

Abstract
ANCA-associated vasculitis (AAV) is a highly inflammatory condition in which ANCA-activated neutrophils interact with the endothelium, resulting in necrotizing vasculitis. We tested the hypothesis that endothelial NF-κB mediates necrotizing crescentic GN (NCGN) and provides a specific treatment target. Reanalysis of kidneys from previously examined murine NCGN disease models revealed NF-κB activation in affected kidneys, mostly as a p50/p65 heterodimer, and increased renal expression of NF-κB–dependent tumor necrosis factor α (TNF-α). NF-κB activation positively correlated with crescent formation, and nuclear phospho-p65 staining showed NF-κB activation within CD31-expressing endothelial cells (ECs) in affected glomeruli. Therefore, we studied the effect of ANCA on NF-κB activation in neutrophil/EC cocultures in vitro. ANCA did not activate NF-κB in primed human neutrophils, but ANCA-stimulated primed neutrophils activated NF-κB in ECs, at least in part via TNF-α release. This effect increased endothelial gene transcription and protein production of NF-κB–regulated interleukin-8. Moreover, upregulation of endothelial NF-κB promoted neutrophil adhesion to EC monolayers, an effect that was inhibited by a specific IKKβ inhibitor. In a murine NCGN model, prophylactic application of E-selectin–targeted immunoliposomes packed with p65 siRNA to downregulate endothelial NF-κB significantly reduced urine abnormalities, renal myeloid cell influx, and NCGN. Increased glomerular endothelial phospho-p65 staining in patients with AAV indicated that NF-κB is activated in human NCGN also. We suggest that ANCA-stimulated neutrophils activate endothelial NF-κB, which contributes to NCGN and provides a potential therapeutic target in AAV.
Keywords: ANCA, endothelium, transcription factors, vasculitis, glomerulonephritis
Vascular inflammation is a hallmark of ANCA-associated vasculitis (AAV), including necrotizing crescentic GN (NCGN).1 ANCA-activated neutrophils and their adhesion to, and interaction with, the endothelium are initial steps culminating in necrotizing vasculitis.2 Signal transduction pathways have been identified that control inflammatory neutrophil priming and subsequent ANCA-induced neutrophil activation. Protein kinase C,3 the mitogen-activated protein kinases (MAPK) p38 MAPK, the extracellular signal-regulated kinase,4 phosphatidylinositol 3-kinase γ (PI3Kγ),5,6 and the spleen tyrosine kinase7 mediate either neutrophil priming or ANCA-induced neutrophil activation. These signaling pathways not only allow mechanistic insight in the activation process, but also provide potential therapeutic targets for ANCA-induced NCGN. For example, myeloid PI3Kγ deficiency, pharmacologic PI3Kγ, and p38 MAPK inhibition reduced ANCA-induced neutrophil activation in vitro, as well as glomerular necrosis and crescent formation in murine MPO-ANCA mouse models in vivo.8,9
ANCA-activated neutrophils cause endothelial cell (EC) activation and damage in vitro.2,10–12 Moreover, patients with active AAV show increased endothelial activation and injury markers,13–15 and circulating detached ECs provide damage markers that correlate with disease activity.16 In contrast to neutrophils, the signal transduction pathways that mediate EC activation and injury in the ANCA context are not well characterized. NF-κB signaling controls gene expression during inflammation and immunity,17–19 and accumulating evidence suggests that NF-κB signaling contributes to renal diseases, including GN.20–24 NF-κB consists of five Rel protein family members, namely p50, p65, p52, c-Rel, and RelB that form homo- and heterodimers. Both pro- and anti-inflammatory NF-κB effects exist, and inhibiting the wrong subunit at the wrong time can be deleterious.25,26 Thus, the exact NF-κB contributions need to be determined for each disease type. We tested the hypothesis that endothelial NF-κB is activated by ANCA-stimulated neutrophils, mediates glomerular necrosis and crescent formation, and provides a potential treatment target.
Results
NF-κB Is Activated and Correlates with Disease Severity in Murine Anti-MPO Antibody-Induced NCGN
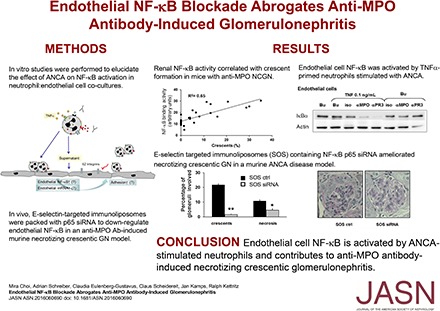
To gain insight into NF-κB activation in NCGN by ANCA, we retrospectively analyzed kidneys from our previous bortezomib study where we had induced NCGN by MPO-immunization of MPO-deficient mice followed by transplantation with wild-type bone marrow.27 Bortezomib, a proteasome inhibitor known for its NF-κB blocking effect, provided varying degrees of protection in this NCGN model. Mice in this study were either treated or not with an early or a late bortezomib protocol.27 We prepared nuclear kidney extracts that were subjected to electrophoretic mobility shift assay (EMSA). Extracts from an animal with 35% crescents showed strong NF-κB DNA binding compared with extracts from a bortezomib-treated animal without any crescent formation (Figure 1A). Supershift experiments with antibodies directed against NF-κB family members determined p50 and p65 as the prevailing components of the NF-κB heterodimeric complex. When we correlated the ODs of p50/p65 as a measure of NF-κB activation with the percentage of crescents, we observed a strong and significant positive correlation (Figure 1B). To corroborate this finding, we assessed NF-κB–dependent TNF-α mRNA expression in the same samples by quantitative RT-PCR and found a significant positive correlation with crescent formation (Figure 1C). To eliminate treatment effects, we restricted our analysis to mice that had not received active treatment (six from the bortezomib and eight from another study28) and again observed a significant correlation between the OD of the p50/p65 complex and crescent formation (Figure 1D). We also analyzed nuclear kidney extracts from two additional previous studies that used protective approaches. We found that the combination of glucocorticoid and cyclophosphamide,27 as well as dipeptidyl peptidase 1 deficiency in myeloid cells,28 reduced both crescent formation and renal NF-κB activity, suggesting that protection from renal inflammation by any means is accompanied by reduction of NF-κB activators (Supplemental Figure 1, A and B).
Figure 1.

NF-κB activity correlates with glomerular crescent formation in mice with anti-MPO antibody-induced NCGN. (A and B) Nuclear kidney extracts from untreated (n=6) or bortezomib (BTZ)-treated mice (n=22) in which we had induced NCGN using an anti-MPO antibody disease model were analyzed by EMSA. (A) Examples from a BTZ-treated mouse with 0% crescent formation (BTZ 0%) and an untreated mouse with 35% crescents (no 35%) are shown. Using the latter sample, we performed competition with a cold probe (comp) and supershift experiments using antibodies to p50, p65, c-Rel, and RelB as indicated. Note that longer exposure was needed to detect the p65 shift (p65*). (B) OD of the p50/p65 heterodimer was assessed in each sample using arbitrary units, and plotted against the percentage of glomerular crescents (n=28, correlation coefficient R2=0.65). (C) mRNA was prepared from the kidney of each mouse shown in (B), TNF-α mRNA expression was analyzed by quantitative RT-PCR and plotted against the percentage of glomerular crescents (n=28, correlation coefficient R2=0.77). (D) OD of the p50/p65 heterodimer was assessed in samples from NCGN mice that had not received active treatments and plotted against the percentage of glomerular crescents (n=14, correlation coefficient R2=0.60). The insert shows significantly less NF-κB activity in mice with <10% crescents (<10%) compared with those with >10% crescents (>10%). (E) Costaining using fluorescence-labeled antibodies to the EC marker CD31 (red) and phospho-p65 (green), together with DAPI staining of nuclei (blue) was performed in renal tissue from untreated mice with NCGN (NCGN) versus BTZ-protected mice (BTZ-protected). Weak phospho-p65 staining occurred in glomeruli from protected mice and strong signals in crescentic glomeruli of NCGN mice. Overlay images with magnifications in the very right panels indicate that some of the intense nuclear phospho-p65 staining in crescentic glomeruli was located within CD31-positive ECs. Low-powered images with scale bars, 50 μm; original magnification, ×40. A typical example is shown. **P<0.01.
