

Abstract
Islet transplantation is associated with early ischaemia/reperfusion, localized coagulation and redox‐sensitive endothelial dysfunction. In animal models, islet cytoprotection by activated protein C (aPC) restores islet vascularization and protects graft function, suggesting that aPC triggers various lineages. aPC also prompts the release of endothelial MP that bear EPCR, its specific receptor. Microparticles (MP) are plasma membrane procoagulant vesicles, surrogate markers of stress and cellular effectors. We measured the cytoprotective effects of aPC on endothelial and insulin‐secreting Rin‐m5f β‐cells and its role in autocrine and paracrine MP‐mediated cell crosstalk under conditions of oxidative stress. MP from aPC‐treated primary endothelial (EC) or β‐cells were applied to H2O2‐treated Rin‐m5f. aPC activity was measured by enzymatic assay and ROS species by dihydroethidium. The capture of PKH26‐stained MP and the expression of EPCR were probed by fluorescence microscopy and apoptosis by flow cytometry. aPC treatment enhanced both annexin A1 (ANXA1) and PAR‐1 expression in EC and to a lesser extent in β‐cells. MP from aPC‐treated EC (eMaPC) exhibited high EPCR and annexin A1 content, protected β‐cells, restored insulin secretion and were captured by 80% of β cells in a phosphatidylserine and ANXA1‐dependent mechanism. eMP activated EPCR/PAR‐1 and ANXA1/FPR2‐dependent pathways and up‐regulated the expression of EPCR, and of FPR2/ALX, the ANXA1 receptor. Cytoprotection was confirmed in H2O2‐treated rat islets with increased viability (62% versus 48% H2O2), reduced apoptosis and preserved insulin secretion in response to glucose elevation (16 versus 5 ng/ml insulin per 10 islets). MP may prove a promising therapeutic tool in the protection of transplanted islets.
Keywords: Islets transplantation, microvesicles, activated protein C, annexin A1, endothelium, β‐cells, beta cells
Introduction
Protein C is the circulating zymogen of aPC, an anticoagulant serine protease generated at the endothelial surface by thrombin‐mediated cleavage of the bound PC 1. aPC exerts its dual property in the preservation of vascular integrity and function not only by limiting thrombin generation through the proteolytic inactivation of factors Va and VIIIa but also by acting as an endothelial cytoprotector triggering the protease‐activated receptor 1 (PAR‐1)‐mediated anti‐inflammatory and anti‐apoptotic pathways 2. When bound to its specific endothelial receptor (EPCR), aPC behaves as a PAR‐1‐biased ligand through a specific cleavage at arginine 46.
Because endothelial damage is a prime sensor of ischaemia–reperfusion and a potential inducer of procoagulant and pro‐inflammatory responses, several pre‐clinical and clinical studies have investigated the interest of aPC in organ failure and examined the mechanism by which aPC protects the vessel and therefore the organ perfusion and function 3. Notably, new engineered aPC molecules, lacking anticoagulant properties but with high cytoprotective abilities, have entered a clinical trial aiming at showing neuroprotective effects in ischaemic brain stroke 4, 5.
Pancreatic islet transplantation by portal vein infusion is a cell therapy often associated with an early and acute form of ischaemia–reperfusion termed IBMIR (Instant Blood‐Mediated Inflammatory Reaction) that causes the destruction of ~70% of the graft within the first 48 hrs 6. Ischaemia is the consequence of the islet isolation that abolishes blood supply, while islet infusion through the portal vein triggers reperfusion damages before islets’ full engraftment at the extremity of the secondary liver vessels 7, 8. Infused islets and residual exocrine cells from the islet preparation trigger platelet activation, thrombin generation, the initiation of the complement cascade, the production of ROS and cytokines by leukocytes recruited at the vicinity of the islets 7, 9, 10, 11. Under such pro‐inflammatory conditions, the expression of tissue factor (TF), the cellular initiator of coagulation, is induced in leukocytes, endothelial and also insulin‐secreting cells that all shed MP bearing the active form of TF thereby promoting coagulation close to the islets 10, 12, 13. MP are plasma membrane vesicles released in response to a variety of cellular stress like inflammation or apoptosis 14. MP expose or contain active proteins, lipids, mRNAs and act as cellular effectors between vascular cells or pancreatic cells 15, 16. Regardless of the eventual presence of TF, all MP are procoagulant because they expose phosphatidylserine, an anionic phospholipid that constitutes the catalytic surface for blood coagulation complexes and that potentiates TF activity 14.
The initial interactions of the islets with the hepatic endothelium and vascular cells are crucial to islet engraftment 17, 18. Indeed, the oxygen pressure decreases along the liver lobule making liver endothelial cells (EC) highly sensitive to oxidative stress and endothelial barrier exchanges pivotal for the maintenance of the microvessel function 19. After transplantation, the restoration of the endothelial lining of the intra‐islet capillary, which is mainly supported by the recipient's endothelial and progenitor cells, is key to islets’ revascularization and function. Indeed, the islet capillary network presents an important proportion of highly fenestrated microvascular ECs that are involved not only in blood supply to endocrine cells, but that also affect adult β‐cells function, that is insulin secretion 17, 18.
MP from progenitor ECs favour endothelial cell recruitment within the islet 20. The possibility of a MP‐driven cell crosstalk between endothelial and insulin‐secreting β‐cells within the liver vessels is supported by recent in vitro data showing that a suspension of MP and exosomes harvested from isolated islets modifies endothelial cell responses 17.
Most studies have examined the noxious MP properties and very few investigated their eventual beneficial effects. Interestingly, neutrophil and endothelial‐derived MP were identified as shuttles for annexin A1 (ANXA1), an anti‐inflammatory lipocortin possibly involved in MP‐driven cytoprotection 21, 22. ANXA1 is a 37‐kD member of the Ca2+ and phospholipid binding protein, superfamily that when secreted mainly binds to its formyl peptide receptors (FPR) 23, 24, 25. Interestingly, MP released from aPC‐treated ECs were reported cytoprotective. They bear the EPCR, the specific receptor of aPC, and deliver aPC to target ECs, thereby protecting them from pro‐apoptotic and inflammatory mediators released during septic shock 26, 27.
Previous studies have underlined the interest of aPC in the preservation of islets from ischaemia–reperfusion during transplantation 28, 29 and underline a possible contribution of MP to the aPC‐mediated beta cell cytoprotection. The mechanisms of aPC‐mediated beta cell protection within the islet, which can be considered as the smallest functional architecture of the pancreas, remain yet unknown.
The aims of this study were (i) to evaluate the protective effect of aPC on beta cells in a model of ischaemia/oxidative stress under conditions mimicking IBMIR, (ii) to evaluate the effects of endothelial‐ and β‐cell‐derived MP released upon aPC treatment on naive beta cell function and survival (iii) to identify the eventual underlying mechanism of aPC‐mediated beta cell cytoprotection.
Materials and methods
Rat β Cell culture
Rat β cells, Rin‐m5f (CRL‐11605™; ATCC, Manassas, VA, USA), were seeded at 125,000 cells/cm2 in RPMI 1640 (PAN™ Biotech GmbH, Aidenbach, Germany) medium containing 4.5 g/l glucose, 10 mM HEPES, 2 mM glutamine, 1 mM sodium pyruvate and supplemented with 10% foetal bovine serum (Gibco, Saint Aubin, France) and 20 μg/ml gentamycin (Lonza, Basel, Switzerland). Cells were cultured in 12‐well culture plates at 37°C and 5% CO2 in a humidified atmosphere.
Islets isolation
Rat islets were isolated by 1 mg/ml collagenase treatment (type XI; Sigma‐Aldrich, L'isle d'Abeau Chesnes, France) followed by density gradient separation (Ficoll; Sigma‐Aldrich) as previously described 30.
Harvested islets were washed in RPMI‐1640 medium containing 2 g/l glucose (PAN™ Biotech GmbH) and hand‐picked. For optimal recovery of functional islets, each hundred recovered was maintained for 12 hrs in RPMI‐1640 supplemented with 10% foetal calf serum, penicillin/streptomycin (100 UI/ml), in a Petri dish placed under humid 5% CO2 atmosphere at 37°C before any experimental procedure.
Primary coronary artery EC
Pig hearts were collected from the local slaughterhouse (COPVIAL, Holtzheim, France), and primary coronary ECs were isolated from the left circumflex coronary arteries as previously described 31. Briefly, left circumflex coronary arteries were excised from fresh heart, cleaned of adhesive conjunctive tissues, and the remaining blood was flushed with cold phosphate‐buffered saline (PBS) without calcium. ECs were isolated by collagenase treatment (type I, Worthington Biochemicals Corp., Lakewood, NJ, USA) at 1 mg/ml for 12 min. at 37°C. ECs were cultured in Petri dishes containing MCDB 131 medium (Life Technologies SAS, St Aubin, France) and 15% foetal calf serum supplemented with penicillin (100 U/ml), streptomycin (100 U/ml), amphotericin B (250 mg/ml) and L‐glutamine (2 mM, all from Lonza, Levallois‐Perret, France) and grown for 48–72 hrs (passage 0).
