

Abstract
Parkinson's disease (PD) is a neurodegenerative disorder, characterized by the loss of dopaminergic neurons in the substantia nigra and their projections to the striatum. Several processes have been described as potential inducers of the dopaminergic neuron death, such as inflammation, oxidative stress, and mitochondrial dysfunction. However, the death of dopaminergic neurons seems to be multifactorial, and its cause remains unclear. ATP-activating purinergic receptors influence various physiological functions in the CNS, including neurotransmission. Purinergic signaling is also involved in pathological scenarios, where ATP is extensively released and promotes sustained purinergic P2X7 receptor (P2X7R) activation and consequent induction of cell death. This effect occurs, among other factors, by oxidative stress and during the inflammatory response. On the other hand, peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) is involved in energy metabolism and mitochondrial biogenesis. Expression and activity upregulation of this protein has been related with reduction of oxidative stress and neuroprotection. Therefore, P2X7R and PGC-1α are potential targets in the treatment of PD. Here hemiparkinsonism was induced by unilateral stereotactic injection of 6-OHDA in a rat model. After 7 days, the establishment of PD was confirmed and followed by treatment with the P2X7R antagonist Brilliant Blue G (BBG) or PGC-1α agonist fenofibrate. BBG, but not fenofibrate, reverted hemiparkinsonian behavior accompanied by an increase in tyrosine hydroxylase immunoreactivity in the substantia nigra. Our results suggest that the P2X7R may be a therapeutic target in Parkinson's disease.
Keywords: Parkinson's disease (PD), P2X7 receptor, Brilliant Blue G (BBG), PGC-1α, Fenofibrate
Introduction
Parkinson's disease (PD) is a neurodegenerative disease, characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta and their projections to the striatum, resulting in motor deficits1. Therapies with dopaminergic drugs are widely used; however, lately, strategies for dopaminergic cell therapy and endogenous neuroprotection are being explored. Purinergic receptors are divided into P1 and P2 receptors, the latter classified into two subtypes, the ionotropic P2X and metabotropic P2Y receptors. While P2X receptors consist of mainly nonselective ion channels activated by 5′-adenosine triphosphate (ATP) and its derivatives, P2Y receptors are coupled to G proteins and are activated by a range of agonists such as ATP, ADP, UTP, and UDP and derivatives of these nucleotides2.
Purinergic receptors also play a role under pathophysiological conditions. When the brain is injured or neurodegeneration occurs, ATP is extensively released into the extracellular matrix, triggering apoptosis or cell survival pathways, depending on the activation of specific P2 receptor subtypes3. The P2X7 subtype (P2X7R) expressed by a range of cells, including neural cells, is activated by ATP and its derived molecules, such as α,β-meATP and BzATP. ATP release occurring due to cell death in a neurodegenerative state promotes sustained P2X7R activation, inducing the formation of large pores in the plasmatic membrane, disrupting ion balance, and triggering cell death4.
One of the mechanisms involved in P2X7R cell death induction is stimulation of oxidative stress. The peroxisome proliferator-activated receptor-γ (PPARγ) coactivator 1α (PGC-1α) is a protein that interacts with nuclear receptors, such as PPARγ, and negatively regulates transcription of NF-κB, inhibiting inflammatory responses. PGC-1α acts by mediating interactions between transcription factors and RNA polymerases, altering the expression of mitochondrial proteins. The modulation of PGC-1α occurs by phosphorylation of different sites through a range of phosphokinases, including p38 mitogen-activated kinase (MAPK) and Akt, whose combination can stimulate or inhibit PGC-1α expression5. A study showed that the use of the P2X7R antagonist Brilliant Blue G (BBG) attenuated apoptosis of neuronal cells in subarachnoid hemorrhage through MAPK inhibition6, indicating modulation of the MAPK pathway and a possible interference in PGC-1α activity. Moreover, Nagasawa and colleagues indicated that nuclear receptors for PPARγ may control P2X7R activity in astrocyte culture7. Therefore, activation of PGC-1α could be a novel therapeutic target in the treatment of diseases related to changes in mitochondrial functions. For instance, the genes expressed in response to PGC-1α stimulation in PD are less abundant, indicating a decrease in cytoprotective pathway activation. Recently, it was demonstrated that PGC-1α activation prevents the loss of dopaminergic neurons in cellular models of PD, indicating that this protein could be a potential target in PD treatment8.
