

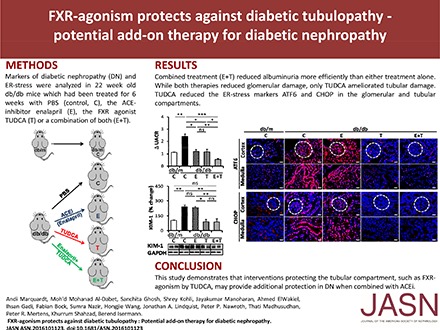
Abstract
Established therapies for diabetic nephropathy (dNP) delay but do not prevent its progression. The shortage of established therapies may reflect the inability to target the tubular compartment. The chemical chaperone tauroursodeoxycholic acid (TUDCA) ameliorates maladaptive endoplasmic reticulum (ER) stress signaling and experimental dNP. Additionally, TUDCA activates the farnesoid X receptor (FXR), which is highly expressed in tubular cells. We hypothesized that TUDCA ameliorates maladaptive ER signaling via FXR agonism specifically in tubular cells. Indeed, TUDCA induced expression of FXR-dependent genes (SOCS3 and DDAH1) in tubular cells but not in other renal cells. In vivo, TUDCA reduced glomerular and tubular injury in db/db and diabetic endothelial nitric oxide synthase–deficient mice. FXR inhibition with Z-guggulsterone or vivo-morpholino targeting of FXR diminished the ER-stabilizing and renoprotective effects of TUDCA. Notably, these in vivo approaches abolished tubular but not glomerular protection by TUDCA. Combined intervention with TUDCA and the angiotensin-converting enzyme inhibitor enalapril in 16-week-old db/db mice reduced albuminuria more efficiently than did either treatment alone. Although both therapies reduced glomerular damage, only TUDCA ameliorated tubular damage. Thus, interventions that specifically protect the tubular compartment in dNP, such as FXR agonism, may provide renoprotective effects on top of those achieved by inhibiting angiotensin-converting enzyme.
Keywords: tudca ER-stress diabetes, diabetic nephropathy, FXR, ACE inhibitors
Around one third of patients with diabetes develop diabetic nephropathy (dNP) and dNP contributes to >40% of newly diagnosed ESRD cases.1 Besides blood glucose control, inhibition of the renin-angiotensin-aldosterone system (RAAS) is the only available therapy for dNP. However, both approaches failed to prevent dNP and typically the disease progression is only delayed.1 Thus, there is an unmet medical need for new therapeutic approaches to target dNP.
RAAS inhibition is thought to primarily provide glomerular protection and therapies targeting pathomechanism specifically in tubular cells are lacking. Recent preclinical studies suggest a pivotal role of maladaptive endoplasmic reticulum (ER) signaling in the pathogenesis of dNP.2 Importantly, the chemical chaperone tauroursodeoxycholic acid (TUDCA) alleviated maladaptive ER signaling in experimental dNP and in glucose-stressed glomerular cells in vitro,3,4 suggesting that this bile acid derivative may be a safe and efficacious therapy in dNP. Whether TUDCA targets a pathomechanism independent of RAAS inhibition and hence provides an added value on top of RAAS inhibition in dNP remains unknown. In addition, although the ER-stabilizing effect of TUDCA is established, the receptors involved remain unknown. Like other bile acid derivatives, TUDCA acts as an agonist of the farnesoid X receptor (FXR, NR1H4), which is highly expressed in renal tubular cells.5 Hence, we hypothesized that TUDCA targets maladaptive ER stress signaling not only in glomerular but additionally in tubular cells via FXR agonism, ameliorating dNP through a mechanism independent and on top of RAAS inhibition.
First, we ascertained the expression of FXR (NR1H4) and FXR-dependent genes in the kidney. On the basis of the Nephroseq database (www.nephroseq.org), FXR and the FXR-dependent genes suppressor of cytokine signaling 3 (SOCS3) and dimethylarginine dimethylaminohydrolase 1 (DDAH1)6,7 are predominantly expressed in the tubulointerstitium (Figure 1A). Furthermore, SOCS3 and DDAH1 were readily detectable in tubular cells, but not within glomeruli of human and mouse kidney (Figure 1, B and C). Analyses of human proximal tubular epithelial cells, endothelial cells, podocytes, and mesangial cells without or with TUDCA treatment revealed an induction of SOCS3 and DDAH1 only in tubular cells (Figure 1D). The necessity of FXR expression for TUDCA-dependent SOCS3 and DDAH1 expression was confirmed by short hairpin RNA-mediated knockdown of FXR (Figure 1E). These results demonstrate that FXR mediates TUDCA-dependent effects in tubular cells.
Figure 1.