MP generation, harvest and quantification
MP were harvested under sterile conditions from the supernatants of either young P1 ECs (eMPaPC) or RIN‐m5f β cells (βMP) submitted to a 70 nM aPC treatment during 24 hrs (Xigris®, Lilly). Floating apoptotic cells and debris were first discarded (800 g 15 min.) and MP washed in Hanks balanced salt solution (HBSS) and concentrated by a double‐centrifugation step (14,000 g, 1 hr). Washed MP were kept at 4°C for not more than 3 weeks. For control purposes, endothelial MP (eMPCTRL) were harvested from the supernatant of untreated ECs and isolated using the same procedure.
Total MP concentration was determined by prothrombinase assay as previously described. Briefly, MP captured onto insolubilized annexin A5 were incubated with blood clotting factors (FXa, FVa, FII) and CaCl2. Conversion of prothrombin to thrombin was revealed by a chromogenic substrate, using a spectrophotometric reader set at 405 nm. Results were expressed as nanomolar PhSer equivalent (nM PhSer eq.) by reference to a standard curve constructed using liposomes of known concentration and PhSer eq. proportion 14.
Endothelial and β cell MP‐mediated crosstalk under oxidative stress in cells and islets
Rin‐m5f rat insulin‐secreting cells were chosen as an adequate model for the study of the β cell response to prolonged oxidative stress and hyperglycaemia. Indeed, although not responsive to a short metabolic raise by glucose stimulation, Rin‐m5f develop apoptosis after prolonged exposure to H2O2 32.
When endothelial or Rin‐m5f cells reached 70% of confluency, they were treated by 100 μM H2O2 to mimic the conditions of oxidative stress during islet ischaemia (Sigma‐Aldrich), as reported elsewhere 13. Cell supernatants were collected 24 hrs after induction of the oxidative stress, MP isolated as above and the MP‐depleted supernatant was kept at 4°C for less than 1 month under sterile conditions for control purposes.
In MP‐mediated crosstalk cellular models, endothelial or β‐cell‐derived washed MP were pre‐incubated with Rin‐m5f during 6 hrs before H2O2 treatment (100 μM). In some experiments, pre‐treatment by 50 nM aPC was performed 4 hrs before application of the oxidative stress eventually combined to eMP.
MP‐mediated crosstalk within the islets was assessed 12 hrs after their isolation and culture in RPMI containing 2 g/l glucose and supplemented with 10% foetal calf serum. 20 nM PhtdSer eq. eMP or eMPAPC were added to each 20 islet suspension, 12 hrs before addition of 100 μM H2O2,.
In some crosstalk experiments, signalling pathways were inhibited by pharmacological treatment of the MP‐targeted cells. Phosphoinositide 3 kinase A (PI3K) was inhibited by 10 μM LY294002 (Tocris, Lille, France) incubated 1 hr prior addition of endothelial‐derived MP (20 nM PhSer eq.). Similarly, PAR‐1 was blocked by pre‐incubation with 10 μg/ml ATAP2 antibody (Santa Cruz, Dallas, Texas, USA) as previously described 33. Inhibition of the ALX/Formyl peptide receptor 2 (FPR2) was performed by continuous exposure to 10 μM WRW4 (Tocris).
Quantification of apoptosis in cell lineages and islets
Rinm‐5F cells were washed and permeabilized by a 70% ethanol solution at 4°C for at least 24 hrs. After three washing steps, cells were resuspended in a solution containing I‐A RNase A (Sigma‐Aldrich) for 15 min. at 37°C. Saturating concentration of propidium iodide (Sigma‐Aldrich) was applied (0.1 mg/ml) and the degree of apoptosis evaluated by the quantification of hypodiploid DNA by flow cytometry (Guava; Merck‐Millipore, Molsheim, France). Necrosis, early apoptosis and late apoptosis were defined by annexin A5/PI+, annexin A5/PI− and annexin A5/PI+, respectively. A total of 5000 cells were acquired for each individual sample.
In some experiments, apoptosis was measured in isolated islets using a modified procedure. Briefly, 20 islets were treated by 100 μM H2O2 for 4 hrs. Islets were harvested and centrifuged (800 g, 5 min.) and further submitted to trypsin (Lonza) during 10 min. at 37°C before inactivation by addition of FCS allowing the complete dissociation of the islets’ and recovery of the constitutive cells. Islet cells were further pelleted and resuspended in RPMI medium without foetal serum. Double staining by FITC‐annexin A5 and propidium iodide (PI) was performed in the dark at 25°C for 15 min.(BD Biosciences, Franklin Lakes, NJ, USA). Apoptosis was measured by flow cytometry (Guava, Merck‐Millipore) using parameters set at linear gain as above.
Insulin measurement
Insulin released in the conditioned medium of Rin‐m5F cells or in islet suspension was assessed by ELISA according to the supplier recommendations using the Matrix protocol when foetal calf serum was present in the medium (ELISA Kit Rat/Mouse Insulin; Millipore).
Kinetics of the endothelial MP capture by target beta cells and EPCR probing
Endothelial MP isolated from aPC‐treated ECs (eMPAPC) were stained using the PKH26 red fluorescence lipid probe (Sigma‐Aldrich) as described elsewhere, 13 and washed. 20 nM PKH26‐stained MP were added to growing cells at 70 % confluency in fresh medium and incubated during 1–24 hrs. Cells were then washed, fixed in paraformaldehyde 4% and kept at 4°C before EPCR labelling or assessment of PKH26‐ stained target cells by flow cytometry (see below).
After three washings, a biotinylated anti‐EPCR antibody (SantaCruz; dilution: 1:100, 1 hrs, RT) was added to the suspension. Washed cells were incubated with FITC‐streptavidin (Sigma‐Aldrich; dilution: 1:150, 1 hr, RT) and DAPI solution (5 min., 300 nM) control conditions were untreated β‐cells (CTRL) and β‐cells incubated with the secondary antibody (data not show). After washing and strip mounting, cells were observed by fluorescent microscopy (Leica FW 4000, ×40 objective). At least nine random fields were analysed by sample. Results are expressed as the percentage of PKH26‐labelled β‐cells exhibiting the red MP fluorescence and as the green fluorescence intensity/β‐cells reflecting the degree of EPCR expression on β‐cells. Analysis was performed using ImageJ software (National Institute of Health, Bethesda, Maryland, USA).
Kinetics of the PKH26‐stained MP capture by target cells was also assessed by measurement of red fluorescence in cells using a flow cytometer set at logarithmic gain (Becton‐Dickinson, Pont‐de‐Claix, France). At least 5000 events were recorded for each sample.
Pharmacological modulation of the interaction between MP and target cells
PKH26‐stained MP (20 nM) were incubated for 1 hr at 37°C with 10 μg/ml annexin A5, 20 μg/ml antibody against annexin A1, or vehicle and washed before their addition to target cells (70 % confluency) eventually pre‐incubated for 30 min. with 10 μg/ml WRW4, an inhibitor of FPR2. Red cell fluorescence was measured after 6 hrs. At least 5000 cells were acquired for each individual sample.
Protein expression in target cells
After treatment, cells were washed twice with PBS and then lysed in TRIS buffer containing protease inhibitors (5 μg/ml leupeptin, 5 mM benzamidine) and 2% Triton® X‐100 on ice. Total proteins (30 μg) were separated by electrophoresis on 10% SDS–polyacrylamide (Sigma‐Aldrich) gels as previously described (4). Blotting membranes (nitrocellulose, GE Healthcare, Amersham, UK, USA) were incubated with the different primary antibodies directed against mouse PAR‐1 (SantaCruz; 1:1000 dilution), rabbit cleaved caspase 3, mouse iNOS, mouse annexin A1 (Cell Signaling Technology, Danvers, MA, USA, 1:1000 dilution), FPR2/ALX (Abcam, cambridge USA, 1:1000) overnight at 4°C. Detection of β‐actin (Sigma‐Aldrich; 1:5000 dilution) or beta tubulin (Abcam; 1:1000) was used for normalization. After washing, membranes were incubated with the secondary anti‐mouse IgG antibody (Cell Signaling Technology, 1:10 000 dilution) at room temperature for 60 min. Pre‐stained markers (Invitrogen™, Carlsbad, CA, USA) were used for molecular mass determinations. Immunoreactive bands were detected by enhanced chemiluminescence (Amersham, GE Healthcare). Density analysis was performed using ImageQuant LAS 4000 imager (GE Healthcare).
Measurement of aPC activity at cell and MP surface
The activity of aPC bound to the cell membrane or borne by the isolated MP and concentrated from the supernatant was assessed using a specific chromogenic substrate (S2366, Cryopep, Montpellier, France) as previously described 27. In brief, cells were washed three times and incubated with 0.75 mM S2366 and absorbance was measured at 37°C using a spectrophotometer equipped with a kinetics software (Versamax, molecular Device, UK). aPC activity was calculated by reference to a standard curve (Xigris®, 0.2–30 nM). Results are expressed as nM aPC/100,000 cells and nM aPC / 10 nM MP.
Measurement of oxidative stress in beta cells
The intracellular ROS concentration was detected by the redox‐sensitive fluorescent probe dihydroethidium (DHE, Thermo Fischer, Illkirch, France), chosen for its high specificity for mitochondrial O2 −. Cells were stained with DHE (5 μM) at 37°C in dark for 30 min. and examined by a fluorescence‐activated cell sorter (FACScan; BD Biosciences) with excitation length at 480–535 nm and emission at length 590–610 nm. ROS‐positive cells were detected by red fluorescence probing. At least 5000 cells were acquired for each individual sample 34.
Islets viability
Twenty islets maintained in cell RPMI supplemented with 10 % SVF were treated during 6 hrs by 100 μM H2O2, then washed by centrifugation (500 g, 5 min.) and incubated with fluorescein diacetate (0.67 μM) and propidium iodide (4 μM) before observation by fluorescent microscopy (Leica FW 4000, ×20 objective). Nine random fields were analysed per sample. Fluorescence intensity was analysed using the ImageJ software. Results are expressed as green intensity/islet surface unit. Data were obtained from four different islet preparations.
Assessment of β‐cells function within the islets
Islets function was assessed by measuring insulin secreted in the suspension of 20 isolated islets challenged by a 10‐fold elevation in glucose concentration. Islets were pre‐incubated for 2 hrs in Krebs solution containing 100 nM HEPES, 0.5 mg/ml human albumin, 2.5 mM glucose. Aliquots from the incubation medium were taken to measure the insulin secreted at low glucose concentration. Islets were thereafter pelleted and re‐suspended in Krebs solution containing 25 mM glucose and medium aliquots were taken after 1 hr 30. All samples were stored at −20°C until insulin measurement by ELISA. Data are expressed as ng/ml of insulin per 10 islets.
Statistical analysis
Data are expressed as mean ± standard error of mean (S.E.M.) and analysed using GraphPad Prism5® Prism5 Graphpad Company, La Jolla, CA, USA. Statistical analysis between two groups was carried out using unpaired Student's t‐test. A P value <0.05 was considered significant. Experiments were performed at least in three separate experiments.
Results
aPC promotes the release of endothelial MP able to protect β‐cells against oxidative stress
An incubation with 20 nM endothelial MP (eMPaPC) harvested from aPC‐treated ECs (ECs) prevented the H2O2‐induced β‐cells apoptosis. The degree of apoptosis was reduced by threefolds (18.7 ± 3.6% versus 5.1 ± 1.2 %, Fig. 1A). The eMPaPC‐mediated cytoprotective effect was confirmed by the prevention of the H2O2‐induced drastic drop in insulin secretion, concentrations in supernatant returning to significantly higher values from 0.7 ± 0.1 ng/ml/100,000 cells in H2O2‐treated β‐cells to 10 ± 0.5 ng/ml/100,000 cells (P < 0.001, n = 4, Fig. 1B). Of note, 20 nM eMPaPC were sufficient to mediate a cytoprotective effect that was not observed in β‐cells treated by aPC alone (Fig. 1C).. Furthermore, 50 nM aPC had no additive effect to eMPaPC, suggesting a specific eMPaPC‐mediated cytoprotection. Importantly, aPC was not toxic to the β‐cells (unchanged viability and absence of apoptosis, data not shown).
Figure 1.
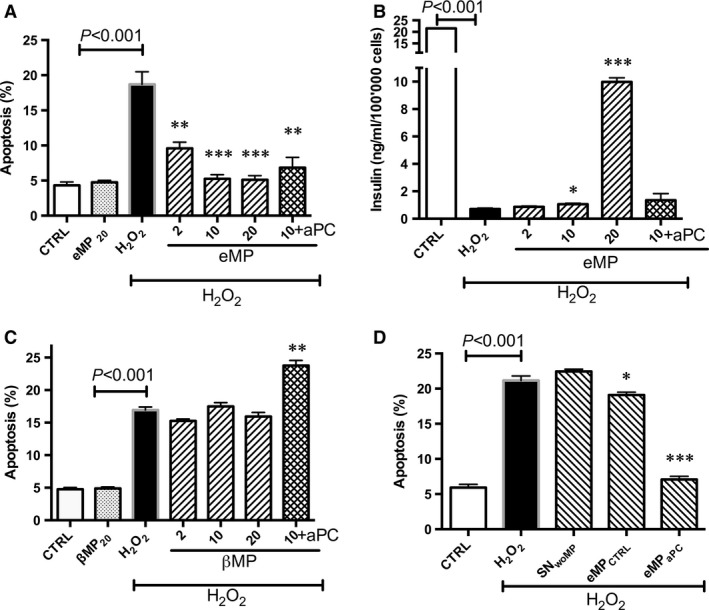
Effect of aPC alone or of aPC‐generated microparticles on β‐cells submitted to oxidative stress. (A–B) β‐cells were pre‐treated by aPC (70nM, 4 hrs) or endothelial cell‐derived MP treated by aPC (eMPaPC, 6 hrs) before the 24 h‐H2O2 treatment. Apoptosis was assessed by hypodiploid DNA labelling (A, n = 4). Insulin secreted in supernatant was measured by ELISA (B, n = 4). (C) β‐cells cells were pre‐treated with β‐cells‐derived MP (ßMP) during 6 hrs before treatment by oxidative stress, in the absence or presence of 50 nM aPC (aPC n = 4). (D) β‐cells were treated by the supernatant of control untreated endothelial cells (SNwoMP) or by MP harvested from untreated resting endothelial cells (MPCTRL) during 6 hrs prior addition of H2O2.. Data expressed as mean ± S.E.M. (aPC, activated protein C; CTRL, untreated cells; eMP, microparticles isolated from aPC‐treated endothelial cells; βMP, microparticles from β‐cells treated by aPC; PhSer eq., Phosphatidylserine equivalent. *P < 0.05 versus H2O2; **P < 0.01 versus H2O2; ***P < 0.001 versus H2O2).
Because the effector abilities of MP vary with the cell and the agonist that have initiated their generation, we examined whether beta cell‐derived MP (βMPaPC) generated by aPC treatment could also behave as autocrine effectors of β‐cells. Conversely to eMPaPC, βMPaPC had no protective effect (Fig. 1C). Interestingly, the comparison between endothelial MP harvested from naive ECs (eMPCTRL) and those generated from APC‐treated ECs showed that eMPaPC were potent inhibitors of apoptosis, whereas prevention by eMPCTRL remained negligible (18.2 ± 1% apoptosis in eMPCTRL‐treated versus 21.2 ± 0.7% in untreated H2O2‐stimulated cells, Fig. 1D). Interestingly, treatment of β‐cells by eMP‐depleted supernatants obtained after extensive centrifugation (14,000 g, 1 hr) did not lead to cytoprotection (MP+H2O2 :21. 2 ± 1.6% versus SN+H2O2 :19.1 ± 1), thereby excluding the effect of a truly soluble effector, such as cytokines or exosomes, eventually present in the conditioned medium, and confirming a specific MP‐driven response.
Altogether, these data indicate that endothelial‐derived MP are specific effectors of β‐cell cytoprotection and that treatment by aPC greatly enhances their ability.
Kinetics of eMPaPC integration by β‐cells and transfer of functional EPCR
PKH26‐labelled eMPaPC were incubated with β‐cells during 1, 6 or 24 hrs. Their integration by the cell was probed by red fluorescence, and EPCR expression at β‐cells surface was simultaneously followed using an anti‐EPCR‐biotinylated antibody. After 6‐hrs incubation with eMPaPC, 32 % of the β‐cells layer area showed red fluorescence, reaching a significant 74% plateau after 24 hrs that represented the maximal capture of the MP by β‐cells (n = 4, P < 0.001 versus MP‐untreated cells, Fig. 2B). EPCR expression, revealed by green fluorescence at β‐cells surface, increased as early as 6 hrs after the addition of eMPaPC (n = 4, P < 0.001 versus untreated cells, Fig. 2C). Green and red fluorescences followed a similar time‐curve, suggesting that eMPaPC transfer EPCR to the target cell or early prompt its expression (Fig. 2).
Figure 2.
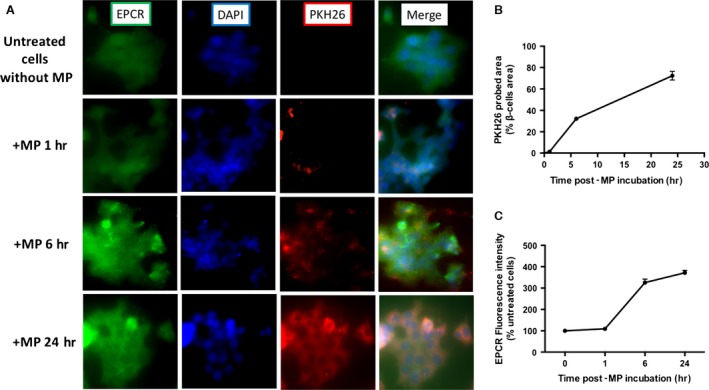
Integration of eMPaPC by β‐cells and EPCR expression. β‐cells were incubated during 1, 6, 24 hrs with PKH26‐labelled endothelial eMPaPC generated by aPC treatment, fixed and labelled with EPCR antibody and nuclei were labelled with DAPI. Representatives images of at least four independent experiments ×40 objective (A). (B) Integration of microparticles in β‐cells. Results are expressed as the percentage of PKH+ cells (n = 4). (C) EPCR expression by fluorescence intensity using FITC labelling EPCR antibody in β‐cells. Results expressed as fluorescence intensity/β‐cells normalized with control of untreated cells without MP. Data expressed as mean ± S.E.M. (n = 4). (aPC, Activated protein C; EPCR, Endothelial protein C Receptor; MP, Microparticles; PFA, paraformaldehyde).
At β‐cells surface, the presence of detectable amounts of a functional EPCR able to bind aPC was supported by the measurement of a low but concentration‐dependent aPC activity that remained, however, 10–15 times less than that quantified at the ECs surface (0.4 ± 0.1 eq nM aPC/100,000 β‐cells versus 10.5 ± 0.4 nM aPC/100,000 ECs, P < 0.001, n = 3, Fig. 3A). Similarly, a functional EPCR borne by eMPaPC was evidenced. Compared to eMPCTRL harvested from aPC‐untreated cells, MPaPC bore a significantly ~ 20‐fold greater aPC activity (eMPCTRL 0.2 ± 0.05 nM aPC versus eMPaPC 1.46 ± 0.27 nM aPC, P < 0.001, n = 5, Fig. 3B).
Figure 3.
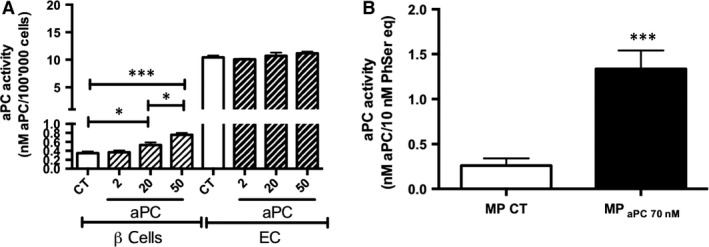
aPC activity at endothelial and β‐cell surface after 24 hrs treatment (A) and by aPC‐generated endothelial microparticles (B). Endothelial cells and β‐cells were treated by aPC during 24 hrs. Harvested endothelial MP were washed and concentrated and their quantity measured by prothrombinase assay. aPC activity was assessed using a S2366 chromogenic substrate. (A) aPC activity expressed as nM aPC/100,000 cells (n = 3‐4). (B) aPC activity expressed as nM aPC/10 nM PhSer eq. (n = 5). Data expressed as mean ± S.E.M. (aPC, activated protein C (Xigris®); CT, untreated cells; βMP, β‐cells; EC, endothelial cells; eMP, endothelial MP; MP, microparticles; PhSer eq., phosphatidylserine equivalent. *P < 0.05; ***P < 0.001).
Interaction between eMPaPC and β‐cells is a phosphatidylserine‐ and annexin A1‐dependent mechanism
To better understand the mechanisms supporting eMPaPC and β‐cells interactions, PKH26‐labelled eMPaPC were pre‐treated with annexin A5, a protein with high affinity for phosphatidylserine, WRW4, an antagonist to FPR2/ALX or with an antibody directed against annexin A1, and further incubated with beta cells (Fig. 4) . MP capture probed by the red fluorescence of beta cells was reduced by annexin A5 or by anti‐annexin A1 antibodies, whereas the FPR2/ALX antagonist had no effect (annexin A5: 22.5 ± 1 %; anti‐ANXA1: 29 ± 2 %, WRW4: 38 ± 1 % of PKH26+cells versus 40.5 ± 2 in control untreated cells, n = 4). These data are strongly suggestive of phosphatidylserine‐ and ANXA1‐dependent mechanisms governing the eMP integration in the target beta cells.
Figure 4.
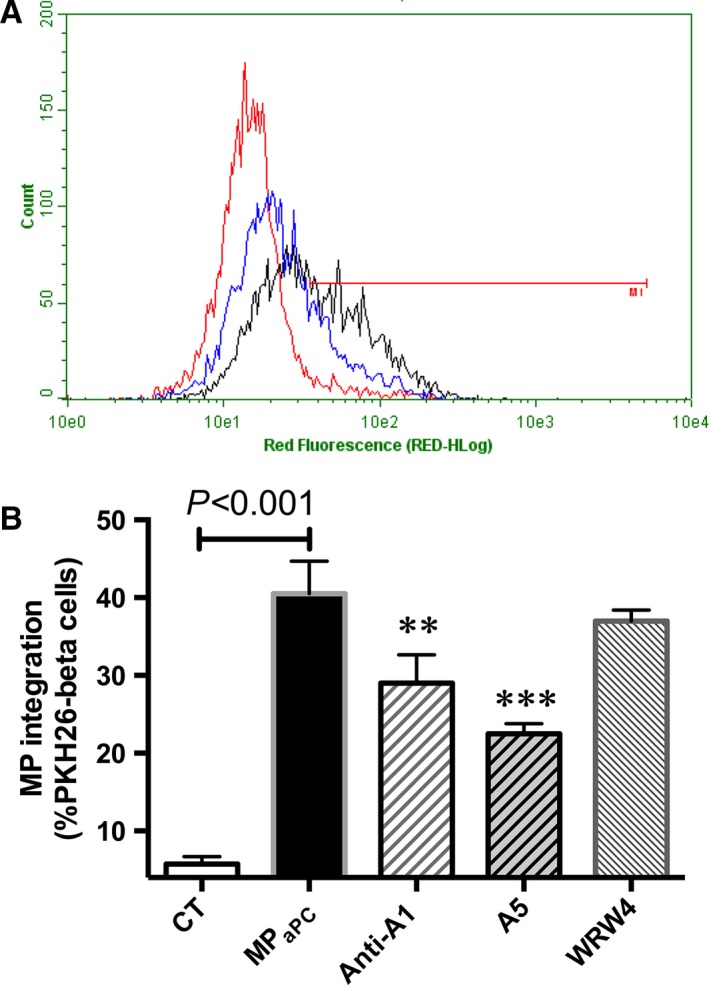
Mechanism of eMPaPC integration by β‐cells. β‐cells pre‐incubated with or without WRW4 (10 μM 1 hr), a FPR2 antagonist were incubated during 6 hrs with PKH26‐labelled eMPaPC pre‐treated or not by 10 μg/ml annexin A5 or 20 μg/ml annexin A1 antibody for 1 hr. Incorporation was assessed by flow cytometry. (A) Representative cytogram of beta cells without MP (red), with MP (black) or with annexin A5 (blue). (B) Data expressed as mean ± S.E.M., n = 4. (CT, untreated cells; eMPaPC, cells treated by endothelial microparticles released after aPC treatment. **P < 0.01 versus eMPaPC; ***P < 0.001 versus eMPaPC; †P < 0.001 versus H2O2+ eMPaPC).
Interestingly, the basal expression of ANXA1 was barely detectable by Western blot in beta cells but was significantly higher in ECs. aPC up‐regulated the endothelial ANXA1 by about 20% (Fig. 5B, P < 0.05). The eMPaPC ANXA1 content was significantly higher than that of eMPCTRL (Fig. 5D, n = 3). At the opposite, the basal PAR‐1 expression in beta and ECs was comparable and was slightly enhanced by aPC. The PAR‐1 content of eMPaPC and eMPCTRL was similar.
Figure 5.
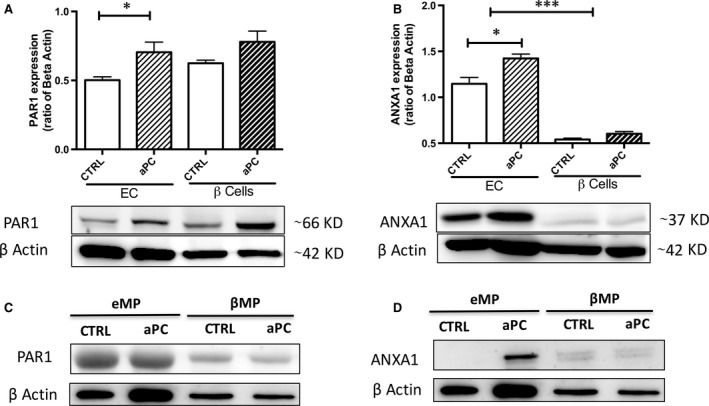
Expression of annexin A1 and PAR‐1 in eMPaPC. After 70 nM aPC treatment during 24 hrs, endothelial cells and β cell‐derived MP were harvested from supernatants, washed and concentrated. Expression of PAR‐1 (A, C) and annexin A1 (B, D) were assessed by Western blot in cell (A, B) (n = 4) and MP lysates (C, D) (n = 3). Data expressed as mean ± S.E.M. (aPC, activated protein C (Xigris®); CTRL, untreated cells; βMP, β‐cell derived MP; EC, endothelial cells; eMP, endothelial MP; MP, microparticles; PhSer eq., phosphatidylserine equivalent. *P < 0.05 versus CTRL ***P < 0.001 versus CTRL).
eMP protect beta cells by EPCR/PAR‐1 and ANXA1/FPR2 pathways
To further investigate the pathways involved in eMP‐mediated beta cell cytoprotection, the cells were treated with different modulators (Fig. 6). Pre‐incubation with a PAR‐1 neutralizing antibody (ATAP2, SantaCruz, USA) partially reversed the eMPaPC‐driven protective effect, and led to enhanced apoptosis (eMPaPC :7.1 ± 1% versus 13 ± 2.1% in eMPaPC+ATAP2) suggesting a PAR1‐dependent pathway. Similarly, pre‐incubation of β‐cells with WRW4 (10 μM) partially reversed the eMPaPC cytoprotective effect. In addition, LY294002, a PI3 kinase inhibitor, totally abolished the eMPaPC‐driven cytoprotection with a degree of apoptosis similar to the values observed in H2O2‐challenged cells. These data suggest that PI3 kinase is a common step to the PAR‐1 and annexin A1‐mediated effects (eMPaPC : 7.1 ± 1%; eMPaPC+ LY294002: 17.6 ± 1.7%; H2O2‐treated cells: 21.2 ± 0.7%).
