

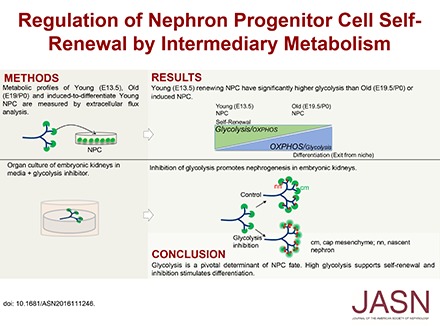
Abstract
Nephron progenitor cells (NPCs) show an age-dependent capacity to balance self-renewal with differentiation. Older NPCs (postnatal day 0) exit the progenitor niche at a higher rate than younger (embryonic day 13.5) NPCs do. This behavior is reflected in the transcript profiles of young and old NPCs. Bioenergetic pathways have emerged as important regulators of stem cell fate. Here, we investigated the mechanisms underlying this regulation in murine NPCs. Upon isolation and culture in NPC renewal medium, younger NPCs displayed a higher glycolysis rate than older NPCs. Inhibition of glycolysis enhanced nephrogenesis in cultured embryonic kidneys, without increasing ureteric tree branching, and promoted mesenchymal-to-epithelial transition in cultured isolated metanephric mesenchyme. Cotreatment with a canonical Wnt signaling inhibitor attenuated but did not entirely block the increase in nephrogenesis observed after glycolysis inhibition. Furthermore, inhibition of the phosphatidylinositol 3-kinase/Akt self-renewal signaling pathway or stimulation of differentiation pathways in the NPC decreased glycolytic flux. Our findings suggest that glycolysis is a pivotal, cell-intrinsic determinant of NPC fate, with a high glycolytic flux supporting self-renewal and inhibition of glycolysis stimulating differentiation.
Keywords: Cell Signaling, kidney development, Stem Cell Renewal, Glycolysis, Differentiation, PI3K/Akt
Nephron abundance varies among individuals and populations, with demonstrated influence of genetics and maternal nutritional status on nephron number in humans.1–4 Nephron progenitor cell (NPC) availability during kidney development is a major determinant of nephron number at birth. Low nephron endowment results in hypertension and CKD, both clinically significant diseases without a cure. Self-renewal of NPCs ensures a supply of cells for nephrogenesis until the cessation of nephrogenesis at P4 in mice and gestational age of 35 weeks in humans.5,6 Despite the critical importance of NPC availability for renal function across the life course, little is known about the mechanisms controlling NPC self-renewal versus differentiation.
NPCs reside in the nephron progenitor niche, which includes the cap mesenchyme (CM), the terminal ureteric tips and branch, and the surrounding stroma.7 NPCs are identified by their combined capacity for self-renewal to expand the progenitor cell pool, and differentiation to epithelial structures of the nascent nephron (Figure 1).8–10 Supported by the nephrogenic niche as the source for growth factors and nutrients, activation of signaling and metabolic pathways in NPCs engages renewal-specific gene expression that sustains the maintenance of the NPC pool for nephrogenesis. A subset of NPCs gradually exit the self-renewing pool to the transit compartment that undergoes differentiation.11,12 Along with a decreased proliferation rate, NPCs have increased exit rates from the renewing niche as development progresses.13 This shift in behavior is largely cell-intrinsic, demonstrated by heterochronic transplantation of young and old NPCs into young niches.13 The young and old NPCs also exhibit differential growth factor signaling and distinct transcriptional profiles.13 Therefore, the young NPCs from early (E13.5) CM are predominantly self-renewing, whereas old NPCs from late CM (E19.5 and P0) are poised to differentiate en masse.
Figure 1.

Experimental scheme of NPC isolation for metabolic profiling and ddPCR. Cited1+/Six2+ NPCs from different ages were isolated by MACS (see Concise Methods) by negative depletion. Isolated NPCs were expanded and used for extracellular flux analysis or ddPCR. Also see Supplemental Figure 1, A and B. CS, comma-shaped body; ECAR, extracellular acidification rate; OCR, oxygen consumption rate; PA, pretubular aggregate; RV, renal vesicle; UB, ureteric bud.
The self-renewing Cited1+/Six2+ cells are refractory to differentiation signals, whereas the transit Cited1−/Six2+ population is induced by cell-autonomous and nonautonomous differentiation signals to undergo differentiation to Six2low/Wnt4+ cells. These further differentiate to the Wnt4/Lef1+ pretubular aggregates (Figure 1), which undergo epithelialization (to the renal vesicle) and progressive maturation.14 Six2 expression is essential to maintain the progenitor state.15 FGF/PI3K-Akt, Bmp7/MAPK/JNK/Jun-ATF2/Smad, and Wnt9b/β-catenin signaling pathways are required for NPC self-renewal and differentiation, demonstrated by pharmacologic or genetic inactivation of different steps of these pathways, resulting in NPC renewal and differentiation defects.10,14,16,17
Bioenergetic pathways have emerged as important regulators of stem cell fate. Besides supporting the rapidly changing energy needs of the developing embryo, cellular metabolism provides macromolecules required to support self-renewal, proliferation, differentiation, or quiescence. Moreover, several metabolites that are intermediates of cellular metabolism pathways are substrates or cofactors for chromatin modifying enzymes (e.g., acetyl CoA, folate and methyl group donors, or α-ketoglutarate in histone demethylation).18,19 Thus, perturbations in cellular energy pathways and consequently metabolite availability directly influence the epigenome and can have drastic consequences on the ability of the stem cell to self-renew and differentiate, suggesting that the metabolic status of a stem cell may actively dictate cell fate rather than just be a marker of differentiation.20–23
In this study we demonstrate that NPC age and fate are intimately linked to intermediary metabolism pathways. Young NPCs demonstrate a high glycolysis and high energy state compared with old NPCs. Inhibition of the PI3K/Akt self-renewal signaling pathway or stimulation of differentiation in the young NPCs decreased their glycolysis flux. Ex vivo inhibition of glycolysis augmented nephrogenesis, with premature NPC differentiation and depletion independent of ureteric bud branching in E12.5 kidneys. Our data demonstrate an intrinsic difference between young and old NPCs and suggest intermediary metabolism as a balancing mechanism for NPC fate.