When we performed phospho-p65 staining of kidney sections, we observed strong NF-κB activation in crescentic glomeruli of untreated mice and less activation in unaffected glomeruli from bortezomib-protected mice (Figure 1E). Overlays with endothelial CD31 and nuclear DAPI clearly established that some of the NF-κB activation occurred within glomerular ECs. We concluded from these analyses that NF-κB is activated in kidneys from NCGN mice, most prevalent as a p50/p65 heterodimer, correlated with crescent formation, and affecting the glomerular endothelium.
ANCA-Stimulated Neutrophils Activate Endothelial NF-κB in Neutrophil–Endothelial Cocultures
We next tested the hypothesis that ANCA activates NF-κB in neutrophils, ECs, or both cell types in vitro. We first stimulated TNF-α–primed neutrophils with monoclonal anti-MPO and anti-PR3 antibodies. To avoid strong NF-κB activation by TNF-α itself, we reduced the most commonly used 2 ng/ml priming dose to 0.1 ng/ml. This dose still caused detectable NF-κB activation as measured by IκBα degradation (Figure 2A). However, neither mAb resulted in additional IκBα degradation. In the next step we increased complexity and established neutrophil/HUVEC cocultures. TNF-α–primed neutrophils were incubated with mAb to PR3 and MPO and then added to the EC monolayers. Neutrophils were removed by washing and EC were further analyzed. These neutrophils induced endothelial NF-κB as measured by IκBα degradation, whereas the isotype control had no effect (Figure 2B). In contrast, we did not observe endothelial NF-κB activation when TNF-α priming was omitted. Several inflammatory mediators prime neutrophils for subsequent ANCA activation. In addition to TNF-α, we tested low-dose LPS, another NF-κB inducer, and formyl-methionyl-leucyl-phenylalanine (fMLP) that does not activate NF-κB. We observed that priming with all these compounds allowed anti-PR3 and anti-MPO antibody-treated neutrophils to induce significant endothelial NF-κB activation (Figure 2C). Primed neutrophils exposed to human PR3- and MPO-ANCA IgG activated endothelial NF-κB similar to the mAbs (Figure 2D). Our experiments indicate that ANCA do not activate NF-κB in neutrophils, whereas ANCA-stimulated neutrophils activate endothelial NF-κB.
Figure 2.

Primed neutrophils stimulated with mAbs to MPO, PR3, and human ANCA IgG activate endothelial NF-κB in cocultures. A typical example for IκBα immunoblotting and the corresponding ODs (OD as arbitrary units) of the IκBα bands are provided for (A) lysates obtained from neutrophils stimulated with buffer control (Bu) or primed with 0.1 ng/ml TNF-α and subsequently treated for 60 minutes with Bu, an isotype control (iso), or mAbs to MPO (αMPO) and PR3 (αPR3), respectively (n=4); and (B) lysates obtained from ECs after 2 hours coculture with neutrophils that were treated with Bu or primed with 0.1 ng/ml TNF-α and subsequently incubated with Bu, iso, or αMPO and αPR3, respectively (n=8 for conditions shown in lanes 1–5, and n=4 for lanes 6–8). α-Actin served as a loading control. (C and D) OD measurements of the IκBα bands in lysates from ECs cocultured with neutrophils for 2 hours. (C) Neutrophils were either not primed or primed with 0.1 µg/ml LPS (black) or 10−8 M fMLP (gray) as indicated, subsequently incubated with αMPO and αPR3, respectively and added to the EC monolayers (n=6). (D) Neutrophils were either not primed or primed with TNF-α and subsequently incubated with Bu, human control IgG (huCtrl), MPO-ANCA (huMPO), or PR3-ANCA (huPR3), respectively, and added to the EC monolayers (n=5). *P<0.05; **P<0.01.
Direct Cell–Cell Contact Is Not Mandatory for Neutrophil-Mediated Endothelial NF-κB Activation In Vitro and NF-κB–Dependent Gene Expression
To test whether neutrophils need direct EC contact to activate endothelial NF-κB, we generated cellfree supernatants (SN) from primed neutrophils that were subsequently stimulated with both mAbs, respectively. We detected significant IκBα degradation in ECs incubated with these SN, but not with SN from isotype control–stimulated neutrophils (Figure 3A).
Figure 3.

Endothelial NF-κB activation by ANCA-stimulated primed neutrophils does not require direct neutrophil/EC contact and leads to upregulation of IL-8, and increased neutrophil adhesion. (A–D) ECs were incubated with cellfree SN harvested from neutrophils treated for 5 hours with buffer (Bu) or primed with 0.1 ng/ml TNF-α and subsequently incubated with Bu, an isotype control (iso), or mAbs to MPO (αMPO) and PR3 (αPR3), respectively. All samples were analyzed on the same SDS gel and membrane. (A) ECs were incubated with cellfree neutrophil SN for 60 minutes and IκBα was assessed by immunoblotting. A typical example and the corresponding ODs (OD as arbitrary units) of the IκBα bands from all experiments are shown (n=5). α-Actin served as a loading control. (B) ECs were incubated with cellfree neutrophil SN for 6 hours and IL-8 mRNA was assessed by quantitative RT-PCR (n=5). (C) ECs were incubated with cellfree neutrophil SN for 18 hours and IL-8 protein was assessed in EC SN by ELISA (n=5). (D) ECs were preincubated with Bu (black) or the specific IKKβ inhibitor BMS (gray) for 60 minutes before cellfree neutrophil SN were added. After 18 hours, freshly isolated neutrophils were added, and neutrophil adhesion was determined after 120 minutes (n=5). *P<0.05; **P<0.01.
IL-8 is an NF-κB–controlled chemokine with strong effects on neutrophils. We found that ECs incubated with SN from ANCA-treated primed neutrophils not only caused IκBα degradation, but also upregulated IL-8 mRNA expression by quantitative RT-PCR (Figure 3B) and released IL-8 protein by ELISA (Figure 3C). Thus, no direct cell contact is needed for endothelial NF-κB activation by ANCA-activated neutrophils. Moreover, NF-κB activation resulted in increased transcription and protein production of NF-κB–dependent genes exemplified by IL-8.
Endothelial NF-κB Activation by ANCA-Stimulated Neutrophils Promotes Adhesion
We hypothesized that upregulation of endothelial NF-κB promotes inflammation by attracting new neutrophils to the endothelium. To test this assumption, we incubated the EC monolayer with cellfree SN from primed neutrophils that were stimulated with mAbs to PR3 and MPO, respectively. Thereafter, isolated neutrophils were added to the EC monolayer. We observed enhanced neutrophil adhesion on ECs incubated with SN from ANCA-treated neutrophils and this effect was blocked by EC pretreatment with the specific IKKβ inhibitor BMS (Figure 3D). Thus, endothelial NF-κB induced by ANCA-stimulated neutrophils provides an inflammatory amplification loop by increasing neutrophil adhesion.
