

Abstract
We present a platform strategy that offers diverse flexibility in tailoring the structure and properties of core–shell plasmonic nanoparticles with built-in nanogaps. Our results have demonstrated that polydopamine serves multiple concerted functions as a nanoscale spacer to afford controllable nanogap sizes, a redox-active coating to promote metal shell growth, and a reactive scaffold to exclusively lock molecular probes inside the nanogap for surface-enhanced Raman scattering (SERS). More interestingly, the universal adhesion of polydopamine on diverse colloidal substrates allows for customized synthesis of multishell plasmonic nanogapped nanoparticles (NNPs) and multifunctional hybrid NNPs containing different cores (i.e., magnetic nanoparticles), which are not readily accessible by conventional methods. Internally coupled plasmonic NNPs with broadly tunable spectroscopic properties, highly active SERS, and multifunctionality hold great promise for emerging fields, such as sensing, optoelectronics, and theranostics, as demonstrated by the ultrasensitive SERS detection and efficient photothermal killing of food-borne pathogens here.
Keywords: nanogap, plasmonic nanostructures, polydopamine, surface-enhanced spectroscopy, multifunctionality
Graphical Abstract
The optical properties of plasmonic nanomaterials, originating from localized surface plasmon resonance (LSPR), are of tremendous potential across many disciplines spanning chemistry, materials science, photonics, and medicine.1–5 The development of plasmonic nanostructures with precisely controlled spectroscopic properties and/or multifunctional characteristics is key to their use in diverse applications. In particular, tailored LSPR of plasmonic nanostructures allows for spatially confining photons at subwavelength scales and controlling light–molecule interactions at specific wavelengths, forming the fundamental basis of their functions in surface-enhanced spectroscopy and optoelectronics.6–9 The promise of multifunctional plasmonic nanoparticles, in which the structural integration of plasmonic materials and complementary counterparts gives rise to synergistic properties, is evident from recent progress in emerging fields such as sensing,10 theranostic nanomedicine,11,12 and plasmon-enhanced photochemical reactions.13
The strong dependence of the LSPR wavelength on interparticle coupling of plasmonic nanostructures has stimulated widespread interest in nanoparticle assemblies with defined nanogaps between the building blocks.14–16 Core–shell nanogapped nanoparticles (NNPs) with a built-in dielectric gap separating core and shell have emerged as a class of internally coupled plasmonic nanostructures.17 The nanogap size plays a key role in tailoring the plasmonic coupling of core and shell toward broadly tunable LSPR across the visible and near-infrared (NIR) spectral range.18 Additionally, the nanogap can act as an electromagnetic hot-spot, giving rise to enormously amplified optical signals for surface-enhanced Raman scattering (SERS) when molecular probes are positioned inside the nanogap.19–21 Considerable efforts have been made in engineering the nanogap in terms of both nanogap sizes and optical encoding, using silica, DNA, polymer, and small molecules as dielectric spacers.22–26 However, it remains challenging for a single strategy to simultaneously achieve tailored nanogap engineering and structural integration toward multifunctional NNPs.
We have recently developed SERS-encoded plasmonic NNPs via nanoparticle-templated self-assembly of amphiphilic block copolymers consisting of a Raman dye-tagged hydrophobic block and a redox-active hydrophilic block, in which the hydrophobic and hydrophilic blocks define the nanogap and metallic shell, respectively.24 Nevertheless, it is necessary to customize well-defined block copolymers of functional monomers by living polymerization techniques for controlled nanoparticle encapsulation and NNP synthesis, which creates a barrier to make the NNPs available for broader communities.
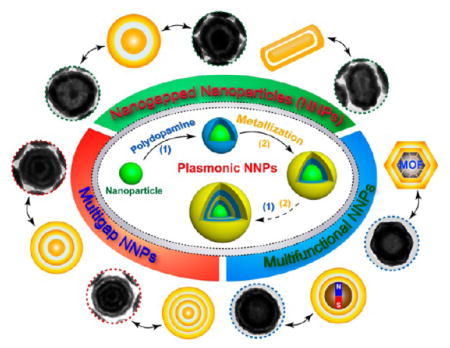
Here we present a broadly applicable platform strategy based on the use of mussel-inspired polydopamine (PDA) to realize customized control over the structure and functionality of the NNPs. As illustrated in Figure 1, PDA serves multiple concerted functions supported by an array of physicochemical properties. First, PDA deposits from aqueous solution onto virtually any solid substrate, forming a rigid conformal coating with precisely controlled thickness in the nanometer scale,27 as a result of self-polymerization of dopamine. Second, the high density of catechol groups imparts reducing activity to PDA, facilitating in situ nucleation and deposition of a metallic layer. Third, the spontaneous Michael addition and/or Schiff base reactions of quinone groups in PDA with nucleophilic thiol and amino groups make it possible to encode the nanogaps with molecular probes for SERS. Importantly, the universal adhesion of PDA enables conveniently building up multiple concentric metallic shells (Figure 1) on diverse inorganic, organic, and hybrid functional cores of different sizes, shapes, and chemical composition, such as spherical Au nanoparticles, anisotropic Au nanorods (AuNRs), metal–organic frameworks (MOFs), and magnetic polymer nanoparticles (MagNPs). In a proof-of-concept study, we have demonstrated that bioconjugated, SERS-encoded magnetoplasmonic NNPs led to efficient magnetic separation, ultrasensitive Raman detection, and effective photothermal killing of a common food-borne pathogen, E. coli. O157:H7.
Figure 1.

Schematic of the synthesis of versatile plasmonic nanogapped nanoparticles based on polydopamine coating.