Results
Distinct Energy Metabolism Profiles of Young and Old NPCs
Glycolysis
To unravel the underlying mechanism of the recently documented differential cellular behavior of young (E13.5) versus old NPCs (P0) in the nephrogenic niche,13 we hypothesized on the basis of stem cell literature that young NPCs preferentially utilize glycolysis, an established driver of self-renewal in embryonic stem cells, to maintain their self-renewing state in contrast to old NPCs that are predominantly exiting the self-renewing niche. We therefore undertook an assessment of the metabolic profiles of young versus old NPCs. Using magnetic-activated cell sorting (MACS) as described by Brown et al.,11,14 NPCs were isolated to near 100% purity for extracellular flux measurements (Supplemental Figure 1). Isolated cells were cultured in the defined NPC expansion medium (NPEM11), which simulates nephron progenitor niche conditions for maintaining undifferentiated progenitor proliferation and self-renewal. Comparable levels of Cited1 and Six2 expression (Supplemental Figure 1B) validate the successful isolation and expansion of the same subpopulations of undifferentiated NPCs from E13.5 and P0 kidneys. Moreover, young and old NPCs demonstrate similar cell growth metrics (Supplemental Table 1).
As old time points, we chose E19.5 and P0 as time of harvest to evaluate whether prenatal and postnatal NPCs exhibit differences in extracellular metabolic flux. Glycolysis is significantly higher in E13.5 NPCs than E19.5 and P0 NPCs (P<0.05), shown by increased extracellular acidification rate (ECAR) after the addition of a saturating dose (10 mM) of glucose (Figure 2, B and C). Pharmacologic modulation of glycolysis allowed calculation of the glycolytic flux and glycolytic capacity. As expected, addition of oligomycin to inhibit mitochondrial respiration further augments the glycolytic flux as the cells try to maintain cellular ATP homeostasis, revealing the cellular glycolytic capacity (Figure 2D). In line with the higher flux, the glycolytic capacity and reserve of younger cells is also higher than that of older cells (Figure 2, D and E). Addition of glucose analog 2-deoxyglucose completely inhibits glucose-dependent ECAR increase (Figure 2, A and B), to specifically allow measurement of glycolysis-mediated acidification of the media. To ascertain that the glycolysis flux difference between young and old NPCs is not an artifact of cell expansion, we measured glycolysis in primary E13.5 and E19.5 NPCs that were not passaged after isolation. Glycolysis is significantly higher in E13.5 than in E19.5 NPCs (Figure 2F), as with expanded cells. Therefore, high glycolysis appears to be a distinguishing metabolic feature of young NPCs.
Figure 2.

Young (E13.5) NPCs have a significantly higher glycolytic rate and capacity than old (E19.5 and P0) NPCs. (A) Graph to explain basal, maximal, and reserve glycolysis capacity measurements. (B–E) Measurement of ECARs in real time in E13.5, E19.5, and P0 NPCs. Each data point is a biologic replicate. (B) Graph plot of a representative ECAR measurement from E13.5, E19.5, and P0 NPCs. (C) Basal glycolysis rate, measured after addition of glucose, is significantly lower in P0 NPCs. *P<0.05 (D) Maximal glycolytic capacity, measured after inhibiting mitochondrial oxygen consumption by oligomycin addition, is significantly lower in old than young NPCs. *P<0.05; ***P<0.001. (E) Glycolysis reserve (excess glycolytic capacity) is significantly higher in E13.5 young NPCs than old NPCs. **P<0.01. (F) Glycolysis in primary E13.5 and E19.5 NPCs that were not passaged after isolation. Glycolysis is significantly higher in E13.5 than in E19.5 NPCs. n=4 biologic replicate experiments. Paired t test, *P<0.05. Plotted values were obtained from three to five biologic replicate experiments. Error bars indicate SEM. P values were obtained by performing t test (C) or one-way ANOVA (D and E).
Oxidative Phosphorylation
Next, we evaluated oxidative phosphorylation (OxPhos) in E13.5, E19.5 and P0 NPCs by measuring their oxygen consumption rate (OCR) and mitochondrial function by employing the mitochondrial stress test (Figure 3). Addition of specific inhibitors of mitochondrial function during the test revealed basal and maximal mitochondrial respiration, the spare respiratory capacity, ATP-linked and -unlinked (proton leak) respiration, and nonmitochondrial respiration (Figure 3, A and B). Younger NPCs registered significantly higher baseline OCR than older NPCs (Figure 3C). Maximal OxPhos capacity was also significantly higher in younger NPCs (Figure 3D). Higher ECAR and OCR are suggestive of higher ATP production in E13.5 NPCs. Accordingly, 30% higher ATP levels were recorded in younger NPCs (Figure 3E). However, there was a significantly higher OCR/ECAR ratio in E19.5 NPCs, showing that old NPCs are more dependent on OxPhos than glycolysis (Figure 3F).
Figure 3.

Young (E13.5) NPCs have a significantly higher basal and maximal respiratory capacity than old (E19.5 and P0) NPCs. (A) Measurement of OCR in real time in young and old NPC, under basal conditions (before oligomycin) and in response to the indicated mitochondrial inhibitors. (B) Sequential addition of indicated mitochondrial inhibitors reveal different aspects of mitochondrial respiration. Addition of the protonophore FCCP reveals the maximal respiratory capacity of the cell. (C and D) Basal respiration decreases significantly during NPC aging (C), as does the maximal respiratory capacity (D). *P<0.05. (E) Young NPCs have significantly higher ATP levels than old NPCs. Luminescence was recorded as counts per second (CPS). ****P<0.001. (F) OCR/ECAR ratio is significantly higher in E19.5 NPCs. *P<0.05. OCR measurements are from two to five biologic replicates. ATP measurements are from three biologic replicates with three to six technical replicates per assay. Error bars indicate SEM. P values were obtained by performing one-way ANOVA (C) or paired t test (D–F).