TNF-α Release from ANCA-Activated Primed Neutrophils Mediates Endothelial NF-κB Activation
To gain initial insight into the mechanisms by which ANCA-stimulated neutrophils mediate endothelial NF-κB activation, we tested several candidates. We excluded neutrophil-derived microparticles (Figure 4A) and NADPH oxidase-dependent mediators, but observed that heat inactivation abrogated the NF-κB stimulating properties of SN (Figure 4B). This observation suggested the presence of a soluble mediator. When we treated the SN with neutralizing antibodies to TNF-α, we observed significantly reduced NF-κB activation in ECs, which was not observed with neutralizing antibodies to IL-8 (Figure 4C). An inhibitory effect of neutralizing anti–IL-8 antibodies could be demonstrated on IL-8–induced neutrophil migration in parallel experiments (data not shown). Thus, TNF-α released from ANCA-activated neutrophils mediates, at least in part, endothelial NF-κB activation.
Figure 4.

TNF-α released from ANCA-activated primed neutrophils contributes to endothelial NF-κB activation. (A) ECs were incubated for 60 minutes with cellfree SN harvested from neutrophils treated for 5 hours with buffer (Bu), or primed with 0.1 ng/ml TNF-α and subsequently incubated with Bu, an isotype control (iso), or mAb to MPO (αMPO), respectively (lanes 1–3). Microparticles were isolated from these SN, and either the microparticles or the microparticle-free SN were added to the ECs for 60 minutes (lane 4–9). IκBα was assessed by immunoblotting. A typical example and the corresponding ODs (OD as arbitrary units) of the IκBα bands from all experiments are shown (n=3). α-Actin served as a loading control. (B) Neutrophil SN preparation and EC treatment for samples in lane 1–3 was as described in (A). When indicated, SN were heat-inactivated or generated from 10 μM DPI-treated neutrophils (n=3). (C) Neutrophil SN preparation and EC treatment for samples in lane 1–3 were as described in (A). When indicated, SN were treated for 60 minutes with control IgG (IgG) or neutralizing antibodies for TNF-α (αTNF, left panel) or IL-8 (αIL-8, right panel). Note, fMLP was used as the priming agent in these experiments (n=4). *P<0.05; **P<0.01.
E-Selectin–Targeted Downregulation of the Endothelial p65 NF-κB Subunit Abrogates NCGN
The p50 subunit has anti-inflammatory properties, whereas p65 is mainly proinflammatory. As a proof of principle study, we used siRNA in a murine ANCA vasculitis model to downregulate p65 in the inflamed endothelium. We previously showed that liposomes conjugated with antibodies to E-selectin deliver the cargo to the activated glomerular endothelium.29 We packed cationic SAINT lipid containing anti–E-selectin liposomes (SAINT-O-Somes; SOS)30 with p65 siRNA and used a murine anti-MPO antibody transfer model for NCGN induction, as previously reported.31 We first injected mice with TNF-α to upregulate endothelial E-selectin followed by the application of SOS p65 siRNA. An “empty” SOS vector conjugated with E-selectin antibodies served as control treatment. At 48 hours thereafter, anti-MPO antibodies were administered followed by an LPS priming dose. Seven days after disease induction, we found marked erythrocyturia, leukocyturia, and proteinuria by dipstick analysis (Figure 5A) and albuminuria by ELISA (Figure 5B) in control-treated mice. Urinary abnormalities were reduced with p65 siRNA. Histology revealed that mice in the control-treated group developed substantial glomerular crescents and necrosis. The percentage of both was significantly diminished in mice that received p65 siRNA (Figure 5, C and D). We reasoned that less endothelial NF-κB activation reduced inflammatory cell recruitment into the kidneys. Flow cytometry revealed a strong influx of macrophages/monocytes and, to a lesser extent, neutrophils into the kidneys of control-treated mice that was significantly reduced with p65 siRNA (Figure 5, E and F). EMSA showed representative glomerular nuclear extracts from kidneys of two control- and two p65 siRNA–treated mice (Figure 6A) and analyzed glomerular nuclear extracts of all animals (Figure 6B). The data show significantly lower NF-κB DNA binding activity with p65 siRNA compared with the control treatment. The fact that p65 siRNA reduced NF-κB binding activity in the band shift supports the p50/p65 prevalence, as concluded from the data shown in Figure 1A. Moreover, less NF-κB activity was paralleled by lower NF-κB–dependent TNF-α mRNA and this mRNA expression correlated with crescent formation (Figure 6C). Costaining of kidney sections with phospho-p65, CD31, and DAPI together with quantitative assessment showed significantly decreased NF-κB activation in glomerular ECs of siRNA- versus control-treated mice (Figure 6D). In an alternative approach, we administered p65 siRNA and control liposomes without prior TNF-α priming, and after LPS and anti-MPO antibody induction. We again observed nephritic urine, albuminuria, and glomerular necrosis, as well as crescents in the control group; however, we observed no significant protection by p65 siRNA (Supplemental Figure 1C).
Figure 5.

E-selectin targeted downregulation of the p65 NF-κB subunit using SOS p65 siRNA abrogates anti-MPO antibody-induced NCGN in mice. (A) Semiquantitative dipstick urine analysis from anti-MPO antibody-treated mice that received a control compound (black bars) or SOS p65 siRNA (gray bars). (B) Albuminuria was quantified by ELISA. (C) Histologic analysis of glomerular crescents formation and necrosis using periodic acid–Schiff staining, with a typical example given in (D), original magnification, ×40. (E and F) Flow cytometry of kidney tissue suspension to assess monocytes/macrophage and neutrophil influx into the kidneys of control-treated versus SOS p65 siRNA-treated mice. n=5 mice in each treatment group; *P<0.05; **P<0.01.
Figure 6.

Preemptive SOS p65 siRNA treatment of mice inhibited glomerular NF-κB activation by EMSA and NF-κB–dependent TNF-α mRNA expression by quantitative RT-PCR that correlated with crescent formation. (A) Glomeruli were prepared from two control-treated (SOS ctrl) and two SOS p65 siRNA-treated (SOS siRNA) mice and NF-κB was assessed by EMSA. (B) Nuclear kidney extracts from SOS ctrl (black, n=5) and SOS siRNA (gray, n=5) mice were subjected to EMSA and the OD of the p50/65 heterodimer was assessed. (C) mRNA was prepared from the kidney of each mouse shown in (B), TNF-α mRNA expression was determined by qRT-PCR and plotted against the percentage of crescents. (D) Costaining using fluorescence-labeled antibodies to the EC marker CD31 (red), phospho-p65 (green), and DAPI staining for nuclei (blue) was performed in renal tissue from SOS ctrl– and SOS siRNA–treated mice. A typical microscopy example (original magnification, ×40) together with the corresponding quantitative assessment of nuclear phospho-p65 staining within CD31-positive glomerular ECs using Image J is depicted. **P<0.01.
NF-κB Is Activated in Glomeruli of Patients with AAV and Active Lesions
Finally, to test the relevance of our findings to the human disease we performed phospho-p65 staining in renal biopsies from patients with AAV using immunohistochemistry. For comparison, we used normal tissue from nonaffected parts of surgically resected tumor nephrectomies and from disease controls with anti-GBM GN, hypertensive and membranous nephropathy (Figure 7, A–D). We observed enhanced nuclear phospho-p65 staining within the glomerular compartments of AAV and anti-GBM disease. When we compared AAV biopsy samples, we observed that glomeruli with crescents or necrosis to glomeruli that appeared normal by light microscopy or showed sclerosis had significantly high numbers of phospho-p65–positive nuclei/glomerulus in glomeruli with active lesions (Figure 7E). Costaining with phospho-p65, CD31, and DAPI, together with quantitative assessment also indicated increased NF-κB activation in the endothelium of crescentic compared with normal glomeruli from tumor nephrectomies (Figure 7F). Additional NF-κB activation was observed in crescents, presumably in macrophages. These data support the notion that glomerular NF-κB activation occurs not only in a murine disease model but also in patients with AAV, and that NF-κB activation includes the glomerular endothelium.
Figure 7.

NF-κB is activated in glomeruli of patients with AAV and active lesions. (A–D) Immunohistochemistry for phospho-p65 was performed in renal tissue from patients with tumor nephrectomy (labeled as normal tissue, n=5), ANCA-NCGN (n=5), anti-GBM NCGN (n=3), and hypertensive (n=3) and membranous nephropathy (n=2) as disease controls, respectively. (E) The corresponding quantifications are depicted. Glomeruli with active lesions (crescents/necrosis), normal appearance by light microscopy, and glomerular sclerosis were separately assessed in biopsies from patients with ANCA-NCGN. (F) Costaining using fluorescence-labeled antibodies to the EC marker CD31 (red), phospho-p65 (green), and DAPI staining for nuclei (blue) was performed in ANCA-GN biopsy samples (NCGN) and in normal tissue from tumor nephrectomies. A typical microscopy example (original magnification, ×40; scale bars = 50 μm) together with the corresponding quantitative assessment of nuclear phospho-p65 staining within CD31-positive glomerular ECs using Image J is depicted. **P<0.01.
Discussion
Our study indicates that glomerular NF-κB is activated in a murine AAV disease model, most abundantly as a p50/p65 heterodimer, and positively correlates with glomerular necrosis and crescent formation. ANCA-stimulated neutrophils induced endothelial NF-κB in vitro, thereby promoting neutrophil recruitment to the endothelial monolayer. As a proof of principle, an endothelium-targeted p65 siRNA approach inhibited glomerular NF-κB, reduced myeloid cell influx and, most importantly, abrogated NCGN in a murine AAV model. Increased phospho-p65 staining in active glomerular lesions from patients with AAV indicates that NF-κB is also activated in the human disease condition and should be further explored as a therapeutic target.
Endothelial injury by ANCA-activated neutrophils is a pivotal event in necrotizing vasculitis. However, current treatments are neither specifically directed to the neutrophil nor to the EC. Conceivably, characterization of signaling pathways that cause endothelial inflammation by ANCA-activated neutrophils, and subsequently NCGN, will identify potential targets for more cell-specific treatment approaches. Ewert et al.32 were the first to describe that ANCA-stimulated primed neutrophils damage human EC in vitro, a finding that was corroborated in subsequent studies.2 Moreover, clinical studies established EC activation markers,13,14 markers of antiangiogenesis,33 endothelial microparticels34,35 and even circulating detached ECs16 in patients with active AAV.
NF-κB is a key transcription factor that controls various cellular functions by forming Rel family member dimers that bind to κB DNA motifs. Some of these dimers, such as p50/p65, activate gene transcription, whereas others, such as p50/p50, are thought to function as repressors. NF-κB is important in induction of inflammation and also in its resolution. We previously described NF-κB activation by inflammatory mediators in human neutrophils and its role in apoptosis.36 However, NF-κB activation has also been reported in renal diseases,23 including experimental anti-GBM crescentic GN.20,26,37 The starting point of our NF-κB study in AAV was to reanalyze kidneys from a previous study, where we had induced NCGN by anti-MPO antibodies and treated some of the mice with bortezomib.27 This study was particularly informative because the proteasome inhibitor bortezomib reduces IκBα degradation and thereby NF-κB activation. Bortezomib provided protection from NCGN in our mouse model. In retrospect, we found that NF-κB was activated in renal tissue obtained from NCGN mice and that the complex contained mostly p50/p65 heterodimers. Bortezomib reduced both renal NF-κB and crescent formation. However, other interventions that abrogated NCGN but did not directly target ubiquitination also inhibited NF-κB activation, indicating that protection from renal inflammation by any means is accompanied by abrogated NF-κB activation. We had additional tissue available in this mouse study to perform EMSA on isolated glomeruli and costaining that clearly established glomerular NF-κB activation within CD31-positive ECs. Substantial NF-κB activation was also observed within crescents, presumably in macrophages. Thus, although glomerular NF-κB activation strongly correlated with crescent formation, this correlation could be pure association or indicate a causative relationship.
We then studied NF-κB–dependent mechanisms in an in vitro coculture system consisting of ANCA-treated primed neutrophils and ECs and observed that ANCA did not activate neutrophil NF-κB, but that ANCA-treated neutrophils activated endothelial NF-κB. Neutrophil priming was important for this effect and it did not matter whether a TNF-α–dependent or –independent primer was used. Direct neutrophil and EC contact was not necessary because cellfree SN from ANCA-treated neutrophils was sufficient to activate endothelial NF-κB. We found that TNF-α release from ANCA-activated neutrophils mediated, at least in part, endothelial NF-κB activation, whereas neutrophil-derived microparticles and reactive oxygen species did not. A mechanistic role for TNF-α was demonstrated in previous murine and rat models of AAV.38,39 We observed that, as a consequence of endothelial NF-κB activation, neutrophil adhesion to endothelial monolayers and IL-8 production were increased in vitro. Depleting experiments established that neutrophils are important for glomerular necrosis and crescents in a murine disease model,40 and our data support the notion that endothelial NF-κB participates in neutrophil recruitment. In addition, IL-8 produced by ECs may keep neutrophils within the intravascular compartment and contribute to glomerular damage.41
With these data we considered the possibility that endothelial NF-κB provides a treatment target for ANCA-induced NCGN. To test this hypothesis, we selected p65 over p50 because p50 is not only important to induction but also to resolution of inflammation.42–46 Panzer et al.25 showed that p50-deficient mice showed prolonged renal inflammation in LPS and Thy-1 models. Because p65 mediates transcriptional activation of proinflammatory cytokines and because of the abundance of p50/p65 complexes in our NCGN disease model, we focused on p65 inhibition. We recently used p65 siRNA to downregulate p65 in TNF-α– and LPS-treated HUVECs in vitro and in TNF-α– and LPS-treated mice.30 p65 siRNA inhibited endothelial p65 mRNA and protein, and NF-κB–dependent proinflammatory cytokines and adhesion molecules. We packed p65 siRNA into liposomes that are based on the cationic amphiphile SAINT-C18. We conjugated these SOS with E-selectin antibodies because E-selectin is specifically expressed by the inflamed endothelium and E-selectin–targeted immunoliposomes were used previously to deliver biologically active agents to the endothelium, including to the microvascular glomerular endothelium.29,47–50 However, the vasculature of lungs, skin, and even aorta showed also activated NF-κB (Supplemental Figure 1, D and E) that would possibly also be reduced by such a treatment. As a proof of principle, p65 siRNA downregulated glomerular NF-κB. This approach strongly reduced renal neutrophil and monocyte influx as well as NCGN. We pretreated mice with TNF-α to ensure that the immunoliposomes were delivered to the endothelial target. Conceivably, such treatment is unnecessary in patients with AAV. We suggest that activated endothelium, a hallmark of vasculitis, is already present when clinical disease manifestation and treatment opportunities occur. Nevertheless, omitting TNF-α priming and application of SOS p65 siRNA after disease induction with anti-MPO antibodies did not provide disease protection, most likely because of the short-term study period. Our 7-day model may not be optimal for testing siRNA treatment at later time points because approximately 48 hours are necessary to downregulate p65.30 Further optimization of our treatment protocol will be addressed in future studies, e.g., by use of the bone-marrow transfer model of anti-MPO NCGN, which first requires extended long-term kinetic studies of SOS siRNA. The fact that p65 is activated in patients with AAV, and particularly in active lesions, suggests its relevance to the human disease. The fact that crescentic glomeruli from patients with anti-GBM disease also showed activated NF-κB suggests that the importance of this signaling pathway is not restricted to AAV.
In summary, we provide strong evidence of the crucial involvement of the NF-κB signaling pathway in ANCA-induced NCGN. The mechanism involves endothelial NF-κB activation by ANCA-stimulated primed neutrophils. Downregulating endothelial p65 NF-κB with siRNA-packed E-selectin immunoliposomes could offer a more specific treatment strategy in AAV.
Concise Methods
Materials
Human and mouse TNF-α, LPS (O26:B6), fMLP, the IKKβ inhibitor BMS 345541, diphenyleneiodonium, and Ficoll–Hypaque were from Sigma (Deisenhofen, Germany). Dextran was from Amersham Pharmacia (Amsterdam, The Netherlands); the α-actin antibody (C-2) and the polyclonal rabbit antibodies against IκBα (C-21), p65 (Rel-A) and phospho-p65, Rel-B, and c-Rel were from Santa Cruz Biotechnology (Santa Cruz, CA); p50 antibodies were from Rockland and Upstate Biotechnology (Lake Placid, NY); anti-CD31 was from Abcam (Cambridge, UK); the mAb to MPO (clone 2C7) was from Acris (Herford, Germany); PR3 (mAb 12.8) was from CLB (Amsterdam, The Netherlands); and the isotype control (clone 11711) was from R&D Systems (Wiesbaden-Nordenstadt, Germany). Neutralizing antibodies against TNF-α, IL-8, and isotype controls were from R&D Systems (Minneapolis, MN). For immunofluorescence staining, the phospho-p65 antibody was from SAB (College Park, MD), anti-CD31 was from Dianova (Hamburg, Germany), anti-rat Cy3 was from Jackson ImmunoResearch Laboratories (Baltimore, PA), anti-rabbit Alexa Fluor 488 was from Thermo Fisher Scientific (Rockford, IL), and DAPI mounting medium was from VECTASHIELD (Peterborough, UK). Endotoxin-free reagents and plastic disposables were used in all experiments. Lipids, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly-ethylene glycol)-2000]-maleimide (DSPE-PEG2000-Mal), and 2-distearoyl-sn-glycero-3-phosphoethanolamine-N [methoxy(polyethylene glycol)-2000] (DSPE-PEG2000) were purchased from Avanti Polar Lipids (Alabaster, AL). The cationic lipid 1-methyl-4-(cis-9-dioleyl)methyl-pyridinium-chloride (SAINT-C18) was obtained from Synvolux Therapeutics (Groningen, The Netherlands). Cholesterol and N-succinimidyl-S-acetylthioacetate (SATA) were from Sigma (St. Louis, MO).
Preparation of Human Neutrophils, ECs, and EC Coincubation with Neutrophils or Cellfree Neutrophil SN
Neutrophils from healthy human donors and HUVECs were obtained after due approval by Charité and governmental authorities and after written informed consent. We isolated neutrophils by density gradient centrifugation and assessed cell viability as described previously.36 Cell viability by trypan blue exclusion was >99%. Neutrophils at 2×106 were treated with buffer control or primed with inflammatory mediators for 30 minutes. Priming was achieved with 0.1 ng/ml TNF-α, 0.1 μg/ml LPS, and 10−8 M fMLP as indicated. Buffer-treated or primed neutrophils were then activated with 10 µg/ml mAbs to PR3 or MPO, with 150 µg/ml human ANCA-IgG, or appropriate control antibodies in a total reaction volume of 400 µl HBSS++ in polypropylene tubes. Samples were incubated for 60 minutes to assess NF-κB activation in neutrophils, or added to the EC monolayer for 2 hours to assess endothelial NF-κB activation, or incubated for 5 hours to produce cellfree neutrophil SN, respectively. In the case of the latter experiments, FCS was added to achieve 1% final FCS concentration. Where indicated, neutrophil microparticles were separated by SN centrifugation at 20,000×g for 30 minutes, or SN was heat-inactivated at 90°C for 5 minutes. In other experiments, SN was generated in the presence of an NADPH inhibitor (10 μM diphenyleneiodonium) or treated for 60 minutes with neutralizing anti–TNF-α (0.5 μg/ml) or anti–IL-8 (5 μg/ml) antibodies or isotype controls, respectively.
Primary HUVECs were used after two to four passages. Confluent ECs in 24-well plates were washed with PBS and incubated either for 60 minutes with cellfree neutrophil SN to assess IκBα degradation, for 6 hours to measure IL-8 mRNA expression, and for 18 hours to measure IL-8 protein release by ECs.
Neutrophil Adhesion Assay
Adhesion experiments were performed with minor modifications as described previously.51 After overnight coincubation, freshly prepared neutrophils (2×105) were incubated with 100 nM IL-8 for 30 minutes at 37°C and added to ECs. After 120 minutes, wells were flicked dry, washed three times with PBS, and adherent cells were estimated using the MPO assay.51
Preparation of Human Neutrophils and Human IgG
Normal IgG and ANCA IgG were prepared from the blood of normal healthy volunteers and patients with AAV as described previously.4
Immunoblotting and ELISA
IκBα degradation was analyzed by immunoblotting using 10 μg of cytoplasmic neutrophil extracts or whole-cell extracts for ECs, respectively. Samples were incubated for 5 minutes at 95°C in loading buffer. After SDS-PAGE and transfer to a nitrocellulose membrane, membranes were developed with antibodies to IκBα or α-actin. IL-8 secretion from ECs was assessed by ELISA (R&D Systems).
Nuclear Extract Preparation and EMSA
Nuclear extracts from renal tissue were prepared analogous to the preparation of nuclear extracts from human neutrophils as described previously with minor modifications.36 Briefly, kidney or enriched fractions of glomerular cells after sieving were homogenized in a tissue homogenizer (Precellys; Bertin Technologies, Erlangen, Germany). Lysates, generated by treatment with hypotonic HEPES buffer and addition of 0.1% NP40, were vortexed, pelleted, and the supernatant containing the cytoplasmic fraction was stored at −80°C. The pellet was resuspended with the hypertonic, high-salt HEPES buffer together with protease inhibitors. After centrifugation the supernatant was collected and EMSA was performed as described.36
Quantitative RT-PCR
Total RNA was isolated according to Qiagen (Hilden, Germany) including DNase treatment. Reverse transcription was performed according to the Superscript protocol (Thermo Fisher Scientific Inc., Waltham, MA) and TaqMan RT-PCR using the TaqMan Fast Universal PCR Master Mix (Applied Biosystems, Weiterstadt, Germany). Quantitative RT-PCR was performed as described previously.36 The forward primer for human IL-8 was 5′-GCCTTCCTGATTTCTGCAGC-3′, the reverse primer was 5′-TGCACTGACATCTAAGTTCTTTAGCA-3′, and the TaqMan probe was FAM-5′-TGTGTGAAGGTGCAGTTTTGCCAAGG-3′-TAMRA. The forward primer for mouse TNF-α was 5′-TCTCTTCAAGGGACAAGGCTG-3′, the reverse primer was 5′-ATAGCAAATCGGCTGACGGT-3′, and the TaqMan probe was FAM-5′-CCCGACTACGTGCTCCTCACCCA-3′-TAMRA. Quantification was checked using the primer and probe for human GAPDH (item number 4310884E; Applied Biosystems) or mouse GAPDH with forward primer 5′-GGCAAATTCAACGGCACAGT-3′ and reverse primer 5′-AGATGGTGATGGGCTTCCC-3′, and the TaqMan probe FAM-5′-AAGGCCGAGAATGGGAAGCTTGTCATC-3′-TAMRA. Data were analyzed using SDS 7500 software and ΔΔCt comparative analysis as described by Applied Biosystems.
Passive Model of Anti-MPO NCGN
Mice were bred in the Max-Delbrück Center animal facility under pathogen-free conditions. Local authorities approved all animal experiments, which followed the ARRIVE guidelines. Polyclonal anti-MPO IgG was obtained by isolation of total IgG from murine MPO-immunized MPO-deficient mice as described previously.31 GN was induced in wild-type C57BL/6 mice (aged 8–10 weeks) by intravenous injection of anti-MPO IgG (50 μg/g body wt), followed by intraperitoneal injection of LPS (5 μg/g body wt; Escherichia coli, serotype O26:B6) 1 hour later. At 50 hours before anti-MPO IgG, mice received 200 ng TNF-α intravenously to upregulate E-selectin, followed by intravenous application of 1 μmol total lipid with SOS siRNA or an empty SOS control preparation 2 hours later (n=5 mice per group). For therapeutic application of SOS siRNA after disease induction, TNF-α was omitted and 1 μmol total lipid with SOS siRNA or empty SOS control preparation was injected 4 hours after anti-MPO IgG and LPS injection (n=5 mice per group). All mice were euthanized after 7 days.
Urine Evaluation in Mice
Mice were placed in metabolic cages on the day before euthanasia at day 7, and urine was collected for 16 hours. Urine was tested by dipstick (Roche Diagnostics, Almere, The Netherlands) for hematuria, leukocyturia, and proteinuria, and the extent is expressed as the mean on a scale of 0 (none) to 4 (severe) for leukocyturia and hematuria, and 0–3 for proteinuria. Albuminuria was quantified using an ELISA kit (R&D Systems).
Histologic Examinations of Renal Injury and NF-κB Activation
Kidneys was collected at the time of euthanasia, fixed in 10% formalin, and embedded in paraffin using routine protocols. Coronal section (2-µm thick) were stained with hematoxylin and eosin or periodic acid–Schiff (PAS) and evaluated by light microscopy. Glomerular crescents and necrosis were expressed as the mean percentage of glomeruli with crescents and necrosis in each animal as previously described.27
Histologic stains for phospho-p65 and CD31 were performed on 2-μm thick paraffin-embedded sections. Briefly, sections were dewaxed in xylene and rehydrated through graded ethanols. Antigen retrieval was performed by heating sections in 10 mM sodium citrate buffer in a microwave oven for 5 minutes at 800 W, followed by 20 minutes at 200 W. Sections were incubated with peroxidase block for 5 minutes using Envision+ System-HRP (Dako, Hamburg, Germany). For immunohistochemistry, sections were incubated at 4°C with the indicated antibodies in a humidified chamber overnight. Incubation with secondary antibody and substrate development was carried out as recommended by the manufacturer. Nuclei were counterstained with hematoxylin. Visualization and quantification was done using a Zeiss Axioplan-2 imaging microscope (Carl Zeiss GmbH, Jena, Germany). For immunofluorescence costaining, sections were incubated with phospho-p65, followed by the anti-CD31 antibody (from Dianova) at 4°C overnight, followed by anti-rabbit Alexa Fluor 488 and anti-rat Cy3 secondary antibodies. Nuclei were counterstained with DAPI. Staining intensity of phospho-p65–positive glomerular ECs was measured using ImageJ software. Sections were transformed into an RGB (red, green, blue) number triplet and background was subtracted to reduce autofluorescence signals. From each section five glomeruli were selected and nuclear mean phospho-p65 staining intensity (green) from 10 to 15 glomerular ECs was calculated. The DAPI staining (blue) was used to define the nuclear ROI. The CD31 staining (red) was used to define ECs. Quantitative data were exported into Microsoft Excel software for further analysis. For phospho-p65 quantification in vascular beds of organs other than kidneys, three to four regions from each section were analyzed.
Flow Cytometry To Assess Neutrophil and Monocyte/Macrophage Influx into Kidneys
Freshly harvested kidney tissue was weighed, minced, digested, and passed through a 70-μm filter (BD Biosciences, Heidelberg, Germany). Suspensions were incubated with fluorochrome-labeled mouse antibodies to CD11b (eBioscience, Frankfurt, Germany), Ly6G, Ly6C (BioLegend, London, UK), and CD45 (Beckman Coulter, Krefeld, Germany) for 15 minutes on ice. Neutrophils were identified by expression of CD45, CD11b, Ly6C, and Ly6G, respectively. TruCount beads (BD Biosciences) were added to the samples before acquisition and measurements were performed on a BD FACS CANTO II. Data were analyzed with the FlowJo Software (Tree Star, Inc., Ashland, OR).
Preparation of siRNA Containing SOS
SOS were prepared as described previously with slight modifications.52 In brief, lipids from stock solutions of POPC, SAINT-C18, Chol, DSPE-PEG2000, and DSPE-PEG2000-Mal in chloroform/methanol (9:1) were mixed in a molar ratio of 37:18:40:4:1. SOS were prepared containing siRNA specific for RelA (Mm_Rela_3 FlexiTube siRNA, 5′-CACCATCAAGATCAATGGCTA-3′). siRNA was dissolved according to the protocol of the manufacturer and mixed with dried lipids at a ratio of 1 nM siRNA per 1 μM total lipid. After extrusion through polycarbonate filters (50 nm pore size) nonencapsulated siRNA was removed by ion exchange chromatography on a DEAE Sepharose CL-6B column (Sigma) using HN buffer (135 mM NaCl, 10 mM HEPES; pH 6.7) as an eluent. The concentration of siRNA encapsulated in liposomes was measured using the Quant-iT Ribo-Green assay (Invitrogen, Breda, The Netherlands). The monoclonal rat anti-mouse E-selectin antibody (MES-1, kindly provided by Dr. D. Brown; UCB Celltech, Slough, UK) was thiolated by means of SATA and coupled to a maleimide group at the distal end of the polyethylene glycol chain by sulfhydryl-maleimide coupling, as described before for albumin.53 The SOS were stored at 4°C under argon gas and used within 4 weeks.
Statistical Analyses
Results are given as mean±SEM. Comparisons between multiple groups were done using ANOVA and appropriate post hoc tests. Comparisons between two groups were done by a two-sided paired t test. Differences were considered significant if P<0.05.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank Susanne Rolle, Sylvia Lucke and Tanja Filipowski for excellent technical assistance.
This work was supported by Deutsche Forschungsgemeinschaft grants KE 576/8-1, CH 299/2-1, and SCHR 771/6-1, an Experimental and Clinical Research Center (ECRC) grant to R.K., and an ECRC grant to A.S.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016060690/-/DCSupplemental.
References
- 1.Davies DJ, Moran JE, Niall JF, Ryan GB: Segmental necrotising glomerulonephritis with antineutrophil antibody: Possible arbovirus aetiology? Br Med J (Clin Res Ed) 285: 606, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halbwachs L, Lesavre P: Endothelium-neutrophil interactions in ANCA-associated diseases. J Am Soc Nephrol 23: 1449–1461, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radford DJ, Lord JM, Savage CO: The activation of the neutrophil respiratory burst by anti-neutrophil cytoplasm autoantibody (ANCA) from patients with systemic vasculitis requires tyrosine kinases and protein kinase C activation. Clin Exp Immunol 118: 171–179, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kettritz R, Schreiber A, Luft FC, Haller H: Role of mitogen-activated protein kinases in activation of human neutrophils by antineutrophil cytoplasmic antibodies. J Am Soc Nephrol 12: 37–46, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Ben-Smith A, Dove SK, Martin A, Wakelam MJ, Savage CO: Antineutrophil cytoplasm autoantibodies from patients with systemic vasculitis activate neutrophils through distinct signaling cascades: Comparison with conventional Fcgamma receptor ligation. Blood 98: 1448–1455, 2001 [DOI] [PubMed] [Google Scholar]
- 6.Kettritz R, Choi M, Butt W, Rane M, Rolle S, Luft FC, Klein JB: Phosphatidylinositol 3-kinase controls antineutrophil cytoplasmic antibodies-induced respiratory burst in human neutrophils. J Am Soc Nephrol 13: 1740–1749, 2002 [DOI] [PubMed] [Google Scholar]
- 7.Hewins P, Williams JM, Wakelam MJ, Savage CO: Activation of Syk in neutrophils by antineutrophil cytoplasm antibodies occurs via Fcgamma receptors and CD18. J Am Soc Nephrol 15: 796–808, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Schreiber A, Rolle S, Peripelittchenko L, Rademann J, Schneider W, Luft FC, Kettritz R: Phosphoinositol 3-kinase-gamma mediates antineutrophil cytoplasmic autoantibody-induced glomerulonephritis. Kidney Int 77: 118–128, 2010 [DOI] [PubMed] [Google Scholar]
- 9.van der Veen BS, Chen M, Müller R, van Timmeren MM, Petersen AH, Lee PA, Satchell SC, Mathieson PW, Saleem MA, Stegeman CA, Zwerina J, Molema G, Heeringa P: Effects of p38 mitogen-activated protein kinase inhibition on anti-neutrophil cytoplasmic autoantibody pathogenicity in vitro and in vivo. Ann Rheum Dis 70: 356–365, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Lu X, Garfield A, Rainger GE, Savage CO, Nash GB: Mediation of endothelial cell damage by serine proteases, but not superoxide, released from antineutrophil cytoplasmic antibody-stimulated neutrophils. Arthritis Rheum 54: 1619–1628, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Jerke U, Hernandez DP, Beaudette P, Korkmaz B, Dittmar G, Kettritz R: Neutrophil serine proteases exert proteolytic activity on endothelial cells. Kidney Int 88: 764–775, 2015 [DOI] [PubMed] [Google Scholar]
- 12.Hong Y, Eleftheriou D, Hussain AA, Price-Kuehne FE, Savage CO, Jayne D, Little MA, Salama AD, Klein NJ, Brogan PA: Anti-neutrophil cytoplasmic antibodies stimulate release of neutrophil microparticles. J Am Soc Nephrol 23: 49–62, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boehme MW, Schmitt WH, Youinou P, Stremmel WR, Gross WL: Clinical relevance of elevated serum thrombomodulin and soluble E-selectin in patients with Wegener’s granulomatosis and other systemic vasculitides. Am J Med 101: 387–394, 1996 [DOI] [PubMed] [Google Scholar]
- 14.Monach PA, Tomasson G, Specks U, Stone JH, Cuthbertson D, Krischer J, Ding L, Fervenza FC, Fessler BJ, Hoffman GS, Ikle D, Kallenberg CG, Langford CA, Mueller M, Seo P, St Clair EW, Spiera R, Tchao N, Ytterberg SR, Gu YZ, Snyder RD, Merkel PA: Circulating markers of vascular injury and angiogenesis in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 63: 3988–3997, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirsch T, Woywodt A, Klose J, Wyss K, Beese M, Erdbruegger U, Grossheim M, Haller H, Haubitz M: Endothelial-derived thrombospondin-1 promotes macrophage recruitment and apoptotic cell clearance. J Cell Mol Med 14: 1922–1934, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woywodt A, Streiber F, de Groot K, Regelsberger H, Haller H, Haubitz M: Circulating endothelial cells as markers for ANCA-associated small-vessel vasculitis. Lancet 361: 206–210, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Hayden MS, Ghosh S: Shared principles in NF-kappaB signaling. Cell 132: 344–362, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Hatada EN, Krappmann D, Scheidereit C: NF-kappaB and the innate immune response. Curr Opin Immunol 12: 52–58, 2000 [DOI] [PubMed] [Google Scholar]
- 19.Karin M: The NF-kappa B activation pathway: Its regulation and role in inflammation and cell survival. Cancer J Sci Am 4[Suppl 1]: S92–S99, 1998 [PubMed] [Google Scholar]
- 20.Tomita N, Morishita R, Lan HY, Yamamoto K, Hashizume M, Notake M, Toyosawa K, Fujitani B, Mu W, Nikolic-Paterson DJ, Atkins RC, Kaneda Y, Higaki J, Ogihara T: In vivo administration of a nuclear transcription factor-kappaB decoy suppresses experimental crescentic glomerulonephritis. J Am Soc Nephrol 11: 1244–1252, 2000 [DOI] [PubMed] [Google Scholar]
- 21.Sakai N, Wada T, Furuichi K, Iwata Y, Yoshimoto K, Kitagawa K, Kokubo S, Kobayashi M, Takeda S, Kida H, Kobayashi K, Mukaida N, Matsushima K, Yokoyama H: p38 MAPK phosphorylation and NF-kappa B activation in human crescentic glomerulonephritis. Nephrol Dial Transplant 17: 998–1004, 2002 [DOI] [PubMed] [Google Scholar]
- 22.López-Franco O, Suzuki Y, Sanjuán G, Blanco J, Hernández-Vargas P, Yo Y, Kopp J, Egido J, Gómez-Guerrero C: Nuclear factor-kappa B inhibitors as potential novel anti-inflammatory agents for the treatment of immune glomerulonephritis. Am J Pathol 161: 1497–1505, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanz AB, Sanchez-Niño MD, Ramos AM, Moreno JA, Santamaria B, Ruiz-Ortega M, Egido J, Ortiz A: NF-kappaB in renal inflammation. J Am Soc Nephrol 21: 1254–1262, 2010 [DOI] [PubMed] [Google Scholar]
- 24.Brähler S, Ising C, Hagmann H, Rasmus M, Hoehne M, Kurschat C, Kisner T, Goebel H, Shankland S, Addicks K, Thaiss F, Schermer B, Pasparakis M, Benzing T, Brinkkoetter PT: Intrinsic proinflammatory signaling in podocytes contributes to podocyte damage and prolonged proteinuria. Am J Physiol Renal Physiol 303: F1473–F1485, 2012 [DOI] [PubMed] [Google Scholar]
- 25.Panzer U, Steinmetz OM, Turner JE, Meyer-Schwesinger C, von Ruffer C, Meyer TN, Zahner G, Gómez-Guerrero C, Schmid RM, Helmchen U, Moeckel GW, Wolf G, Stahl RA, Thaiss F: Resolution of renal inflammation: A new role for NF-kappaB1 (p50) in inflammatory kidney diseases. Am J Physiol Renal Physiol 297: F429–F439, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Gotot J, Piotrowski E, Otte MS, Tittel AP, Linlin G, Yao C, Ziegelbauer K, Panzer U, Garbi N, Kurts C, Thaiss F: Inhibitor of NFkappaB kinase subunit 2 blockade hinders the initiation but aggravates the progression of crescentic GN. J Am Soc Nephrol 27: 1917–1924, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bontscho J, Schreiber A, Manz RA, Schneider W, Luft FC, Kettritz R: Myeloperoxidase-specific plasma cell depletion by bortezomib protects from anti-neutrophil cytoplasmic autoantibodies-induced glomerulonephritis. J Am Soc Nephrol 22: 336–348, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schreiber A, Pham CT, Hu Y, Schneider W, Luft FC, Kettritz R: Neutrophil serine proteases promote IL-1β generation and injury in necrotizing crescentic glomerulonephritis. J Am Soc Nephrol 23: 470–482, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asgeirsdóttir SA, Kamps JA, Bakker HI, Zwiers PJ, Heeringa P, van der Weide K, van Goor H, Petersen AH, Morselt H, Moorlag HE, Steenbergen E, Kallenberg CG, Molema G: Site-specific inhibition of glomerulonephritis progression by targeted delivery of dexamethasone to glomerular endothelium. Mol Pharmacol 72: 121–131, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Kowalski PS, Zwiers PJ, Morselt HW, Kuldo JM, Leus NG, Ruiters MH, Molema G, Kamps JA: Anti-VCAM-1 SAINT-O-Somes enable endothelial-specific delivery of siRNA and downregulation of inflammatory genes in activated endothelium in vivo. J Control Release 176: 64–75, 2014 [DOI] [PubMed] [Google Scholar]
- 31.Schreiber A, Xiao H, Falk RJ, Jennette JC: Bone marrow-derived cells are sufficient and necessary targets to mediate glomerulonephritis and vasculitis induced by anti-myeloperoxidase antibodies. J Am Soc Nephrol 17: 3355–3364, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Ewert BH, Jennette JC, Falk RJ: Anti-myeloperoxidase antibodies stimulate neutrophils to damage human endothelial cells. Kidney Int 41: 375–383, 1992 [DOI] [PubMed] [Google Scholar]
- 33.Le Roux S, Pepper RJ, Dufay A, Néel M, Meffray E, Lamandé N, Rimbert M, Josien R, Hamidou M, Hourmant M, Cook HT, Charreau B, Larger E, Salama AD, Fakhouri F: Elevated soluble Flt1 inhibits endothelial repair in PR3-ANCA-associated vasculitis. J Am Soc Nephrol 23: 155–164, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erdbruegger U, Grossheim M, Hertel B, Wyss K, Kirsch T, Woywodt A, Haller H, Haubitz M: Diagnostic role of endothelial microparticles in vasculitis. Rheumatology (Oxford) 47: 1820–1825, 2008 [DOI] [PubMed] [Google Scholar]
- 35.Bertram A, Lovric S, Engel A, Beese M, Wyss K, Hertel B, Park JK, Becker JU, Kegel J, Haller H, Haubitz M, Kirsch T: Circulating ADAM17 level reflects disease activity in proteinase-3 ANCA-associated vasculitis. J Am Soc Nephrol 26: 2860–2870, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi M, Rolle S, Wellner M, Cardoso MC, Scheidereit C, Luft FC, Kettritz R: Inhibition of NF-kappaB by a TAT-NEMO-binding domain peptide accelerates constitutive apoptosis and abrogates LPS-delayed neutrophil apoptosis. Blood 102: 2259–2267, 2003 [DOI] [PubMed] [Google Scholar]
- 37.Kim JH, Ha IS, Hwang CI, Lee YJ, Kim J, Yang SH, Kim YS, Cao YA, Choi S, Park WY: Gene expression profiling of anti-GBM glomerulonephritis model: The role of NF-kappaB in immune complex kidney disease. Kidney Int 66: 1826–1837, 2004 [DOI] [PubMed] [Google Scholar]
- 38.Huugen D, Xiao H, van Esch A, Falk RJ, Peutz-Kootstra CJ, Buurman WA, Tervaert JW, Jennette JC, Heeringa P: Aggravation of anti-myeloperoxidase antibody-induced glomerulonephritis by bacterial lipopolysaccharide: Role of tumor necrosis factor-alpha. Am J Pathol 167: 47–58, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Little MA, Bhangal G, Smyth CL, Nakada MT, Cook HT, Nourshargh S, Pusey CD: Therapeutic effect of anti-TNF-alpha antibodies in an experimental model of anti-neutrophil cytoplasm antibody-associated systemic vasculitis. J Am Soc Nephrol 17: 160–169, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Xiao H, Heeringa P, Liu Z, Huugen D, Hu P, Maeda N, Falk RJ, Jennette JC: The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. Am J Pathol 167: 39–45, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cockwell P, Brooks CJ, Adu D, Savage COS: Interleukin-8: A pathogenetic role in antineutrophil cytoplasmic autoantibody-associated glomerulonephritis. Kidney Int 55: 852–863, 1999 [DOI] [PubMed] [Google Scholar]
- 42.Cao S, Zhang X, Edwards JP, Mosser DM: NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J Biol Chem 281: 26041–26050, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wessells J, Baer M, Young HA, Claudio E, Brown K, Siebenlist U, Johnson PF: BCL-3 and NF-kappaB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J Biol Chem 279: 49995–50003, 2004 [DOI] [PubMed] [Google Scholar]
- 44.Mizgerd JP, Lupa MM, Kogan MS, Warren HB, Kobzik L, Topulos GP: Nuclear factor-kappaB p50 limits inflammation and prevents lung injury during Escherichia coli pneumonia. Am J Respir Crit Care Med 168: 810–817, 2003 [DOI] [PubMed] [Google Scholar]
- 45.Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA: Possible new role for NF-kappaB in the resolution of inflammation. Nat Med 7: 1291–1297, 2001 [DOI] [PubMed] [Google Scholar]
- 46.Oakley F, Mann J, Nailard S, Smart DE, Mungalsingh N, Constandinou C, Ali S, Wilson SJ, Millward-Sadler H, Iredale JP, Mann DA: Nuclear factor-kappaB1 (p50) limits the inflammatory and fibrogenic responses to chronic injury. Am J Pathol 166: 695–708, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spragg DD, Alford DR, Greferath R, Larsen CE, Lee KD, Gurtner GC, Cybulsky MI, Tosi PF, Nicolau C, Gimbrone MA Jr: Immunotargeting of liposomes to activated vascular endothelial cells: A strategy for site-selective delivery in the cardiovascular system. Proc Natl Acad Sci U S A 94: 8795–8800, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adrian JE, Morselt HW, Süss R, Barnert S, Kok JW, Asgeirsdóttir SA, Ruiters MH, Molema G, Kamps JA: Targeted SAINT-O-Somes for improved intracellular delivery of siRNA and cytotoxic drugs into endothelial cells. J Control Release 144: 341–349, 2010 [DOI] [PubMed] [Google Scholar]
- 49.Kowalski PS, Kuninty PR, Bijlsma KT, Stuart MC, Leus NG, Ruiters MH, Molema G, Kamps JA: SAINT-liposome-polycation particles, a new carrier for improved delivery of siRNAs to inflamed endothelial cells. Eur J Pharm Biopharm 89: 40–47, 2015 [DOI] [PubMed] [Google Scholar]
- 50.Kułdo JM, Ásgeirsdóttir SA, Zwiers PJ, Bellu AR, Rots MG, Schalk JA, Ogawara KI, Trautwein C, Banas B, Haisma HJ, Molema G, Kamps JA: Targeted adenovirus mediated inhibition of NF-κB-dependent inflammatory gene expression in endothelial cells in vitro and in vivo. J Control Release 166: 57–65, 2013 [DOI] [PubMed] [Google Scholar]
- 51.Choi M, Salanova B, Rolle S, Wellner M, Schneider W, Luft FC, Kettritz R: Short-term heat exposure inhibits inflammation by abrogating recruitment of and nuclear factor-kappaB activation in neutrophils exposed to chemotactic cytokines. Am J Pathol 172: 367–777, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kowalski PS, Lintermans LL, Morselt HW, Leus NG, Ruiters MH, Molema G, Kamps JA: Anti-VCAM-1 and anti-E-selectin SAINT-O-Somes for selective delivery of siRNA into inflammation-activated primary endothelial cells. Mol Pharm 10: 3033–3044, 2013 [DOI] [PubMed] [Google Scholar]
- 53.Kamps JA, Swart PJ, Morselt HW, Pauwels R, De Béthune MP, De Clercq E, Meijer DK, Scherphof GL: Preparation and characterization of conjugates of (modified) human serum albumin and liposomes: Drug carriers with an intrinsic anti-HIV activity. Biochim Biophys Acta 1278: 183–190, 1996 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.