RESULTS AND DISCUSSION
Dopamine undergoes consecutive oxidation, intramolecular cyclization, and oligomerization/self-assembly in alkaline conditions, leading to highly cross-linked adhesive PDA that is able to form a conformal coating on colloidal particles of diverse surface composition.28 We found that the deposition of PDA on citrate-stabilized Au nanoparticles can be controlled by the starting concentration of dopamine. Transmission electron microscopy (TEM) images (Figures 2a–c and S1) clearly reveal that monodisperse PDA-coated Au nanoparticles (Au@ PDA) with a PDA thickness of 2 to 13 nm were produced after 8 h reaction in bicine buffer (pH 8.5) with a dopamine concentration from 0.025 to 0.2 mg/mL. Note that, although the PDA thickness in one cycle of reaction culminates up to 13 nm, it can be further increased to tens of nanometers by applying multiple coating cycles (Figure S2).
Figure 2.

TEM images of 20 nm Au nanoparticles with a PDA coating of 2 (a), 5 (b), and 13 nm (c). TEM images of NNPs with a built-in nanogap of 2 (d), 5 (d), and 13 nm (f). TEM image of NNPs (50 nm Au nanoparticles as cores) with single (g), double (h), and triple (i) shells.
PDA is known to carry a high density of catechol groups, which can induce localized reduction of metal precursors.29,30 Our results show that successive addition of KAuCl4 and NH2OH in the presence of Au@PDA at 50 °C gave rise to well-defined Au NNPs (Figure 2d–f). A key finding here is that the nanogap size of the NNPs matches the thickness of the PDA coating in Au@PDA, which together with the flexibly tunable PDA thickness makes it possible to systematically tailor the nanogap size in a broad range. Apparently, complexation and reduction of AuCl4− ions by catechol groups facilitates in situ nucleation, which confines the subsequent growth of a Au shell on the surface of the PDA layer. This analysis is supported by the observation of rapid completion of colorimetric changes within 1 min during the reaction. Scanning electron microscopy (SEM) observation (Figure S3) reveals that the continuous growth of isolated domains during this process resulted in a complete shell when an increasing amount of KAuCl4 precursor was introduced. Notably, Nam and co-workers recently reported that HAuCl4 causes degradation of PDA instead of experiencing localized reduction.31 We reason that their use of HAuCl4 rather than KAuCl4 induced an acidic environment, in which catechol groups have weak reducing power.29
More interestingly, our strategy affords access to multigap NNPs consisting of multiple concentric nanoshells surrounding the core. NNPs are first prepared on 50 nm Au nanoparticles with a 13 nm nanogap (Figures 2g and S4). Simply by repeating the cycles of PDA coating and metallization, NNPs with two or three plasmonic shells were obtained, as shown in Figures 2h, i and S5. Such multigap NNPs were theoretically predicted to function as a series of optical condensers to direct light toward the center of their structures, inducing a dramatically amplified local field in the gap between the nanoparticle core and its adjacent shell.32 Our PDA-based approach therefore provides interesting opportunities for in-depth experimental studies by offering flexible control over the structure of multigap NNPs in terms of gap size and the number of shells.
The LSPR of plasmonic nanostructures is highly sensitive to changes in structural parameters and local dielectric environment.33 Au nanoparticles of 20 nm with an original LSPR centered at 522 nm experienced a gradual red-shift to 530, 538, and 548 nm (Figure 3a) for PDA coating thicknesses of 2, 5, and 13 nm, respectively, due to the larger refractive index of PDA in comparison with that of water. Au NNPs with a 2 nm gap showed a further spectral shift to 575 nm (Figure 3b) because of the strong coupling of closely arranged core and shell. When the gap expanded to 5 and 13 nm, a new resonance peak around 750 nm appeared and became dominant in the NNPs with a 13 nm gap due to a greater extinction coefficient of the larger Au shell. Similarly, in the case of the NNPs structured with a 50 nm core, 13 nm gap, and 15 nm shell, hybridization of the core and shell plasmon modes gave rise to two separate peaks at 610 and 823 nm (Figure 3c), which further red-shifted for the double-shell NNPs and eventually leveled off in the triple-shell NNPs to cover almost the entire visible and NIR spectral range. In line with the SEM observation (Figure S3), when the Au shell gradually closes up during the growth, a weak shoulder at 823 nm (Figure S6) emerges and evolves to a distinct strong peak.
Figure 3.

(a) UV–vis spectra of 20 nm Au nanoparticles coated with PDA. (b) UV–vis spectra of NNPs with different gap sizes. (c) UV–vis spectra of 50 nm Au nanoparticles, Au nanoparticle coated with PDA, and NNPs with different shell number. (d) SERS spectra of different Au nanostructures with RhB tags positioned on the PDA layer.
The confined electromagnetic field surrounding the plasmonic nanostructures plays a leading role in SERS of the molecules in their close proximity.19,34,35 Strongly coupled plasmonic nanostructures separated by a nanogap junction of less than 5 nm lead to a dramatically amplified local field in the nanogap, representing an efficient SERS hot-spot. While a number of chemical and self-assembly methods have been proposed to generate nanogaps,36–40 most of the previous methods lack the ability to precisely position molecular probes inside the hot-spots, instead relying on random diffusion of the probes, which becomes a major challenge for using SERS nanoprobes in quantitative detection. In our design, spontaneous covalent coupling of nucleophilic thiol and amine groups with quinone groups in PDA (Figure S7) provides an opportunity for stable, quantitative molecular fixation inside the SERS-active nanogap.27 Rhodamine B (RhB) carrying a primary amine group was selected as a model Raman probe to be tagged on the PDA layer prior to the deposition of the Au nanoshell. The number of RhB molecules anchored can be controlled by the feeding ratio of RhB and the nanoparticle core. Figure 3d shows the Raman spectra of NNPs and control samples containing an average of about 200 RhB molecules under the excitation of a 633 nm laser. The excellent SERS activity of the NNPs with a 2 nm nanogap was confirmed by an enhancement factor of 8.8 × 107, which dropped to 2.0 × 107 and 9.6 × 106 for the 5 and 13 nm gap, respectively. Incubating of the as-prepared SERS-encoded NNPs in aqueous medium did not lead to any obvious change of Raman intensity over time, indicating that the covalent linkage and the complete Au shell locked the RhB tags inside the nanogap. Also important is that Raman intensity shows linear dependence on the number of RhB molecules attached (Figure S8), offering the possibility of tailoring the Raman signal quantitatively. Moreover, Raman intensity of the as-prepared probes remains constant in aqueous medium (Figure S9), suggesting that the Raman tags are locked inside the nanogap by the covalent linkage and the complete Au shell. In contrast, the NNPs without the RhB probe loaded in the nanogap showed only a featureless background spectrum (Figure S10) at the same condition. Control nanoparticles, i.e., the RhB-tagged Au@PDA nanoparticles without the nanoshell, also exhibit negligible signals (Figure 3d), highlighting the significance of the nanogap hot-spots.