Inhibition of Glycolysis Promotes NPC Differentiation in Embryonic Kidneys
Older NPCs (P0) demonstrate an increased predilection to exit self-renewal and differentiate than younger (E13.5) NPCs.13 Because young NPCs demonstrate increased glycolysis compared with old NPCs (Figure 2), we hypothesized that decreasing the glycolysis flux would switch E13.5 NPC behavior from young to old, i.e., from predominantly self-renewing toward differentiation. To test this idea, we added glycolysis inhibitor YN1 (Figure 4A) to culture media. Dose-dependent reduction in glycolysis rate was achieved, monitored as a decrease in ECAR measurements (data points after glucose addition, Supplemental Figure 2A) after 18 hours of treatment with YN1. Addition of YN1 to organ culture media for 24–72 hours enhanced nephrogenesis (Figure 4, B, C and E, Supplemental Figure 2C). The increase in nephrogenesis was observed with up to 25 µM YN1, whereas high-dose (100 µM) YN1 treatment proved to be lethal (Supplemental Figure 2B). As our aim was to achieve inhibition of glycolysis and not complete repression, with the expectation that this would result in lethality, we used 5–25 µM YN1 to achieve approximately 30% reduction in glycolysis (see below, and Supplemental Figure 2A). Note that the increase in nascent nephrons (Lhx1+) is not accompanied by an increase in ureteric bud tip number after 24 hours of YN1 treatment (Figure 4, C and D, Supplemental Figure 2C). The induced nephrons continue to mature, demonstrated by the significant increase of proximal tubule marker lotus tetragonolobus lectin staining 72 hours after YN1 treatment (Figure 4, E and F). The CM in YN1-treated kidneys appeared diffused and diminished, and exhibited ectopic Lhx1 staining (Figure 5A). Markers for cell proliferation (phospho-Histone 3) and apoptosis (PARP) were unchanged between control kidneys and those with 24 hours of YN1 treatment (Figure 5, B and C). Lack of change in proliferation rate after 24 hours of YN1 treatment was confirmed by flow cytometry–based cell-cycle analysis of Six2GFP+ NPCs from E12.5 kidneys (Supplemental Figure 3). Quantification showed a decrease in Six2+ cell number that is statistically significant after 48 hours but not 24 hours of YN1 treatment (Figure 5, D and E). As neither proliferation nor survival of NPCs was altered after 24 hours of YN1 treatment, the gradual depletion of the CM is likely from increased differentiation (exit) of NPCs from the renewing pool.
Figure 4.

Inhibition of glycolysis promotes nephrogenesis in embryonic kidneys. (A) YN1 inhibits PFKFB3, the enzyme required for generation of F-2, 6-bisP, the potent stimulator of glycolysis via PFK1.45 (B) Addition of YN1 to organ culture media for 24 hours enhanced nephrogenesis, visualized by Lhx1 immunostaining. Number of Lhx1+ structures are indicated. (C) Significant increase in Lhx1(+) nascent nephrons after 24 hours of YN1 treatment; ***P<0.001. (D) Ureteric bud tip number did not show a significant change after 24 hours of YN1 treatment. (E) The induced nephrons continue to mature, demonstrated by increased staining for proximal tubule marker lotus tetragonolobus lectin (LTL) detected after 72 hours of YN1 treatment. (F) Increased LTL staining in (E) is statistically significant. *P<0.05. Error bars indicate SEM. P values were obtained by performing paired t test. 4× magnification. Also see Supplemental Figure 2.
Figure 5.

Increased nephrogenesis depletes the CM in YN1-treated kidneys. (A) The CM (Six2+, green) exhibits ectopic Lhx1 (white arrow) staining after 24 hours of YN1 treatment of E12.5 kidneys. 60× magnification. (B and C) No significant difference in proliferation or apoptosis rate was observed in CM of YN1-treated kidneys. (B) Proliferation was quantified by counting phosphohistone 3(+) immunostained cells in Six2+ CM. (C) Apoptosis was quantified by counting PARP(+) cells in Six2(+) CM. Each data point shows number of positive cells per 1000 Six2+ cells, from n=3–4 biologic replicates. (D and E) Quantification by flow cytometry of Six2GFP+ cells from kidneys treated with YN1 for 24 hours (n=9 kidney pairs) or 48 hours (n=6 kidney pairs). A significant decrease in Six2+ cell number 48 hours after YN1 treatment, but not at 24 hours. Each data point represents GFP+ cells per kidney. Error bars indicate SEM. P values were obtained by performing paired t test. *P<0.05.
Wnt/β-catenin signaling induces the Cited1−/Six2+ NPCs to undergo nephrogenesis.24 Increased foci of Wnt4 mRNA expression in E12.5 kidneys accompanied the increase of Lhx1+ nascent nephrons after YN1 treatment, suggesting that the increased differentiation is β-catenin–dependent (Figure 6B). To test this implication, we cotreated embryonic kidney cultures with glycolysis and canonical Wnt signaling inhibitors, YN1 and IWR1, respectively. Addition of IWR1 reduced YN1-induced nephrogenesis (Figure 6, C and D), suggesting Wnt/β-catenin signaling is a likely mediator of increased nephrogenesis. However, the significant increase in Lhx1+ nascent nephrons on addition of YN1 to IWR1-treated kidneys in comparison to IWR1-only treatment suggests differentiation may occur via a β-catenin–independent pathway as well.
Figure 6.

Inhibition of glycolysis stimulates β-catenin–dependent differentiation of NPCs. (A and B) Increased Wnt4 mRNA expression accompanies the increased nephrogenesis observed after 24 hours of YN1 treatment in E12.5 kidneys, shown by whole mount in situ hybridization. (C and D) Cotreatment of E12.5 kidneys with YN1 and β-catenin inhibitor IWR1 for 24 or 48 hours. Glycolysis inhibitor YN1-mediated nephron induction is abrogated by addition of 2 μM IWR1. Lhx1+ nascent nephrons were counted after indicated treatments and mean number is shown in the bar graph. Data from at least two independent experiments, with at least n=3 kidneys per treatment. Error bars indicate SEM. P values were obtained by performing the paired t test. *P<0.05; **P<0.01. 10× magnification.
To determine whether the decrease in glycolysis is sufficient to stimulate differentiation gene expression in isolated NPCs, we assessed expression of renewal and differentiation genes by droplet digital PCR (ddPCR)(Supplemental Figure 4). Although addition of YN1 to E13.5 NPCs growing in NPEM decreased glycolysis, differentiation gene expression was not induced (data not shown). Media supporting renewal and inhibiting differentiation is not conducive to induction of differentiation genes. Therefore, we added YN1 to DMEM/F12 without (DMEM–CHIR) or with CHIR (DMEM+CHIR). Cited1 and Six2 expression did not show an additional decrease with YN1 than that observed with DMEM–CHIR (Figure 7A). However, β-catenin target gene expression (Wnt4 and Lef1) is stimulated comparably to that with low-dose 0.1 µM CHIR (Figure 7B). Simultaneous addition of 10 µM YN1 and low-dose CHIR (0.1 µM) further upregulated Wnt4, Lef1, Lhx1, and Jag1 expression than with either treatment alone, indicating an additive effect of YN1 and low-dose CHIR on differentiation gene expression in isolated NPCs. Maximal expression of the four differentiation marker genes was obtained with low-dose 0.1 µM CHIR with 10 µM YN1, and is even greater than expression with 0.5 µM CHIR treatment.
