

Abstract
To examine the in vivo responses of promyelocytic leukemia protein (PML) to arsenic, rats (male, 6 weeks old, Sprague Dawley) were administered a single intraperitoneal dose of 5 mg/kg arsenic trioxide (ATO). The protein was examined in the heart, lung, liver, and brain 6 and 48 hours after administration: a significant response of PML was observed in the brain. Oxidative DNA modification was also observed in the brain as revealed by increased immunoreactivity to anti-8-hydroxy-2’-deoxyguanosine (8-OHdG) antibody. In contrast, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) stain reactivity was only slightly increased, suggesting oxidative cellular stress without apoptotic cell death in the ATO-administered rat brain. Among the DNA damage response pathways, the ATR-Chk1 axis was activated, while the ATM-Chk2 axis was not, implying that the PML response is associated with activation of the ATR-Chk1 DNA repair pathway in the brain.
Keywords: arsenic trioxide, brain, DNA damage, promyelocytic leukemia protein, ataxia telangiectasia and rad3 related
Arsenic is an environmental risk factor for humans as it is a frequent contaminant in drinking water, especially in developing countries1,2,3. Arsenic trioxide (ATO) is one of the most toxic species among arsenic substances, as trivalent arsenic (As3+) acts as a bridge between vicinal cysteine residues within and/or between proteins.
The promyelocytic leukemia (PML) protein is a multifunctional protein involved in inflammation, aging, and cancer4. PML is a highly cysteine-rich protein and, therefore, is extraordinarily sensitive to ATO5. The in vivo response of this protein is the basis for the efficacy of ATO as a medication for the treatment of acute promyelocytic leukemia (APL), which is caused by the generation of the oncogenic PML-retinoic acid receptor (RAR) fusion protein6, 7. Binding to ATO causes PML to undergo conformational changes that lead to its degradation by cellular proteases.
PML undergoes a complicated posttranscriptional modification process that includes the addition of small ubiquitin-like modifier (SUMO) and SUMO-dependent ubiquitination by RING finger protein 44. The addition of SUMO to PML is necessary not only for its degradation but also for its oligomerization and the subsequent formation of PML-nuclear bodies, which are implicated in the cellular response against oxidative stresses8. Both the PML-RAR oncogenic fusion protein and the wild type PML protein have been shown to be extremely sensitive to arsenic in vitro5, but their sensitivity in vivo has so far received little attention in the literature.
Oxidative DNA damage is a form of oxidative cellular stress and leads to two distinct, but mutually interacting, cellular responses: 1) when the DNA damage is relatively serious, such as a double-strand DNA break, ATM (ataxia telangiectasia mutated) and its downstream target Chk2 (check point kinase2) are activated, thus leading to apoptosis; 2) when the DNA damage is relatively mild, such as an oxidative DNA modification or nick formation, ATR (ATM and rad3 related) and its downstream Chk1 are activated to repair the DNA damage9. In the response to double-strand DNA break, histone H2AX is phosphorylated by ATM10. The aim of this study was to examine the in vivo effect of ATO on the PML protein and the possible pathophysiological implication of the PML response to ATO.
The anti-PML antibody was obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA), and the antibodies against ATR, p-ATR (Ser428-phosphorylated), p-ATM (Ser1981-phosphorylated), p-Chk1 (Ser345-phosphorylated), p-Chk2 (Thr68-phosphorylated), p-H2AX (Ser139-phosphorylated), and cleaved caspase-3 antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-actin and anti-8-hydroxy-2’-deoxyguanosine (8-OHdG) antibodies were from Sigma-Aldrich (St. Louis, MO, USA) and JaICA (Shizuoka, Japan), respectively. Animal experiment protocols were approved by the Institutional Animal Care and Use Committee of the Tokyo Medical and Dental University. Sprague-Dawley rats (male, six-weeks-old, n = 4/group) received a single intraperitoneal injection of ATO (Sigma, St. Louis, MO, USA; 1 mg/mL solution prepared in PBS, pH 7.4) at a dose of 5 mg/kg body weight (ATO group) or PBS (control group). The rats were sacrificed 6 or 48 hours after injection, the organs were extracted, and the tissues were fixed immediately in formalin and embedded in paraffin for histological and immunohistochemical analysis. Tissue lysates were also extracted and stored at −80°C until used for western blot analysis. Western blot analysis was performed as described previously11, 12. In brief, tissue lysates were electrophoresed, blotted to PVDF membranes (Millipore), and probed with the abovementioned primary antibodies, followed by incubation with appropriate secondary antibodies conjugated with horseradish peroxidase. For immunohistochemical analysis, paraffin-embedded tissue sections were deparaffinized, rehydrated, and retrieved with their antigenicity by microwave exposure in 10 mM sodium citrate buffer. The samples were then incubated with anti-8-OHdG antibody overnight at 4°C, and the antigens were visualized using a Histofine Simple Stain MAX PO kit (Nichirei Biosciences, Tokyo, Japan) with diaminobenzidine (DAB) as a substrate and examined under a light microscope (Olympus AX80). For terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) analysis, an Apoptosis in situ detection kit (Wako, Osaka, Japan) was used.