The universal adhesion of PDA offers the possibility of growing Au shells on nanoparticles of different sizes, shapes, and compositions. Figure 4 shows the NNPs templated by AuNRs and MOF nanocrystals.41 The rigid and conformal nature of the PDA coating gives rise to anisotropic NNPs that retain the shape of the nanocrystal cores. When elongated AuNRs are used as the core, ellipsoidal NNPs can be easily produced (Figures 4a–c and S11). We also investigated the synthesis of hybrid analogues of Au NNPs with nonmetallic cores, i.e., MOF nanocrystals with well-defined shapes. When octahedral UiO-66 nanocrystals (Figure 4d), formed by 1,4-benzenedicarboxylic acid (H2BDC) as organic linkers and zirconium(IV) as metal nodes,42 are chosen as the MOF core, an integral octahedral Au shell can be readily formed on the MOF core (Figure 4e). Furthermore, a plasmonic nanogap between two adjacent metallic nanoshells (Figures 4f and S11) is generated after another cycle of PDA coating and metallization, demonstrating the flexibility of our strategy in nanogap engineering and structural integration.
Figure 4.

TEM images of AuNR (a), AuNR@PDA (b), and AuNR@PDA@Au (c). TEM images of UiO-66 (d), UiO-66@PDA@Au (UiO-66@ single shell) (c), and UiO-66@PDA@Au@PDA@Au (UiO-66@double shell) (f). Insets: Corresponding samples at higher magnification; scale bar = 100 nm.
The compatibility of a PDA coating with diverse core materials further encouraged us to develop multifunctional NNPs with a magnetic core and a double-shell plasmonic nanogap. The uses of magnetic nanomaterials in bioseparation and bioimaging are representative examples of translation bionanotechnology.43 Imparting magnetic properties to NNPs leads to magnetoplasmonic nanostructures of considerable interest for biosensing, theranostic, and catalytic applications.44–47 TEM images in Figures 5 and S12 confirm that our synthesis protocol is also applicable for magnetic polystyrene nanoparticles. The growth of two consecutive layers of 15 nm Au nanoshells led to a uniform nanogap around the magnetic core (Figure 5a, b). The resulting NNPs retained the magnetic response of the core and can be easily collected by an external magnet, as shown in the inset of Figure 5b. MagNPs and PDA-coated MagNPs show a broad absorbance in the visible–NIR region (Figure 5c). When the first Au shell was built, a strong LSPR peak appeared at 834 nm. A PDA coating on the Au shell resulted in a red-shift to 868 nm. After the second Au shell was grown, the LSPR became even broader to cover the entire visible and NIR spectral range between 400 and 1100 nm (Figure 5c). Introducing a Raman tag, i.e., 4-nitrothiophenol (NTP), in the sub-5 nm nanogap gave rise to Raman signals 5.4-fold stronger than that of the MagNP@ PDA@Au with only one Au shell (Figure 5d), which also led to strong signal amplification because NTP is able to anchor on the Au nanoshell via the Au–S bond. Note that locking Raman tags inside the nanogap insulates them from interfering factors from the surrounding environment, which is essential for quantitative detection.
Figure 5.

TEM images of MagNP@PDA@Au (a) and magnetic NNPs (b). Inset: Photograph of magnetic separation of the magnetic NNPs. (c) UV–vis spectra of magnetic NNPs at different stages of growth. (d) SERS spectra of NTP-encoded magnetic NNPs and the control nanoparticles.
SERS-encoded magnetoplasmonic NNPs offer the possibility of combining magnetic separation, Raman spectroscopy for ultrasensitive detection, and photothermal transduction. As a proof of concept, we applied the NNPs for the quantitative immunoassay of a common food-borne bacterial pathogen, i.e., E. coli O157:H7 (Figure 6). Two antibodies specific to E. coli O157:H7 were attached to PEGylated NNPs and a 96-well microtiter plate blocked by bovine serum albumin, respectively. In the assay (Figure 6a), antibody-conjugated magnetic NNPs were first introduced in the samples containing spiked E. coli O157:H7, which were magnetically captured and enriched afterward upon the binding of the NNPs on the bacteria (Figure S13). The labeled bacteria were then exposed to the antibody-coated substrates. Finally, the substrates with immobilized bacteria were subjected to Raman detection of the SERS-encoded NNP probes. Our results (Figure 6b) show that the SERS intensity gradually increases in a bacterial concentration range of 10 to 108 CFU/mL with a high sensitivity (~102 CFU/mL). As summarized in Figure 6c, both buffer controls (PBS and Luria–Bertani culture medium) and other bacteria such as E. coli O6, S. enterica ATCC 13311, P. aeruginosa PA01, and E. faecalis ATCC 29212 of the same concentration gave rise to negligible signals. Apparently, the high specificity of the antibodies and surface blocking strategies gave rise to excellent selectivity in the immune sandwich assay. The magnetoplasmonic NNPs are also highly efficient photothermal transducers that lead to a rapid temperature increase of 39.5 °C upon 5 min of laser irradiation, as shown in Figure S14. Live/dead analysis with the BacLight kit utilizing a mixture of SYTO 9 and propidium iodide (PI) for fluorescence staining shows that only dead bacteria labeled with red PI dye are observed after laser irradiation of 15 min (Figure S15), confirming the nearly 100% bacterial killing by the photothermal effect of the magnetic NNPs. In comparison with platforms reported previously,48–50 magnetoplasmonic NNPs in this work not only allow for highly sensitive detection of pathogens by collective magnetic enrichment and excellent SERS activity but also lead to effective killing of the separated pathogen by the photothermal effect of the NNPs. The easy structural integration of diverse functional cores in the NNPs makes it possible for developing multifunctional plasmonic nanostructures, which are of particular interest for emerging applications in theranostic nanomedicine.11,12
Figure 6.