Figure 7.

Glycolysis inhibition reduces CHIR requirement to induce differentiation gene expression. (A and B) Quantitation of mRNA by ddPCR on RNA isolated from NPCs cultured in differentiation medium. YN1 was added to media without (DMEM–CHIR) or with indicated amount of CHIR (DMEM+CHIR). Data show mean±SD from technical replicates from pooled kidneys. Also see Supplemental Figure 3. (C, a–f) Isolated E11.5 mesenchyme cultured for 48 hours as indicated. Immunostaining for Six2 (red) and Lhx1 (green). 10× magnification.
To evaluate whether YN1 treatment reduces the requirement for CHIR for epithelial induction, E11.5 mesenchyme cultures were treated with YN1 and/or CHIR. Lhx1 expression was used as a marker of induction because Lhx1 expression precedes epithelialization. Mesenchyme cultures treated with YN1 alone demonstrated Lhx1 immunostaining after 48 hours of YN1 treatment, which was enhanced in cultures cotreated with YN1 and CHIR (low dose, 0.01 and 0.1 µM) (Figure 7C). Addition of low-dose CHIR alone did not induce Lhx1 expression, whereas high-dose CHIR (1 µM) induced Lhx1 expression (data not shown). Therefore, YN1 treatment facilitates differentiation by reducing the requirement for CHIR for epithelial induction of the metanephric mesenchyme.
Inhibition of PI3K/Akt Signaling or Activation of Differentiation Programs Reduces the Glycolytic Flux in NPCs
FGF/PI3K and BMP/MAPK pathways maintain NPCs in a self-renewing state, whereas decreased PI3K or activation of Bmp/Smad signaling promotes NPC differentiation,11,14,25,26 similar to our treatments with glycolysis inhibitor YN1. We therefore hypothesized that self-renewing signaling pathways stimulate glycolysis for NPC maintenance; thus, inhibition of these self-renewal signals in NPCs should result in decreased glycolysis. Indeed, addition of PI3K inhibitor LY294002 to NPCs significantly decreased glycolysis, similar to the rate decrease observed with addition of glycolysis inhibitor YN1 (Figure 8). Decreased P-Akt immunostaining confirmed inhibition of PI3K activity after LY294002 addition to culture media (Supplemental Figure 5, A and B). The ability of LY-treated NPCs to mount a compensatory response to metabolic stress and basal OCR comparable to YN1- or Wnt9b-treated NPCs confirmed the viability of NPCs treated with 10 µM LY294002 (Supplemental Figure 5, C and D). Differentiation of LY-treated NPCs was confirmed by induction of Lhx1 expression (Supplemental Figure 5E), as demonstrated by Lindstrom et al.26 In contrast to the inhibition, activation of PI3K/Akt pathway by addition of phosphatase and tensin homolog (PTEN) inhibitor bpV(Phen) to NPEM increased ECAR (Figure 8, A and B). PTEN inhibits the PI3K signaling pathway via its lipid phosphatase activity, which prevents the phosphorylation and activation of Akt by PIP3 and PDK1.27 Thus, the analogous effect of PI3K inhibition or activation on glycolysis levels suggests that the glycolytic flux in NPCs is downstream to PI3K/Akt signaling.
Figure 8.

Inhibition of PI3K/Akt signaling or activation of differentiation programs decreases glycolysis in NPCs. (A) Pharmacologic inhibition or activation of the PI3K/Akt pathway by LY294002 or bpV(Phen), respectively. (B) Addition of 10 µM LY294002 to NPEM for 20 hours decreased glycolysis (ECAR) significantly in E13.5 NPCs. In contrast, addition of 30–60 µM bpV(Phen) (Phen, PTEN inhibitor) significantly increased ECAR. Growth in differentiation media (DMEM+CHIR) also significantly reduced NPC glycolysis. YN1 was added to NPEM as positive control for monitoring decrease in ECAR measurement. Error bars indicate SEM. *P<0.05; **P<0.005, t test. (C) Maximal glycolysis capacity of NPCs decreased after culture in NPEM with PI3K inhibitor LY294002 (10 µM) or YN1 (5 or 20 µM), whereas addition of PTEN inhibitor bpV(Phen) significantly increased the glycolytic capacity. Replacement of NPEM by DMEM+CHIR differentiation medium showed a comparable decrease in glycolytic capacity as with LY or YN1. Error bars indicate SEM. All changes relative to ECAR measured in NPCs cultured in NPEM. *P<0.05; **P<0.01 by t test. Also see Supplemental Figure 4.
We next examined whether NPC growth in differentiation inducing DMEM/F12 plus 3 μM CHIR14 media altered glycolysis. We confirmed decrease in Cited1 and Six2 expression and increase of Wnt4 expression (Supplemental Figure 6). In addition to decreasing the basal glycolysis flux (Figure 8B), induction of differentiation by DMEM–CHIR or PI3K inhibition also decreased the maximal glycolysis capacity of NPCs (Figure 8C). Note that old NPCs (E19.5 and P0) demonstrate a lower glycolytic capacity than younger NPCs (E13.5) (Figure 2D).
Discussion
NPCs have a limited life span. The mechanistic aspects of what determines NPC aging are unknown. The dissimilar behavior patterns of young and old NPCs, namely decreased proliferation and increased exit rate from the progenitor niche of the old NPC, is reflected in the differential transcript profiles and utilization of different signaling pathways.13 Here, we report that native (freshly harvested) young and old NPCs have distinct metabolic profiles, with young NPCs displaying a higher energy state as a result of significantly higher glycolysis and mitochondrial respiration rates. Higher glycolysis rate in young NPCs appears to be a cell-intrinsic function as it persists when both young and old NPCs were cultured in the same NPEM. Moreover, manipulating intermediary metabolism shifts the balance between stem cell self-renewal and differentiation. Interestingly, comparison of transcriptomes of embryonic and postnatal kidneys by microarray analysis revealed decreased expression of several glycolytic pathway genes between P0 and P2 mouse kidneys, but not at earlier ages.28 It is possible that the decrease in glycolysis we observe in functional assays between young and old embryonic age NPCs is from post-translational regulation, leaving the pathway open to modulation by niche signals. However, postnatally toward the cessation of nephrogenesis as the progenitors differentiate the reduction in enzyme expression is likely permanent and achieved transcriptionally.
