

Abstract
Liddle syndrome is an autosomal dominant form of hypokalemic hypertension due to mutations in the β- or γ-subunit of the epithelial sodium channel (ENaC). Here, we describe a family with Liddle syndrome due to a mutation in αENaC. The proband was referred because of resistant hypokalemic hypertension, suppressed renin and aldosterone, and no mutations in the genes encoding β- or γENaC. Exome sequencing revealed a heterozygous, nonconservative T>C single-nucleotide mutation in αENaC that substituted Cys479 with Arg (C479R). C479 is a highly conserved residue in the extracellular domain of ENaC and likely involved in a disulfide bridge with the partner cysteine C394. In oocytes, the C479R and C394S mutations resulted in similar twofold increases in amiloride-sensitive ENaC current. Quantification of mature cleaved αENaC in membrane fractions showed that the number of channels did not increase with these mutations. Trypsin, which increases open probability of the channel by proteolytic cleavage, resulted in significantly higher currents in the wild type than in C479R or C394S mutants. In summary, a mutation in the extracellular domain of αENaC causes Liddle syndrome by increasing intrinsic channel activity. This mechanism differs from that of the β- and γ-mutations, which result in an increase in channel density at the cell surface. This mutation may explain other cases of patients with resistant hypertension and also provides novel insight into ENaC activation, which is relevant for kidney sodium reabsorption and salt-sensitive hypertension.
Keywords: ENaC, hypertension, hypokalemia, genetic renal disease, electrophysiology
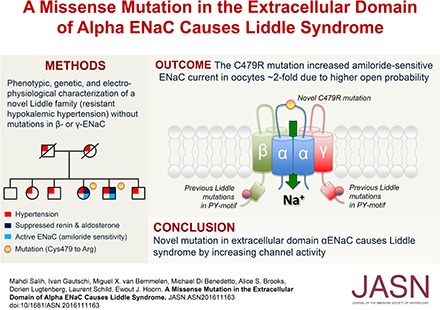
Hypertension is one of the most common noncommunicable disorders worldwide and a major risk factor for stroke, myocardial infarction, heart failure, and ESRD.1 Primary or essential hypertension is a complex genetic trait that is also influenced by other risk factors, such as dietary sodium and potassium intake, obesity, and diabetes.2,3 In contrast, monogenic forms of hypertension are very rare but have been instrumental in revealing the molecular pathways contributing to primary hypertension.4 The majority of these pathways point toward a role for increased sodium reabsorption by the kidneys, especially in the aldosterone-sensitive distal nephron.5 Indeed, several monogenic forms of hypertension are caused by mutations increasing sodium reabsorption in this segment through the sodium chloride cotransporter or the epithelial sodium channel (ENaC).6 In 1963, Liddle et al.7 reported a “familial renal disorder simulating primary aldosteronism but with negligible aldosterone secretion.” Liddle syndrome or pseudoaldosteronism (OMIM 177200) is now known as an autosomal dominant form of salt-sensitive hypertension that is further characterized by suppressed plasma renin and aldosterone, hypokalemia, and metabolic alkalosis.8 The syndrome was linked to mutations in the SCNN1B or SCNN1G gene, encoding the β- or γ-subunit of ENaC.9,10 Mutations in SCNN1B or SCNN1G delete or modify the intracellular PY motifs in ENaC in such a way that Nedd4–2 fails to ubiquitylate the channel, leading to a retention of active ENaC at the cell surface.11,12 Here, we report a family with Liddle syndrome due to a gain of function mutation in the extracellular domain of the α-subunit of ENaC (SCNN1A) that predominantly increases channel open probability (Po) but not channel surface density.
Results
Clinical and Genetic Characteristics of a Novel ENaC Mutation
The proband was referred because of resistant hypertension, hypokalemia, metabolic alkalosis, and suppressed levels of plasma renin and aldosterone. Despite a positive family history for hypertension (Figure 1A), no mutations in SCNN1B or SCNN1G were identified. Diagnostic exome sequencing revealed a novel heterozygous, nonconservative T>C single-nucleotide mutation that results in the substitution of cysteine 479 to arginine (C479R) in αENaC [c.1435T>C(p.(Cys479Arg))] (Figure 1B). The mutation is reported at a very low frequency in a large database collecting >60,000 exomes as proxy for variant allele frequencies in the general population (seven times heterozygously in >100,000 alleles; Exome Aggregation Consortium). The ENaC blocker triamterene normalized BP and serum potassium in the proband. Genotyping of the five siblings also identified the novel C479R mutation in subject II-4. The mutation segregated with suppressed plasma renin and aldosterone but not with hypertension (Figure 1C). Whole-exome sequencing in the proband did not identify additional mutations to explain the hypertensive trait in this family. Subject II-4 had mild hypertension (average ambulatory BP of 138/88 mmHg) that was sensitive to sodium chloride supplementation (145/91 mmHg) and also improved with triamterene (121/71 mmHg). In a standardized diuretic test, the natriuretic response to triamterene in the proband and II-4 was in the high range or increased compared with the response in healthy volunteers (Figure 1D).13 Thus, two siblings (the proband and II-4) show a clinical picture compatible with Liddle syndrome and carry the C479R missense mutation.
Figure 1.

The novel αENaC mutation is characterized clinically by hypertension, suppressed plasma renin and aldosterone, and an exaggerated natriuretic response to an ENaC blocker. (A) Pedigree showing three generations of the family with Liddle syndrome. Generation II was analyzed by genotyping and biochemical profiling. The arrow indicates the proband. (B) Sequence chromatogram. (C) The C479R mutation segregated with suppressed plasma renin and aldosterone but not with hypertension. Renin and aldosterone were measured in the absence of interfering drugs. Dashed lines represent lower limits of normal. HT, hypertension; NT, normotension; WT, wild type. (D) Results of a standardized diuretic test showing the natriuretic response to a single dose of the ENaC blocker triamterene in the proband and subject II-4 in comparison with healthy volunteers.13 *Proband; #subject II-4.
C479 Is Located in the Extracellular Domain of ENaC
The ultimate proof of Liddle syndrome, however, is the demonstration that the mutation results in a gain of function of ENaC. The DNA variant that encodes the C479R mutant has so far never been described. The C479 is a highly conserved Cys residue that belongs to the second cysteine-rich domain (CRD2) of the extracellular domain of ENaC that is likely involved in disulfide bridges.14 The human αENaC subunit (hαENaC) C479 is conserved among not only the ENaC subunits and ENaC homologs but also the Acid-Sensing Ion Channel 1 (ASIC1) orthologs (Figure 2A). The crystal structure of chicken ASIC1 reveals that the cASIC1 C366 forms a disulfide bond with another highly conserved cysteine, C291 in CRD2, that corresponds in hαENaC to a disulfide bond between C479 and the C394 (Figure 2B).15 Therefore, we analyzed not only the consequences of the C479R mutation on hENaC function in Xenopus laevis oocytes but also, the functional effects of the mutation of the partner Cys C394S involved in the disulfide bond. In addition, because C479 is a highly conserved Cys, we performed a similar functional analysis of the corresponding Cys mutations C507S and C422S in rat αENaC.
Figure 2.

C479 is a highly conserved Cys that forms a disulfide bond with C394. (A) Sequence comparison of hαENaC and rat αENaC (SCAA), βENaC (SCAB), and γENaC (SCAG) subunit isoforms with human hASIC1 and chicken cASIC1. (B) Crystal structure of a cASIC1 subunit with the disulfide bonds in the extracellular domain labeled in green for the first cysteine-rich domain (CRD1) and yellow for CRD2. The Cys366 (red) corresponding to Cys479 in the human αENaC (hSCAA) makes a disulfide bond with Cys291 (purple) corresponding to C394 in hSCAA (inset).
C479R Increases ENaC Current in Oocytes
Both C479R and C394S result in a similar approximately twofold increase in amiloride-sensitive ENaC current (Figure 3A). These results strongly suggest that the channel gain of function is due to the disruption of the disulfide bond between the two Cys. Because the proband is heterozygous for the C479R mutation, we replicated this condition in vitro by coinjecting ENaC wild type and C479R in a 1:1 ratio and observed still a significant increase in ENaC current, but this effect was reduced by one half compared with C479R expressed alone (Figure 3B). To provide further evidence for a gain of function due to the disruption of the disulfide bridge, we tested the corresponding mutations in rat αENaC subunit and found that the C507S and C422S mutations have comparable stimulatory effects on ENaC activity as C479R and C394S in hαENaC (Figure 3C). These results are consistent with previous observations that disruption of particular disulfide bonds in the CRD2 of the extracellular domain of rat ENaC results in a channel gain of function.14 Furthermore both C479Arg and C507Ser substitutions in hαENaC and rat αENaC subunit have comparable stimulatory effects on ENaC current, indicating that the effect does not depend on the substituting amino acid. Together, these observations support the idea that the disruption of the C479-C394 disulfide bridge by the C479R substitution is likely the primary cause of the observed gain of function in ENaC.
Figure 3.

αENaC C479R is a gain of function mutation. (A) Amiloride-sensitive current increase of αC479R ENaC mutant and the Cys partner, αC394S, mutant. Current values were normalized for the average INa of the wild-type (wt) control obtained in oocytes of each independent batch (n≥4). Bars represent mean±SD for hαENaC wt (n=225), hαENaC C479R (n=231), and hαENaC C394S (n=18). (B) Normalized amiloride-sensitive current values as in A with hαENaC wt (n=14) and hαENaC C479R expressed alone (n=13) or together with hαENaC wt at a cRNA weight ratio of 1:1 (n=13). (C) Normalized amiloride-sensitive current values for rat αENaC wt (n=12), C507S (n=12), and C422S (n=12) corresponding to C479R and C394S in the human αENaC sequence. *P < 0.05.
Surface Density of C479R Channel Mutant
The increase in ENaC activity due to the C479R mutation can result from an increase in channel Po, single-channel conductance, or the number of channels at the cell surface. To test the latter possibility, we analyzed on Western blot the cleaved (CL) forms of the wild type and C479R α- and γENaC subunits (Figure 4, A and B), expressed in the whole oocyte, at the cell surface (Figure 4, C and D, Supplemental Figure 1) and analyzed urinary extracellular vesicles (Supplemental Figure 2).16 It is now well established that the CL forms of α and γ represent the mature ENaC subunits that are incorporated in the functional channel complex present at the cell surface.17 The full length (FL) of αENaC (93 kD) was detected in oocytes expressing α-subunit alone or αβγENaC wild type and C479R mutant (Figure 4A). The CL form of αENaC (69 kD) was detected only for the αβγENaC wild type and the αC479Rβγ mutant. The CL form of γ-subunit (76 kD) was detected for both αβγENaC wild type and αC479Rβγ mutant. Quantification of the intensities of the CL forms of α- and γ-subunits from the αC479Rβγ ENaC complex relative to those of α and γ in the αβγENaC complex did not show any significant difference. Subsequently, cell surface expression of ENaC wild type and mutant was assessed by biotinylation of surface proteins and followed by affinity purification on Neutravidin-agarose beads. The representative immunoblot in Figure 4C shows that, in oocytes expressing comparable amounts of αENaC wild type and mutant under their FL and CL forms (Figure 4C, left panel) (CL, 69 kD; FL, 97 kD), the CL band is the main detected form at the cell surface at similar amounts for both channel types (Figure 4C, right panel). We quantified the FL + CL band intensities in total membrane and cell surface fractions of wild-type and C479R mutant ENaC-expressing oocytes (Figure 4D). The data obtained from four independent experiments show that, for a comparable expression of αENaC wild type and C479R mutant, the amounts of wild-type and C479R mutant ENaC functional channels at the cell surface are similar. Similarly, the αENaC C479R mutation did increase γENaC levels at the cell surface of injected oocytes (Supplemental Figure 1). On the basis of these data, we can conclude that the approximately twofold higher ENaC current measured for the hαC479R mutant is not correlated with an increase in the mature channel density at the cell surface. Consistently, no differences in the abundance of CL αENaC in urinary extracellular vesicles were detected in the two subjects carrying the mutation (Supplemental Figure 2).16
Figure 4.

C479R does not increase channel surface density. (A) Anti-HA tag (red; left panel) and anti-γENaC (green; right panel) Western blot analysis of Triton-soluble fractions from Xenopus oocytes noninjected (n.i.) or injected with cRNAs for hα-HA alone or with β- and γENaC cRNAs together with either the wild type (wt) or C479R mutant hα-HA. (B) The intensities of the bands corresponding to CL hα- and hγENaC were normalized to the amount of 2,2,2-Trichloroethanol–labeled total protein obtained for each lane on the blot. The ratios between the thus-calculated values for hα- and hγENaC in the hαwtβγENaC and those for hαC479RβγENaC are shown in the graph. Data correspond to mean±SEM (ten blots from seven independent experiments); differences are NS. (C, right panel) Anti-HA immunoblot analysis of neutravidin-bound fractions isolated from control (−) or cell surface biotinylated (+) Xenopus oocytes n.i. or injected with either αwt-HA/β/γ or αC479R-HA/β/γ cRNAs. (C, left panel) Inputs corresponding to 1% of the Triton-soluble preparations from biotinylated oocytes used in the pull-down experiments. (D) Values (mean±SD) of CL + FL band intensities of inputs or neutravidin-bound fractions corresponding to experiments shown in C. Results from four blots with samples of four independent experiments. Differences are NS.
Intrinsic Activity of C479R Mutant
To test the possibility of a gain of function C479R mutation due to an increase in channel Po, we used trypsin, which proteolytically cleaves ENaC and activates the channel by increasing the Po.18 We reasoned that, if the C479R channel mutant has a higher Po than the ENaC wild type, then it should show a higher current but a lower sensitivity to activation by trypsin. The magnitude of the currents in the absence of trypsin was significantly higher for the C479R and C394S mutants compared with the wild type (2.1- and 2.4-fold increases, respectively). After trypsin treatment, the currents expressed by the mutant forms were no longer different from those of ENaC wild type, although a trend toward higher currents was observed for the mutants (Figure 5A, Supplemental Figure 3 shows original traces). Shown in Figure 5B for individual oocytes, the wild-type hENaC has a significantly lower current than either of the mutants, but the trypsin-induced increase in current is higher than that for the C479R and C394S mutants. This smaller effect of trypsin shown for the C479R and C394S gain of function mutants is consistent with a higher basal Po. It is interesting to note that, even at high ENaC baseline currents, such as those observed for the mutants, the trypsin effect plateaued at a twofold increase in INa. This suggests that the baseline Po of C479R and C394S is below or equal to 0.5, whereas the baseline Po of wild-type hENaC is below or equal to 0.27. An alternative but less likely explanation is that the C479R and the C394S mutations decrease the efficiency of ENaC cleavage by trypsin, despite a gain of function effect of the mutation. An alternative explanation for the lower sensitivity to trypsin of the C479R and C394S mutants is an apparent saturable expression of the ENaC current because of the limited capacity of the oocyte to face large inward Na+ currents.19 The latter possibility was, however, excluded in experiments testing the effect of trypsin on wild-type, C479R, and C394S hENaC at different levels of current expression by injecting increasing amounts of cRNAs ranging from 0.1 to 10 ng per oocyte (Figure 5C). The slope of this linear relation was approximately twofold lower for the ENaC mutants than for the wild type. This lower response to trypsin for the gain of function ENaC mutants is consistent with a higher intrinsic activity of the channel with a higher Po. We then verified these observations with the corresponding Cys mutations, C507S and C422S, of rat ENaC and also included the rat βENaC Liddle mutant Y618A (Figure 6). Y618A disrupts the PY motif and increases the number of active channels at the cell surface.20 Wild-type ENaC and the Y618A mutant show a significantly higher response to trypsin compared with the C507S and C422S mutants, despite the fact that the Y618A baseline current was significantly higher than that of the wild type and the Cys mutants (Figure 6A). The linear relationship between currents with or without trypsin was similar for wild-type and Y618C rat ENaC but approximately twofold smaller for the C507S and C422S rat ENaC mutants (Figure 6B). Taken together, these results suggest that the ENaC gain of function mutations C479R and C507S are essentially due to an increased intrinsic channel activity.
Figure 5.

Lower trypsin sensitivity of C479R and C394S than wild type αENaC suggests higher intrinsic channel activity. (A) Amiloride-sensitive current (microampere) of hENaC wild-type (wt; n=17), hαENaC C479R (n=17), and hαENaC C394S (n=18) in the absence (−) or presence (+) of trypsin. The magnitude of the currents in the absence of trypsin was significantly higher for C479R and C394S compared with wt (P<0.01 by one-way ANOVA), whereas the currents in oocytes expressing the mutant forms were no longer significantly higher after trypsin treatment. (B) Relationship between baseline INa in the absence of trypsin and fold increase in INa after the addition of trypsin in wt and mutant human ENaC (C479R and C394S). Current values for a single oocyte (filled symbols) and means±SD (open symbols) are shown (P<0.01 by ANOVA). (C) Correlation between current INa (microampere) values in the absence and presence of trypsin. Linear regression analysis gives the following best fit values for the slopes hαwt (4.08; 95% confidence interval, 3.82 to 4.35), hαC479R (2.28; 95% confidence interval, 2.14 to 2.42), and hαC394S (2.23; 95% confidence interval, 2.03 to 2.42). Supplemental Figure 3 shows original traces. INa, Na+ current.
Figure 6.

Rat αENaC mutations C507S and C422S are also less sensitive to trypsin than wild type and the βENaC Liddle mutation Y618C. (A) Relationship between the baseline INa in the absence of trypsin and the fold increase in INa after the addition of trypsin for wild-type (wt) rat ENaC, two αENaC mutants (C507S and C422S), and the βENaC Liddle mutant Y618A. Current values for a single oocyte (filled symbols) and means±SD (open symbols) are shown (P<0.01 by ANOVA). (B) Correlation between current INa values (microampere) in the absence and presence of trypsin. Linear regression analysis gives best fit values for the slopes corresponding to rαwt/rβ/rγ, rαC507S/rβ/rγ, rαC422S/rβ/rγ, and rαwt/rβ/rγY618A, and the values were 4.07 (95% confidence interval, 3.92 to 4.23), 2.09 (95% confidence interval, 2.0 to 2.18), 1.92 (95% confidence interval, 1.74 to 2.09), and 4.04 (95% confidence interval, 3.79 to 4.30), respectively. INa, Na+ current.
Discussion
Here, we report a mutation in αENaC associated with Liddle syndrome. Our functional investigations of the C479R mutation show that this Cys mutation in the second cysteine-rich domain of the extracellular domain of αENaC is a gain of function mutation. Our data are consistent with a higher intrinsic ENaC activity due to an increase in Po, likely resulting from the disruption of the disulfide bonds between Cys479 and Cys394. The ENaC channel opener trypsin induces a lower response for the gain of function mutants hαC479R and rαC507S, consistent with a higher channel Po. This functional effect of the disruption of the Cys479-Cys394 disulfide bond is conserved in both rat and human ENaC channel isoforms. Our data are remarkably consistent with the mutational analysis of cysteine-rich domains of ENaC previously reported by Firsov et al.14 They showed that the αC16S mutation in rat αENaC (corresponding to the C479R and C507S mutations in our work) resulted in a threefold increase in amiloride-sensitive current without changing channel surface expression. Furthermore, mutation of the partner cysteine involved in the disulfide bridge (αC7S) had the same effects. Of interest, mutations of the cysteines in the extracellular cysteine-rich domains can result in either channel loss or gain of function. Indeed, mutation of the human Cys133 into a tyrosine causes the mirror image of Liddle syndrome, pseudohypoaldosteronism type 1, a severe salt-losing syndrome in neonates.14,21
The majority of the previously reported Liddle mutations affect the intracellular PY motif of β- or γENaC, impairing channel degradation by Nedd4–2.9,10 A mutation in αENaC causing Liddle syndrome has only been reported once previously and was also located in the PY motif.22 Therefore, this report on an αENaC mutation causing Liddle syndrome contains two novel aspects, including the location in the extracellular domain and the effect on Po rather than surface expression. Of note, αENaC gain of function mutations have been identified previously in patients with cystic fibrosis–like symptoms, but it is not known whether these mutations also caused hypertension.23,24 In addition, some of the mutations in α- or βENaC causing atypical cystic fibrosis were located outside the PY motif and increased channel Po.24,25 A gain of function mutation (N530) in the putative extracellular domain of γENaC causing Liddle syndrome has also been reported previously.26 However, structural models of ENaC on the basis of the crystal structure of the homolog ASIC1 predict that the N530 residue is located in the second transmembrane α-helix of γENaC and is not located in the extracellular domain of the channel. Finally, single-nucleotide polymorphisms of the αENaC gene locus correlated with salt-sensitive hypertension in one Chinese study.27
Of interest, the proband had the classic Liddle syndrome phenotype, whereas his sibling had a milder degree of salt-sensitive hypertension and no hypokalemia. Such differences between affected family members, including the absence of hypokalemia, have been observed previously.28 The phenotypic differences between the proband and II-4 suggest variable expressivity, possibly related to sex, environmental factors, or genetic modifiers. Alternatively, the proband and his hypertensive siblings without the mutation (subjects II-2, II-3, and II-6) (Figure 1A) may have an additional genetic variant predisposing to hypertension, although we did not identify this using whole-exome sequencing. Thus, in this family, the mutation segregated with suppressed renin and aldosterone but not hypertension. In addition, our family seemed to have a milder phenotype compared with previous Liddle kindreds.9,10,28 This may also relate to the molecular mechanism of the αENaC mutation.
In vivo ENaC channel Po determined by the patch clamp technique ranged from 0.3 to 0.7.29 In Xenopus oocytes, ENaC Po under comparable conditions was estimated to be 0.3.19 Assuming that trypsin stabilizes ENaC in the open conformation with a Po of one, then the slope current relations for the ENaC wild type and mutants suggest that, under our experimental conditions, the Po of wild-type ENaC is around 0.25 and that the Po of the mutants is around 0.5. Such an increase in Po, estimated from the trypsin response, represents the main component of the higher ENaC current expressed by the C479R and the related mutants. Such a gain of function resulting from an increased Po is certainly limited by the functional characteristics of the channels. Because ENaC likely never functions physiologically with a Po of one, a gain of function due to an increased channel gating would reasonably not exceed three- to fourfold. In contrast, Liddle gain of function mutation resulting from an increased ENaC retention at the cell surface may potentially enhance sodium transport to a much greater extent, because the capacity of the membrane surface to accommodate a high density of ENaC channels is less restricted than the capacity of ENaC to increase in its Po. Along this line, we observed that the Y618A mutation, affecting channel interaction with Nedd4–2, is more efficient than C507S in increasing ENaC-mediated current (Figure 6). Although an increase in single-channel conductance could theoretically explain the C479R channel gain of function, we know from previous structure-function studies on ENaC that such mutations are restricted to the second transmembrane domain, a region that participates in the ion channel pore and selectivity filter.30
In summary, we report a mutation in αENaC associated with Liddle syndrome. This represents the first ENaC gain of function mutation in the extracellular domain causing a higher intrinsic channel activity, a mechanism different from the previously reported mutations in β or γ associated with Liddle syndrome.9,10 This study raises the question about the necessity of genotyping patients with unexplained cases of hypertension associated with suppressed plasma renin and aldosterone and a poor response to standard antihypertensive therapy. Furthermore, this mutation provides novel insight into ENaC activation and potentially, distal nephron sodium reabsorption and salt-sensitive hypertension.
Concise Methods
Additional information can be found in Supplemental Material.
Studies in Patients
The patient studies were performed in accordance with the Declaration of Helsinki. All patients provided written informed consent, and the ethics committee approved the study. Plasma renin concentration and plasma aldosterone were measured by enzyme-kinetic assay and LC/MS, respectively. These measurements were performed in the absence of interfering drugs (renin-angiotensin system inhibitors or diuretics). The triamterene test was on the basis of the thiazide test.31 Results were compared with a historic cohort of healthy subjects receiving 100 mg triamterene in a similar setting.13
Exome Sequencing
The analyses of the exome data were divided into two steps: the renal gene panel analysis and the exome analysis. In the renal gene panel analysis, an in silico enrichment of genes associated with genetic renal disorders was performed (version: DGD141114). After the patient consented for the second step, exome analysis, likely pathogenic variants in all coding genes were analyzed. Exome sequencing was performed using an Illumina HiSeq2000TM sequencer at BGI-Europe (Copenhagen, Denmark). Read alignment to the human reference genome (GrCH37/hg19) and variant calling were performed at BGI-Europe using BWA and GATK software, respectively. Variant annotation was performed using a custom designed in-house annotation and variant prioritization pipeline.
Site-Directed Mutagenesis and Expression in X. laevis Oocytes
Mutant forms of the wild type of human α-, β-, and γENaC subunits had been cloned in the pBSK(+)_Xglob vector.32 In these vectors, the ENaC cDNAs are flanked by sequences corresponding to the 5′ and 3′ noncoding stretches of Xenopus β-globin, which boosts protein expression when injected into Xenopus oocytes.33 Plasmids suitable for in vitro transcription of wild-type and mutant forms of the hαENaC were generated as described in Supplemental Material. All constructs were verified by sequencing. Subsequently, healthy stage 5 and 6 X. laevis oocytes were pressure injected with mixes containing equal amounts of cRNAs encoding α-, β-, and γ-subunits for a total of (unless stated otherwise) 1 or 3 ng cRNA for human and rat ENaC, respectively. Mutant and control cRNAs were prepared in parallel, and greater than or equal to three independent batches of cRNA were used.
Electrophysiology
Electrophysiologic measurements were made 24–32 hours after injection with the standard two-electrode voltage clamp technique using a TEV-200A voltage clamp amplifier (Dagan, Minneapolis, MN), an ITC-16 digitizer interface (Instrutech Corp., Elmont, NY), and the PatchMaster data acquisition and analysis package (HEKA Elektronik Dr. Schulze GmbH, Ludwigshafen/Rhein, Germany). The amiloride-sensitive currents were measured in the presence of 10 μM of this blocker adjusted in a separate solution. Inward Na+ currents were generated by switching from the amiloride-containing perfusion solution to that without amiloride. In the experiment with proteases, the oocytes were perfused for 2 minutes with the amiloride-free solution supplemented with 2 μg/ml trypsin (Sigma-Aldrich Chemie GmbH, Buchs, Switzerland). All INa values were normalized in all experiments to the mean of the amiloride-sensitive currents measured for wild-type ENaC with the same oocyte batch.
Isolation of ENaC-Enriched Fractions
To isolate membrane fractions, 15–30 oocytes were disrupted by pipetting in 1.5 ml membrane isolation buffer followed by centrifugation through cell shredders. The membrane pellets obtained after 30 minutes of centrifugation at 20,000×g (4°C) were resuspended in membrane solubilization solution. Proteins in samples were resolved along with prestained molecular weight markers by SDS-PAGE on 5%–15% acrylamide gradient minigels supplemented with 0.5% (vol/vol) of 2,2,2-Trichloroethanol for subsequent in-gel fluorescence labeling of proteins.34 Total protein per lane was assessed densitometrically from these images using ImageJ. Band intensities were assessed with the Odyssey v2.1 software and normalized with the amount of total 2,2,2-Trichloroethanol–labeled protein in the corresponding lanes.
Analyses of Cell Surface Biotinylated Fractions
Control or injected oocytes (approximately 25 per condition) were incubated for 15 minutes on ice in 1 ml Biotinylation buffer. Biotinylated fractions were isolated from ENaC-enriched fractions that had been purified as described before.35 Neutravidin-bound fractions and Triton-soluble fractions (1% of total) were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and blocked as described before. To account for nonspecific binding to Neutravidin beads, band intensity values from control, nonbiotinylated samples were subtracted from the values of the corresponding biotinylated samples.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism software (GraphPad Software). Differences between groups were assessed by one-way ANOVA and the Tukey post hoc test. P<0.05 was considered statistically significant.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank the proband and his family members who participated in this study and the referring physician Dr. Arie T.J. Lavrijssen. We also thank Usha Musterd-Bhaggoe for technical support and Dr. Jan Loffing (University of Zürich) for epithelial sodium channel antibodies.
This study was supported by Swiss National Science Foundation grant 310030_135378 (to L.S.) and Dutch Kidney Foundation grant KSP-14OK19 (to E.J.H.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016111163/-/DCSupplemental.
References
- 1.Poulter NR, Prabhakaran D, Caulfield M: Hypertension. Lancet 386: 801–812, 2015 [DOI] [PubMed] [Google Scholar]
- 2.Mente A, O’Donnell MJ, Rangarajan S, McQueen MJ, Poirier P, Wielgosz A, Morrison H, Li W, Wang X, Di C, Mony P, Devanath A, Rosengren A, Oguz A, Zatonska K, Yusufali AH, Lopez-Jaramillo P, Avezum A, Ismail N, Lanas F, Puoane T, Diaz R, Kelishadi R, Iqbal R, Yusuf R, Chifamba J, Khatib R, Teo K, Yusuf S; PURE Investigators : Association of urinary sodium and potassium excretion with blood pressure. N Engl J Med 371: 601–611, 2014 [DOI] [PubMed] [Google Scholar]
- 3.Padmanabhan S, Caulfield M, Dominiczak AF: Genetic and molecular aspects of hypertension. Circ Res 116: 937–959, 2015 [DOI] [PubMed] [Google Scholar]
- 4.Lifton RP, Gharavi AG, Geller DS: Molecular mechanisms of human hypertension. Cell 104: 545–556, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Meneton P, Loffing J, Warnock DG: Sodium and potassium handling by the aldosterone-sensitive distal nephron: The pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol 287: F593–F601, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Scheinman SJ, Guay-Woodford LM, Thakker RV, Warnock DG: Genetic disorders of renal electrolyte transport. N Engl J Med 340: 1177–1187, 1999 [DOI] [PubMed] [Google Scholar]
- 7.Liddle GW, Bledsoe T, Coppage WS: A familial renal disorder simulating primary aldosteronism but with negligible aldosterone secretion. Trans Assoc Am Phys 76: 199–213, 1963 [Google Scholar]
- 8.Gennari FJ, Hussain-Khan S, Segal A: An unusual case of metabolic alkalosis: A window into the pathophysiology and diagnosis of this common acid-base disturbance. Am J Kidney Dis 55: 1130–1135, 2010 [DOI] [PubMed] [Google Scholar]
- 9.Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, Lifton RP: Hypertension caused by a truncated epithelial sodium channel gamma subunit: Genetic heterogeneity of Liddle syndrome. Nat Genet 11: 76–82, 1995 [DOI] [PubMed] [Google Scholar]
- 10.Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR, Ulick S, Milora RV, Findling JW, Canessa CM, Rossier BC, Lifton RP: Liddle’s syndrome: Heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 79: 407–414, 1994 [DOI] [PubMed] [Google Scholar]
- 11.Abriel H, Loffing J, Rebhun JF, Pratt JH, Schild L, Horisberger JD, Rotin D, Staub O: Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle’s syndrome. J Clin Invest 103: 667–673, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schild L, Canessa CM, Shimkets RA, Gautschi I, Lifton RP, Rossier BC: A mutation in the epithelial sodium channel causing Liddle disease increases channel activity in the Xenopus laevis oocyte expression system. Proc Natl Acad Sci U S A 92: 5699–5703, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Möhrke W, Knauf H, Mutschler E: Pharmacokinetics and pharmacodynamics of triamterene and hydrochlorothiazide and their combination in healthy volunteers. Int J Clin Pharmacol Ther 35: 447–452, 1997 [PubMed] [Google Scholar]
- 14.Firsov D, Robert-Nicoud M, Gruender S, Schild L, Rossier BC: Mutational analysis of cysteine-rich domains of the epithelium sodium channel (ENaC). Identification of cysteines essential for channel expression at the cell surface. J Biol Chem 274: 2743–2749, 1999 [DOI] [PubMed] [Google Scholar]
- 15.Jasti J, Furukawa H, Gonzales EB, Gouaux E: Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 449: 316–323, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Salih M, Fenton RA, Zietse R, Hoorn EJ: Urinary extracellular vesicles as markers to assess kidney sodium transport. Curr Opin Nephrol Hypertens 25: 67–72, 2016 [DOI] [PubMed] [Google Scholar]
- 17.Frindt G, Ergonul Z, Palmer LG: Surface expression of epithelial Na channel protein in rat kidney. J Gen Physiol 131: 617–627, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleyman TR, Carattino MD, Hughey RP: ENaC at the cutting edge: Regulation of epithelial sodium channels by proteases. J Biol Chem 284: 20447–20451, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anantharam A, Tian Y, Palmer LG: Open probability of the epithelial sodium channel is regulated by intracellular sodium. J Physiol 574: 333–347, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamura H, Schild L, Enomoto N, Matsui N, Marumo F, Rossier BC: Liddle disease caused by a missense mutation of beta subunit of the epithelial sodium channel gene. J Clin Invest 97: 1780–1784, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang SS, Grunder S, Hanukoglu A, Rösler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, Rossier BC, Lifton RP: Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet 12: 248–253, 1996 [DOI] [PubMed] [Google Scholar]
- 22.Goehl K, Haerteis S, Nelson-Williams C, Lifton RP, Korbmacher C, Rauh R: Functional characterization of a novel mutation in the α-subunit of the epithelial sodium channel (ENaC) found in Liddle’s syndrome [Abstract]. Acta Physiol 198: P-TUE-59, 2010 [Google Scholar]
- 23.Azad AK, Rauh R, Vermeulen F, Jaspers M, Korbmacher J, Boissier B, Bassinet L, Fichou Y, des Georges M, Stanke F, De Boeck K, Dupont L, Balascáková M, Hjelte L, Lebecque P, Radojkovic D, Castellani C, Schwartz M, Stuhrmann M, Schwarz M, Skalicka V, de Monestrol I, Girodon E, Férec C, Claustres M, Tümmler B, Cassiman JJ, Korbmacher C, Cuppens H: Mutations in the amiloride-sensitive epithelial sodium channel in patients with cystic fibrosis-like disease. Hum Mutat 30: 1093–1103, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Rauh R, Diakov A, Tzschoppe A, Korbmacher J, Azad AK, Cuppens H, Cassiman JJ, Dötsch J, Sticht H, Korbmacher C: A mutation of the epithelial sodium channel associated with atypical cystic fibrosis increases channel open probability and reduces Na+ self inhibition. J Physiol 588: 1211–1225, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rauh R, Soell D, Haerteis S, Diakov A, Nesterov V, Krueger B, Sticht H, Korbmacher C: A mutation in the β-subunit of ENaC identified in a patient with cystic fibrosis-like symptoms has a gain-of-function effect. Am J Physiol Lung Cell Mol Physiol 304: L43–L55, 2013 [DOI] [PubMed] [Google Scholar]
- 26.Hiltunen TP, Hannila-Handelberg T, Petäjäniemi N, Kantola I, Tikkanen I, Virtamo J, Gautschi I, Schild L, Kontula K: Liddle’s syndrome associated with a point mutation in the extracellular domain of the epithelial sodium channel gamma subunit. J Hypertens 20: 2383–2390, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Xu H, Li NF, Hong J, Zhang L, Zhou L, Li T, Ou Yang WJ, Cheng QY: [Relationship between four single nucleotide polymorphisms of epithelial sodium channel alpha subunit gene and essential hypertension of Kazakhs in Xinjiang.] Zhongguo Yi Xue Ke Xue Yuan Xue Bao 31: 740–745, 2009 [DOI] [PubMed] [Google Scholar]
- 28.Botero-Velez M, Curtis JJ, Warnock DG: Brief report: Liddle’s syndrome revisited--A disorder of sodium reabsorption in the distal tubule. N Engl J Med 330: 178–181, 1994 [DOI] [PubMed] [Google Scholar]
- 29.Nesterov V, Dahlmann A, Krueger B, Bertog M, Loffing J, Korbmacher C: Aldosterone-dependent and -independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. Am J Physiol Renal Physiol 303: F1289–F1299, 2012 [DOI] [PubMed] [Google Scholar]
- 30.Kellenberger S, Gautschi I, Pfister Y, Schild L: Intracellular thiol-mediated modulation of epithelial sodium channel activity. J Biol Chem 280: 7739–7747, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Colussi G, Bettinelli A, Tedeschi S, De Ferrari ME, Syrén ML, Borsa N, Mattiello C, Casari G, Bianchetti MG: A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin J Am Soc Nephrol 2: 454–460, 2007 [DOI] [PubMed] [Google Scholar]
- 32.Dirlewanger M, Huser D, Zennaro MC, Girardin E, Schild L, Schwitzgebel VM: A homozygous missense mutation in SCNN1A is responsible for a transient neonatal form of pseudohypoaldosteronism type 1. Am J Physiol Endocrinol Metab 301: E467–E473, 2011 [DOI] [PubMed] [Google Scholar]
- 33.Krieg PA, Melton DA: Formation of the 3′ end of histone mRNA by post-transcriptional processing. Nature 308: 203–206, 1984 [DOI] [PubMed] [Google Scholar]
- 34.Ladner CL, Yang J, Turner RJ, Edwards RA: Visible fluorescent detection of proteins in polyacrylamide gels without staining. Anal Biochem 326: 13–20, 2004 [DOI] [PubMed] [Google Scholar]
- 35.Michlig S, Harris M, Loffing J, Rossier BC, Firsov D: Progesterone down-regulates the open probability of the amiloride-sensitive epithelial sodium channel via a Nedd4-2-dependent mechanism. J Biol Chem 280: 38264–38270, 2005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.