(a) Schematic of the immune assay of using SERS-encoded magnetic NNPs for bacterial detection. (b) SERS spectra of different concentrations of E. coli O157:H7. Inset: Raman intensity at 1341 cm−1 vs logarithm of the corresponding E. coli O157:H7 concentration. (c) Raman intensity at 1341 cm−1 of the substrates from the assays of control buffers and various types of bacteria (106 CFU/mL).
CONCLUSION
In summary, we have developed an enabling platform technology that offers extraordinary flexibility in tailoring the optical properties and structural diversity of plasmonic NNPs. We demonstrated that the adhesive and reactive nature of the PDA coating allows for rational designs of a broad spectrum of NNPs with customized combinations of functional cores and optically encoded nanogaps with desired gap sizes. The resulting multigap NNPs represent excellent model systems that support plasmon hybridization. More importantly, optically active multifunctional NNPs are of great potential in surface-enhanced spectroscopy, biosensing, nanomedicine, and photocatalysis.51,52
METHODS
Synthesis of 20 nm Au Nanoparticles
Au nanoparticles of 20 nm were prepared by citrate reduction of HAuCl4 in aqueous phase. Typically, a sodium citrate (92 mg) DI-water solution (3 mL) was rapidly injected into a boiling aqueous HAuCl4 (8 mg in 80 mL of water) solution under vigorous stirring. After boiling for 30 min, the solution was cooled to room temperature.
Synthesis of PDA-Coated 20 nm Au Nanoparticles (Au@ PDA)
Typically, as-synthesized 20 nm Au nanoparticles were centrifuged at 7000 rcf for 15 min. Then, the pellets were redispersed in 2 mL of H2O. A 500 μL sample of the concentrated AuNPs was dispersed in 16 mL of bicine buffer (pH 8.5), followed by adding different amounts of dopamine to achieve the corresponding PDA thickness. The reaction solution was stirred for 8 h. The purple product was purified by centrifugation and was stored in 2 mL of H2O at 4 °C for further use.
Synthesis of 50 nm Au Nanoparticles
AuNPs of 50 nm were prepared using a seeded-growth method. Briefly, 50 mL of water was added into a 100 mL round-bottom flask. A 2 mL amount of seed AuNP solution and 200 μL of 0.2 M NH2OH·HCl were added into this flask consecutively. Afterward, 3 mL of 0.1 wt % HAuCl4 was added dropwise into the solution under vigorous stirring followed by 30 min reaction at room temperature. A gradual color change from light red to dark red was observed. Finally, the concentration of sodium citrate was adjusted to 1 mM. After reacting for another 2 h, the nanoparticle dispersion was stored at 4 °C for further use.
Synthesis of PDA-Coated 50 nm Au Nanoparticles (Au(50 nm)@PDA)
Typically, 50 nm Au nanoparticles were centrifuged at 1200 rcf for 15 min. Then, the pellets were redispersed in 1 mL of H2O. The concentrated AuNPs was dispersed in 16 mL of bicine buffer (pH 8.5), followed by adding dopamine to achieve the required PDA thickness. The reaction solution was stirred for 8 h, and the purple product was purified by centrifugation.
Synthesis of Au Nanogapped Nanoparticles (Au NNPs)
Typically, 80 μL of Au@PDA (0.6 nM) was added into 2 mL of H2O at 50 °C. After stirring for 2 min, 100 μL of 2.5 mM KAuCl4 was injected, followed by 50 μL of 0.2 M NH2OH·HCl. The color of the solution changed from light red to dark purple immediately. The reaction solution was stirred for 2 min. After cooling, 50 μL of PEG-SH (5 kDa, 10 mg/mL) was added into the solution to further stabilize the NNPs. Finally, the product was purified by centrifugation. For NNPs of different PDA thickness, the amount of Au precursor was changed accordingly.
Synthesis of Multishell NNPs
For double-shell NNPs, we used the single-shell NNPs as a core and repeated the procedures as described above for the growth of the Au shell. Typically, Au(50 nm) @Single Shell was dispersed in 4 mL of bicine buffer (pH 8.5), followed by adding dopamine (0.1 mg/mL). The reaction solution was stirred for 8 h, and the resultant Au(50 nm)@Single Shell@PDA was purified by centrifugation. Next, the obtained product was added into 2 mL of H2O at 50 °C. After stirring for 2 min, 120 μL of 2.5 mM KAuCl4 was injected, followed by 60 μL of 0.2 M NH2OH·HCl. The reaction solution was stirred for 2 min, and 50 μL of PEG-SH (10 mg/mL) was added into the solution to further stabilize the double-shell NNPs. Finally, the product (Au(50 nm)@Double Shells) was purified by centrifugation. In the synthesis of triple-shell NNPs, the double-shell NNPs were used as the cores.
Synthesis of Raman Dye-Tagged Au NNPs
Typically, Au@ PDA nanoparticles were dispersed in 2 mL of bicine buffer (pH 8.5) under continuous stirring, followed by adding 0.5 mg/mL RhB-NH2 solution. After reacting for 24 h, Au@PDA-RhB nanoparticles were collected by centrifuge and washed with DI water three times. The number of conjugated dyes was determined by the fluorescence intensity of unbound RhB molecules in the supernatant and can be controlled by the feeding ratio of RhB and Au@PDA. For example, in case of Au@PDA-2, a conjugation efficiency of 68% was achieved when the feeding ratio was 300:1. These Raman dye-tagged Au@PDA-RhB nanoparticles were further used as the cores to construct Au NNPs (Au@PDA-RhB@Au) for SERS detection.