FXR and FXR-dependent genes (SOCS3 and DDAH1) are expressed in tubular cells. (A) Predominant tubulointerstitial expression of FXR (NR1H4), SOCS3, and DDAH1 in the Woroniecka cohort of the Nephroseq database. (B and C) Predominant tubular expression of the FXR-dependent genes SOCS3 and DDAH1; immunofluorescence analyses of SOCS3 and DDAH1 (red; nuclear DAPI counterstain, blue) in nondiabetic human kidney biopsy samples (B) and murine kidney sections [decibels per meter (C)]. (D) Expression of SOCS3 and DDAH1 is readily detectable at baseline (control, C) and is further increased by TUDCA (T; 500 μM, 24 hours) specifically in human tubular cells (HKC-8) but not in endothelial cells (EA.hy926), podocytes (h.Podo), or mesangial cells (h.MC). (E) shRNA lentiviral–mediated knockdown of FXR expression (FXRKD) in tubular cells (HKC-8) averts the TUDCA-mediated induction of FXR-dependent genes (SOCS3 and DDAH1) compared with nontransduced cells (C) or cells transduced with scrambled shRNA (Scr). Representative immunoblots (bottom, D and E) and bar graphs (top, D and E) summarizing results. Bar graphs showing mean±SEM obtained from three independent repeat experiments each with three disjunct replicates. Scale bar, 20 μm (B and C); glomeruli indicated by white dashed circles (B and C); *P<0.05; **P<0.01 (one-way ANOVA).
To scrutinize the in vivo relevance of FXR agonism by TUDCA, we treated 16-week-old db/db mice with TUDCA in the absence or presence of the FXR inhibitor Z-guggulsterone.8,9
As Z-guggulsterone may have off-target effects, we ascertained the role of FXR using vivo-morpholinos (FXR-MO). Treatment of mice with FXR-MO efficiently reduced renal FXR expression (Supplemental Figure 1A) and treatment with Z-guggulsterone efficiently abolished the TUDCA-mediated induction of SOCS3 and DDAH1 (Supplemental Figure 1B). Furthermore, the TUDCA-mediated reduction of activating transcription factor 6 (ATF6) and CCAAT-enhancer-binding homologous protein (CHOP) expression was abrogated by Z-guggulsterone (Supplemental Figure 1C). These results demonstrate the efficacy of the chosen in vivo approaches. Blood glucose level did not differ among the experimental groups (Supplemental Figure 1D). TUDCA treatment reduced albuminuria (Figure 2A) and improved histologic glomerular injury, as reflected by extracellular matrix accumulation (determined as fractional mesangial area) and glomerular diameter (Figure 2, B, C, and F). However, although Z-guggulsterone or FXR-MO largely abolished TUDCA’s protective effect on albuminuria, these interventions did not impede TUDCA-mediated protection from glomerular injury (Figure 2, B, C, and F). These data suggest that TUDCA ameliorates glomerular damage in dNP independent of FXR while protecting the tubular compartment via FXR. Indeed, TUDCA induced SOCS3 and DDAH1 expression specifically in tubular cells via FXR agonism (Supplemental Figure 1, E and F), which is congruent with the in vitro effects. Furthermore, TUDCA reduced hallmarks of tubular injury in dNP, such as tubular dilation (Figure 2, D and F), expression of kidney injury molecule-1 (KIM-1; Figure 2E), or tubulointerstitial inflammation and fibrosis (Figure 2G, Supplemental Figure 1G) in db/db mice. These tubular protective effects of TUDCA were lost after concomitant treatment with Z-guggulsterone or in vivo FXR knockdown (Figure 2, D–F), corroborating that TUDCA conveys its tubular protective effects in dNP via FXR agonism. Even prolonged FXR agonism with Z-guggulsterone (10 weeks) did not abolish glomerular protection by TUDCA (Supplemental Figure 2). This suggests that TUDCA protects from glomerular damage in dNP independent of FXR agonism even at later disease stages, which is congruent with the lack of glomerular FXR expression (Figure 1). The glomerular and tubular protective effects of TUDCA were confirmed using the well established model of dNP in eNOS knockout (eNOS−/−) mice.10 Again, TUDCA ameliorated both glomerular and tubular damage (Supplemental Figure 3), corroborating the fact that TUDCA protects both the glomerular and tubular compartment.
Figure 2.

TUDCA protects the tubular compartment in db/db mice via FXR. (A) TUDCA (T) reduces albuminuria (ΔUACR; reflecting the fold change of albuminuria from baseline) in db/db mice as compared with PBS-treated control mice (db). This effect is largely abolished by Z-guggulsterone (T+Gu) or in vivo knockdown of FXR using morpholino (T+FXR-MO). (B and C) TUDCA (T) reduces (B) extracellular matrix accumulation (fractional mesangial area [FMA]) and (C) glomerular diameter (Glom. diameter) in db/db treated mice as compared with PBS-treated control mice (db). Concomitant treatment with Z-guggulsterone (T+Gu) or in vivo knockdown of FXR using morpholino (T+FXR-MO) does not impede TUDCA-mediated improvement of glomerular injury markers. (D and E) TUDCA (T) reduces (D) tubular diameter (Tub. diameter) and (E) KIM-1 expression (bottom: representative immunoblots), reflecting tubular injury, in db/db mice as compared with PBS-treated control mice (db). This effect is largely abolished by concomitant Z-guggulsterone treatment (T+Gu) or in vivo knockdown of FXR using morpholino (T+FXR-MO). (F) Representative histologic sections (PAS staining) showing glomerular and tubular morphology in db/db control mice (db), db/db mice with TUDCA (T), db/db mice with concomitant TUDCA and Z-guggulsterone (T+Gu) treatment, or TUDCA treatment paralleled by in vivo knockdown of FXR using morpholino (T+FXR-MO). (G) Expression of inflammatory (IL-6, TNF-α) and fibrotic (collagen 1α2) markers is reduced in TUDCA-treated (T) db/db mice as compared with PBS-treated db/db control mice (db). This effect is abrogated by concomitant treatment with Z-guggulsterone (T+Gu). Representative RT-PCR images (bottom) and bar graph summarizing results. Bar graphs reflecting mean±SEM (A–E and G) of at least six mice per group (number of mice per group: db, 15; T, 13; T+Gu, 12; T+FXR-MO, 6); scale bar, 20 μm (F); *P<0.05; **P<0.01; ***P<0.001 (one-way ANOVA).
The above data demonstrate that TUDCA targets the tubular and glomerular compartments through FXR-dependent and -independent mechanisms, respectively, in dNP. As RAAS inhibition and TUDCA appear to target different pathomechanisms in dNP, we next evaluated whether TUDCA provides protection from dNP on top of RAAS inhibition. Sixteen-week-old db/db mice were treated with PBS (control), ACE inhibitor enalapril, TUDCA, or a combination of both. Neither single nor combined treatment had an effect on blood glucose levels (Supplemental Figure 4A).
Enalapril normalized angiotensin II plasma levels, whereas TUDCA had no significant effect (Supplemental Figure 4B). Treatment with TUDCA or enalapril for 6 weeks comparably diminished albuminuria (Figure 3A) and glomerular extracellular matrix accumulation and diameter (Figure 3, B–D). Importantly, combined treatment (enalapril and TUDCA) ameliorated albuminuria and fractional mesangial area even further (Figure 3, A and B). However, compared with other glomerular injury markers, the combined treatment did not provide additional protection (Figure 3, C, E, and F), suggesting that the efficacious reduction of albuminuria by combined enalapril and TUDCA treatment cannot be attributed to additional glomerular protection. Considering TUDCA’s tubuloprotective effects observed above we analyzed tubular injury markers. TUDCA but not enalapril ameliorated tubular damage, as reflected by tubular diameter (Figure 3, D and G) and KIM-1 expression (Figure 3H). Thus, TUDCA provides additional renal protection on top of enalapril, apparently by protecting the tubulointerstitial compartment.
Figure 3.

TUDCA provides add-on protection on top of enalapril in db/db mice. (A) Combined treatment with enalapril and TUDCA (E+T) over 6 weeks reduces the increase of albuminuria (ΔUACR, fold change of the urine albumin-to-creatinine ratio) in db/db mice more efficaciously than treatment with enalapril (E) or TUDCA (T) alone. (B–F) Enalapril (E), TUDCA (T), and the combination of enalapril and TUDCA (E+T) reduce glomerular damage, as reflected by extracellular matrix accumulation (fractional mesangial area [FMA]) (B and D), glomerular diameter (C and D), or glomerular basement membrane (GBM) thickness (E and F) to an overall comparable extent. Representative PAS-stained histologic sections (D) and transmission electron microscope (E) images. (G and H) TUDCA (T), but not enalapril (E), reduces tubular damage, as reflected by tubular diameter (G, see also D) and KIM-1 expression (H). Bar graphs reflecting mean±SEM (A–C and F–H) of at least six mice in each group (number of mice per group: C, db/m, 6; C, db/db, 10; E, 6; T, 8; E+T, 6); representative immunoblots (H, bottom); scale bar, 20 μm (D) or 1 µm (E); *P<0.05; **P<0.01; ***P<0.001 (one-way ANOVA).
To ascertain whether TUDCA targets maladaptive ER signaling in the tubular compartment and whether this distinguishes TUDCA treatment from enalapril treatment we determined expression of ATF6 and CHOP, two mediators of maladaptive ER stress. Renal expression of ATF6 and CHOP was reduced in TUDCA-treated db/db mice; however, in agreement with previous results,11 enalapril alone failed to do so (Figure 4, A and B). Immunofluorescence analyses revealed that the TUDCA-mediated reduction of ATF6 and CHOP expression was detectable in the glomerular compartment (Figure 4, C and D), which corroborates previous findings,3,12 but it was also detected in the renal cortex and medulla (Figure 4, C and D). Intriguingly, Z-guggulsterone specifically reversed TUDCA-mediated ATF6 and CHOP suppression in the tubular but not the glomerular compartment (Figure 4, E and F), suggesting that TUDCA ameliorates maladaptive ER signaling in tubular cells specifically via FXR. This tubular-specific effect of TUDCA provides a rationale for the renoprotective effect provided by TUDCA on top of enalapril.
Figure 4.

TUDCA but not enalapril ameliorates ER stress in tubular cells. (A and B) TUCDA but not enalapril reduces the ER stress markers ATF6 (A) and CHOP (B) in renal extracts. The combined treatment of enalapril plus TUDCA (E+T) has no additive effect on ER stress markers. Bar graphs reflecting mean±SEM (A and B, top) of at least six mice in each group (number of mice per group: C, db/m, 6; C, db/db, 10; E, 6; T, 8; E+T, 6); representative immunoblots (A and B, bottom); *P<0.05; **P<0.01; ***P<0.001 (one-way ANOVA). (C and D) Immunofluorescence analyses of ATF6 (C) and CHOP (D) expression in outer renal cortex (top, glomeruli indicated by the white dashed circles) and in inner renal tissue (medulla, bottom) in control decibels per meter (C) and diabetic (db/db) without treatment (C), enalapril (E), TUDCA (T), or enalapril plus TUDCA (E+T) treatment; scale bar, 20 μm (C and D). (E and F) Immunofluorescence analyses of ATF6 (E) and CHOP (F) expression in outer renal cortex (top, glomeruli indicated by the white dashed circles) and in inner renal tissue (medulla, bottom) in control diabetic (db/db) mice without treatment (db), TUDCA (T), or concomitant treatment with Z-guggulsterone (T+Gu), scale bar, 20 μm (E and F). (G) Proposed model showing the tubular specific effect of TUDCA via FXR agonism. TUDCA ameliorates ER stress in the glomerular and tubular compartment. Within the tubular compartment alleviation of ER stress depends on FXR, whereas other bile acid receptors (BARs) in the glomerular compartment remain to be identified. TUDCA may provide additional tubular protective effects via FXR-dependent regulation of SOCS3, DDAH1, or other genes.
A mechanistic relevance of maladaptive ER signaling for glomerulopathy in dNP has recently been established and amelioration of maladaptive ER signaling improved dNP in animal models.3,4,13 Here, we show that the ER stress chaperone TUDCA ameliorates both glomerular and tubulointerstitial damage in two murine models of dNP. Intriguingly, FXR agonism specifically protects the tubular compartment and is sufficient to reduce albuminuria. This finding is congruent with the close correlation of tubular damage and interstitial fibrosis with albuminuria and progression of dNP.14,15 Accordingly, a primary causative function of proximal tubule injury in dNP has been proposed.16 Furthermore, lower markers of tubular damage are associated with regression of microalbuminuria in patients with diabetes, underscoring a therapeutic potential of tubuloprotection.17
These findings support a pathogenic role of tubulointerstitial damage in dNP and, importantly, implies that therapies specifically targeting tubular damage may provide additional renoprotective effects on top of ACE inhibition.
Although the tubuloprotective effect of TUDCA depends on FXR, the receptors through which TUDCA targets the glomerular compartment remain unknown. Of note, TUDCA targets other bile acid receptors, including GPCR-TGR5, which has been shown to protect from dNP.18 Further studies are needed to identify the receptors mediating the glomeruloprotective effect of TUDCA in dNP (Figure 4G).
In addition to amelioration of maladaptive ER signaling, TUDCA induced expression of FXR-dependent genes (e.g., SOCS3 and DDAH1), suggesting that TUDCA targets additional pathways via FXR agonism in dNP. SOCS3 overexpression protects rats from dNP and reverses glucose-induced activation of JAK/STAT signaling and expression of STAT-dependent genes, including growth factors, chemokines, and extracellular matrix proteins.19 In addition, DDAH1 inhibits asymmetric dimethylarginine and Ng-monomethyl-L-arginine, two inhibitors of nitric oxide synthase and hence of nitric oxide production.20 As eNOS deficiency markedly aggravates dNP in mice21 and is suggested to contribute to dNP in humans,22 induction of DDAH1 via FXR may endow TUDCA with additional FXR-dependent renoprotective effects in dNP (Figure 4G). Accordingly, TUDCA may protect from dNP not only by ameliorating maladaptive ER signaling, but additionally by targeting FXR- or TGR5-dependent pathophysiologic mechanisms.
Collectively, this study demonstrates that TUDCA-mediated FXR agonism specifically protects the tubular compartment from hyperglycemia-induced renal damage, thus identifying a tubular-specific pathogenic but therapeutically amendable pathway in dNP. Considering that TUDCA has been in clinical use for many years and is generally well tolerated, clinical studies evaluating the combined effect of TUDCA and RAAS inhibition in patients with dNP seem to be warranted.
Concise Methods
For further details please see Supplemental Material.
Mice
Male db/db mice (C57BL/KSJRj-db) were obtained from Janvier Labs (Le Genest-Saint-Isle, France). Male Nos3−/− (also known as eNOS−/−) mice were obtained from Jackson Laboratories (Bar Harbor, ME). All mouse experiments were conducted following standards and procedures approved by the local Animal Care and Use Committee (Landesverwaltungsamt Halle, Germany).
dNP Models
In this study, we utilized two distinct mouse models of dNP. We analyzed 16-week-old (db/db) mice, representing type 2 diabetes mellitus, and eNOS−/− mice with 10 weeks of persistent hyperglycemia after streptozotocin (STZ) injection, representing type 1 diabetes mellitus.10 eNOS−/− mice received STZ (60 mg/kg body wt administered intraperitoneally, freshly dissolved in 0.05 M sterile sodium citrate, pH 4.5) for 5 consecutive days.23 Persistent hyperglycemia was ensured by demonstrating blood glucose levels >300 mg/dl from 2 weeks after the last STZ injection.23 Blood glucose levels were determined in tail vein blood samples using ACCU-CHEK glucose strips. In the first 3 weeks after the onset of hyperglycemia, blood glucose values were measured two times per week, and thereafter once per week. Mice with blood glucose levels >500 mg/dl received 1–2 U of insulin (Lantus) to avoid excessive and potentially lethal hyperglycemia.23
In Vivo Interventions
Male db/db mice were randomly assigned to five experimental groups at 16 weeks of age: db/db control mice (n=15); db/db mice with enalapril treatment (ACE inhibitor, 50 mg/L administered orally via the drinking water24; n=6); TUDCA (150 mg/kg body wt once daily, administered intraperitoneally3; n=13); enalapril and TUDCA (n=8); TUDCA and Z-guggulsterone (catalogue no. CAS 39025–23–5, 10 mg/kg body wt, once daily, administered intraperitoneally; n=12). Z-guggulsterone was prepared in 100 mM DMSO, and diluted in 1% methylcellulose.25 A subset of the db/db control mice (n=10) received 100 mM DMSO diluted in 1% methylcellulose, whereas the other mice received PBS as a control. Results in PBS– and DMSO methylcellulose–injected db/db mice did not differ. Additionally, db/m (nondiabetic) mice were used as nondiabetic controls (db/m-C, n=6). eNOS−/− mice with 10 weeks persistent hyperglycemia were randomly assigned to two experimental groups: eNOS−/−diabetic with PBS injection group (n=5), and diabetic with TUDCA injection group (n=5; 150 mg/kg body wt once daily, administered intraperitoneally5). After 6 or 10 weeks of treatment, mice were euthanized, perfused first with ice-cold PBS, followed by 4% paraformaldehyde buffered in PBS. Kidneys were extracted as previously described.3,23,24
Transmission Electron Microscopy
Ultrastructural images of the glomerular filtration barrier were obtained by transmission electron microscopy as previously described.3 Renal tissues were fixed with a mix of 2.5% glutaraldehyde, 2.5% polyvidone 25, and 0.1 M sodium cacodylate (pH 7.4). After washing with 0.1 M sodium cacodylate buffer (pH 7.4), samples were postfixed in the same buffer containing 2% osmium tetroxide and 1.5% potassium ferrocyanide for 1 hour, washed in water, contrasted en bloc with uranyl acetate, dehydrated using an ascending series of ethanol, and embedded in glycidyl ether 100-based resin. Ultrathin sections were cut with a Reichert Ultracut S ultramicrotome (Leica Microsystems, Wetzlar, Germany), contrasted with uranyl acetate and lead citrate, and were viewed with an EM 10 CR electron microscope (Carl Zeiss NTS, Oberkochen, Germany). Thickness of the glomerular basement membrane was analyzed using ImageJ software. For each image, the basement membrane thickness was determined at 15 adjacent and evenly distributed locations.
Angiotensin II Analysis
Blood samples were obtained from the inferior vena cava of 22-week-old anticoagulated mice (500 U of unfractionated heparin via intraperitoneal injection before blood sampling24). Heparinized plasma was obtained by centrifugation of blood samples for 10 minutes at 2000×g at room temperature. Plasma samples were stored at −80°C until analyses. Angiotensin II was determined using a mouse Angiotensin II ELISA (Sigma Aldrich) according to the manufacturer’s instructions.
Cell Culture
Podocytes were routinely grown in RPMI 1640 media. The culture plates were coated with collagen type 1 at 33°C in the presence of IFN-γ (10 U/ml) to enhance expression of a thermosensitive T antigen.25 Under these conditions, cells proliferate and remain undifferentiated. To induce differentiation, podocytes were grown at 37°C in the absence of IFN-γ for 12–14 days. Experiments were performed after 12 days of differentiation. Differentiation was confirmed by determining expression of synaptopodin and Wilms tumor 1 protein. Human proximal tubular epithelial cells (HKC-8) were grown in DMEM/F12 (1:1) media in the presence of 10% FCS and 1% ITS.26 Cells were grown to confluent monolayers at 37°C and then trypsinized and passaged for the planned experiments. Human endothelial hybrid cells (EA.hy926) were maintained in DMEM low glucose media at 37°C.27 Immortalized human mesangial cells were cultured as previously described, with minor modifications.28 In the subset of in vitro experiments the cells were treated with TUDCA (500 μM in PBS for 24 hours) on reaching 80% confluency.
In Vitro Knockdown
Knockdown of the NR1H4 (FXR) gene in HKC-8 cells was achieved using lentiviral transduction. Short hairpin RNA constructs (pLKO1-NR1H4) targeting human NR1H4 (ATTATAGTGGTATCCAGAGGC) and scrambled nonsilencing RNA (ATGTCCGTAATTCAGTCAGGC) were purchased from Dharmacon (Lafayette, CO). Lentiviral particles were generated and concentrated from HEK-293T cells as previously described, with minor modifications.29 In brief, HEK-293T cells were transduced with pLKO1-NR1H4 together with the packaging plasmid psPAX2 and VSV-G expressing plasmid (pMD2.g; Addgene). Lentiviral particles were harvested from the supernatant after 36 and 48 hours post-transduction. The lentiviral supernatant was concentrated and then added to HKC-8 cells. Knockdown efficiency was confirmed by immunoblotting.
RT-PCR
Kidney tissues were thawed on ice and transferred directly into TRIzol (Life Technologies, Darmstadt, Germany) for isolation of total RNA following the manufacturer’s protocol. Quality of total RNA was ensured on agarose gel and by analyses of the A260/280 ratio. The reverse transcription reaction was conducted using 1 μg total RNA after treatment with DNase (5 U/5 μg RNA) followed by reverse transcription using RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Semiquantitative polymerase chain reactions were performed and gene expression was normalized to β-actin. Reactions lacking reverse transcription served as negative controls. The PCR primer sequences were as follows: IL-6, forward 5′-TCTCTGCAAGAGACTTCCATCC-3′, reverse 5′-GGAAATTGGGGTAGGAAGGACTA-3′; TNF-α, forward 5′-ACAGAAAGCATGATCCGCGA-3′, reverse 5′-TCCACTTGGTGGTTTGCTACG-3′; collagen 1α2, forward 5′-GCTGGTGTAATGGGTCCTCC-3′, reverse 5′-ATCCGATGTTGCCAGCTTCA-3′; β-actin, forward 5′- CTAGACTTCGAGCAGGAGATGG-3′, reverse 5′-GCTAGGAGCCAGAGCAGTAATC-3′. PCR products were separated on a 1.8% agarose gel and visualized by ethidium bromide staining.
Vivo-Morpholinos Oligomer Treatment
The oligonucleotides sequence 5′-TTCATCTTGGCTACCACTCCAACTT-3′ against FXR (nuclear receptor subfamily 1, group H, member 4; NR1H4), blocking the translation of transcript variant 2, was synthesized as morpholinos (Gene Tools). MO were dissolved in PBS (100 μl; 6 mg morpholinos per kg body wt)30 and were injected intraperitoneally (every other day for 6 weeks) into a subset (n=6) of TUDCA-treated mice.
Statistical Analyses
The data are summarized as mean±SEM. Statistical analyses were performed with paired t test or ANOVA, as appropriate, and post hoc comparison with the method of Tukey. The Kolmogorov–Smirnov test or D’Agostino–Pearson Normality test was used to determine whether the data are consistent with a Gaussian distribution. StatistiXL (www.statistixl.com) and Prism 5 (www.graphpad.com) software were used for statistical analyses. Statistical significance was accepted at values of P<0.05.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank Kathrin Deneser, Julia Judin, Juliane Friedrich, René Rudat, and Rumiya Makarova for excellent technical support.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (IS 67/5-3, IS 67/8-1, IS 67/11-1, SFB-845/B26N, and TH 1789/1-1 to T.M.; BI 1281/3-1 and SFB-1118 to P.P.N.; WA 3663/2-1 to H.W.; SFB854/A01, Me1365/7-2, and 1365/9-1 to P.R.M.; LI-1031/4-1 to J.A.L.; and SH 849/2-1 to K.S.) and Deutscher Akademischer Austauschdienst scholarships to M.M.A.-D. and A.E.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016101123/-/DCSupplemental.
References
- 1.Rosolowsky ET, Skupien J, Smiles AM, Niewczas M, Roshan B, Stanton R, Eckfeldt JH, Warram JH, Krolewski AS: Risk for ESRD in type 1 diabetes remains high despite renoprotection. J Am Soc Nephrol 22: 545–553, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gnudi L, Coward RJ, Long DA: Diabetic nephropathy: Perspective on novel molecular mechanisms. Trends Endocrinol Metab 27: 820–830, 2016. [DOI] [PubMed] [Google Scholar]
- 3.Madhusudhan T, Wang H, Dong W, Ghosh S, Bock F, Thangapandi VR, Ranjan S, Wolter J, Kohli S, Shahzad K, Heidel F, Krueger M, Schwenger V, Moeller MJ, Kalinski T, Reiser J, Chavakis T, Isermann B: Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat Commun 6: 6496, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inagi R: Endoplasmic reticulum stress as a progression factor for kidney injury. Curr Opin Pharmacol 10: 156–165, 2010 [DOI] [PubMed] [Google Scholar]
- 5.Zhang X, Huang S, Gao M, Liu J, Jia X, Han Q, Zheng S, Miao Y, Li S, Weng H, Xia X, Du S, Wu W, Gustafsson JA, Guan Y: Farnesoid X receptor (FXR) gene deficiency impairs urine concentration in mice. Proc Natl Acad Sci U S A 111: 2277–2282, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo F, Xu Z, Zhang Y, Jiang P, Huang G, Chen S, Lyu X, Zheng P, Zhao X, Zeng Y, Wang S, He F: FXR induces SOCS3 and suppresses hepatocellular carcinoma. Oncotarget 6: 34606–34616, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu T, Chouinard M, Cox AL, Sipes P, Marcelo M, Ficorilli J, Li S, Gao H, Ryan TP, Michael MD, Michael LF: Farnesoid X receptor agonist reduces serum asymmetric dimethylarginine levels through hepatic dimethylarginine dimethylaminohydrolase-1 gene regulation. J Biol Chem 281: 39831–39838, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Mencarelli A, Renga B, Palladino G, Distrutti E, Fiorucci S: The plant sterol guggulsterone attenuates inflammation and immune dysfunction in murine models of inflammatory bowel disease. Biochem Pharmacol 78: 1214–1223, 2009 [DOI] [PubMed] [Google Scholar]
- 9.Guan B, Li H, Yang Z, Hoque A, Xu X: Inhibition of farnesoid X receptor controls esophageal cancer cell growth in vitro and in nude mouse xenografts. Cancer 119: 1321–1329, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brosius FC 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T; Animal Models of Diabetic Complications Consortium : Mouse models of diabetic nephropathy. J Am Soc Nephrol 20: 2503–2512, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartner A, Cordasic N, Klanke B, Menendez-Castro C, Veelken R, Schmieder RE, Hilgers KF: Renal protection by low dose irbesartan in diabetic nephropathy is paralleled by a reduction of inflammation, not of endoplasmic reticulum stress. Biochim Biophys Acta 1842: 558–565, 2014 [DOI] [PubMed] [Google Scholar]
- 12.Chen YM, Zhou Y, Go G, Marmerstein JT, Kikkawa Y, Miner JH: Laminin β2 gene missense mutation produces endoplasmic reticulum stress in podocytes. J Am Soc Nephrol 24: 1223–1233, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qi W, Mu J, Luo ZF, Zeng W, Guo YH, Pang Q, Ye ZL, Liu L, Yuan FH, Feng B: Attenuation of diabetic nephropathy in diabetes rats induced by streptozotocin by regulating the endoplasmic reticulum stress inflammatory response. Metabolism 60: 594–603, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Ziyadeh FN, Goldfarb S: The renal tubulointerstitium in diabetes mellitus. Kidney Int 39: 464–475, 1991 [DOI] [PubMed] [Google Scholar]
- 15.Rodríguez-Iturbe B, Johnson RJ, Herrera-Acosta J: Tubulointerstitial damage and progression of renal failure. Kidney Int Suppl 68: S82–S86, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Bonventre JV: Can we target tubular damage to prevent renal function decline in diabetes? Semin Nephrol 32: 452–462, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaidya VS, Niewczas MA, Ficociello LH, Johnson AC, Collings FB, Warram JH, Krolewski AS, Bonventre JV: Regression of microalbuminuria in type 1 diabetes is associated with lower levels of urinary tubular injury biomarkers, kidney injury molecule-1, and N-acetyl-β-D-glucosaminidase. Kidney Int 79: 464–470, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang XX, Edelstein MH, Gafter U, Qiu L, Luo Y, Dobrinskikh E, Lucia S, Adorini L, D’Agati VD, Levi J, Rosenberg A, Kopp JB, Gius DR, Saleem MA, Levi M: G protein-coupled bile acid receptor TGR5 activation inhibits kidney disease in obesity and diabetes. J Am Soc Nephrol 27: 1362–1378, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ortiz-Muñoz G, Lopez-Parra V, Lopez-Franco O, Fernandez-Vizarra P, Mallavia B, Flores C, Sanz A, Blanco J, Mezzano S, Ortiz A, Egido J, Gomez-Guerrero C: Suppressors of cytokine signaling abrogate diabetic nephropathy. J Am Soc Nephrol 21: 763–772, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding H, Wu B, Wang H, Lu Z, Yan J, Wang X, Shaffer JR, Hui R, Wang DW: A novel loss-of-function DDAH1 promoter polymorphism is associated with increased susceptibility to thrombosis stroke and coronary heart disease. Circ Res 106: 1145–1152, 2010 [DOI] [PubMed] [Google Scholar]
- 21.Nakagawa T, Sato W, Glushakova O, Heinig M, Clarke T, Campbell-Thompson M, Yuzawa Y, Atkinson MA, Johnson RJ, Croker B: Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol 18: 539–550, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Huo P, Zhang D, Guan X, Mei Y, Zheng H, Feng X: Association between genetic polymorphisms of ACE & eNOS and diabetic nephropathy. Mol Biol Rep 42: 27–33, 2015 [DOI] [PubMed] [Google Scholar]
- 23.Shahzad K, Bock F, Dong W, Wang H, Kopf S, Kohli S, Al-Dabet MM, Ranjan S, Wolter J, Wacker C, Biemann R, Stoyanov S, Reymann K, Söderkvist P, Groß O, Schwenger V, Pahernik S, Nawroth PP, Gröne HJ, Madhusudhan T, Isermann B: Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int 87: 74–84, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shahzad K, Bock F, Al-Dabet MM, Gadi I, Kohli S, Nazir S, Ghosh S, Ranjan S, Wang H, Madhusudhan T, Nawroth PP, Isermann B: Caspase-1, but Not Caspase-3, Promotes Diabetic Nephropathy. J Am Soc Nephrol 27: 2270–2275, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isermann B, Vinnikov IA, Madhusudhan T, Herzog S, Kashif M, Blautzik J, Corat MA, Zeier M, Blessing E, Oh J, Gerlitz B, Berg DT, Grinnell BW, Chavakis T, Esmon CT, Weiler H, Bierhaus A, Nawroth PP: Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med 13: 1349–1358, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Racusen LC, Monteil C, Sgrignoli A, Lucskay M, Marouillat S, Rhim JG, Morin JP: Cell lines with extended in vitro growth potential from human renal proximal tubule: Characterization, response to inducers, and comparison with established cell lines. J Lab Clin Med 129: 318–329, 1997 [DOI] [PubMed] [Google Scholar]
- 27.Edgell CJ, McDonald CC, Graham JB: Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci U S A 80: 3734–3737, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banas B, Luckow B, Möller M, Klier C, Nelson PJ, Schadde E, Brigl M, Halevy D, Holthöfer H, Reinhart B, Schlöndorff D: Chemokine and chemokine receptor expression in a novel human mesangial cell line. J Am Soc Nephrol 10: 2314–2322, 1999 [DOI] [PubMed] [Google Scholar]
- 29.Kashif M, Hellwig A, Hashemolhosseini S, Kumar V, Bock F, Wang H, Shahzad K, Ranjan S, Wolter J, Madhusudhan T, Bierhaus A, Nawroth P, Isermann B: Nuclear factor erythroid-derived 2 (Nfe2) regulates JunD DNA-binding activity via acetylation: A novel mechanism regulating trophoblast differentiation. J Biol Chem 287: 5400–5411, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grande MT, Sánchez-Laorden B, López-Blau C, De Frutos CA, Boutet A, Arévalo M, Rowe RG, Weiss SJ, López-Novoa JM, Nieto MA: Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med 21: 989–997, 2015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.