Metabolism has emerged as an active regulator of stem cell fate, and niche-directed (nutrients, oxygen tension, growth factors) changes in metabolic flux and redox homeostasis direct cell fate decisions from renewal to differentiation. Methods promoting a metabolic shift toward glycolysis facilitate stemness maintenance and are a critical step in reprogramming somatic cells to induced pluripotent stem cells.20,21,23 In contrast, promoting mitochondrial biogenesis and OxPhos in embryonic and somatic stem cells accelerates their commitment to a more differentiated state.23 Low oxygen concentrations (1%–5%) are conducive to stem cell proliferation and maintenance of pluripotency, whereas higher levels promote differentiation.29 Nutrient availability in the niche also influences cellular metabolic pathway usage. How does intermediary metabolism direct cell fate? In addition to ATP, metabolism produces metabolites that are critical substrates or cofactors for essential processes. A direct link was demonstrated between glycolysis-driven acetyl CoA production and histone acetylation in human and mouse embryonic stem cells.30 Inhibition of glycolysis leads to histone deacetylation and differentiation, whereas decrease in NAD+, a cofactor for the sirtuin class of HDACs, inhibits histone deacetylation.19,30,31 O-linked N-acetylglucosamine protein modification directly reflects changes in ambient glucose levels; occurrence of this modification on histones is another link to nutrient availability–directed chromatin modification by intermediary metabolism.32 Hence, the metabolite milieu directly influences epigenetic modifications and associated changes in transcription and cell fate. The documented heterogeneity of the NPC pool and changes in signaling pathways may well result from focal variations in microniche biochemistry. In line with the Barker hypothesis on the in utero origin of adult disease,33–35 changes in gestational metabolism that accompany maternal nutrition habits are recognized to affect the metabolic status of stem cell populations in the developing fetus.
NPC self-renewal is dependent on PI3K activity.26 The PI3K/Akt pathway positively regulates glycolysis at multiple points in the pathway: it facilitates glucose uptake36,37; stimulates hexokinase activity38; stimulates PFK1, the rate-determining step of glycolysis38; and activates expression and activity of rate-regulating pyruvate kinase isoform M2 (PKM2), which is expressed exclusively in embryonic, proliferating, and cancer cells via the mTOR/Hif1a/cMyc pathway.39 On the basis of our data, we propose a model whereby a high glycolysis flux is an essential intermediary of growth factor PI3K signaling–mediated NPC self-renewal (Figure 9A). PI3K and glycolysis downregulation switches the NPC to a Wnt/β-catenin–dependent (26 and this study) or –independent differentiation program in embryonic kidneys. As PI3K inhibition results in decreased glycolysis, we posit that the PI3K pathway potentiates NPC renewal by promoting glycolysis (Figure 9, A and B). Single-cell transcriptomics shows changes in mTOR, growth factor signaling, and extracellular matrix pathway genes, as well as in chromatin modifiers between young and old NPCs.13 As mTOR activity may also support mitochondrial respiration,40 an increase in mTOR pathway gene expression in P0 (old) NPCs relative to young (E13.5) NPCs may restrict the old cells to a lower glycolytic flux.
Figure 9.

Role of glycolysis in NPC self-renewal. (A) Schematic illustration of NPC fate specification by glycolysis. Per our model, we propose a high glycolysis flux is an essential mediator of PI3K signaling–driven NPC self-renewal. Stimulation of PI3K/Akt signaling increases glycolysis (green box arrow). High glycolysis flux in self-renewing NPCs may inhibit Wnt-induced differentiation that occurs on glycolysis inhibition. MAPK, cMyc, and mTOR signaling are also known stimulators of glycolysis and may contribute to the glycolysis flux (not tested in this study), denoted by blue arrows. (B) Both PI3K and glycolysis downregulation switches the NPC to a Wnt-dependent differentiation program in embryonic kidneys. As PI3K inhibition results in decreased glycolysis, we posit that the PI3K pathway potentiates NPC renewal by promoting glycolysis.
A dissociated response in expression of renewal and differentiation genes was observed with both CHIR and YN1 treatment of NPCs in that the mRNA of renewal genes did not decrease although differentiation gene expression (Wnt4, Lef1, Lhx1, and Jag1) increased (Figure 7, A and B). Whereas in vivo more subtle gradations of signaling and metabolic pathways influence the temporal and spatial pattern of gene expression, the manipulations here are tools to demonstrate a proof of principle and do not necessarily reflect the physiologic situation. It would be of interest to compare the metabolic profiles of NPCs in different expansion or differentiation media, and in response to other critical regulatory signaling pathways in the NPC (for example, Bmp7/MAPK, YAP/TAZ, or Wnt17,41) and extend these analyses to human induced pluripotent stem cells and NPCs. Strategies to expand NPCs in vivo or in vitro will benefit greatly from understanding how cellular metabolic processes influence the balance between NPC renewal and differentiation. For example, glycolysis downmodulation may be beneficial in the treatment of Wilms’ tumor, where stimulation of cellular differentiation may be a desirable outcome. In contrast, glycolysis activators or inhibitors of OxPhos may favor NPC expansion.
While this article was under revision two articles were published that address the role of a glycolysis gradient in body axis elongation42 and presomitic mesoderm differentiation.43 In the former publication, the authors demonstrate a gradient of glycolysis enzymes regulated by FGF signaling in the mouse and chick embryo tail bud, and that inhibition of glycolysis reduces cell motility, Wnt signaling, and changes cell fate decisions. The focus to date on genetic signals has allowed mapping growth factor signaling and transcriptional networks that regulate NPC renewal and differentiation. This study adds another dimension that needs to be considered when describing the self-renewing versus differentiating potential of NPCs.
Concise Methods
MACS and Expansion of NPCs for Extracellular Flux Measurements and RNA Isolation
E13.5, E19.5, or P0 CD1 mouse kidneys were dissected into HBSS (Invitrogen) and treated with 1% pancreatin/0.25% collagenase mixture to collect the suspension of nephrogenic zone cells as described.11 NPCs were enriched by negative bead depletion with MACS–Phycoerythrin-conjugated antibodies against endothelial progenitors (anti-CD105), RBCs and erythroid cells (anti-TER119), cortical interstitium (anti-CD140a), and renal epithelial cells (anti-CD326) following the manufacturer’s (Miltenyi Biotec) and published protocol.11 Isolated cells were cultured on Matrigel-coated plates in a monolayer in the APEL-based (Stem Cell Technologies) NPEM exactly as described.11 The NPCs were expanded for two to three passages for all experiments, and split at 60%–70% confluence. Animal protocols utilized in this study were approved by and in strict adherence to guidelines established by the Tulane University Institutional Animal Care and Use Committee.