Calculation of Enhancement Factor (EF)
The EF of NNPs was determined by computing the ratio of SERS to normal Raman scattering of RhB using the following equation: EF = (ISERS × CNormal)/(INormal × CSERS), where ISERS and INormal are the Raman intensities at 1647 cm−1 for NNPs and pure RhB solution, and CSERS and CNormal the concentrations of RhB on NNPs and in pure solution. CSERS was calculated using the equation CSERS = N × CAu, where N is the number of RhB in the NNPs.
Synthesis of Au Nanorods (AuNRs)
A seed-mediated method was used to prepare the AuNRs. Typically, two steps were included. First, gold seeds were synthesized as reported previously. A HAuCl4 solution (250 μL of 10 mM) was added to the cetyltrimethylammonium bromide (CTAB) solution (9.75 mL, 0.1 M); then, under vigorous stirring, a freshly prepared NaBH4 solution (0.6 mL, 0.01 M) was injected. The solution color changed immediately from yellow to dark brown. After stirring for 5 min, the mixture solution, as seed solution, was kept for at least 1 h at room temperature before it was used in the next step. Second, AuNRs were synthesized in a growth solution. HAuCl4 solution (500 μL of 10 mM) was added to 9.5 mL of the CTAB solution. The mixture solution was incubated at 40 °C for 10 min. Then a AgNO3 solution (0.1 M), dopamine hydrochloride solution (0.2 M), and seed solution were added sequentially. The resulting growth solution was mixed thoroughly and kept undisturbed in a water bath set at 40 °C for 3 h.
Synthesis of UiO-66 Nanoparticles
Nanosized UiO-66 particles were prepared by dissolving 4 mM ZrCl4 and 4 mM H2BDC in a mixture of dimethylformamide (DMF) and EtOH containing acetic acid. The reaction vial was capped and placed into an oven preheated at 100 °C for 12 h. The product was collected by centrifugation and then washed three times with DMF and MeOH, respectively. The product was suspended in MeOH.
Synthesis of MagNPs
Polystyrene-trapped magnetic iron oxide nanoparticles were prepared by emulsion polymerization.41 In brief, FeCl3·6H2O (2.4 g) and FeCl2·4H2O (0.982 g) were used to prepare magnetite nanoparticles first. Magnetite nanoparticles (0.5 g) were added into 12 mL of water containing 10 mg of sodium dodecyl sulfate (SDS) to obtain a miniemulsion of magnetite nanoparticles. Meanwhile, a styrene emulsion was prepared using 5 mL of styrene, 50 mg of SDS, 40 mL of water, and 0.033 mL of tetradecane. Finally, a styrene emulsion was added into the miniemulsion of magnetite nanoparticles containing potassium persulfate (KPS) in a three-neck flask. The flask was placed in an 80 °C water bath and maintained for 20 h to obtain MagNPs.
Synthesis of Magnetic NNPs
Briefly, 50 μL of MagNP was dispersed in 16 mL of bicine buffer (pH 8.5), followed by adding dopamine to achieve the required PDA thickness. After 8 h of reaction, the product (MagNP@PDA) was purified by centrifugation and stored in 1 mL of H2O. To fabricate the first Au shell, 100 μL of MagNP@ PDA was added into 10 mL of H2O at 50 °C. After stirring for 2 min, 1.2 mL of 2.5 mM KAuCl4 was injected, followed by 1.2 mL of 0.2 M NH2OH·HCl. The obtained product (MagNP@PDA@Au) was collected and further dispersed in bicine buffer to undergo another cycle of PDA coating and metallization. Eventually, the color of the solution changed from brown to green. The result magnetic NNPs were surface modified with bifunctional HOOC-PEG-SH (3.4 kDa).
Surface Modification of Magnetic NNPs
The magnetic NNPs were collected by centrifuge and dispersed in 5 mL of 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 5.5). To activate the carboxylic acid group on the surface of these particles, 0.2 mL of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC, 5 mg/mL) and sulfo-N-hydroxysuccinimide (NHS, 5 mg/mL) were added to the solution and incubated for 30 min. The excess EDC and NHS were removed by centrifuge. Then, the detection monoclonal antibody (8B1-C2-B1) (30 μg/mL) in borate saline buffer (pH 8.0) was quickly added to the activated particles with gentle stirring for 3 h at room temperature. Finally, 1 mL of bovine serum albumin (BSA) solution (2 mg/mL) was added to the mixture to block the unreacted sites for 1 h. The free reactants were removed by centrifuge. The bioconjugated magnetic NNPs were stored at 4 °C before use.
Bacterial Culture
E. coli O157:H7 (ATCC 43888) and other bacteria were cultured in Luria–Bertani medium for 20 h at 37 °C before use. The number of viable cells was determined by plate count. The cells were treated with 0.3% formaldehyde for 24 h to kill all bacteria. The inactivated bacteria were collected by centrifugation at 4000 rpm and resuspended in 0.01 M phosphate-buffered saline (PBS) (pH 7.4). Finally, these bacteria were serially diluted to the desired concentrations with 0.01 M PBS (pH 7.4) for further use.
Detection and Photothermal Killing of E. coli O157:H7 Using Magnetic NNPs
A 25 μL amount of antibody-conjugated magnetic NNPs (0.5 mg/mL) was added to 1 mL samples containing 10, 102, 103, 104, 105, 106, 107, and 108 CFU/mL of E. coli O157:H7 or other bacteria. The mixture was gently shaken for 30 min and placed in a magnetic field for 10 min to separate the immune complex of E. coli O157:H7 and magnetic NNPs. The complex in 50 μL of PBS was added to the capture antibody (10C5-H3-B6)-immobilized 96-well microtiter plate and incubated for 30 min at room temperature. The plate was then washed three times with 0.01 M PBS (pH 7.4) containing 0.05% Tween 20. Then the plate was placed under Raman microscopy for spectral collection in the range of 800–1800 cm−1 using 50 mW of laser power. The calibration curve was plotted using the peak intensities of NTP at 1341 cm−1 vs the concentration of E. coli O157:H7 (10–108 CFU/mL). The photothermal treatment of the captured bacteria was conducted by exposure to an 808 nm laser (1 W/cm2) for 15 min. The temperature was monitored by an infrared camera. The bacteria were stained by LIVE/DEAD BacLight bacterial viability kits in the dark for 15 min and then imaged using laser scanning confocal microscopy.