We first examined the responses of the PML protein to ATO administration in the major organs of rats. The rats were treated with ATO or PBS (control), and western blot analysis was performed to examine the status of the PML protein in the heart, lung, liver, and brain (Fig. 1A). In accordance with the presence of various PML isoforms that are generated from a single gene through tissue-dependent alternative splicing13, western blot analysis showed different band patterns among the organs: while no apparent changes in the PML proteins were observed in the heart or liver, numerous changes were observed in the lung and brain (Fig. 1A). In the ATO-treated lung, ~55 kDa and 100 kDa species of PML seemed to be upregulated and downregulated, respectively (Fig. 1A). In the brain, two bands (bands A and B) showed clear and statistically significantly different patterns between ATO-treated and control samples (Fig. 1 A and B). In vitro experiments have indicated that prior to PML degradation, SUMO- and/or ubiquitin-modified high molecular weight PML species accumulate transiently in ATO-treated cells13. Thus, this high molecular PML (band A) might be SUMO/ubiquitin-modified PML, but for the time being, we cannot draw a definitive conclusion concerning the exact nature of this PML species. Since the changes in the brain seem to be the most obvious response of PML, we further examined the brains of ATO-administered rats.
Fig. 1.

(A) Western blot analysis of PML protein in the heart, lung, liver, and brain of rats at the indicated times following the administration of arsenic trioxide (ATO). White and black arrowheads indicate protein bands that decreased and increased, respectively, in response to ATO treatment in the lung. (B) Bands A and B indicate protein bands that increased and decreased, respectively, in response to ATO treatment in the brain. Representative western blot images and a densitometric analysis of the levels of bands A and B relative to actin are shown (n = 4). N.D., not detected.
Although PML has been implicated in various cellular activities, it has been stressed that PML plays a crucial role in the DNA damage response8, 14. As arsenic causes oxidative cellular stress, including oxidative DNA damage, we performed an immunohistochemical analysis of 8-OHdG, a marker of DNA oxidation. Immunoreactivity against an anti-8-OHdG antibody was observed even in the cerebral cortex of rats in the control group, suggesting the development of 8-OHdG during tissue fixation in formalin (Fig. 2A). However, substantially increased immunoreactivity was observed 48 hours after ATO treatment compared with in the case of no treatment, showing the development of oxidative DNA damage during ATO treatment (Fig. 2A). We further checked the status of DNA damage pathway molecules in the ATO-administered rat brain. Western blot analysis indicated the activation of ATR as revealed by the phosphorylation of ATR (Fig. 2 B and E). ATR activation was also supported by the phosphorylation of Chk1, a direct target of ATR (Fig. 2 C and E). In contrast to the activation of the ATR-Chk1 axis, neither phosphorylated ATM nor phosphorylated Chk2 was detected (Fig. 2D). The absence of ATM-Chk2 axis activation was also supported by the constant phosphorylation levels of H2AX, which is generally activated in parallel with ATM-Chk29 (Fig. 2C).
Fig. 2.

Oxidative DNA damage and the activation of the ATR-Chk1 axis of the DNA damage response pathway in the brain. Whole brains were excised 6 or 48 hours after the administration of ATO. (A) Immunohistochemical analyses of 8-OHdG in the brain. Immunoreactivities to 8-OHdG in the cerebral cortex are shown. (−) Tissue sections stained without anti-8-OHdG antibody (negative control). (B–E) Western blot analyses for p-ATR, ATR, p-Chk1, p-H2AX, p-ATM, p-Chk2, and actin. Representative results (n = 4) are shown. Densitometric analysis of the levels of p-ATR and p-Chk1 relative to actin are shown (n = 4). *p<0.05.