Extracellular Flux Measurements
Kidneys were pooled from multiple litters for E13.5 NPC isolation. Cells were seeded 18–20 hours before the assay, at 150,000 cells per well, on Matrigel-coated Seahorse microplates for flux measurements on the XFe24 Extracellular Flux Analyzer (Agilent Seahorse Technologies). For flux measurements after media manipulations or glycolysis or signaling pathway inhibition, cells were plated 40 hours before the assay in NPEM, and media changed 18–20 hours before the assay with the indicated treatments. Optimal cell density was determined empirically, on the basis of attaining flux measurements in the linear range. To probe glycolysis and OxPhos beyond basal measurements, glycolysis and mitochondrial stress tests, respectively, were done. The stress tests require sequential addition of specific pathway inhibitors to query various aspects of these two metabolic pathways. All metabolic inhibitors were titrated to obtain maximal response in the absence of toxicity. The XF sensor cartridges were hydrated overnight at 37°C without carbon dioxide in the Seahorse XF calibrant solution, as recommended in the manufacturer’s protocol. For glycolysis rate measurements, NPEM was replaced with the XF assay medium (unbuffered DMEM [pH 7.4] with 2 mM glutamine) and the microplate with cells was placed in a 37°C incubator without carbon dioxide. Glucose (10 mM, final), oligomycin (2 µg/ml), and 2-deoxyglucose (0.1 M final) were added to the ports at concentrations to achieve appropriate final dilutions. For OCR measurements, the assay medium was Seahorse XF base medium (unbuffered DMEM) supplemented with 3–5 mM pyruvate, 5 mM D-glucose, and 2 mM L-glutamine (pH 7.4). Oligomycin (1 µg/ml), FCCP (1.5 µM), rotenone (0.1 µM), and antimycin (1 µM) were added to the ports. Both ECAR and OCR measurements were performed at 8-minute intervals (2 minutes mixing, 2 minutes recovery, and 4 minutes measuring) for 3 hours, using the Seahorse XF24 analyzer. Measurements were recorded from three to five biologic replicates with three technical replicates per experiment from young and old NPCs. All data are normalized to cell number.
Embryonic Kidneys and Metanephric Mesenchyme Culture and Immunofluorescence Staining
E12.5 kidneys were collected from CD1 mice for culture in DMEM/F12 complete media as previously described.44 Glycolysis inhibitor YN1 (Sigma) was added at 5–20 µM for the times indicated. YN1 was developed using a structure-based approach and has high specificity to PFKFB3 over the other isoenzymes, specifically targeted toward the fructose 6-phosphate–binding site.45,46 This dose was determined after titration of 1–100 µM YN1, added to media for 24 hours culture. IWR1 [4-(1,3,3a,4,7,7a-hexahydro-1,3-dioxo-4,7-methano-2H-isoindole-2-yl)-N-8-quinolinyl-benzamide], LY294002, and potassium bisperoxo(1,10-phenanthroline) oxovanadate [bpV(Phen)] were all purchased from Sigma and added as indicated. Kidneys were fixed in 4% paraformaldehyde and processed for immunofluorescence staining in Tris-Saponin buffer. For mesenchyme cultures, E11.5 mesenchymes were manually dissected from whole kidneys after gentle trypsinization and neutralization in complete media.14,26 Per experiment, pooled mesenchymes in dissection media from 12 to 15 kidney pairs were equally divided for individual treatments and placed on transwell filters. Cultures were incubated in NPEM with or without YN1 as indicated for 0–24 hours, and then in DMEM/F12 with additions for 24–48 hours as noted in Figures 4–7. At 48 hours, cultures were fixed per whole-mount kidney protocol for immunofluorescence staining. For kidney sections, YN1-treated kidneys were fixed in 10% formalin and processed for paraffin embedding and sectioning as previously described.47 Antibodies against the following proteins were used: Lhx1 (4F2; Developmental Studies Hybridoma Bank), Pax2 (Invitrogen), Six2 (Proteintech), and phosphohistone 3 (Ser10) (Cell Signaling).
RNA Isolation and ddPCR
E13.5 NPCs were isolated and cultured in NPEM. After 24 hours of YN1 treatment, total RNA was isolated using a commercially available RNA isolation kit (Qiagen). RNA concentration was quantified using Nanodrop 2000 (Thermo Scientific). In addition, β-actin levels were detected by ddPCR before analysis of target genes to adjust for minor differences in sample RNA concentrations. ddPCR was performed on a Bio-Rad ddPCR system to determine Cited1, Six2, Osr1, Wnt4, Lef1, Lhx1, and Jag1 gene expression. Primers, probes, and reagents for the One-step RT-ddPCR system were purchased from Bio-Rad to generate cDNA and quantify gene expression. After droplet generation and PCR amplification, droplets were analyzed on the QX200 droplet reader and target cDNA concentration was determined using the QuantaSoft analysis software (Bio-Rad). For each target gene, the amount of total RNA in a PCR reaction was determined by a pilot ddPCR, using serially diluted total RNA, in which the determined amount is in a linear range. Primer sequences (Supplemental Table 2) and a representative standard curve (Supplemental Figure 4) are provided in the Supplemental Material. Data are expressed as copy numbers of target gene in 1 µl PCR reaction. Experimental and biologic replicates were applied.
Measurement of ATP Levels
ATPlite (Perkin Elmer) luminescence assay was performed as per the manufacturer’s instructions as previously described.47 This assay is on the basis of light produced by conversion of luciferin by luciferase to oxyluciferin in an ATP-dependent reaction. The emitted light is proportional to ATP concentration in the cell lysate. NPCs (2.5×104 per well) were seeded onto an ATP-free microplate. At 18–20 hours after seeding, cells were lysed in the well, and the luminescence was measured after the addition of substrate. Values were normalized to cell number. Measurements were recorded from three biologic replicates (n=3), with three to six technical replicates per experiment from young and old NPCs.
Statistical Analyses
For extracellular flux analysis (ECAR and OCR), paired two-tailed t test was used to calculate P values. Values of P<0.05 are considered significant. Error bars represent ±SEM for biologic replicates from pooled kidneys from three to five mouse litters per age. Each data point represents a biologic replicate with NPCs from distinct isolations and cultures. For Lhx1+ nascent nephron counting in organ culture experiments, 19 paired kidneys from four litters were used for 24-hour treatment and control. Paired two-tailed paired t test was used to calculate P values, with P<0.05 considered significant. For ddPCR analysis, error bars represent ±SD for technical replicates. RNA was isolated from pooled kidneys (10–12 pairs) from two litters, with two biologic replicates. Cell quantification by flow cytometry or manual counts was done on multiple kidney pairs, as indicated in Figure 5 legend. Data points represent experimental replicates, and error bars denote ±SEM unless noted otherwise. All experiments utilized kidneys harvested from two or more different litters.