The activation of P2X7R plays a pivotal role in PD. P2X7R activity and consequent ion influx lead to the death of nigrostriatal dopaminergic neurons by IL-1β release, contributing to worsening of the degenerative state9. It was demonstrated that the P2X7R antagonist A-438079 prevented dopamine store depletion in the 2,4,5-trihydroxyphenethylamine (6-OHDA) animal model of PD without counteracting dopaminergic cell loss10. A recent study using Wistar rats lesioned with 6-OHDA striatal injections that received intraperitonial (IP) BBG administration on the same day, that is, before lesion establishment and therefore indicating a preventive characteristic, resulted in a neuroprotective effect by partial prevention of dopaminergic loss in the striatum and substantia nigra11. Altogether, these data indicate that P2X7R inhibition could improve PD injury. Altogether, studies of P2X7R activity control in PD shall provide a key piece for understanding regeneration mechanisms in this neurodegenerative disease.
Based on these data, the aim of this study was to evaluate the neuroprotective/regenerative effects triggered through blocking P2X7R activity by BBG or through activation of PGC-1α by fenofibrate in an animal model of PD.
Materials and Methods
Animals
Male Sprague-Dawley rats that were 8 weeks old at the beginning of treatment were provided by the animal facility of the Institute of Chemistry, University of Sao Paulo, and housed there with free access to food and water and on a light/dark cycle of 12:12 h. All procedures were previously approved by the local ethics committee and complied with the Guidelines of the Brazilian College of Animal Experimentation (COBEA) and National Institutes of Health (NIH) Guide for Care and Use of Laboratory Animals.
Model Induction
For induction of unilateral lesion of the nigrostriatal pathway, the animals were anesthetized with ketamine (90 mg/kg, IP; Ceva, Paulínia, Brazil) and xylazine (13 mg/kg, IP; Ceva) and placed in the stereotaxic frame (KOPF, Los Angeles, CA, USA). The skull was perforated with a drill (1 mm in diameter), and 3 μl of freshly prepared 6-OHDA (7 μg/μl; Sigma-Aldrich, St. Louis, MO, USA), calculated as free base, dissolved in 0.9% saline solution containing 0.02% ascorbic acid (Sigma-Aldrich), was injected into the right hemisphere of the animal brains12, following stereotactic coordinates from the bregma: AP, −4.4; ML, −1.2; DV, −8.213. The needle was kept at the injection site for 3 min before and after injection14. After surgery, the animals received 1 ml of glycosylated saline (Merck-Millipore, Darmstadt, Germany) and 200 μl of veterinary pentabiotic (Fort Dodge, Campinas, Brazil) by subcutaneous and intramuscular injection, respectively.
Brilliant Blue G (BBG) and Fenofibrate Administration
Both treatments were initiated following 7 days of injury induction, schemed as in Figure 1. Treatment with BBG (100 mg/ml dissolved in 0.9% saline; Sigma-Aldrich) was performed by IP administration for 7 days at a dose of 50 mg/kg daily15,18. Saline injections were performed for control experiments. Fenofibrate (0.2%) was added to the food of the animals during manufacturing by the Rhoster company (Araçoiaba da Serra, Brazil). An identical diet, but without fenofibrate, was used for the control group. The animals were kept in individual boxes for 14 days. The amount in grams of food consumed was measured daily, allowing the monitoring of the quantity of fenofibrate that each animal received. The effectiveness of treatment with fenofibrate at a concentration of 0.2% in the diet has already been described19.
Figure 1.

Evaluation and treatment schedules. The black arrow indicates the course of the experiments (time unit: day). On day 1, animals were lesioned by unilateral 2,4,5-trihydroxyphenethylamine (6-OHDA) injection. Seven days later, animals underwent rotational tests to confirm hemiparkinsonism, and treatments were started. The animals were treated for 7 days with BBG and then submitted to rotational testing on days 14 and 21 (A). Animals treated with fenofibrate for 14 days were submitted to a second round of rotational testing on day 21 (B). For immunohistochemistry or Western blotting analysis, animals were sacrificed, and samples were collected on the last day of treatment. BT, behavioral test; BBG, Brilliant Blue G.
Behavioral Test: Rotational Behavior Induced by Apomorphine
Animals underwent rotational tests 7 days after the lesion of the nigrostriatal pathway with 6-OHDA, and then 7 and 14 days following treatment with BBG and 14 days after fenofibrate administration. For rotational tests, animals were weighed and injected with apomorphine hydrochloride (Sigma-Aldrich) at a dose of 0.5 mg/kg (10 mg/ml, IP) in 0.9% sterile saline supplemented with 0.02% ascorbic acid. The number of rotations in a counterclockwise direction, that is, to the side opposite to the lesion, was recorded for 30 min and shown as rotations/min. This test was used to confirm the lesion and analyze treatment effects19. Only animals showing rotational behavior 7 days following 6-OHDA were included in the study.