We finally evaluated apoptosis in the ATO-treated rat brain. TUNEL-positive nuclei were scarcely observed in the ATO-treated rat brain (Fig. 3A). Only a subtle increase in the levels of cleaved-caspase-3, the activated form of caspase-3, was observed in the ATO-treated brain as compared with the control brain (Fig. 3B). Taking the results shown in Figs. 2 and 3 together, we conclude that ATO at this dose activates the ATR-Chk1 axis of the DNA damage response, which might lead to DNA repair in neuronal cells rather than apoptosis.
Fig. 3.
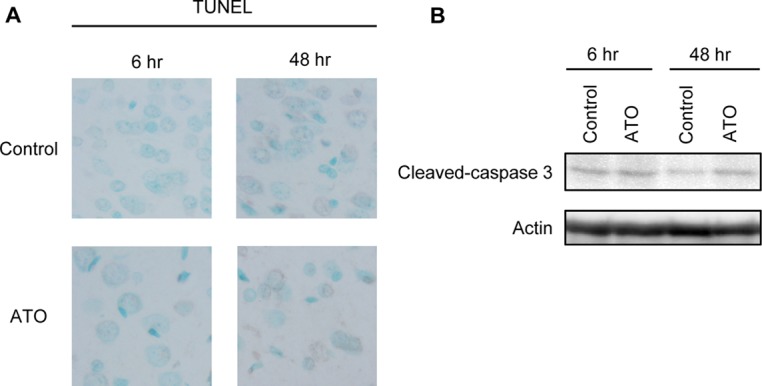
Neuronal cell death is scarcely observed in the cerebral cortex of the ATO-administered rat brain. Whole brains were excised 6 or 48 hours after the administration of ATO. (A) TUNEL analysis in the cerebrum cortex. Nuclei were also counterstained with methylene blue. Representative images (n = 4) are shown. (B) Cell lysates extracted from whole brain were subjected to western blot analysis using anti-cleaved-caspase-3 and actin antibodies. Representative results (n = 4) are shown.
In the current study, we observed the response of PML to ATO in the brain but not in the liver or heart (Fig. 1A). The apparent lack of a PML response in the liver is somewhat surprising, but this might be due to the ability of this organ to efficiently detoxify ATO15,16,17. It has been suggested that ATO might penetrate the blood-brain barrier and reach the central nervous system: it has been reported that ATO concentrations in the cerebrospinal fluid of humans treated orally with ATO reach approx. 17–18% of that of the blood concentration18. Indeed, there are some reports of coma and encephalopathy arising from ATO19. A report has suggested oxidative DNA damage in the brain of mice administered ATO orally20.
To the best of our knowledge this is the first report demonstrating the in vivo response of PML to ATO in the brain. Activation of the ATR-Chk1 system in response to ATO was also demonstrated. PML is one of the targets of ATR, and the nucleolar localization of PML is dependent on its phosphorylation by ATR21. On the other hand, PML participates in the activation of Chk1 in response to DNA damage, at least in a certain situation22. Thus, ATO might activate ATR-Chk1 in an oxidative DNA damage-dependent manner. ATO-dependent changes in PML dynamics might be also involved in the Chk1 activation. Although at present we cannot draw a definitive conclusion concerning the precise cause-and-effect relationship among them, all of these pathways should converge at the activation Chk1. Given the established importance of PML in oxidative stress and the DNA damage response in cells, the ATR-PML response to ATO in the brain demonstrated in this study should be important for understanding the pathophysiology of arsenic intoxication.
Acknowledgments
This work was supported in part by a Grant-in-Aid from the Japan Society for the Promotion of Science (16K09201 to Ko.U.).
Footnotes
Disclosure of Potential Conflicts of Interest: The authors declare that there are no conflicts of interest.
