

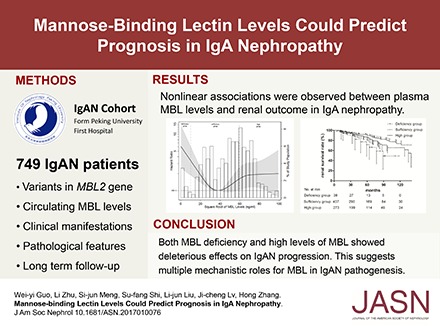
Abstract
IgA nephropathy (IgAN) is characterized by infections followed by episodic gross hematuria. Deficiency of mannose-binding lectin (MBL) is associated with recurrent infection in many diseases, but controversy exists regarding the role of MBL in IgAN. Here, we measured MBL2 variants and MBL levels in 749 patients with IgAN and 489 healthy controls. Overall, 5.2% (39 of 749) of patients with IgAN had MBL deficiency (MBL levels <100 ng/ml), among whom LYPB/LYPB and LXPA/LYPB were the predominant MBL2 haplotypes (82%; 32 of 39). We found a nonlinear association between MBL levels and renal outcome in IgAN. Patients with IgAN and MBL deficiency had a higher incidence of prodromic infections and gross hematuria than those with sufficient MBL levels (100–3540 ng/ml). Moreover, MBL deficiency independently associated with poor renal outcome in IgAN after multiple adjustments (hazard ratio, 5.18; 95% confidence interval, 2.50 to 10.72; P<0.001). Patients with high MBL levels (>3540 ng/ml) had more severe proteinuria and a higher proportion of crescents, although the association with IgAN progression did not reach statistical significance after adjustments. In conclusion, MBL deficiency and MBL excess may both have deleterious effects on IgAN progression, which suggests that MBL contributes to IgAN pathogenesis through multiple mechanisms.
Keywords: IgA nephropathy, MBL, renal outcome
IgA nephropathy (IgAN) is the most common primary GN1 with unclear pathogenesis.2 The classic presentation in patients with IgAN is episodic gross hematuria after mucosal infections. In a recent genome-wide association study of IgAN, multiple genes involved in immunity against intestinal mucosal infection were identified, suggesting the involvement of mucosal infections in IgAN,3 although further studies are needed to reveal the underlying mechanisms.
Mannose-binding lectin (MBL) is an important protein in innate immunity.4,5 Multiple factors are identified to influence MBL levels. First, several regulatory variants of its coding gene, MBL2, are reported to influence MBL expression, including c.-550G>C, c.-427A>C, c.-349A>G, c.-336A>G, c.-324_-329delAAAGAG, c.-221G>C, c.-70C>T, and c.4C>T.6–8 Second, three coding variants (Arg52Cys, Gly54Asp, and Gly57Glu) in exon 1 of MBL2 result in four alleles: the A (52Arg/54Gly/57Gly), B (52Arg/54Asp/57Gly), C (52Arg/54Gly/57Glu), and D (52Cys/54Gly/57Gly) alleles.7,8 Among them, A allele was the wild-type allele, whereas the others (B/C/D alleles), which are called O allele, led to a reduction of functional MBL. The combined effects of these variants result in the most common immune defect: MBL deficiency (MBL levels <100 ng/ml). Third, three IL-6–responsive elements have been found in the MBL2 promoter, and they confer increased acute-phase protein levels during severe inflammatory conditions.9
MBL functions as a pattern recognition molecule to recognize carbohydrate patterns of micro-organisms and activate complement via the lectin pathway. Complement activation is widely regarded as a double-edged sword. MBL-initiated complement activation causes both pathogen opsonization and tissue injury. Therefore, MBL deficiency shows opposite (both protective and risk) effects with different diseases. Individuals with MBL deficiency are more susceptible to recurrent infections,10,11 but it is protective in ischemia-reperfusion injury.12,13
Controversy exists regarding the role of MBL in IgAN. Ohsawa et al.14 found a tendency toward improved proteinuria and renal function in MBL-deficient patients with IgAN, whereas two other groups identified an association between low MBL levels and genetic variants with abundant immune deposits15 and IgAN progression.16 However, these studies were performed in relatively small populations (61–160 patients). Perhaps limited by the population size, they focused solely on MBL deficiency, and no further attention was paid to differing MBL levels in patients without MBL deficiency. To solve the puzzle, we enrolled 749 patients with IgAN with regular follow-up and 489 controls to evaluate the exact role of MBL in IgAN. Phenotypic characteristics of enrolled patients are summarized in Table 1.