Figure 6.
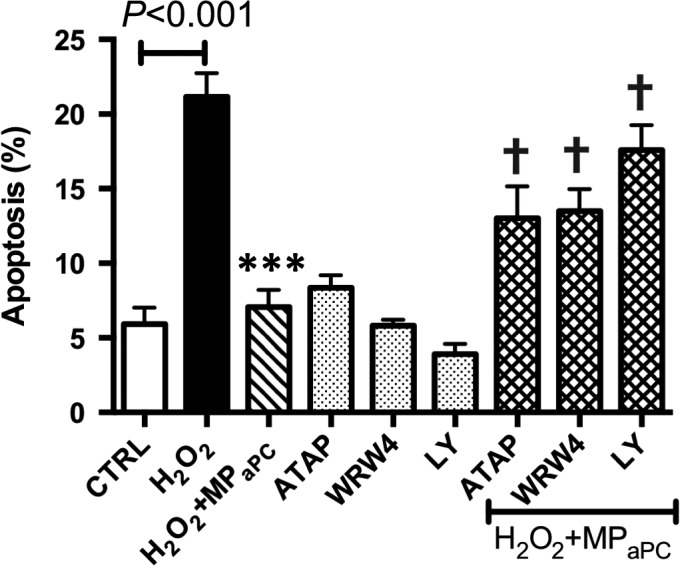
The protective effects of eMPaPC are mediated through the PAR‐1/EPCR and ANXA1/FPR2 pathways. Apoptosis of β‐cells submitted or not to oxidative stress and MP was assessed by flow cytometry. β‐cells were pre‐incubated with a PI3K inhibitor (LY 294002, 10 μM, 1 hr) or FPR2 antagonist (WRW4, 10 μM, 1 hr) or a PAR‐1 antibody (ATAP2, 10 μg/ml) before a 6‐hrs treatment by MP and H2O2 stimulation. Data expressed as mean ± S.E.M., n = 4. (CTRL, untreated cells; MPaPC, endothelial microparticles released by aPC treatment. ***P < 0.001 versus H2O2; †P < 0.001 versus H2O2+ MPaPC).
eMPaPC up‐regulate FPR2/ALX expression in β‐cells
To confirm the contribution of FPR2/ALX, we analysed its expression in H2O2‐treated β‐cells after 6 hrs. Whereas FPR2/ALX expression was low in control untreated and H2O2‐treated β‐cells, Western blots showed that 20 nM eMPaPC prompted a significant sevenfold up‐regulation of FPR2/ALX, suggesting a MP‐driven effect (P < 0.01, n = 4, Fig. 7).
Figure 7.
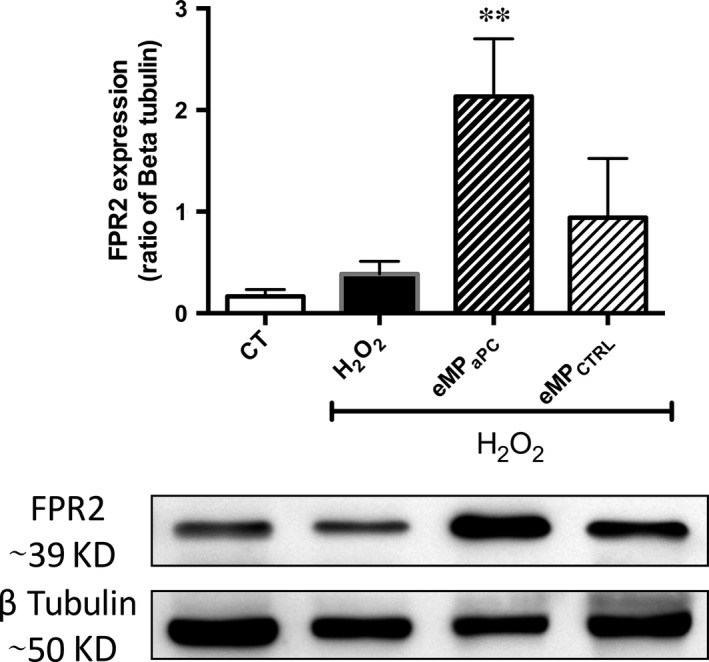
Expression of FPR2/ALX in eMPaPC ‐ and eMPCTRL‐treated β‐cells submitted to H2O2. β‐cells were pre‐incubated for 12 hrs with 20 nM eMPaPC or eMP harvested from untreated cells before addition of H2O2. FPR2/ALX expression was evidenced by Western blot in cell lysates after 24 hrs. Data expressed as mean ± S.E.M. (CTRL, untreated cells; FPR2, formyl peptide receptor 2; eMPaPC, endothelial microparticles released by aPC treatment; eMPCTRL, endothelial microparticles harvested from unstimulated cells. **P < 0.01 versus H2O2).
eMPaPC reduce markers of oxidative stress and apoptosis in β‐cells
We examined the oxygen species (ROS) content in eMPaPC‐treated cells, in the presence of H2O2. The amount of ROS by DHE fluorescence was decreased by 20 nM eMPaPC (149.4 ± 2.5 in H2O2‐treated cells versus 120 ± 4.7 in H2O2+ MP treated cells (A.U. P < 0.01, n = 4, Fig. 8A). Similarly, the expression of inducible NO synthase (iNOS) assessed by Western blot returned to baseline, suggesting its prime role in the eMPaPC‐driven counteracted ROS generation. In addition, pro caspase‐3 activation prompted by H2O2. was limited in the presence of eMPaPC, as shown by its reduced cleavage into active caspase 3 (n = 4, P < 0.001, Fig. 8C).
Figure 8.
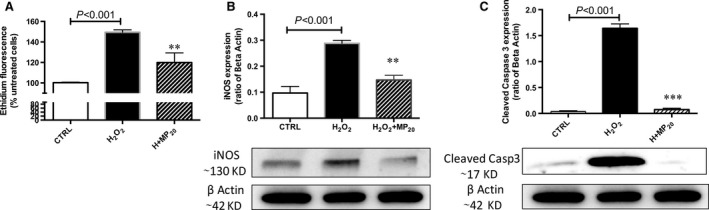
Effects of eMPaPC on H2O2‐induced β‐cells apoptosis and oxidative stress. (A) β‐cells were pre‐incubated with 20 nM eMPaPC during 12 hrs before addition of H2O2. Cells were incubated with the red probe dihydroxylethidium during 30 min. and red fluorescence was analysed by flow cytometry (n = 4). (B, C) β‐cells were pre‐incubated with 20 nM eMPaPC during 12 hrs before addition of H2O2 (n = 4). iNOS (B) and cleaved caspase 3 (C) were evidenced by Western blot in cell lysates harvested after 24 hrs. Data expressed as mean ± S.E.M. (CTRL, untreated cells; iNOS, inducible nitric oxide synthase; MPaPC, endothelial microparticles released after aPC treatment. **P < 0.01 versus H2O2; ***P < 0.001 versus H2O2).
eMPaPC protect islets against oxidative stress‐induced apoptosis and restore islet function
The β‐cell cytoprotection by eMPaPC observed above in Rinm‐5f was further confirmed in isolated islets incubated with 100 μM H2O2 during 4 hrs. eMPaPC pre‐treatment limited the H2O2‐induced apoptosis measured by PI/AnV double staining of the dissociated constitutive islet cells using flow cytometry (H2O2: 17.2 ± 4.7 % versus H2O2+MP: 3.8 ± 2.8 %, unstimulated islets: 1 ± 0.3%, P = 0.03 versus H2O2, n = 4). Accordingly, increased viability using PI/FDA double labelling was also measured (H2O2: 47.8 ± 2.8%, H2O2+MP: 61.8 ± 2%, unstimulated islets: 97.5 ± 1 %, P = 0.007 versus H2O2, n = 4). Importantly, when H2O2‐treated islets were challenged by high glucose concentration (25 mM), their ability to secrete insulin was restored by eMPaPC (H2O2: 5.3 ± 1; H2O2+MP: 16.1 ± 4.8; unstimulated: 25.2 ± 1.9, ng/ml insulin /10 islets, P = 0.04, n = 4, Fig. 9).
Figure 9.
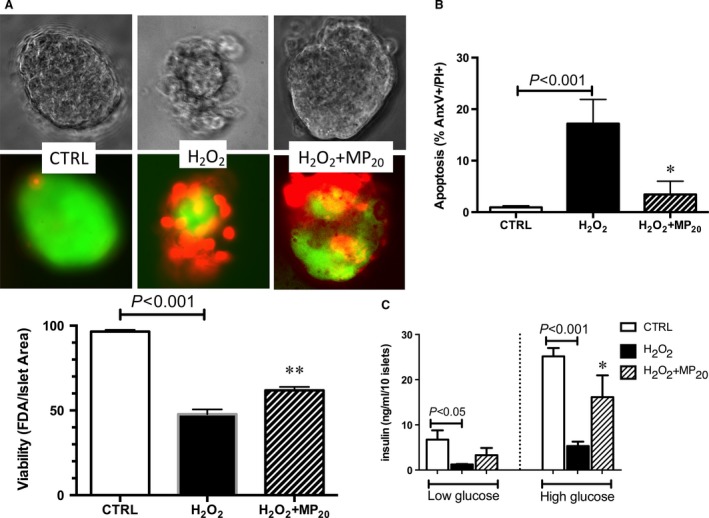
Effects of MP on islet viability, apoptosis and function. (A) Viability of islets. Rat islets were pre‐incubated with MPaPC during 12 hrs before treatment by H2O2 during 6 additional hours a : Representative photographies of one islet per each condition. Following incubation, islets were stained by green FDA probe and red propidium iodide fluorescent probes. Red and green fluorescences were analysed after dissociation of the islet constitutive cells by flow cytometry following trypsin treatment. (B) Apoptosis : Cells of islets were stained by propidium iodide and annexin A5. Data expressed as mean ± S.E.M. (C) islets function assessed by insulin secretion after glucose challenge: Islets were pre‐incubated for 2 hrs in Krebs solution containing 2.5 mM glucose before incubation in Krebs buffer containing 25 mM glucose (high glucose). (CTRL, untreated cells; FDA, fluorescein diacetate; MPaPC, endothelial microparticles released by aPC treatment. *P < 0.05 versus H2O2; **P < 0.01 versus H2O2).
Discussion
Our work demonstrates the protective effects of aPC‐generated endothelial MP on β‐cell function. The key finding of our data is that eMPaPC convey EPCR together with aPC and act as paracrine β‐cell cytoprotective effectors, in line with previous observations of their autocrine cytoprotective effects 27. In the present study, we show that eMPaPC deliver EPCR to target β‐cells and prompt an EPCR‐dependent PAR‐1 pathway within β‐cells. Interestingly, we evidenced that eMPaPC cargo annexin‐A1 and active aPC and that MP interactions with the target cell are mediated through phosphatidylserine and ANXA1‐dependent mechanisms22. In addition, EPCR and FPR2/ALX expressions in target cells were up‐regulated by eMPaPC. The preservation of β‐cell survival and function by eMPaPC was confirmed in isolated islets submitted to H2O2 to mimic the oxidative stress associated with ischaemia. Altogether, the study demonstrates that endothelial MP act as true mediators in cell crosstalk within the islets and confirms the key contribution of the endothelium for islet engraftment, survival and function of prime importance in islet‐transplanted patients 35.
aPC favours the generation of endothelial MP able to prevent β‐cell dysfunction and apoptosis
Several studies have suspected a possible MP‐mediated effect on β‐cell function and survival. Figliolini et al. studying a suspension of MP and exosomes, also termed extracellular vesicles, proposed that they would contribute to the crosstalk between endothelial and β‐cells within the islets through the delivery of specific miRNA involved in β‐cell function and endothelial angiogenesis 17. We previously reported that MP shed from β‐cells in response to oxidative or cytokine stress induce the apoptosis of naive β‐cells 13 and that eMP isolated from aPC‐treated septic rats restored the vascular tone and protect cardiac and vascular tissues from NF‐κB and cyclooxygenase‐2 activation 26. Extracellular vesicles derived from endothelial progenitor cells were also reported protective in an islet model 20. They also reduced ischaemia–reperfusion injury in rats by transferring pro‐angiogenic miRNA 36, 37.
In the present study, we accordingly evidenced limited apoptosis in β‐cells treated by endothelial‐derived MP that was highly enhanced when MP were harvested from aPC‐treated cells (Fig. 1). These observations of the eMPaPC‐mediated effects were confirmed in eMPaPC‐treated islets submitted to H2O2 (Fig. 9) and were independent from the presence of exosomes (Fig. 1D).
eMPaPC deliver EPCR and mediate PAR‐1‐dependent cytoprotective signalling
Our data extend the previous observation that eMPaPC transfer of EPCR and convey aPC to target ECs 27 (Fig. 2). We showed that eMPaPC also target β‐cells cytoprotection and relies at least in part on PAR‐1‐ and PI3‐kinase‐dependent pathways. Specific inhibition of PI3 kinase totally abolished cytoprotection (Fig. 6), in line with the reported pI3K/Akt‐dependent cytoprotective pathway initiated by the aPC‐driven cleavage of raft‐localized PAR‐1 38. In our model, the transfer of EPCR/aPC may account for the MP‐mediated beneficial effects (Figs 8 and 9). However, the eventual role of a truly soluble form of the EPCR/aPC complex eventually cleaved from eMPaPC by proteases present at the vicinity of the transplanted islet remains to be investigated in vivo. Indeed, in response to inflammatory and oxidative stress, endothelial EPCR is cleaved by TACE and shed as a soluble form in the perivascular environment, thereby blunting the cellular action of circulating aPC 39, 40. Under conditions of oxidative stress, our data indicate that eMPaPC are beneficial actors still able to rescue damaged ECs through the delivery of the functional EPCR, aPC, PAR‐1 complex and of ANXA1, of eventual interest in the preservation of the intra‐islet endothelium (Fig. 1).
Endothelial and β‐cell‐derived MP have distinct properties in response to aPC treatment
In our study, we could not detect any β‐cell cytoprotection against oxidative stress by aPC alone, although low EPCR expression was detectable as reported previously 41. In addition, βMPaPC did not either protect β‐cells and 50 nM aPC even slightly increased apoptosis (Fig. 1).
The fact that Contreras et al. demonstrated aPC efficacy in the preservation of islets and liver endothelium after islet transplantation questions the underlying mechanisms 28. In our study, despite the fact that aPC prompted PAR‐1 expression in both β‐cells and ECs, EPCR concentration and ANXA1 remained poorly expressed in β‐cells and derived‐MP compared to their endothelial counterparts (Figs 3 and 5).
Conversely, we evidenced, enhanced aPC‐driven annexin A1 expression in ECs. In line with our data, the expression of various cytoprotective proteins as a Rac1, a PI3K‐dependent EPCR downstream event has been reported 42. Furthermore, an EPCR/PAR‐1‐dependent down‐regulation of sPLA2 was described by Bae et al. 43. One explanation could be brought by our observation of the EPCR‐driven up‐regulation of ANXA1, a well‐known PLA2 inhibitor 43. Therefore, ANXA1‐enriched eMPaPC could account at least in part for the anti‐inflammatory and anti‐oxidative responses in target β‐cells and islets.
Other observations of the preservative action of aPC on β‐cells through the fine‐tuning of immunosuppressive Treg in type 1 diabetes were associated with the up‐regulation of EPCR on beta cells 41. Our data showing a direct eMPaPC‐driven modulation of the pro‐inflammatory and pro‐apoptotic responses of β‐cells are in line with these.
eMPaPC contain annexin A1 and mediate FPR2/ALX‐dependent pathways in β‐cells
As previously described in ECs, we found that PAR‐1 inhibition only leads to the partial loss of the eMPaPC cytoprotective effect in β‐cells 27. In addition, Perez‐Casal showed that aPC harboured by EPCR+‐eMP was more efficient than aPC alone in cardiovascular cytoprotection and angiogenesis 42, an observation that was confirmed by our finding that eMPaPC expose higher aPC activity and greater amounts of EPCR (Figs 2 and 3) 21, 22, 44, 45, 46
Our data bring a bunch of evidences strengthening the hypothesis of an alternate eMPaPC‐mediated ANXA1‐dependent pathway in β‐cells (Figs 4, 5, 6, 7). Indeed, eMPaPC reduced apoptosis, enhanced viability and restored insulin secretion under conditions of oxidative stress (Fig. 9), in line with the reported observation that ANXA1 directly enhances islet β‐cell secretory function 47, 48. Our data indeed confirm previous reports describing MP‐embedded annexin A1 as a contributor to the MP capture by target cells and to downstream events 22. Indeed, ANXA1‐ containing endothelial MP were proven beneficial through the transfer of either proteins, RNAs or miRNA to target cells 22, 35.
The previous observation that ANXA1 effects persisted after its removal from the culture medium 46 is consistent with our data showing that eMPaPC exert β‐cell cytoprotection through the up‐regulation of FPR2/ALX and PI3‐kinase, a downstream event previously described in the inflamed endothelium (Fig. 6) 45. In our hands, the ANXA1 content of endothelial MP was negligible when they were harvested from unstimulated ECs, whereas it was greatly enhanced by aPC treatment (Fig. 5).
Because FPR2 inhibition by the WRW4 antagonist only partially limited the eMPaPC‐driven protection, it is tempting to speculate that ANXA1 exerts indirect effects through multiple mechanisms, among which ANXA1 binding to the phospholipids of the β‐cell plasma membrane, as already reported in the presence of exogenous soluble ANXA1 23, 24, 25. However, our data demonstrate the up‐regulation of FPR2/ALX by eMPaPC in target β‐cells that would also have amplify the cell response to eMPaPC 49.
Our demonstration of an eMP‐driven cytoprotection that is FPR2/ALX and phosphatidylserine dependent is consistent with a pioneered work showing that ANXA1 binds to pancreatic islets in a calcium‐dependent and independent manner, highly suggestive of both receptor‐ and phospholipid‐mediated mechanisms 47.
Altogether, in our hands, ANXA1 appeared pivotal in the eMPaPC‐driven β‐cell cytoprotection through multiple pathways, namely enhancement of eMP capture, FPR2/ALX activation and its up‐regulation 21, 22.
Nevertheless, our data remain to be confirmed in animal models although several clues of the importance of ANXA1 in cytoprotection can be found in the literature. Indeed, ANXA1 is localized into insulin secretory granules of the islets and was suggested to modulate insulin secretion in an autocrine or paracrine manner 22, 46, 50. Under physiopathological conditions, the initial exposure of phosphatidylserine by apoptotic cells and MP shedding could be viewed as a protective response enabling the binding of the secreted ANXA1 51 of benefit through an anti‐inflammatory MP‐mediated crosstalk, eventually prompted by miRNA transfer 22, 35. Such a paracrine ANXA1 action was demonstrated for neutrophil‐derived MP that exert a powerful anti‐inflammatory effect on ECs via ANXA1 delivery 21.
In summary, this study characterizes a new cytoprotective action exerted by endothelial MP on insulin‐secreting β‐cells that could be significantly enhanced by aPC with great potency in isolated islets submitted to major oxidative stress (Fig. 10). In our model, aPC protected both β‐cells and pancreatic islets through the release of endothelial aPC/EPCR+‐MP that behaved as cellular effectors in the preservation of islet function. Two different and non‐exclusive pathways initiated by such MP have been identified: one dependent on EPCR/PAR‐1 and the other pertaining to the delivery of ANXA1 and FPR2/ALX‐mediated responses. In the context of transplantation, MP may prove a promising therapeutic tool in the pre‐ and post‐conditioning of islet grafts, especially in view of the new engineered protein C that retain cytoprotective effects with minimal anticoagulant properties 4. Our data strongly support the ability of endothelial MP to convey endogenous or cytoprotective proteins that preserve islet function, limit the graft cell loss and favour islet re‐vascularization and engraftment.
Figure 10.
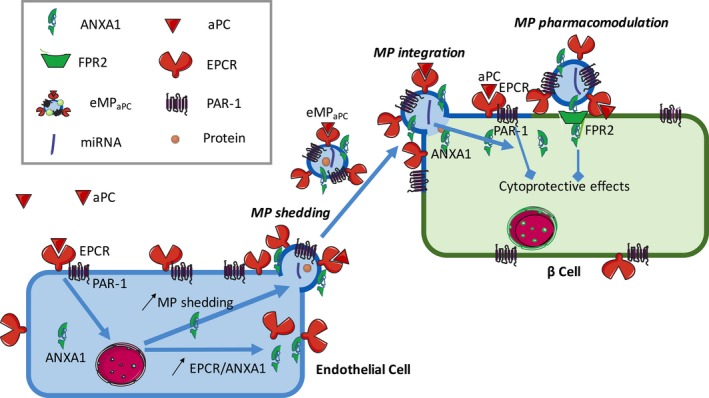
Hypothetical cytoprotective mechanisms triggered by endothelial microparticles released from aPC‐treated cells. The binding of aPC to its receptor, EPCR, causes the specific cleavage of PAR‐1 and activation of its dependent cytoprotective pathway in endothelial cell, thereby causing the up‐regulation of annexin A1 and the release of EPCR and annexin A1 enriched‐MP. These endothelial microparticles act by autocrine and paracrine pathways via the activation of the annexin A1 receptor, FPR2/ALX. They bind to target β‐cells in a phosphatidylserine and FPR2‐dependent manner and exert cytoprotection through the transfer of EPCR / PAR‐1 and downstream via the activation of FPR2/ALX and PAR‐1‐mediated pathways.
Author Contributions
GK wrote the manuscript and performed the main part of the experimental study. MK contributed to the design of improved islet isolation procedures and performed part of the islet experiments and realized the image analysis. AEH performed part of the ECs experiments and Western blot and PB contributed to the production of MP. MA and LA helped in the extraction of primary endothelial cell and performed Western blot and DHE measurements. BY measured the MP, and FZ and LA gave technical support for flow cytometry. FT, FZ and JBH gave support for the design of aPC activity measurement in MP and cells. JP contributed to flow cytometry analysis. FT, GUS and LK designed the study and FT, GUS and LK corrected the manuscript.
Disclosure
The authors of this manuscript have no conflict of interest to disclose.
Acknowledgements
We are indebted to Romain Vauchelles from the platform PIQ (Plateforme d'Imagerie Quantitative), UMR 7213 CNRS, Laboratory of Biophotonics and pharmacology, University of Strasbourg, who gave expert advices in the analysis of fluorescent cytometry images. This study was supported by the ASDIA (Assistance Service DIAbète) company. GK received grant from the University Hospital of Strasbourg.
References
- 1. Griffin JH, Zlokovic BV, Mosnier LO. Protein C anticoagulant and cytoprotective pathways. Int J Hematol. 2012; 95: 333–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007; 109: 2. [DOI] [PubMed] [Google Scholar]
- 3. Ranieri VM, Thompson BT, Barie PS, et al PROWESS‐SHOCK: Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012; 366: 2055–64. [DOI] [PubMed] [Google Scholar]
- 4. Mosnier LO, Zlokovic BV, Griffin JH. Cytoprotective‐selective activated protein C therapy for ischaemic stroke. Thromb Haemost. 2014; 7–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Y, Zhao Z, Chow N, et al Activated protein C analog promotes neurogenesis and improves neurological outcome after focal ischemic stroke in mice via protease activated receptor 1. Brain Res. 2013; 1507: 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eich T, Eriksson O, Lundgren T. Visualization of Early Engraftment in Clinical Islet Transplantation by Positron‐Emission Tomography. N Engl J Med. 2007; 356: 2754–5. [DOI] [PubMed] [Google Scholar]
- 7. Emamaullee JA, Shapiro AMJ. Factors influencing the loss of beta‐cell mass in islet transplantation. Cell Transplant. 2007; 16: 1–8. [PubMed] [Google Scholar]
- 8. Bruni A, Gala‐Lopez B, Pepper AR, et al Islet cell transplantation for the treatment of type 1 diabetes: recent advances and future challenges. Diabetes Metab Syndr Obes. 2014; 7: 211–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goto M, Tjernberg J, Dufrane D, et al Dissecting the instant blood‐mediated inflammatory reaction in islet xenotransplantation. Xenotransplantation 2008(1); 15: 225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moberg L, Johansson H, Lukinius A, et al Production of tissue factor by pancreatic islet cells as a trigger of detrimental thrombotic reactions in clinical islet transplantation. Lancet. 2002; 360: 2039–45. [DOI] [PubMed] [Google Scholar]
- 11. Özmen L, Ekdahl KN, Elgue G, et al Inhibition of thrombin abrogates the instant blood‐mediated inflammatory reaction triggered by isolated human islets: possible application of the thrombin inhibitor Melagatran in clinical islet transplantation. Diabetes. 2002; 51: 1779–84. [DOI] [PubMed] [Google Scholar]
- 12. Ma X, Ye B, Gao F, et al Tissue factor knockdown in porcine islets: an effective approach to suppressing the instant blood‐mediated inflammatory reaction. Cell Transplant. 2012; 21: 61–71. [DOI] [PubMed] [Google Scholar]
- 13. Gleizes C, Constantinescu A, Abbas M, et al Liraglutide protects Rin‐m5f β cells by reducing procoagulant tissue factor activity and apoptosis prompted by microparticles under conditions mimicking Instant Blood‐Mediated Inflammatory Reaction. Transpl Int. 2014; 27: 733–40. [DOI] [PubMed] [Google Scholar]
- 14. Hugel B, Martínez MC, Kunzelmann C, et al Membrane microparticles: two sides of the coin. Physiology (Bethesda). 2005; 20: 22–7. [DOI] [PubMed] [Google Scholar]
- 15. Buzas EI, Gy”orgy B, Nagy G, et al Emerging role of extracellular vesicles in inflammatory diseases. Nat Rev Rheumatol. 2014; 10: 356–64. [DOI] [PubMed] [Google Scholar]
- 16. Morel O, Toti F, Morel N, et al Mircroparticles in endothelial cell and vascular homeostasis: are they really noxious? Haematologica. 2009; 94: 313–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Figliolini F, Cantaluppi V, De Lena M, et al Isolation, characterization and potential role in beta cell‐endothelium cross‐talk of extracellular vesicles released from human pancreatic islets. PLoS One. 2014; 9: e102521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zanone MM, Favaro E, Camussi G. From endothelial to beta cells: insights into pancreatic islet microendothelium. Curr Diabetes Rev. 2008; 4: 1–9. [DOI] [PubMed] [Google Scholar]
- 19. Poisson J, Lemoinne S, Boulanger C, et al Liver sinusoidal endothelial cells: physiology and role in liver diseases. J Hepatol. 2016; 66(1): 212–227 doi:10.1016/j.jhep.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 20. Cantaluppi V, Biancone L, Figliolini F, et al Microvesicles derived from endothelial progenitor cells enhance neoangiogenesis of human pancreatic islets. Cell Transplant. 2012; 21: 1305–20. [DOI] [PubMed] [Google Scholar]
- 21. Dalli J, Norling LV, Renshaw D, et al Annexin 1 mediates the rapid anti‐inflammatory effects of neutrophil‐derived microparticles. Blood. 2008; 112: 2512–9. [DOI] [PubMed] [Google Scholar]
- 22. Jansen F, Yang X, Hoyer FF, et al Endothelial microparticle uptake in target cells is annexin I/phosphatidylserine receptor dependent and prevents apoptosis. Arterioscler Thromb Vasc Biol. 2012; 32: 1925–35. [DOI] [PubMed] [Google Scholar]
- 23. Sugimoto MA, Vago JP, Teixeira MM, et al Annexin A1 and the Resolution of Inflammation: modulation of Neutrophil Recruitment, Apoptosis, and Clearance. J Immunol Res. 2016; 2016: 8239258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. D'Acquisto F, Perretti M, Flower RJ. Annexin‐A1: a pivotal regulator of the innate and adaptive immune systems. Br J Pharmacol. 2008; 155: 152–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perretti M, Dalli J. Exploiting the Annexin A1 pathway for the development of novel anti‐inflammatory therapeutics. Br J Pharmacol. 2009; 158: 936–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boisramé‐Helms J, Delabranche X, Degirmenci S‐E, et al Pharmacological modulation of procoagulant microparticles improves haemodynamic dysfunction during septic shock in rats. Thromb Haemost. 2014; 111(1): 154–64. [DOI] [PubMed] [Google Scholar]
- 27. Perez‐Casal M, Downey C, Cutillas‐Moreno B, et al Microparticle‐associated endothelial protein C receptor and the induction of cytoprotective and anti‐inflammatory effects. Haematologica. 2009; 94: 387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Contreras JL, Eckstein C, Smyth CA, et al Activated protein C preserves functional islet mass after intraportal transplantation: a novel link between endothelial cell activation, thrombosis, inflammation, and islet cell death. Diabetes. 2004; 53: 2804–14. [DOI] [PubMed] [Google Scholar]
- 29. Akima S, Hawthorne WJ, Favaloro E, et al Tirofiban and activated protein c synergistically inhibit the Instant Blood Mediated Inflammatory Reaction (IBMIR) from allogeneic islet cells exposure to human blood. Am J Transplant. 2009; 9: 1533–40. [DOI] [PubMed] [Google Scholar]
- 30. Gotoh M, Maki T, Satomi S, et al Reproducible high yield of rat islets by stationary in vitro digestion following pancreatic ductal or portal venous collagenase injection. Transplantation. 1987; 43: 725–30. [DOI] [PubMed] [Google Scholar]
- 31. Auger C, Kim J‐H, Chabert P, et al The EGCg‐induced redox‐sensitive activation of endothelial nitric oxide synthase and relaxation are critically dependent on hydroxyl moieties. Biochem Biophys Res Commun. 2010; 393: 162–7. [DOI] [PubMed] [Google Scholar]
- 32. Gleizes C, Kreutter G, Abbas M, et al β cell membrane remodelling and procoagulant events occur in inflammation‐driven insulin impairment: a GLP‐1 receptor dependent and independent control. J Cell Mol Med. 2016; 20: 231–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ortiz‐Stern A, Deng X, Smoktunowicz N, et al PAR‐1‐dependent and PAR‐independent pro‐inflammatory signaling in human lung fibroblasts exposed to thrombin. J Cell Physiol. 2012; 227: 3575–84. [DOI] [PubMed] [Google Scholar]
- 34. Khemais‐benkhiat S, Idris‐khodja N, Ribeiro TP, et al The Redox‐sensitive Induction of the Local Angiotensin System Promotes Both Premature and Replicative Endothelial Senescence : preventive Effect of a Standardized Crataegus Extract. J Gerontol Biol Sci. 2015; 1–10. [DOI] [PubMed] [Google Scholar]
- 35. Jansen F, Yang X, Baumann K, et al Endothelial microparticles reduce ICAM‐1 expression in a microRNA‐222‐dependent mechanism. J Cell Mol Med. 2015; 19: 2202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cantaluppi V, Gatti S, Medica D, et al Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia‐reperfusion injury by microRNA‐dependent reprogramming of resident renal cells. Kidney Int. 2012; 82: 412–27. [DOI] [PubMed] [Google Scholar]
- 37. Bitzer M, Ben‐Dov IZ, Thum T. Microparticles and microRNAs of endothelial progenitor cells ameliorate acute kidney injury. Kidney Int. 2012; 82: 375–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bae J‐S, Yang L, Rezaie AR. Lipid raft localization regulates the cleavage specificity of protease activated receptor 1 in endothelial cells. J Thromb Haemost. 2008; 6: 954–61. [DOI] [PubMed] [Google Scholar]
- 39. Qu D, Wang Y, Esmon NL, et al Regulated endothelial protein C receptor shedding is mediated by tumor necrosis factor‐alpha converting enzyme/ADAM17. J Thromb Haemost. 2007; 5: 395–402. [DOI] [PubMed] [Google Scholar]
- 40. Menschikowski M, Hagelgans A, Eisenhofer G, et al Regulation of endothelial protein C receptor shedding by cytokines is mediated through differential activation of MAP kinase signaling pathways. Exp Cell Res. 2009; 315: 2673–82. [DOI] [PubMed] [Google Scholar]
- 41. Xue M, Dervish S, Harrison LC, et al Activated protein C inhibits pancreatic islet inflammation, stimulates T regulatory cells, and prevents diabetes in non‐obese diabetic (NOD) mice. J Biol Chem. 2012; 287: 16356–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pérez‐Casal M, Thompson V, Downey C, et al The clinical and functional relevance of microparticles induced by activated protein C treatment in sepsis. Crit Care. 2011; 15: R195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bae J‐S, Rezaie AR. Thrombin and activated protein C inhibit the expression of secretory group IIA phospholipase A(2) in the TNF‐alpha‐activated endothelial cells by EPCR and PAR‐1 dependent mechanisms. Thromb Res. 2010; 125: e9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Facio FN, Sena AA, Araújo LP, et al Annexin 1 mimetic peptide protects against renal ischemia/reperfusion injury in rats. J Mol Med. 2011; 89: 51–63. [DOI] [PubMed] [Google Scholar]
- 45. Peshavariya HM, Taylor CJ, Goh C, et al Annexin peptide Ac2‐26 suppresses TNFα‐induced inflammatory responses via inhibition of Rac1‐dependent NADPH oxidase in human endothelial cells. PLoS One. 2013; 8: e60790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rackham CL, Vargas AE, Hawkes RG, et al Annexin A1 is a key modulator of Mesenchymal Stromal Cell mediated improvements in islet function. Diabetes. 2015; 2: db150990. [DOI] [PubMed] [Google Scholar]
- 47. Hong S‐H, Won JH, Yoo S‐A, et al Effect of annexin I on insulin secretion through surface binding sites in rat pancreatic islets. FEBS Lett. 2002; 532: 17–20. [DOI] [PubMed] [Google Scholar]
- 48. Kang N, Won JH, Park YM. Annexin I stimulates insulin secretion through regulation of cytoskeleton and PKC activity. Animal Cells Syst. 13: 17–23. [Google Scholar]
- 49. Belvedere R, Bizzarro V, Popolo A, et al Role of intracellular and extracellular annexin A1 in migration and invasion of human pancreatic carcinoma cells. BMC Cancer. 2014; 14: 961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ohnishi M, Tokuda M, Masaki T, et al Involvement of annexin‐I in glucose‐induced insulin secretion in rat pancreatic islets. Endocrinology. 1995; 136: 2421–6. [DOI] [PubMed] [Google Scholar]
- 51. Blume KE, Soeroes S, Waibel M, et al Cell surface externalization of annexin A1 as a failsafe mechanism preventing inflammatory responses during secondary necrosis. J Immunol. 2009; 183: 8138–47. [DOI] [PubMed] [Google Scholar]