Immunohistochemistry
Immediately after the second rotational test, animals were deeply anesthetized with a mix of ketamine/xylazine and perfused with a peristaltic pump (World Precision Instruments, Sarasota, FL, USA) with 200 ml of phosphate-buffered saline (PBS; Synth, Diadema, Brazil) per animal to remove all blood. Subsequently, tissue fixation with a 250-ml solution of PBS/4% paraformaldehyde (PFA; Sigma-Aldrich) at 4°C was performed. At the end of perfusion, brains were removed and placed in a solution of PBS/30% sucrose (Synth) for 48 h at 4°C. Coronal sections were made in a cryostat (HM 500 OM; MICROM International GmbH, Walldorf, Germany) to obtain sequential sections of 30 μm. Slices were then treated with 0.3% H2O2 (Synth) to eliminate endogenous peroxidase activity and incubated overnight with anti-tyrosine hydroxylase (TH; for 24 h; 1:500; Sigma-Aldrich) and anti-P2X7R (for 48 h; 1:500; Sigma-Aldrich) polyclonal antibodies in PBS containing 0.1% Triton X-100 (Sigma-Aldrich). The slices were washed three times with PBS, followed by 2 h of incubation with peroxidase-conjugated secondary antibody (Jackson Laboratories, Bar Harbor, ME, USA) at a 1:500 dilution. After washing with PBS, the reaction was developed using SIGMAFAST™ 3,3′-diaminobenzidine tablets (Sigma-Aldrich).
Images were obtained using the EVOS XL Cell Imaging System (Thermo-Fisher, Waltham, MA, USA). Opti cal density measurements of photomicrographs of TH-immunostained slices were performed to determine the density of the TH+ fibers in the striatum. The measurement was made using grayscale in ImageJ software (US NIH, Bethesda, MD, USA). The values were normalized using the hemisphere control as 100% to eliminate any difference in the background of each slice. In the substantia nigra, the number of TH+ cells was detected and quantified by automated analysis using the StrataQuest software (TissueGnostics GmbH, Vienna, Austria). Nine slices obtained for each animal were subjected to automated counting of TH+ cells followed by determination of mean values ± standard error (SE) of injured and their respective control hemispheres.
Western Blotting
Animals were anesthetized with ketamine (90 mg/kg, IP) and xylazine (13 mg/kg, IP) and decapitated using a guillotine for removal of the striatum for subsequent protein analysis. The tissue was homogenized in lysis buffer containing 20 mM tris(hydroxymethyl) aminomethane (LGC, Cotia, Brazil), 1 mM ethylenediaminetetraacetic acid (EDTA; Synth), 20% glycerol (Sigma-Aldrich), 0.5% Igepal® (Sigma-Aldrich), and protease inhibitor cocktail (Amresco, Solon, OH, USA), pH 7.5. Total protein contents were quantified using the Bradford reagent (Thermo-Fisher), and 30 μg was separated with the sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) technique using the Owl Dual-Gel Vertical Electrophoresis System (Thermo-Fisher) with a 10% SDS-polyacrylamide gel-containing acrylamide, bis-acrylamide (Sigma-Aldrich), tris(hydroxymethyl)aminomethane (LGC), and SDS (Synth); transferred to polyvinylidene difluoride (PVDF) membranes (Merck-Millipore); and incubated for 16 h with anti-β-actin (1:100; Sigma-Aldrich), anti-TH (1:500; Sigma-Aldrich), and anti-P2X7R (1:500; Sigma-Aldrich) primary antibodies at 4°C. Then membranes were incubated for 2 h at room temperature with a secondary antibody conjugated to Alexa Fluor 555 (1:1,000; Thermo-Fisher) for β-actin and to Alexa Fluor 647 (1:1,000; Thermo-Fisher) for TH and P2X7R visualization. Membranes were scanned with the Typhoon phosphor imager (GE Healthcare, Pittsburgh, PA, USA), and detected bands were subjected to densitometric analysis with the ImageJ software.
Statistical Analysis
Statistical analyses were performed using Prism software (version 288 5.01; GraphPad Software, San Diego, CA, USA). Behavioral rotational immunohistochemistry and Western blotting data were analyzed with two-way analysis of variance (ANOVA) followed by Tukey's multiple comparison test. A significance level of p < 0.05 was assumed. The results shown are mean values of at least three independent experiments. Error bars represent the mean ± standard error of the mean (SEM).