Table 1.
Demographic, clinical, and histologic characteristics of patients with IgAN
| Characteristic | Mean±SD or Median (IQR), n=749 | Deficiency Group, n=39 | Sufficiency Group, n=437 | High Group, n=273 | P Valuea | P Valueb | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | ||||||||||
| Age, yr | 34.7±12.1 | 38.4±11.4 | 35.1±11.9 | 33.3±12.3 | 0.10 | 0.052 | ||||
| Sex (% men) | 362 (48.3) | 13 (33.3) | 193 (44.2) | 156 (57.1) | 0.19 | 0.001 | ||||
| Initial proteinuria, g/d | 1.29 (0.69, 2.51) | 1.73 (0.64, 2.43) | 1.15 (0.67, 2.28) | 1.50 (0.84, 3.25) | 0.31 | <0.001 | ||||
| Prodromic infection (%) | 256 (34.2) | 26 (66.7) | 138 (31.6) | 92 (33.7) | <0.001 | 0.56 | ||||
| Gross hematuria (%) | 213 (28.4) | 16 (41.0) | 115 (26.3) | 82 (30.0) | 0.049 | 0.28 | ||||
| eGFR, ml/min per 1.73 m2 | 83.89±30.64 | 88.52±30.46 | 82.95±30.24 | 84.72±31.34 | 0.27 | 0.46 | ||||
| CKD stages 1/2/3/4–5c (%) | 345 (47.4)/231 (30.8)/141 (18.8)/32 (4.3) | 20 (51.3)/12 (30.8)/5 (12.8)/2 (5.1) | 197 (45.1)/136 (31.1)/87 (19.9)/17 (3.9) | 128 (46.9)/83 (30.4)/49 (17.9)/13 (4.8) | 0.71 | 0.85 | ||||
| HBP, mmHg (%) | 369 (49.3) | 19 (48.7) | 211 (48.3) | 139 (50.9) | 0.96 | 0.50 | ||||
| Oxford classificationd (%) | ||||||||||
| M1 | 609 (82.4) | 31 (79.5) | 357 (82.8) | 221 (82.2) | 0.60 | 0.82 | ||||
| E1 | 368 (49.8) | 22 (56.4) | 211 (49.0) | 135 (50.2) | 0.37 | 0.75 | ||||
| S1 | 481 (65.1) | 28 (71.8) | 277 (64.3) | 176 (65.4) | 0.35 | 0.76 | ||||
| T1/T2 | 220 (29.8)/123 (16.6) | 6 (15.4)/7 (17.9) | 132 (30.6)/72 (16.7) | 82 (30.5)/44 (16.4) | 0.31 | 0.89 | ||||
| C1/C2 | 334 (45.2)/68 (9.2) | 19 (48.7)/1 (2.6) | 192 (44.5)/34 (7.9) | 123 (45.7)/33 (12.3) | 0.54 | 0.05 | ||||
| Follow-up | ||||||||||
| Follow-up interval, mo | 47.0 (26.0, 81.0) | 38.0 (19.0, 69.0) | 44.0 (25.0, 84.0) | 52.0 (27.5, 80.0) | 0.31 | 0.31 | ||||
| Treated with immunosuppressive agents or prednisone (%) | 353 (47.1) | 13 (33.3) | 192 (43.9) | 148 (54.2) | 0.20 | 0.01 | ||||
| RAS blocker (%) | 713 (95.2) | 38 (97.4) | 414 (94.7) | 261 (95.6) | 0.46 | 0.60 | ||||
| Slope, ml/min per 1.73 m2 per year | −2.91 (−5.22, −1.25) | −4.28 (−5.84, −2.24) | −2.85 (−5.13, −1.19) | −2.89 (−5.72, −1.25) | 0.03 | 0.42 | ||||
| Outcome | No. (%) | Per 100 Patient-yr | No. (%) | Per 100 Patient-yr | No. (%) | Per 100 Patient-yr | No. (%) | Per 100 Patient-yr | ||