References
- 1.Hughes MF, Beck BD, Chen Y, Lewis AS, and Thomas DJ. Arsenic exposure and toxicology: a historical perspective. Toxicol Sci. 123: 305–332. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, Hudecova D, Rhodes CJ, and Valko M. Arsenic: toxicity, oxidative stress and human disease. J Appl Toxicol. 31: 95–107. 2011. [DOI] [PubMed] [Google Scholar]
- 3.Jomova K, and Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 283: 65–87. 2011. [DOI] [PubMed] [Google Scholar]
- 4.Ivanschitz L, De Thé H, and Le Bras M. PML, SUMOylation, and Senescence. Front Oncol. 3: 171 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirano S, Watanabe T, and Kobayashi Y. Effects of arsenic on modification of promyelocytic leukemia (PML): PML responds to low levels of arsenite. Toxicol Appl Pharmacol. 273: 590–599. 2013. [DOI] [PubMed] [Google Scholar]
- 6.Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, Chen Y, Zhou L, Fang ZW, Wang YT, Ma J, Zhang P, Zhang TD, Chen SJ, Chen Z, and Wang ZY. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 89: 3354–3360. 1997. [PubMed] [Google Scholar]
- 7.Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ, Si GY, Jin XL, Tang W, Li XS, Xong SM, Shen ZX, Sun GL, Ma J, Zhang P, Zhang TD, Gazin C, Naoe T, Chen SJ, Wang ZY, and Chen Z. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood. 88: 1052–1061. 1996. [PubMed] [Google Scholar]
- 8.Sahin U, Ferhi O, Jeanne M, Benhenda S, Berthier C, Jollivet F, Niwa-Kawakita M, Faklaris O, Setterblad N, de Thé H, and Lallemand-Breitenbach V. Oxidative stress-induced assembly of PML nuclear bodies controls sumoylation of partner proteins. J Cell Biol. 204: 931–945. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matt S, and Hofmann TG. The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell Mol Life Sci. 73: 2829–2850. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burma S, Chen BP, Murphy M, Kurimasa A, and Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 276: 42462–42467. 2001. [DOI] [PubMed] [Google Scholar]
- 11.Okawa S, Unuma K, Yamada A, Aki T, and Uemura K. Lipopolysaccharide induces expression of collagen VI in the rat lung. J Toxicol Pathol. 28: 37–41. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimura-Kojima H, Unuma K, Funakoshi T, Kato C, Komatsu A, Aki T, and Uemura K. Increased MFG-E8 expression and its implications in the vascular pathophysiology of cocaine abuse. J Toxicol Pathol. 29: 131–138. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hands KJ, Cuchet-Lourenco D, Everett RD, and Hay RT. PML isoforms in response to arsenic: high-resolution analysis of PML body structure and degradation. J Cell Sci. 127: 365–375. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dellaire G, and Bazett-Jones DP. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. BioEssays. 26: 963–977. 2004. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe T, and Hirano S. Metabolism of arsenic and its toxicological relevance. Arch Toxicol. 87: 969–979. 2013. [DOI] [PubMed] [Google Scholar]
- 16.Kumagai Y, and Sumi D. Arsenic: signal transduction, transcription factor, and biotransformation involved in cellular response and toxicity. Annu Rev Pharmacol Toxicol. 47: 243–262. 2007. [DOI] [PubMed] [Google Scholar]
- 17.Sumi D, and Himeno S. Role of arsenic (+3 oxidation state) methyltransferase in arsenic metabolism and toxicity. Biol Pharm Bull. 35: 1870–1875. 2012. [DOI] [PubMed] [Google Scholar]
- 18.Au WY, Tam S, Fong BM, and Kwong YL. Determinants of cerebrospinal fluid arsenic concentration in patients with acute promyelocytic leukemia on oral arsenic trioxide therapy. Blood. 112: 3587–3590. 2008. [DOI] [PubMed] [Google Scholar]
- 19.Ratnaike RN. Acute and chronic arsenic toxicity. Postgrad Med J. 79: 391–396. 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piao F, Ma N, Hiraku Y, Murata M, Oikawa S, Cheng F, Zhong L, Yamauchi T, Kawanishi S, and Yokoyama K. Oxidative DNA damage in relation to neurotoxicity in the brain of mice exposed to arsenic at environmentally relevant levels. J Occup Health. 47: 445–449. 2005. [DOI] [PubMed] [Google Scholar]
- 21.Bernardi R, Scaglioni PP, Bergmann S, Horn HF, Vousden KH, and Pandolfi PP. PML regulates p53 stability by sequestering Mdm2 to the nucleolus. Nat Cell Biol. 6: 665–672. 2004. [DOI] [PubMed] [Google Scholar]
- 22.Boichuk S, Hu L, Makielski K, Pandolfi PP, and Gjoerup OV. Functional connection between Rad51 and PML in homology-directed repair. PLoS One. 6: e25814 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]