Results
The effects of P2X7R inhibition and PGC-1α activation were studied in an animal model of hemiparkinsonism. Seven days after the induction of the 6-OHDA lesion, animals presented a rotational behavior after the administration of apomorphine, characteristic for this animal model of PD, confirming lesion establishment. After 7 days of daily treatment with BBG (50 mg/kg, IP) or saline, animals were resubmitted twice to the behavioral test (on days 14 and 21). While saline-treated animals presented a rotational behavior, measured as rotations/min similar to the first trial (12 ± 5.2, first trial; 12.6 ± 6.3, second trial; 14 ± 3.6, third trial), BBG treatment reduced these motor deficits of hemiparkinsonism (13.2 ± 5.1, first trial; 5.1 ± 2.2, second trial; 6.1 ± 1.9, third trial). This difference is significant (p < 0.05) when compared to the first tests (prior to BBG treatment) and when compared to the saline group (Fig. 2A). On the other hand, therapy for 14 days of diet supplemented with 0.2% PGC-1α activator (fenofibrate), which was stably consumed by the rats (data not shown), had no effect on apomorphine-induced rotational profile in this animal model of PD (10.6 ± 0.8, first trial; 14.6 ± 0.5 rotations/min, second trial) (Fig. 2B).
Figure 2.
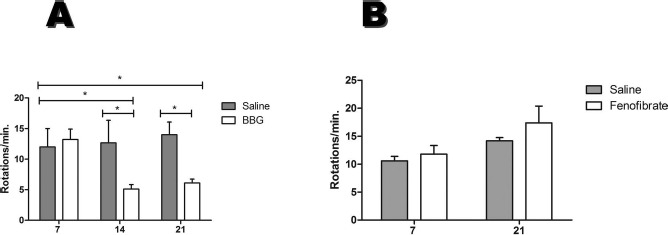
Reversion of rotation behavior following BBG treatment. The bar plots show the comparison between numbers of rotations (A) before and after 7 days of BBG (n = 12) or saline (n = 6) treatment. After the treatment, the animals were submitted to two trials of behavioral test following 7 and 14 days of treatment with BBG (B) before and after 14 days of food without (n = 6) or with fenofibrate (n = 5). Data are shown as mean values ± SEM. *p < 0.05 in relation to the BBG-treated animal group before treatment.
Percentages of TH+ cells measured by immunohistochemical analysis following BBG treatment for 7 days indicated recovery in the 6-OHDA-lesioned hemisphere (86.7% in relation to the unlesioned hemisphere), when compared to the control group treated with saline (19.9% in relation to the unlesioned hemisphere) (Fig. 3A-C). Immunohistochemical analysis of the striatum did not show any significant differences between groups (62.9% in the saline group; 61.5% in the BBG-treated group, in relation to unlesioned hemispheres; images not shown). Western blotting analysis confirmed these data in the substantia nigra (Fig. 3D), although a slight intensity increase was observed in the TH band corresponding to the striatum of the 6-OHDA-lesioned hemisphere, which had been treated with BBG (densitometry results in A.U.: 0.74 ± 0.46, BBG-unlesioned hemisphere; 0.64 ± 0.24, BBG-lesioned hemisphere; 0.85 ± 0.19, saline-unlesioned hemisphere; 0.58 ± 0.23, saline-lesioned hemisphere).
Figure 3.
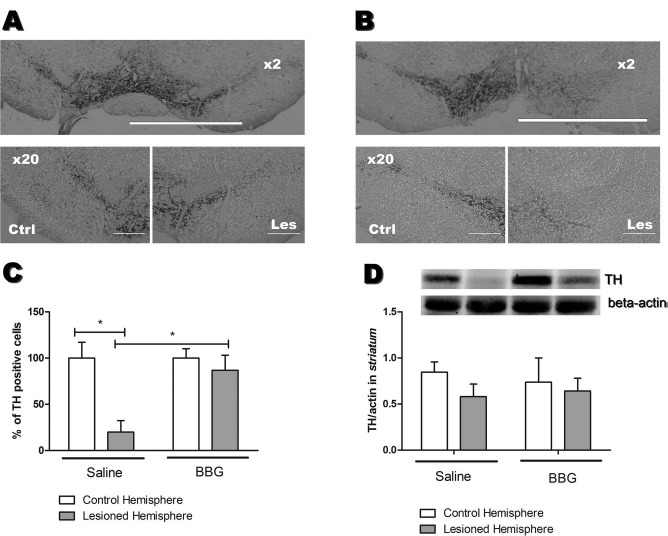
Tyrosine hydroxylase-positive (TH+) cells and protein expression following 7 days of BBG treatment. TH+ cells in the substantia nigra of (les) lesioned and (ctrl) unlesioned control hemispheres of (A) BBG- and (B) saline-treated animals. (C) Percentages of TH+ cells in the substantia nigra (n = 6). Cells were automatically counted using the StrataQuest Software (TissueGnostics). (D) Western blotting analysis of TH protein expression (60 kDa) in the striatum (n = 3). For normalization of relative TH expression levels, β-actin (42 kDa) expression levels were measured, which did not change under the used experimental conditions. *p < 0.05, when compared to the control hemisphere of saline-treated animals. Scale bars: 500 μm (2× amplified image), 50 μm (20× amplified image).