| 50% Decline in eGFR | 102 (13.6) | 3.47 | 8 (20.5) | 6.47 | 47 (10.8) | 2.95 | 47 (17.2) | 3.97 | 0.07 | 0.01 |
| ESRD | 67 (8.9) | 2.27 | 9 (23.1) | 7.29 | 38 (8.7) | 2.37 | 32 (11.7) | 2.70 | 0.04 | 0.19 |
| Compositee | 112 (15.0) | 3.83 | 10 (25.6) | 8.08 | 51 (11.7) | 3.19 | 51 (18.7) | 4.32 | 0.01 | 0.01 |
| MBL | ||||||||||
| Plasma MBL, ng/ml | 2690.3 (961.9, 4227.2) | 0.00 (0.00, 36.2) | 1661.6 (744.5, 2630.1) | 4842.4 (4067.8, 5687.4) | <0.001 | <0.001 | ||||
| Haplotype (frequency) | ||||||||||
| LYPB | 205 (0.137) | 52 (0.667) | 150 (0.171) | 3 (0.006) | <0.001 | <0.001 | ||||
| LXPA | 176 (0.117) | 17 (0.218) | 130 (0.148) | 29 (0.053) | ||||||
| LYPA | 96 (0.064) | 0 (0) | 65 (0.074) | 31 (0.057) | ||||||
| LYQA | 178 (0.119) | 3 (0.038) | 98 (0.112) | 77 (0.142) | ||||||
| HYPA | 827 (0.552) | 6 (0.077) | 421 (0.481) | 400 (0.735) | ||||||
| Rare | 16 (0.011) | 0 (0) | 12 (0.014) | 4 (0.007) | ||||||
| HYPB | 5 (0.003) | 0 (0) | 5 (0.006) | 0 (0) | ||||||
| HYPD | 2 (0.001) | 0 (0) | 2 (0.002) | 0 (0) | ||||||
| HXPA | 5 (0.003) | 0 (0) | 3 (0.003) | 2 (0.004) | ||||||
| HYQA | 3 (0.002) | 0 (0) | 1 (0.001) | 2 (0.004) | ||||||
| LYQC | 1 (0.001) | 0 (0) | 1 (0.001) | 0 (0) | ||||||
Oxford classification: mesangial hypercellularity score (M1>0.5), the presence of endocapillary proliferation (E1: present), segmental glomerulosclerosis/adhesion (S1: present), severity of tubular atrophy/interstitial fibrosis (T1=26%–50%; T2>50%), and presence of crescent (C1: 1%–25%; C2: 26%–100%). IQR, interquartile range; HBP, high BP; RAS, renin-angiotensin system.
P value was used to indicate the difference between the MBL deficiency group and the sufficiency group. A two-tailed P<0.05 was considered statistically significant.
P value was used to indicate the difference between the MBL high group and the sufficiency group. A two-tailed P<0.05 was considered statistically significant.
CKD stages 1–4 denote eGFR≥90, 60–89, 30–59, and 15–29 ml/min per 1.73 m2, respectively, according to the Kidney Disease Outcomes Quality Initiative.
The Oxford classification was developed by the Working Group of the International IgA Nephropathy Network and the Renal Pathology Society. Oxford scores of ten patients were unavailable, because each of them had fewer than eight glomeruli.
Composite outcome included 50% eGFR decline or ESRD, whichever occurred first.
We at first performed genotyping of the 11 previously reported MBL expression–associated variants. The distributions of allele and genotype frequencies at each single variant showed no significant differences between patients with IgAN and healthy controls (Supplemental Table 1), although patients with IgAN presented with a slightly lower frequency of the O allele than healthy controls (14.2% versus 16.9%; P=0.07) (Supplemental Table 2). Among the eight regulatory variants, six (c.-427A>C, c.-349A>G, c.-336A>G, c.-324_-329delAAAGAG, c.-70C>T, and c.4C>T) were completely linked as previously reported.8 Therefore, the combination of the remaining two variants (c.-550G>C [H/L type] and c.-221C>G [X/Y type]) and variant c.4C>T (P/Q type; completely tagged c.-427A>C, c.-349A>G, c.-336A>G, c.-324_-329delAAAGAG, and c.-70C>T) with the four alleles in exon 1 (A/B/C/D) resulted in several haplotypes, including five major haplotypes (frequencies >0.01; LYPB/LXPA/LYPA/LYQA/HYPA) and five minor haplotypes (frequencies <0.01; HYPB/HXPA/HYQA/HYPD/LYQC), in our recruited individuals. When the haplotype frequencies were compared between patients with IgAN and controls, no significant differences were observed (Supplemental Table 3). Although we failed to find any significant genetic associations, which is in accordance with another study in a white population,17 given the limited number of recruited patients and controls and the relative low frequencies of some haplotypes and variants in MBL2, we did not have enough power (Supplemental Table 4). Future replication in large independent populations is needed to make sure that there is no genetic association between IgAN and MBL2 variants.
Next, we detected MBL levels in our recruited individuals with available plasma (749 patients with IgAN and 219 healthy controls). Compared with patients with IgAN, MBL levels in healthy controls were significantly lower (1315.0 ng/ml [433.4, 2651.7] versus 2690.4 ng/ml [962.0, 4227.3]; P<0.001; Supplemental Figure 1). However, the proportions of individuals with MBL deficiency were comparable between patients with IgAN and healthy controls (39 of 749 [5.2%] versus 15 of 219 [6.8%]; P=0.17). Among those with MBL deficiency, 16 (41.0%) patients and six (40.0%) controls were LYPB/LYPB, whereas 15 (38.5%) patients and nine (60%) controls were LXPA/LYPB. Thus, LYPB/LYPB and LXPA/LYPB were the predominant genotypes that contributed to MBL deficiency, accounting for 79.5% (31 of 39) of patients with IgAN and 100% (12 of 12) of healthy controls. Our results confirm the influence of MBL2 haplotype on MBL levels as widely reported before.6–8
To obtain a complete picture of the relationship between MBL levels and renal outcomes in IgAN, we next modeled MBL levels as a continuous variable using restricted cubic splines. Skewed distributed MBL levels were square root transformed to make the data close to a normal distribution. Interestingly, a nonlinear association was observed for IgAN progression (Figure 1). Patients with MBL deficiency showed distinct associations with poor renal outcome. Moreover, although patients with IgAN with MBL levels >3540 ng/ml (square root of MBL levels >59.50 ng/ml) tended to have a less favorable renal survival, the relationship was not statistically significant.
Figure 1.

Nonlinear association between MBL levels and adjusted hazard ratios of 50% eGFR decline or ESRD in patients with IgAN. Four knots at the 10th, 25th, 75th, and 90th percentiles of MBL levels were used to model the association between MBL levels and composite end point, defined as a 50% eGFR decline or ESRD, using restricted cubic splines. The solid line represents the estimated hazard ratio, the shaded area represents the 95% confidence interval, and the histogram represents the distribution of the square root of MBL levels in patients with IgAN. Models were adjusted for age; sex; eGFR; proteinuria; hypertension; Oxford M (mesangial hypercellularity score), E (the presence of endocapillary proliferation), S (segmental glomerulosclerosis/adhesion), T (severity of tubular atrophy/ininterstitial fibrosis), and C (presence of crescent) scores; and corticosteroids/immunosuppressive therapy. Patients with MBL deficiency (MBL<100 ng/ml; square root of MBL levels <10 ng/ml) showed distinctly higher hazard ratios for the composite end point, whereas although patients with higher MBL levels (square root of MBL levels >59.50 ng/ml) tended to have a less favorable renal survival, the relationship was not statistically significant.
To elucidate the clinical implications and underlying mechanisms of MBL levels in IgAN, we simplified our analysis by dividing patients into three groups: deficiency group (<100 ng/ml), sufficiency group (100–3540 ng/ml), and high group (>3540 ng/ml). We found that there were significantly more patients with IgAN in the deficiency group than in the sufficiency group who experienced prodromic infection (26 of 39 [66.7%] versus 138 of 437 [31.6%]; P<0.001) and gross hematuria (16 of 39 [41.0%] versus 115 of 437 [26.3%]; P=0.049) (Table 1). MBL deficiency is well known as a contributor to recurrent infection in toddlers, whose adaptive immune systems are still immature.10,11,18 Therefore, the higher proportion of prodromic infection observed in patients with IgAN with MBL deficiency might share some mechanisms with the recurrent infections in toddlers with MBL defects, which have been attributed to the pathogen opsonization function of MBL. Because episodic gross hematuria within several days of infection is a characteristic clinical feature of IgAN,19 it was unsurprising to also find that a higher proportion of MBL-deficient patients with IgAN presented with gross hematuria and prodromic infection. During follow-up, ten patients (25.6%) in the deficiency group and 51 patients (11.7%) in the sufficiency group reached the composite end point (P=0.01) (Table 1). Kaplan–Meier survival analysis revealed that renal survival for the composite outcome was significantly lower in the deficiency group than in the sufficiency group (P=0.02) (Figure 2). In a disease as complex as IgAN, multiple risk factors are involved in progression. Therefore, we further used Cox proportional hazards model for IgAN prognosis prediction. After adjustment for multiple known risk factors for IgAN progression (including baseline proteinuria, hypertension, eGFR, Oxford scores, and steroids/immunosuppressive therapy), the MBL deficiency group retained a predictive value for poor renal outcome compared with the sufficiency group (hazard ratio, 5.18; 95% confidence interval, 2.50 to 10.72; P<0.001) (Table 2), which indicates MBL deficiency as an independent risk factor for IgAN progression. In accordance with our survival analysis results, patients in the deficiency group showed worse eGFR slopes than those in the sufficiency group (−4.28 [−5.84, −2.24] versus −2.85 [−5.13, −1.19] ml/min per 1.73 m2 per year; P=0.03) (Table 1), which supports the important role of MBL deficiency in IgAN progression. Patients with IgAN often experience disease onset or exacerbation after infection; therefore, we suspected that MBL-deficient patients with IgAN, who are prone to infection, might have more frequent IgAN onset or exacerbation, resulting in rapid progression and poor renal outcome. Future prospective cohort studies designed to reveal the connection among MBL deficiency, infection frequency/type, and disease progression are needed in IgAN.
Figure 2.

Kaplan–Meier renal survival curves of patients with IgAN according to MBL levels. Patients with IgAN were stratified into three groups according to MBL levels: a deficiency group (<100 ng/ml; dash-dot line), a sufficiency group (100–3540 ng/ml; dashed line), and a high group (>3540 ng/ml; solid line). Patients in the deficiency group and the high group had significantly lower renal survival rates than those in the sufficiency group (P=0.02 for the deficiency group; P=0.02 for the high group). The renal survival curves for patients in the deficiency group and the high group showed no significant difference (P=0.08).
Table 2.
Risks of composite end point of MBL levels
| MBL Subgroup | MBL Median (IQR), ng/ml | Hazard Ratio (95% Confidence Interval) and P Value | |||
|---|---|---|---|---|---|
| Unadjusted | Model 1a | Model 2b | Model 3c | ||
| Sufficiency group | 1663 (745.8, 2630.8) | 1 (Reference) | 1 (Reference) | 1 (Reference) | 1 (Reference) |
| Deficiency group | 0.0 (0.0, 3.6) | 2.82 (1.43 to 5.57) | 2.85 (1.44 to 5.68) | 4.06 (1.99 to 8.30) | 5.18 (2.50 to 10.72) |
| P value | 0.003 | 0.003 | <0.001 | <0.001 | |
| High group | 4847.7 (4073.9, 5689.0) | 1.56 (1.06 to 2.30) | 1.52 (1.02 to 2.24) | 1.50 (1.00 to 2.27) | 1.54 (1.02 to 2.33) |
| P value | 0.03 | 0.04 | 0.05 | 0.04 | |
Composite end point was defined as a 50% decline in eGFR or ESRD. IQR, interquartile range.
Model 1 was adjusted for sex and age. Sex was analyzed as dichotomous data.
Model 2 was adjusted for covariates in model 1 plus eGFR, proteinuria, high BP (yes or no), and Oxford M (mesangial hypercellularity), E (the presence of endocapillary proliferation), S (segmental glomerulosclerosis/adhesion), T (severity of tubular atrophy/interstitial fibrosis), and C (presence of crescent) scores. The latter five variables were analyzed as categorical data.
Model 3 was adjusted for covariates in model 2 plus steroids or other immunosuppressive agents use (yes or no).
Moreover, compared with the sufficiency group, patients in the high group presented with more severe proteinuria (1.50 [0.84, 3.25] versus 1.15 [0.67, 2.28] g/d; P<0.001) (Table 1) and higher Oxford C scores (C0/C1/C2: 113 [42.0%]/123 [45.7%]/33 [12.3%] versus 205 [47.6%]/192 [44.5%]/34 [7.9%]; P=0.05) (Table 1). We also found that patients in the high group showed significantly worse renal outcome compared with those in the sufficient group (P=0.02) (Figure 2), although the significance was lost after adjustment for clinical and histologic manifestations using the Cox regression model (Table 2). Because IgAN is a slow progression disease and our patients were only followed for a short time, future studies recruiting patients with IgAN with longer-term follow-up would be required for replication and further evaluation. MBL is an acute-phase protein and can be induced by multiple inflammatory factors.20,21 Several studies have shown renal inflammation in IgAN; thus, active glomerular lesion may be one of the reasons for overproduction of MBL in these patients. Accordingly, we observed more severe IgAN phenotypes in the high group. Because the lectin pathway is observed in approximately 25% of patients with IgAN22 and MBL is an initiator of this pathway, we propose a hypothesis that patients with IgAN with high circulating MBL levels might be prone to lectin pathway–initiated complement activation at renal local, resulting in severe renal tissue injury and a trend of rapid progression. Unfortunately, because of the retrospective design of our study, we did not have data on local renal MBL staining to support our hypothesis.
In conclusion, we found that both MBL deficiency and high levels of MBL are risk factors for poor renal outcome in IgAN. They may be involved in different mechanisms of IgAN pathogenesis.
Concise Methods
Study Population
From 2003 to 2014, 1750 patients participated and registered in the IgAN cohort based at Peking University First Hospital (http://ckd.edc-china.com.cn/). Among them, 1052 patients were followed up for at least 1 year. Finally, 749 patients with IgAN with both available plasma and DNA were recruited in our study. Diagnosis of IgAN was on the basis of the presence of dominant IgA deposition in the mesangial area by immunofluorescence and electron-dense material deposition in the mesangial area by electron microscopy. Patients with Henoch–Schonlein purpura, liver cirrhosis, and other secondary etiologies of IgAN by detailed clinical and laboratory examinations were excluded. In addition, 489 age-, sex-, and geographically matched unrelated healthy individuals were recruited as controls.
For recruited individuals, anticoagulated (EDTA) peripheral venous blood was obtained on the day of renal biopsy (for patients with IgAN) or inclusion in the study (for controls). After the blood samples were centrifuged at 2000×g for 15 minutes at 4°C, plasma was stored in aliquots at −70°C until use, and genomic DNA was extracted by a salting-out procedure.23
The research was in compliance with the principles of the Declaration of Helsinki and approved by the local ethics committees. Informed consent was obtained from all enrolled individuals.
Clinical and Histologic Manifestations
Clinical manifestations at the time of renal biopsy, including age, sex, serum creatinine levels, 24-hour urine protein excretion, history of high BP, prodromic infection, and gross hematuria, were collected from the medical records. High BP was defined as a systolic BP of 140 mmHg or more, a diastolic BP of 90 mmHg or more, or taking antihypertensive medications to prevent hypertension. Prodromic infection was defined as onset of IgAN within 2 weeks after signs and symptoms of infections. The GFR of patients with IgAN was calculated using the Modified Glomerular Filtration Rate Estimating Equation.24 Histologically, the Oxford classification was used for the evaluation of pathologic lesions for those with more than eight glomeruli in biopsy specimens.25 Additionally, crescent scores of C0 (no crescents), C1 (crescents in less than one fourth of glomeruli), and C2 (crescents in over one fourth of glomeruli), which were recently added to the Oxford classification, were also used in our study.26,27 For survival analysis, 50% eGFR decline or ESRD, whichever occurred first, was used as the definition of composite end point.
Genotyping and Haplotypes of the MBL2 Gene
Previously, eight variants in the promoter and 5′ untranslated region (c.-550G>C [rs11003125], c.-427A>C [rs11003124], c.-349A>G [rs7084554], c.-336A>G [36014597], c.-324_-329delAAAGAG [rs10556764], c.-221G>C [rs7096206], c.-70C>T [rs11003123], and c.4C>T [rs7095891]) and three variants in exon 1 (c.223C>T [rs5030737], c.230G>A [rs1800450], and c.239G>A [rs1800451]) of MBL2 were reported to influence MBL expression. Therefore, a 990-bp sequence (from −677 to 313) containing the above 11 variants of MBL2 was amplified by PCR in our recruited 749 patients with IgAN and 489 healthy controls, and genotyping of these variants was performed by direct sequencing.
To determine MBL2 haplotypes, a PCR sequence-specific primer was further applied to individuals with more than one locus showing heterozygous genotypes among six variants, including c.-550G>C, c.-221G>C, c.4C>T, c.223C>T, c.230G>A, and c239G>A, followed by direct sequencing.
All primers used for PCR and the PCR sequence-specific primer were designed using the Primer 3 program (http://bioinfo.ut.ee/primer3-0.4.0/) and are listed in Supplemental Table 5 along with the PCR annealing temperatures. PCR products were sequenced using an ABI 3730 XL Genetic Analyzer (Applied Biosystems, Carlsbad, CA) and aligned to the reference sequence (version NC_000010.11) to determine the genotypes and haplotypes of the MBL2 gene.
MBL Level Detection
Plasma MBL levels of recruited individuals with available plasma (749 patients with IgAN and 219 healthy controls) were detected by ELISA using a commercial MBL Oligomer ELISA kit (BIOPRTO, Hellerup, Denmark) according to the manufacturer’s protocol.
Statistical Analyses
Quantitative variables are presented as the means±SD (for normally distributed data) or medians with interquartile range (for non-normally distributed data). Categorical data are summarized as absolute frequencies and percentages. For continuous variables, an independent samples t test was used if the data were normally distributed; if not, Mann–Whitney or Kruskal–Wallis tests were performed. Categorical variables were compared using the chi-squared test. Cumulative kidney survival curves were derived using the Kaplan–Meier method, and differences between curves were analyzed using a log-rank test. The prognostic factors were determined by multivariate analysis with Cox regression. The results are expressed as hazard ratios with 95% confidence intervals. The statistical software SPSS 11.0 (SPSS, Chicago, IL) was used for mean/median/percentage comparison and survival analysis.
To explore the association of MBL levels as continuous variables with renal outcome using Cox regression, MBL levels were modeled using restricted cubic splines after multivariable adjustment, for which Stata Edition 14.0 (Stata, College Station, TX) was used. Statistical significance was considered as a P<0.05.
Disclosures
None.
Supplementary Material
Acknowledgments
We acknowledge the contributions of Jin-wei Wang at Peking University First Hospital to the statistical analysis.
This work was supported by National Science Foundation of China grants 81470945 and 81670638, Beijing New-Star Plan of Science and Technology grant Z161100004916167, Capital of Clinical Characteristics and the Applied Research Fund grant Z141107002514037, Natural Science Foundation for Innovation Research Group of China grant 81621092, National Key Research and Development Program of China grant 2016YFC0904100, and Training Program of the Major Research Plan of the National Natural Science Foundation of China grant 91642120.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2017010076/-/DCSupplemental.
References
- 1.D’Amico G: The commonest glomerulonephritis in the world: IgA nephropathy. Q J Med 64: 709–727, 1987 [PubMed] [Google Scholar]
- 2.Liu ZH: Nephrology in china. Nat Rev Nephrol 9: 523–528, 2013 [DOI] [PubMed] [Google Scholar]
- 3.Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, Fasel D, Lata S, Prakash S, Shapiro S, Fischman C, Snyder HJ, Appel G, Izzi C, Viola BF, Dallera N, Del Vecchio L, Barlassina C, Salvi E, Bertinetto FE, Amoroso A, Savoldi S, Rocchietti M, Amore A, Peruzzi L, Coppo R, Salvadori M, Ravani P, Magistroni R, Ghiggeri GM, Caridi G, Bodria M, Lugani F, Allegri L, Delsante M, Maiorana M, Magnano A, Frasca G, Boer E, Boscutti G, Ponticelli C, Mignani R, Marcantoni C, Di Landro D, Santoro D, Pani A, Polci R, Feriozzi S, Chicca S, Galliani M, Gigante M, Gesualdo L, Zamboli P, Battaglia GG, Garozzo M, Maixnerová D, Tesar V, Eitner F, Rauen T, Floege J, Kovacs T, Nagy J, Mucha K, Pączek L, Zaniew M, Mizerska-Wasiak M, Roszkowska-Blaim M, Pawlaczyk K, Gale D, Barratt J, Thibaudin L, Berthoux F, Canaud G, Boland A, Metzger M, Panzer U, Suzuki H, Goto S, Narita I, Caliskan Y, Xie J, Hou P, Chen N, Zhang H, Wyatt RJ, Novak J, Julian BA, Feehally J, Stengel B, Cusi D, Lifton RP, Gharavi AG: Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet 46: 1187–1196, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dommett RM, Klein N, Turner MW: Mannose-binding lectin in innate immunity: Past, present and future. Tissue Antigens 68: 193–209, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ip WK, Takahashi K, Ezekowitz RA, Stuart LM: Mannose-binding lectin and innate immunity. Immunol Rev 230: 9–21, 2009 [DOI] [PubMed] [Google Scholar]
- 6.Madsen HO, Garred P, Thiel S, Kurtzhals JA, Lamm LU, Ryder LP, Svejgaard A: Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J Immunol 155: 3013–3020, 1995 [PubMed] [Google Scholar]
- 7.Turner MW, Hamvas RM: Mannose-binding lectin: Structure, function, genetics and disease associations. Rev Immunogenet 2: 305–322, 2000 [PubMed] [Google Scholar]
- 8.Seyfarth J, Garred P, Madsen HO: The ‘involution’ of mannose-binding lectin. Hum Mol Genet 14: 2859–2869, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Naito H, Ikeda A, Hasegawa K, Oka S, Uemura K, Kawasaki N, Kawasaki T: Characterization of human serum mannan-binding protein promoter. J Biochem 126: 1004–1012, 1999 [DOI] [PubMed] [Google Scholar]
- 10.Koch A, Melbye M, Sørensen P, Homøe P, Madsen HO, Mølbak K, Hansen CH, Andersen LH, Hahn GW, Garred P: Acute respiratory tract infections and mannose-binding lectin insufficiency during early childhood. JAMA 285: 1316–1321, 2001 [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Xu Z, Ou X, Wang M, Yang X, Li Q: Mannose-binding lectin polymorphisms and recurrent respiratory tract infection in Chinese children. Eur J Pediatr 168: 1305–1313, 2009 [DOI] [PubMed] [Google Scholar]
- 12.de Vries B, Walter SJ, Peutz-Kootstra CJ, Wolfs TG, van Heurn LW, Buurman WA: The mannose-binding lectin-pathway is involved in complement activation in the course of renal ischemia-reperfusion injury. Am J Pathol 165: 1677–1688, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trendelenburg M, Theroux P, Stebbins A, Granger C, Armstrong P, Pfisterer M: Influence of functional deficiency of complement mannose-binding lectin on outcome of patients with acute ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention. Eur Heart J 31: 1181–1187, 2010 [DOI] [PubMed] [Google Scholar]
- 14.Ohsawa I, Ishii M, Ohi H, Tomino Y: Pathological scenario with the mannose-binding lectin in patients with IgA nephropathy. J Biomed Biotechnol 2012: 476739, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong R, Liu Z, Li L: Mannose-binding lectin gene polymorphism associated with the patterns of glomerular immune deposition in IgA nephropathy. Scand J Urol Nephrol 35: 228–232, 2001 [DOI] [PubMed] [Google Scholar]
- 16.Shi B, Wang L, Mou S, Zhang M, Wang Q, Qi C, Cao L, