]
- 27.Holten-Andersen N, Harrington MJ, Birkedal H, Lee BP, Messersmith PB, Lee KY, Waite JH. pH-Induced Metal-Ligand Cross-Links Inspired by Mussel Yield Self-Healing Polymer Networks with Near-Covalent Elastic Moduli. Proc Natl Acad Sci U S A. 2011;108:2651–2655. doi: 10.1073/pnas.1015862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mozhdehi D, Ayala S, Cromwell OR, Guan Z. Self-Healing Multiphase Polymers via Dynamic Metal–Ligand Interactions. J Am Chem Soc. 2014;136:16128–16131. doi: 10.1021/ja5097094. [DOI] [PubMed] [Google Scholar]
- 29.Krogsgaard M, Behrens MA, Pedersen JS, Birkedal H. Self-Healing Mussel-Inspired Multi-pH-Responsive Hydrogels. Bio-macromolecules. 2013;14:297–301. doi: 10.1021/bm301844u. [DOI] [PubMed] [Google Scholar]
- 30.Basak S, Nanda J, Banerjee A. Multi-Stimuli Responsive Self-Healing Metallo-Hydrogels: Tuning of the Gel Recovery Property. Chem Commun. 2014;50:2356–2359. doi: 10.1039/c3cc48896a. [DOI] [PubMed] [Google Scholar]
- 31.Zvarec O, Purushotham S, Masic A, Ramanujan RV, Miserez A. Catechol-Functionalized Chitosan/Iron Oxide Nano-particle Composite Inspired by Mussel Thread Coating and Squid Beak Interfacial Chemistry. Langmuir. 2013;29:10899–10906. doi: 10.1021/la401858s. [DOI] [PubMed] [Google Scholar]
- 32.Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel-Inspired Surface Chemistry for Multifunctional Coatings. Science. 2007;318:426–430. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee BP, Dalsin JL, Messersmith PB. Synthesis and Gelation of DOPA-Modified Poly (Ethylene Glycol) Hydrogels. Biomacromolecules. 2002;3:1038–1047. doi: 10.1021/bm025546n. [DOI] [PubMed] [Google Scholar]
- 34.Herlinger E, Jameson RF, Linert W. Spontaneous Autoxidation of Dopamine. J Chem Soc, Perkin Trans 2. 1995:259–263. [Google Scholar]
- 35.Menyo MS, Hawker CJ, Waite JH. Versatile Tuning of Supramolecular Hydrogels through Metal Complexation of Oxidation-Resistant Catechol-Inspired Ligands. Soft Matter. 2013;9:10314–10323. doi: 10.1039/C3SM51824H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cathébras N, Collet A, Viguier M, Berret JF. Synthesis and Linear Viscoelasticity of Fluorinated Hydrophobically Modified Ethoxylated Urethanes (F-HEUR) Macromolecules. 1998;31:1305–1311. [Google Scholar]
- 37.Palmer RG, Stein DL, Abrahams E, Anderson PW. Models of Hierarchically Constrained Dynamics for Glassy Relaxation. Phys Rev Lett. 1984;53:958–961. [Google Scholar]
- 38.Li SC, Chu LN, Gong XQ, Diebold U. Hydrogen Bonding Controls the Dynamics of Catechol Adsorbed on a TiO2 (110) Science. 2010;328:882–884. doi: 10.1126/science.1188328. [DOI] [PubMed] [Google Scholar]
- 39.Pinna N, Grancharov S, Beato P, Bonville P, Antonietti M, Niederberger M. Magnetite Nanocrystals: Nonaqueous Synthesis, Characterization, and Solubility. Chem Mater. 2005;17:3044–3049. [Google Scholar]
- 40.Barrett DG, Bushnell GG, Messersmith PB. Mechanically Robust, Negative-Swelling, Mussel-Inspired Tissue Adhesives. Adv Healthcare Mater. 2013;2:745–755. doi: 10.1002/adhm.201200316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mondini S, Ferretti A, Puglisi A, Ponti A. PEBBLES and PEBBLEJUGGLER: Software for Accurate, Unbiased, and Fast Measurement and Analysis of Nanoparticle Morphology from Transmission Electron Microscopy (TEM) Micrographs. Nanoscale. 2012;4:5356–5372. doi: 10.1039/c2nr31276j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.