Supplementary Material
Acknowledgments
This work is supported by Ministry of Education-Singapore (MOE2015-T2-1-112 and MOE2013-T3-1-002). P.B.M. acknowledges support from National Institutes of Health grants R37 DE014193 and R01 EB005772. J.R. is grateful for the financial support from the National Natural Science Foundation of China (81372712).
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.6b05951.
Additional microscopic and spectroscopic characterization of the NNPs and photothermal killing of pathogens (PDF)
References
- 1.Lane LA, Qian XM, Nie SM. SERS Nanoparticles in Medicine: From Label-Free Detection to Spectroscopic Tagging. Chem Rev. 2015;115:10489–10529. doi: 10.1021/acs.chemrev.5b00265. [DOI] [PubMed] [Google Scholar]
- 2.Lal S, Link S, Halas NJ. Nano-Optics from Sensing to Waveguiding. Nat Photonics. 2007;1:641–648. [Google Scholar]
- 3.Sepulveda B, Angelome PC, Lechuga LM, Liz-Marzan LM. LSPR-Based Nanobiosensors. Nano Today. 2009;4:244–251. [Google Scholar]
- 4.Cobley CM, Chen JY, Cho EC, Wang LV, Xia YN. Gold Nanostructures: A Class of Multifunctional Materials for Biomedical Applications. Chem Soc Rev. 2011;40:44–56. doi: 10.1039/b821763g. [DOI] [PubMed] [Google Scholar]
- 5.Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, Van Duyne RP. Biosensing with Plasmonic Nanosensors. Nat Mater. 2008;7:442–453. doi: 10.1038/nmat2162. [DOI] [PubMed] [Google Scholar]
- 6.Halas NJ, Lal S, Chang WS, Link S, Nordlander P. Plasmons in Strongly Coupled Metallic Nanostructures. Chem Rev. 2011;111:3913–3961. doi: 10.1021/cr200061k. [DOI] [PubMed] [Google Scholar]
- 7.Taylor RW, Lee TC, Scherman OA, Esteban R, Aizpurua J, Huang FM, Baumberg JJ, Mahajan S. Precise Subnanometer Plasmonic Junctions for SERS within Gold Nanoparticle Assemblies Using Cucurbit[n]uril Glue. ACS Nano. 2011;5:3878–3887. doi: 10.1021/nn200250v. [DOI] [PubMed] [Google Scholar]
- 8.Hao F, Sonnefraud Y, Van Dorpe P, Maier SA, Halas NJ, Nordlander P. Symmetry Breaking in Plasmonic Nanocavities: Subradiant LSPR Sensing and a Tunable Fano Resonance. Nano Lett. 2008;8:3983–3988. doi: 10.1021/nl802509r. [DOI] [PubMed] [Google Scholar]
- 9.Jain PK, El-Sayed MA. Universal Scaling of Plasmon Coupling in Metal Nanostructures: Extension from Particle Pairs to Nanoshells. Nano Lett. 2007;7:2854–2858. doi: 10.1021/nl071496m. [DOI] [PubMed] [Google Scholar]
- 10.Han XX, Schmidt AM, Marten G, Fischer A, Weidinger IM, Hildebrandt P. Magnetic Silver Hybrid Nanoparticles for Surface-Enhanced Resonance Raman Spectroscopic Detection and Decontamination of Small Toxic Molecules. ACS Nano. 2013;7:3212–3220. doi: 10.1021/nn305892j. [DOI] [PubMed] [Google Scholar]
- 11.Jin YD, Jia CX, Huang SW, O’Donnell M, Gao XH. Multifunctional Nanoparticles as Coupled Contrast Agents. Nat Commun. 2010;1:41. doi: 10.1038/ncomms1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan Z, Shelton M, Singh AK, Senapati D, Khan SA, Ray PC. Multifunctional Plasmonic Shell-Magnetic Core Nanoparticles for Targeted Diagnostics, Isolation, and Photothermal Destruction of Tumor Cells. ACS Nano. 2012;6:1065–1073. doi: 10.1021/nn2045246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christopher P, Xin HL, Linic S. Visible-Light-Enhanced Catalytic Oxidation Reactions on Plasmonic Silver Nanostructures. Nat Chem. 2011;3:467–472. doi: 10.1038/nchem.1032. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez-Fernandez D, Langer J, Henriksen-Lacey M, Liz-Marzan LM. Hybrid Au–SiO2 Core–Satellite Colloids as Switchable SERS Tags. Chem Mater. 2015;27:2540–2545. [Google Scholar]
- 15.Cheng L, Song JB, Yin J, Duan HW. Self-Assembled Plasmonic Dimers of Amphiphilic Gold Nanocrystals. J Phys Chem Lett. 2011;2:2258–2262. [Google Scholar]
- 16.Song JB, Huang P, Duan HW, Chen XY. Plasmonic Vesicles of Amphiphilic Nanocrystals: Optically Active Multifunctional Platform for Cancer Diagnosis and Therapy. Acc Chem Res. 2015;48:2506–2515. doi: 10.1021/acs.accounts.5b00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bardhan R, Mukherjee S, Mirin NA, Levit SD, Nordlander P, Halas NJ. Nanosphere-in-a-Nanoshell: A Simple Nanomatryushka. J Phys Chem C. 2010;114:7378–7383. [Google Scholar]