Immunohistochemical analysis of patterns of the TH+ fibers in the striatum (data not shown) and TH+ cells in the substantia nigra of fenofibrate- (Fig. 4A) and saline-treated animals (Fig. 4B) did not reveal any statistically relevant differences (36.3% in the substantia nigra and 62.3% in the striatum, in relation to unlesioned hemispheres) compared to those animals that had received placebo (52.5% in the substantia nigra and 54.8% in the striatum, in relation to unlesioned hemispheres). Statistically, the numbers of dopaminergic cells (TH+) in the injured hemisphere were different from those observed in the unlesioned control hemispheres in both groups, indicating no recovery (Fig. 4C).
Figure 4.

Effects of fenofibrate on the percentage of TH+ cells in the substantia nigra. Representative slices of TH+ cells in the substantia nigra of (les) lesioned and (ctrl) control hemispheres of hemiparkinsonian rats undergoing 14 days of (A) fenofibrate or (B) saline treatment. (C) Bar plot showing the comparison between percentages of TH+ fibers in the substantia nigra of lesioned and unlesioned control hemispheres of animals receiving food without (n = 6) or with fenofibrate (n = 5). *p < 0.01, when compared to the unlesioned control hemisphere of the respective treatment. Scale bars: 50 μm.
Western blotting analysis of the striatum showed that animals treated with BBG presented a statistically significant decrease in P2X7R expression levels (p < 0.05) in the lesioned hemispheres compared with the unlesioned hemispheres (densitometry results in A.U: 1.03 ± 0.03, BBG-unlesioned hemisphere; 0.4 ± 0.09, BBG-lesioned hemisphere) and with the saline-treated animals (0.8 ± 0.07, saline-unlesioned hemisphere; 0.98 ± 0.04, saline-lesioned hemisphere) (Fig. 5).
Figure 5.
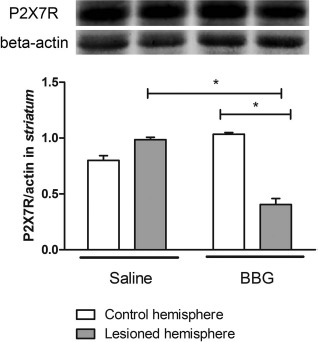
Western blotting analysis of P2X7R expression in the striatum of animals treated with BBG. Analysis of P2X7R expression (75 kDa) in the striatum of hemiparkinsonian rats undergoing 7 days of BBG or saline treatment. For normalization of relative P2X7R expression levels, β-actin (42 kDa) expression was measured, which did not change under the used experimental conditions (n = 3).
Discussion
P2X7R activity has been implicated in various neurodegenerative diseases, including PD, Alzheimer's disease, Huntington's disease, and multiple sclerosis20. As underlying mechanisms, large concentrations of ATP are released from damaged cells, resulting in prolonged exposure of neighboring cells expressing P2X7R to elevated ATP, causing cytoskeletal changes, neuroinflammation, and apoptotic cell death due to overloading of the cytoplasm and mitochondria by uncontrolled calcium influx. BBG, a noncompetitive and blood-brain barrier-permeant antagonist of the P2X7R, blocks the rat receptor subtype at least 1,000-fold more potent than P2X4R. Furthermore, BBG inhibited YO-PRO1 uptake and blebbing of membranes, which are characteristics for P2X7R activity17.
The results presented in this work provide evidence from behavorial and immunohistochemical studies that treatment with the P2X7R antagonist BBG reverses the depletion of dopaminergic neurons in an animal model of PD induced by 6-OHDA injection. 6-OHDA, a structural analog of dopamine, is a neurotoxin, which uses dopamine transporters to reach the cytosol of dopaminergic neurons. The intracellular increase in 6-OHDA concentration results in mitochondrial dysfunction, accumulation of reactive species of oxygen, and consequent cell death18.
Several cell types have their signaling pathways associated with activation of P2X7R, including immune cells that release proinflammatory cytokines, leading to the formation of reactive oxygen species and nitrogen, formation of phagolysosomes, and apoptosis22,24. Moreover, P2X7R is the major physiological regulator of the secretion of IL-125. This microenvironment activates phagocytic functions, which in turn release cytokines and interleukins activating astrocytes and microglia, regulating central nervous system immune responses, modulating survival and migration of neurons, and altering neuronal plasticity26.
Our results show the efficacy of BBG treatment in an animal PD model induced by 6-OHDA injection. While BBG treatment increased the TH immunoreactivity in the lesioned substantia nigra, such regenerative effects were not observed in the striatum. The absence of regeneration in the striatum under our experimental conditions differs from the results observed by Carmo and colleagues, who noted a prevention of TH+ neuron deficit when animals were treated with BBG concomitantly with the 6-OHDA injection11. This might be due to the preventive characteristic of the protocol of Carmo and colleagues11, since the lesion was not established before BBG administration and differs from the protocol presented here, where the dopaminergic deficit was completed before BBG administration.