Disclosures
None.
Supplementary Material
Acknowledgments
We are extremely grateful to Samir El-Dahr for helpful discussions over the course of the project and critical review of the manuscript. We thank the Tulane Hypertension and Renal Centers of Excellence Molecular Core Service. This work was partially funded by grants from National Institute of Diabetes and Digestive and Kidney Diseases of the National Institute of Health (R56DK104779) and the Diabetic Complications Consortium (DiaComp) (DK076169) to Z.S.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “New Insights into Fuel Choices of Nephron Progenitor Cells,” on pages 3133–3135.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016111246/-/DCSupplemental.
References
- 1.Benz K, Amann K: Maternal nutrition, low nephron number and arterial hypertension in later life. Biochim Biophys Acta 1802: 1309–1317, 2010. 10.1016/j.bbadis.2010.03.002 [DOI] [PubMed] [Google Scholar]
- 2.Hoy WEH, Hughson MD, Singh GR, Douglas-Denton R, Bertram JF: Reduced nephron number and glomerulomegaly in Australian Aborigines: A group at high risk for renal disease and hypertension. Kidney Int 70: 104–110, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Luyckx VA, Brenner BM: The clinical importance of nephron mass. J Am Soc Nephrol 21: 898–910, 2010. 10.1681/asn.2009121248 [DOI] [PubMed] [Google Scholar]
- 4.Wlodek ME, Westcott K, Siebel AL, Owens JA, Moritz KM: Growth restriction before or after birth reduces nephron number and increases blood pressure in male rats. Kidney Int 74: 187–195, 2008. 10.1038/ki.2008.153 [DOI] [PubMed] [Google Scholar]
- 5.Hartman HA, Lai HL, Patterson LT: Cessation of renal morphogenesis in mice. Dev Biol 310: 379–387, 2007. 10.1016/j.ydbio.2007.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rumballe BA, Georgas KM, Combes AN, Ju AL, Gilbert T, Little MH: Nephron formation adopts a novel spatial topology at cessation of nephrogenesis. Dev Biol 360: 110–122, 2011. 10.1016/j.ydbio.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Little MH, McMahon AP: Mammalian kidney development: principles, progress, and projections. Cold Spring Harb Perspect Biol 4, 2012. 10.1101/cshperspect.a008300, 6–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyle S, Misfeldt A, Chandler KJ, Deal KK, Southard-Smith EM, Mortlock DP, Baldwin HS, de Caestecker M: Fate mapping using Cited1-CreERT2 mice demonstrates that the cap mesenchyme contains self-renewing progenitor cells and gives rise exclusively to nephronic epithelia. Dev Biol 313: 234–245, 2008. 10.1016/j.ydbio.2007.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP: Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3: 169–181, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karner CM, Das A, Ma Z, Self M, Chen C, Lum L, Oliver G, Carroll TJ: Canonical Wnt9b signaling balances progenitor cell expansion and differentiation during kidney development. Development 138: 1247–1257, 2011. 10.1242/dev.057646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown AC, Muthukrishnan SD, Oxburgh L: A synthetic niche for nephron progenitor cells. Dev Cell 34: 229–241, 2015. 10.1016/j.devcel.2015.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Short KM, Combes AN, Lefevre J, Ju AL, Georgas KM, Lamberton T, Cairncross O, Rumballe BA, McMahon AP, Hamilton NA, Smyth IM, Little MH: Global quantification of tissue dynamics in the developing mouse kidney. Dev Cell 29: 188–202, 2014. 10.1016/j.devcel.2014.02.017 [DOI] [PubMed] [Google Scholar]
- 13.Chen S, Brunskill EW, Potter SS, Dexheimer PJ, Salomonis N, Aronow BJ, Hong CI, Zhang T, Kopan R: Intrinsic age-dependent changes and cell-cell contacts regulate nephron progenitor lifespan. Dev Cell 35: 49–62, 2015. 10.1016/j.devcel.2015.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown AC, Muthukrishnan SD, Guay JA, Adams DC, Schafer DA, Fetting JL, Oxburgh L: Role for compartmentalization in nephron progenitor differentiation. Proc Natl Acad Sci USA 2013;110(12):4640–4645 10.1073/pnas.1213971110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, Oliver G: Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J 25: 5214–5228, 2006. 10.1038/sj.emboj.7601381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown AC, Adams D, de Caestecker M, Yang X, Friesel R, Oxburgh L: FGF/EGF signaling regulates the renewal of early nephron progenitors during embryonic development. Development 138: 5099–5112, 2011. 10.1242/dev.065995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blank U, Brown A, Adams DC, Karolak MJ, Oxburgh L: BMP7 promotes proliferation of nephron progenitor cells via a JNK-dependent mechanism. Development 136: 3557–3566, 2009. 10.1242/dev.036335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Etchegaray JP, Mostoslavsky R: Interplay between metabolism and epigenetics: A nuclear adaptation to environmental changes. Mol Cell 62: 695–711, 2016. 10.1016/j.molcel.2016.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katada S, Imhof A, Sassone-Corsi P: Connecting threads: Epigenetics and metabolism. Cell 148: 24–28, 2012. 10.1016/j.cell.2012.01.001 [DOI] [PubMed] [Google Scholar]
- 20.Shyh-Chang N, Daley GQ, Cantley LC: Stem cell metabolism in tissue development and aging. Development 140: 2535–2547, 2013. 10.1242/dev.091777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Marsboom G, Glick D, Zhang Y, Toth PT, Jones N, Malik AB, Rehman J: Bioenergetic shifts during transitions between stem cell states (2013 Grover Conference series). Pulm Circ 4: 387–394, 2014. 10.1086/677353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teslaa T, Teitell MA: Pluripotent stem cell energy metabolism: An update. EMBO J 34: 138–153, 2015. 10.15252/embj.201490446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wanet A, Arnould T, Najimi M, Renard P: Connecting mitochondria, metabolism, and stem cell fate. Stem Cells Dev 24: 1957–1971, 2015. 10.1089/scd.2015.0117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park JS, Valerius MT, McMahon AP: Wnt/beta-catenin signaling regulates nephron induction during mouse kidney development. Development 134: 2533–2539, 2007. 10.1242/dev.006155 [DOI] [PubMed] [Google Scholar]
- 25.Muthukrishnan SD, Yang X, Friesel R, Oxburgh L: Concurrent BMP7 and FGF9 signalling governs AP-1 function to promote self-renewal of nephron progenitor cells. Nat Commun 6: 10027, 2015. 10.1038/ncomms10027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindström NO, Carragher NO, Hohenstein P: The PI3K pathway balances self-renewal and differentiation of nephron progenitor cells through β-catenin signaling. Stem Cell Rep 4: 551–560, 2015. 10.1016/j.stemcr.2015.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song MS, Salmena L, Pandolfi PP: The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 13(5): 283–296, 2012 [DOI] [PubMed] [Google Scholar]
- 28.Brunskill EW, Lai HL, Jamison DC, Potter SS, Patterson LT: Microarrays and RNA-Seq identify molecular mechanisms driving the end of nephron production. BMC Dev Biol 11: 15, 2011. 10.1186/1471-213X-11-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivanovic Z: Hypoxia or in situ normoxia: The stem cell paradigm. J Cell Physiol 219: 271–275, 2009. 10.1002/jcp.21690 [DOI] [PubMed] [Google Scholar]
- 30.Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, Nemirovski A, Shen-Orr S, Laevsky I, Amit M, Bomze D, Elena-Herrmann B, Scherf T, Nissim-Rafinia M, Kempa S, Itskovitz-Eldor J, Meshorer E, Aberdam D, Nahmias Y: Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab 21: 392–402, 2015. 10.1016/j.cmet.2015.02.002 [DOI] [PubMed] [Google Scholar]
- 31.Cluntun AA, Huang H, Dai L, Liu X, Zhao Y, Locasale JW: The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab 3: 10, 2015. 10.1186/s40170-015-0135-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durning SP, Flanagan-Steet H, Prasad N, Wells L: O-Linked β-N-acetylglucosamine (O-GlcNAc) Acts as a glucose sensor to epigenetically regulate the insulin gene in pancreatic beta cells. J Biol Chem 291: 2107–2118, 2016. 10.1074/jbc.M115.693580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boubred F, Saint-Faust M, Buffat C, Ligi I, Grandvuillemin I, Simeoni U: Developmental origins of chronic renal disease: An integrative hypothesis. Int J Nephrol. 2013:346067, 2013. 10.1155/2013/346067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dorey ES, Pantaleon M, Weir KA, Moritz KM: Adverse prenatal environment and kidney development: implications for programing of adult disease. Reproduction 147: R189–R198, 2014. 10.1530/REP-13-0478 [DOI] [PubMed] [Google Scholar]
- 35.Luyckx VA, Shukha K, Brenner BM: Low nephron number and its clinical consequences. Rambam Maimonides Med J 2: e0061, 2011. 10.5041/RMMJ.10061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simons AL, Kevin PO, Joshua MM, Peter MS, Spitz DR. The role of akt pathway signaling in glucose metabolism and metabolic oxidative stress. In: Oxidative Stress in Cancer Biology and Therapy, edited by Spitz DR, New York, Humana Press, 2012, pp 21–46 [Google Scholar]
- 37.Wieman HL, Wofford JA, Rathmell JC: Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of glut1 activity and trafficking. Mol Biol Cell 18(4):1437–1446, 2007. 10.1091/mbc.E06-07-0593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB: Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol 23: 7315–7328, 2003. 10.1128/mcb.23.20.7315-7328.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, Wang Y, Jing Y, Yang H, Chen R, Chang L, Zhang Y, Goto J, Onda H, Chen T, Wang M-R, Lu Y, You H, Kwiatkowski D, Zhang H: Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci USA 108(10):4129–4134, 2011. 10.1073/pnas.1014769108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schieke SM, Phillips D, McCoy JP Jr, Aponte AM, Shen R-F, Balaban RS, Finkel T: The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem 281: 27643–27652, 2006. 10.1074/jbc.M603536200 [DOI] [PubMed] [Google Scholar]
- 41.Das A, Tanigawa S, Karner CM, Xin M, Lum L, Chen C, Olson EN, Perantoni AO, Carroll TJ: Stromal-epithelial crosstalk regulates kidney progenitor cell differentiation. Nat Cell Biol 15: 1035–1044, 2013. 10.1038/ncb2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oginuma M, Moncuquet P, Xiong F, Karoly E, Chal J, Guevorkian K, Pourquie O: A gradient of glycolytic activity coordinates FGF and wnt signaling during elongation of the body axis in amniote embryos. Dev Cell 40(4):342-353 e10, 2017. 10.1016/j.devcel.2017.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bulusu V, Prior N, Snaebjornsson MT, Kuehne A, Sonnen KF, Kress J, Stein F, Schultz C, Sauer U, Aulehla A: Spatiotemporal analysis of a glycolytic activity gradient linked to mouse embryo mesoderm development. Dev Cell 40(4):331–341 e4, 2017. 10.1016/j.devcel.2017.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saifudeen Z, Dipp S, Stefkova J, Yao X, Lookabaugh S, El-Dahr SS: p53 regulates metanephric development. J Am Soc Nephrol 20: 2328–2337, 2009. 10.1681/ASN.2008121224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seo M, Kim J-D, Neau D, Sehgal I, Lee Y-H: Structure-based development of small molecule PFKFB3 inhibitors: A framework for potential cancer therapeutic agents targeting the Warburg effect. PLoS One 6: e24179, 2011. 10.1371/journal.pone.0024179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schoors S, De Bock K, Cantelmo AR, Georgiadou M, Ghesquière B, Cauwenberghs S, Kuchnio A, Wong BW, Quaegebeur A, Goveia J, Bifari F, Wang X, Blanco R, Tembuyser B, Cornelissen I, Bouché A, Vinckier S, Diaz-Moralli S, Gerhardt H, Telang S, Cascante M, Chesney J, Dewerchin M, Carmeliet P: Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab 19: 37–48, 2014. 10.1016/j.cmet.2013.11.008 [DOI] [PubMed] [Google Scholar]
- 47.Li Y, Liu J, Li W, Brown A, Baddoo M, Li M, Carroll T, Oxburgh L, Feng Y, Saifudeen Z: p53 Enables metabolic fitness and self-renewal of nephron progenitor cells. Development 142: 1228–1241, 2015. 10.1242/dev.111617 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.