- 18.Ayala-Orozco C, Urban C, Knight MW, Urban AS, Neumann O, Bishnoi SW, Mukherjee S, Goodman AM, Charron H, Mitchell T, Shea M, Roy R, Nanda S, Schiff R, Halas NJ, Joshi A. Au Nanomatryoshkas as Efficient Near-Infrared Photothermal Transducers for Cancer Treatment: Benchmarking against Nanoshells. ACS Nano. 2014;8:6372–6381. doi: 10.1021/nn501871d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim DK, Jeon KS, Hwang JH, Kim H, Kwon S, Suh YD, Nam JM. Highly Uniform and Reproducible Surface-Enhanced Raman Scattering from DNA-Tailorable Nanoparticles with 1-nm Interior gap. Nat Nanotechnol. 2011;6:452–460. doi: 10.1038/nnano.2011.79. [DOI] [PubMed] [Google Scholar]
- 20.Ayala-Orozco C, Liu JG, Knight MW, Wang YM, Day JK, Nordlander P, Halas NJ. Fluorescence Enhancement of Molecules Inside a Gold Nanomatryoshka. Nano Lett. 2014;14:2926–2933. doi: 10.1021/nl501027j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin L, Zapata M, Xiong M, Liu ZH, Wang SS, Xu H, Borisov AG, Gu HC, Nordlander P, Aizpurua J, Ye J. Nanooptics of Plasmonic Nanomatryoshkas: Shrinking the Size of a Core-Shell Junction to Subnanometer. Nano Lett. 2015;15:6419–6428. doi: 10.1021/acs.nanolett.5b02931. [DOI] [PubMed] [Google Scholar]
- 22.Prodan E, Radloff C, Halas NJ, Nordlander P. A Hybridization Model for the Plasmon Response of Complex Nanostructures. Science. 2003;302:419–422. doi: 10.1126/science.1089171. [DOI] [PubMed] [Google Scholar]
- 23.Oh JW, Lim DK, Kim GH, Suh YD, Nam JM. Thiolated DNA-Based Chemistry and Control in the Structure and Optical Properties of Plasmonic Nanoparticles with Ultrasmall Interior Nanogap. J Am Chem Soc. 2014;136:14052–14059. doi: 10.1021/ja504270d. [DOI] [PubMed] [Google Scholar]
- 24.Song JB, Duan B, Wang CX, Zhou JJ, Pu L, Fang Z, Wang P, Lim TT, Duan HW. SERS-Encoded Nanogapped Plasmonic Nanoparticles: Growth of Metallic Nanoshell by Templating Redox-Active Polymer Brushes. J Am Chem Soc. 2014;136:6838–6841. doi: 10.1021/ja502024d. [DOI] [PubMed] [Google Scholar]
- 25.Tsai MF, Chang SHG, Cheng FY, Shanmugam V, Cheng YS, Su CH, Yeh CS. Au Nanorod Design as Light-Absorber in the First and Second Biological Near-Infrared Windows for in Vivo Photothermal Therapy. ACS Nano. 2013;7:5330–5342. doi: 10.1021/nn401187c. [DOI] [PubMed] [Google Scholar]
- 26.Gandra N, Portz C, Singamaneni S. Multifunctional Plasmonic Nanorattles for Spectrum-Guided Locoregional Therapy. Adv Mater. 2014;26:424–429. doi: 10.1002/adma.201302803. [DOI] [PubMed] [Google Scholar]
- 27.Wang CX, Zhou JJ, Wang P, He WS, Duan HW. Robust Nanoparticle–DNA Conjugates Based on Mussel-Inspired Polydopamine Coating for Cell Imaging and Tailored Self-Assembly. Bioconjugate Chem. 2016;27:815–823. doi: 10.1021/acs.bioconjchem.6b00021. [DOI] [PubMed] [Google Scholar]
- 28.Zhou JJ, Wang P, Wang CX, Goh YT, Fang Z, Messersmith PB, Duan HW. Versatile Core–Shell Nanoparticle@ Metal–Organic Framework Nanohybrids: Exploiting Mussel-Inspired Polydopamine for Tailored Structural Integration. ACS Nano. 2015;9:6951–6960. doi: 10.1021/acsnano.5b01138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Black KCL, Liu ZQ, Messersmith PB. Catechol Redox Induced Formation of Metal Core-Polymer Shell Nanoparticles. Chem Mater. 2011;23:1130–1135. doi: 10.1021/cm1024487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou JJ, Duan B, Fang Z, Song JB, Wang CX, Messersmith PB, Duan HW. Interfacial Assembly of Mussel-Inspired Au@Ag@Polydopamine Core–Shell Nanoparticles for Recyclable Nanocatalysts. Adv Mater. 2014;26:701–705. doi: 10.1002/adma.201303032. [DOI] [PubMed] [Google Scholar]
- 31.Kumar A, Kumar S, Rhim WK, Kim GH, Nam JM. Oxidative Nanopeeling Chemistry-Based Synthesis and Photodynamic and Photothermal Therapeutic Applications of Plasmonic Core-Petal Nanostructures. J Am Chem Soc. 2014;136:16317–16325. doi: 10.1021/ja5085699. [DOI] [PubMed] [Google Scholar]
- 32.Xu HX. Multilayered Metal Core–Shell Nanostructures for Inducing a Large and Tunable Local Optical Field. Phys Rev B: Condens Matter Mater Phys. 2005;72:073405. [Google Scholar]
- 33.Miller MM, Lazarides AA. Sensitivity of Metal Nanoparticle Surface Plasmon Resonance to the Dielectric Environment. J Phys Chem B. 2005;109:21556–21565. doi: 10.1021/jp054227y. [DOI] [PubMed] [Google Scholar]
- 34.Osberg KD, Rycenga M, Harris N, Schmucker AL, Langille MR, Schatz GC, Mirkin CA. Dispersible Gold Nanorod Dimers with Sub-5 nm Gaps as Local Amplifiers for Surface-Enhanced Raman Scattering. Nano Lett. 2012;12:3828–3832. doi: 10.1021/nl301793k. [DOI] [PubMed] [Google Scholar]