Dopamine deficit in the PD striatum is preceded by a degeneration of these neurons in the substantia nigra, the structure where dopaminergic neurons are located and cast their projections to the striatum. Our results indicate that BBG treatment was sufficient to reverse the loss of dopaminergic neurons in the substantia nigra but not to restore the ramifications that continue until the striatum. In view of that, the moment of BBG effect evaluation following lesion induction becomes important for the extent of regeneration of dopaminergic neurons. It is worthwhile to emphasize that the conditions chosen in our present work better reflect the reality of PD in comparison with those of Carmo and colleagues11, as treatment starts following degeneration of a majority of dopaminergic neurons.
Even without noting the recovery from dopaminergic deficits in the striatum, a decrease in the number of rotations in the apomorphine-induced rotational test was obtained. Apomorphine injection induces rotations in animals that have at least 90% dopamine depletion in one of the hemispheres27. Therefore, a complete reversal of dopaminergic degeneration is not required to observe improvements in the rotational test. In our study, BBG treatment for 7 days (50 mg/kg, IP) reduced the number of apomorphine-induced rotations by almost 70%. Carmo and colleagues11 showed that BBG treatment at a dose of 45 mg/kg every 48 h for 14 days presented around 50% less rotations than the lesioned group, corroborating our behavioral findings, even though their treatment protocol was started prior to lesion establishment. One possible mechanism that requires further study for the P2X7 antagonist repair of dopaminergic neurons may be based on mobilization of neural stem and progenitor cells from the nearby subventricular zone28. In agreement, pharmacological inhibition or gene expression silencing of P2X7R-promoted neurogenesis of stem cell lines in vitro29,30 and functional P2X7R are expressed by adult NPCs in the subventricular zone31. Support for dopaminergic neurogenesis comes from the work of Albright et al.32, who used a nestin+ lineage tracing approach for proving adult neurogenesis for generating new dopaminergic neurons. Future studies may explore this hypothesis.
PGC-1α, a transcriptional coactivator of PPARγ, exerts neuroprotective functions by its anti-inflammatory and antioxidative characteristics. Thus, this protein might be an excellent target for PD treatment, which has in its pathophysiology these two components. However, our data show that the 0.2% fenofibrate-supplemented diet for 14 days was neither able to reverse hemiparkinsonian behavior nor restore dopaminergic levels in either the striatum or the substantia nigra. These data suggest that fenofibrate acts through anti-inflammatory effects rather than antioxidative, since fenofibrate and PPARγ agonists were discovered to be effective in the animal PD model using 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which presents a more accentuated inflammatory element than with a 6-OHDA lesion, whose mechanism of action occurs mainly through energy imbalance19,33. Although fenofibrate per se does not affect all cell death pathways involved in PD, this compound could be studied for cotreatment with other antioxidant agents, thus covering these two mechanisms of cell death.
In summary, P2X7R inhibition following establishment of PD in an animal model partially reversed behavioral deficits, such as apomorphine-induced rotation, foreseeing possible therapeutic applications. Blood-brain barrier-permeable compounds, such as BBG, are especially suitable for such approaches.
Acknowledgments
This work was supported by research grants to H.U. from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP Project No. 2012/50880-4) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq Project Nos. 486294/2012-9 and 467465/2014-2), Brazil. Fellowships were granted by FAPESP to E.G.F. (doctorate; Project No. 2011/09852-4), H.D.N.S. (doctorate; Project No. 2012/20685-5), and I.C.N. (postdoctorate; Project No. 2015/18730-0). A.O.-G. is grateful for a doctorate fellowship granted by CNPq. T.T.S. was supported by fellowships from FAPESP and CNPq. TissueGnostics (Vienna, Austria) is acknowledged for performing automated counting of tyrosine hydroxylase+ cells in the substantia nigra.