- 35.Im H, Bantz KC, Lee SH, Johnson TW, Haynes CL, Oh SH. Self-Assembled Plasmonic Nanoring Cavity Arrays for SERS and LSPR Biosensing. Adv Mater. 2013;25:2678–2685. doi: 10.1002/adma.201204283. [DOI] [PubMed] [Google Scholar]
- 36.Xu LG, Yan WJ, Ma W, Kuang H, Wu XL, Liu LQ, Zhao Y, Wang LB, Xu CL. SERS Encoded Silver Pyramids for Attomolar Detection of Multiplexed Disease Biomarkers. Adv Mater. 2015;27:1706–1711. doi: 10.1002/adma.201402244. [DOI] [PubMed] [Google Scholar]
- 37.Chen G, Wang Y, Yang MX, Xu J, Goh SJ, Pan M, Chen HY. Measuring Ensemble-Averaged Surface-Enhanced Raman Scattering in the Hotspots of Colloidal Nanoparticle Dimers and Trimers. J Am Chem Soc. 2010;132:3644–3645. doi: 10.1021/ja9090885. [DOI] [PubMed] [Google Scholar]
- 38.Schutz M, Schlucker S. Molecularly Linked 3D Plasmonic Nanoparticle Core/Satellite Assemblies: SERS Nanotags with Single-Particle Raman Sensitivity. Phys Chem Chem Phys. 2015;17:24356–24360. doi: 10.1039/c5cp03189c. [DOI] [PubMed] [Google Scholar]
- 39.Wang YL, Schlucker S. Rational Design and Synthesis of SERS Labels. Analyst. 2013;138:2224–2238. doi: 10.1039/c3an36866a. [DOI] [PubMed] [Google Scholar]
- 40.Banholzer MJ, Millstone JE, Qin LD, Mirkin CA. Rationally Designed Nanostructures for Surface-Enhanced Raman Spectroscopy. Chem Soc Rev. 2008;37:885–897. doi: 10.1039/b710915f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meek ST, Greathouse JA, Allendorf MD. Metal–Organic Frameworks: A Rapidly Growing Class of Versatile Nanoporous Materials. Adv Mater. 2011;23:249–267. doi: 10.1002/adma.201002854. [DOI] [PubMed] [Google Scholar]
- 42.Wu H, Yildirim T, Zhou W. Exceptional Mechanical Stability of Highly Porous Zirconium Metal–Organic Framework UiO-66 and Its Important Implications. J Phys Chem Lett. 2013;4:925–930. doi: 10.1021/jz4002345. [DOI] [PubMed] [Google Scholar]
- 43.Lee N, Yoo D, Ling D, Cho MH, Hyeon T, Cheon J. Iron Oxide Based Nanoparticles for Multimodal Imaging and Magnetoresponsive Therapy. Chem Rev. 2015;115:10637–10689. doi: 10.1021/acs.chemrev.5b00112. [DOI] [PubMed] [Google Scholar]
- 44.Zhou JJ, Wang CX, Wang P, Messersmith PB, Duan HW. Multifunctional Magnetic Nanochains: Exploiting Self-Polymerization and Versatile Reactivity of Mussel-Inspired Polydopamine. Chem Mater. 2015;27:3071–3076. [Google Scholar]
- 45.Lim EK, Kim T, Paik S, Haam S, Huh YM, Lee K. Nanomaterials for Theranostics: Recent Advances and Future Challenges. Chem Rev. 2015;115:327–394. doi: 10.1021/cr300213b. [DOI] [PubMed] [Google Scholar]
- 46.Noh MS, Jun BH, Kim S, Kang H, Woo MA, Minai-Tehrani A, Kim JE, Kim J, Park J, Lim HT, Park SC, Hyeon T, Kim YK, Jeong DH, Lee YS, Cho MH. Magnetic Surface-Enhanced Raman Spectroscopic (M-SERS) Dots for the Identification of Bronchioalveolar Stem Cells in Normal and Lung Cancer Mice. Biomaterials. 2009;30:3915–3925. doi: 10.1016/j.biomaterials.2009.03.059. [DOI] [PubMed] [Google Scholar]
- 47.Duan B, Zhou JJ, Fang Z, Wang CX, Wang XJ, Hemond HF, Chan-Park MB, Duan HW. Surface Enhanced Raman Scattering by Graphene-Nanosheet-Gapped Plasmonic Nanoparticle Arrays for Multiplexed DNA Detection. Nanoscale. 2015;7:12606–12613. doi: 10.1039/c5nr02164b. [DOI] [PubMed] [Google Scholar]
- 48.Wang YL, Lee K, Irudayaraj J. Silver Nanosphere SERS Probes for Sensitive Identification of Pathogens. J Phys Chem C. 2010;114:16122–16128. [Google Scholar]
- 49.Wang JF, Wu XZ, Wang CW, Shao NS, Dong PT, Xiao R, Wang SQ. Magnetically Assisted Surface-Enhanced Raman Spectroscopy for the Detection of Staphylococcus aureus Based on Aptamer Recognition. ACS Appl Mater Interfaces. 2015;7:20919–20929. doi: 10.1021/acsami.5b06446. [DOI] [PubMed] [Google Scholar]
- 50.Wang HY, Zhou YF, Jiang XX, Sun B, Zhu Y, Wang H, Su YY, He Y. Simultaneous Capture, Detection, and Inactivation of Bacteria as Enabled by a Surface-Enhanced Raman Scattering Multifunctional Chip. Angew Chem, Int Ed. 2015;54:5132–5136. doi: 10.1002/anie.201412294. [DOI] [PubMed] [Google Scholar]
- 51.Lim J, Majetich SA. Composite Magnetic-Plasmonic Nanoparticles for Biomedicine: Manipulation and Imaging. Nano Today. 2013;8:98–113. [Google Scholar]
- 52.Wang YQ, Yan B, Chen LX. SERS Tags: Novel Optical Nanoprobes for Bioanalysis. Chem Rev. 2013;113:1391–1428. doi: 10.1021/cr300120g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.