References
- 1.Schwindt T.T., Ferrazoli E.G., Ulrich H. Parkinson's Disease: Genetics, mechanisms and diagnosis. In: Hajj G.N.M., editor. Young perspectives for old diseases. Sharjah (UAE): Bentham Science Publishers Ltd.; 2014. p. 155–76. [Google Scholar]
- 2.Abbracchio M.P., Burnstock G., Verkhratsky A., Zimmermann H. Purinergic signalling in the nervous system: An overview. Trends Neurosci. 2009; 32: 19–29. [DOI] [PubMed] [Google Scholar]
- 3.Rathbone M.P., Middlemiss P.J., Gysbers J.W., Andrew C., Herman M.A., Reed J.K., Ciccarelli R., Di Iorio P., Caciagli F. Trophic effects of purines in neurons and glial cells. Prog Neurobiol. 1999; 59: 663–90. [DOI] [PubMed] [Google Scholar]
- 4.Volonté C., Apolloni S., Skaper S.D., Burnstock G. P2X7 receptors: Channels, pores and more. CNS Neurol Disord Drug Targets 2012; 11: 705–21. [DOI] [PubMed] [Google Scholar]
- 5.Róna-Vörös K., Weydt P. The role of PGC-1α in the pathogenesis of neurodegenerative disorders. Curr Drug Targets 2010; 11: 1262–9. [DOI] [PubMed] [Google Scholar]
- 6.Chen S., Ma Q., Krafft P.R., Chen Y., Tang J., Zhang J., Zhang J.H. P2X7 receptor antagonism inhibits p38 mitogen-activated protein kinase activation and ameliorates neuronal apoptosis after subarachnoid hemorrhage in rats. Crit Care Med. 2012; 41: e466–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagasawa K., Miyaki J., Kido Y., Higashi Y., Nishida K., Fujimoto S. Possible involvement of PPAR gamma in the regulation of basal channel opening of P2X7 receptor in cultured mouse astrocytes. Life Sci. 2009; 84(23–24): 825–31. [DOI] [PubMed] [Google Scholar]
- 8.Zheng B., Liao Z., Locascio J.J., Lesniak K.A., Roderick S.S., Watt M.L., Eklund A.C., Zhang-James Y., Kim P.D., Hauser M.A., Grünblatt E., Moran L.B., Mandel S.A., Riederer P., Miller R.M., Federoff H.J., Wüllner U., Papapetropoulos S., Youdim M.B., Cantuti-Castelvetri I., Young A.B., Vance J.M., Davis R.L., Hedreen J.C., Adler C.H., Beach T.G., Graeber M.B., Middleton F.A., Rochet J.C., Scherzer C.R. PGC-1α, a potential therapeutic target for early intervention in Parkinson's disease. Sci Transl Med. 2010; 2: 52ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuxe K., Manger P., Genedani S., Agnati L. The nigrostriatal DA pathway and Parkinson's disease. J Neural Transm. 2006; Suppl. 70: 71–83. [DOI] [PubMed] [Google Scholar]
- 10.Marcellino D., Suárez-Boomgaard D., Sánchez-Reina M.D., Aguirre J.A., Yoshitake T., Yoshitake S., Hagman B., Kehr J., Agnati L.F., Fuxe K., Rivera A. On the role of P2X(7) receptors in dopamine nerve cell degeneration in a rat model of Parkinson's disease: Studies with the P2X(7) receptor antagonist A-438079. J Neural Transm. 2010; 117: 681–7. [DOI] [PubMed] [Google Scholar]
- 11.Carmo M.R., Menezes A.P., Nunes A.C., Pliássova A., Rolo A.P., Palmeira C.M., Cunha R.A., Canas P.M., Andrade G.M. The P2X7 receptor antagonist Brilliant Blue G attenuates contralateral rotations in a rat model of Parkinsonism through a combined control of synaptotoxicity, neurotoxicity and gliosis. Neuropharmacology 2014; 81: 142–52. [DOI] [PubMed] [Google Scholar]
- 12.Annett L.E., Rogers D.C., Hernandez T.D., Dunnett S.B. Behavioural analysis of unilateral monoamine depletion in the marmoset. Brain 1992; 115: 825–56. [DOI] [PubMed] [Google Scholar]
- 13.Paxinos G., Watson C. The rat brain in stereotaxic coordinates. 7th ed. San Diego (CA): Academic Press; 2014. [Google Scholar]
- 14.Ebert A.D., McMillan E.L., Svendsen C.N. Isolating, Expanding, and infecting human and rodent fetal neural progenitor cells. Curr Protoc Stem Cell Biol. 2008; 6: 2D.2.1–2D.2.16. [DOI] [PubMed] [Google Scholar]
- 15.Arbeloa J., Pérez-Samartín A., Gottlieb M., Matute C. P2X7 receptor blockade prevents ATP excitotoxicity in neurons and reduces brain damage after ischemia. Neurobiol Dis. 2012; 45: 954–61. [DOI] [PubMed] [Google Scholar]
- 16.Diaz-Hernandez M., Diez-Zaera M., Sanchez-Nogueiro J., Gomez-Villafuertes R., Canals J.M., Alberch J., MirasPortugal M.T., Lucas J.J. Altered P2X7-receptor level and function in mouse models of Huntington's disease and therapeutic efficacy of antagonist administration. FASEB J. 2009; 23: 1893–906. [DOI] [PubMed] [Google Scholar]
- 17.Jiang L.H., Mackenzie A.B., North R.A., Surprenant A. Brilliant blue G selectively blocks ATP-gated rat P2X(7) receptors. Mol Pharmacol. 2000; 58(1): 82–8. [PubMed] [Google Scholar]
- 18.Ryu J.K., McLarnon J.G. Block of purinergic P2X(7) receptor is neuroprotective in an animal model of Alzheimer's disease. Neuroreport 2008; 19: 1715–9. [DOI] [PubMed] [Google Scholar]
- 19.Kreisler A., Duhamel A., Vanbesien-Mailliot C., Destée A., Bordet R. Differing short-term neuroprotective effects of the fibrates fenofibrate and bezafibrate in MPTP and 6-OHDA experimental models of Parkinson's disease. Behav Pharmacol. 2010; 21(3): 194–205. [DOI] [PubMed] [Google Scholar]
- 20.Tewari M., Seth P. Emerging role of P2X7 receptors in CNS health and disease. Ageing Res Rev. 2015; 24(Pt B): 328–42. [DOI] [PubMed] [Google Scholar]
- 21.Simola N., Morelli M., Carta A.R. The 6-hydroxydopamine model of Parkinson's disease. Neurotox Res. 2007; 11(3–4): 151–67. [DOI] [PubMed] [Google Scholar]
- 22.Di Virgilio F. The therapeutic potential of modifying inflammasomes and NOD-like receptors. Pharmacol Rev. 2013; 65: 872–905. [DOI] [PubMed] [Google Scholar]
- 23.Hewinson J., Mackenzie A.B. P2X(7) receptor-mediated reactive oxygen and nitrogen species formation: From receptor to generators. Biochem Soc Trans. 2007; 35(Pt 5): 1168–70. [DOI] [PubMed] [Google Scholar]
- 24.Miller C.M., Boulter N.R., Fuller S.J., Zakrzewski A.M., Lees M.P., Saunders B.M., Wiley J.S., Smith N.C. The role of the P2X7 receptor in infectious diseases. PLoS Pathog. 2011; 7(11): e1002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muñoz-Planillo R., Kuffa P., Martínez-Colón G., Smith B.L., Rajendiran T.M., Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013; 38: 1142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanisch U.K. Microglia as a source and target of cytokines. Glia 2002; 40: 140–55. [DOI] [PubMed] [Google Scholar]
- 27.Ungerstedt U. Postsynaptic supersensitivity after 6-hydroxy-dopamine induced degeneration of the nigro-striatal dopamine system. Acta Physiol Scand. 1971; Suppl. 367: 69–93. [DOI] [PubMed] [Google Scholar]
- 28.van den Berge S.A., van Strien M.E., Hol E.M. Resident adult neural stem cells in Parkinson's disease—The brain's own repair system? Eur J Pharmacol. 2013; 719: 117–27. [DOI] [PubMed] [Google Scholar]
- 29.Glaser T., de Oliveira S.L., Cheffer A., Beco R., Martins P., Fornazari M., Lameu C., Junior H.M., Coutinho-Silva R., Ulrich H. Modulation of mouse embryonic stem cell proliferation and neural differentiation by the P2X7 receptor. PLoS One 2014; 9(5): e96281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuahasi K.K., Demasi M.A., Tamajusuku A.S., Lenz G., Sogayar M.C., Fornazari M., Lameu C., Nascimento I.C., Glaser T., Schwindt T.T., Negraes P.D., Ulrich H. Regulation of neurogenesis and gliogenesis of retinoic acid-induced P19 embryonal carcinoma cells by P2X2 and P2X7 receptors studied by RNA interference. Int J Dev Neurosci. 2012; 30: 91–7. [DOI] [PubMed] [Google Scholar]
- 31.Messemer N., Kunert C., Grohmann M., Sobottka H., Nieber K., Zimmermann H., Franke H., Nörenberg W., Straub I., Schaefer M., Riedel T., Illes P., Rubini P. P2X7 receptors at adult neural progenitor cells of the mouse subventricular zone. Neuropharmacology 2013; 73: 122–37. [DOI] [PubMed] [Google Scholar]
- 32.Albright J.E., Stojkovska I., Rahman A.A., Brown C.J., Morrison B.E. Nestin-positive/SOX2-negative cells mediate adult neurogenesis of nigral dopaminergic neurons in mice. Neurosci Lett. 2016; 615: 50–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lecca D., Nevin D.K., Mulas G., Casu M.A., Diana A., Rossi D., Sacchetti G., Carta A.R. Neuroprotective and anti-inflammatory properties of a novel non-thiazolidinedione PPARγ agonist in vitro and in MPTP-treated mice. Neurosci. 2015; 302: 23–35. [DOI] [PubMed] [Google Scholar]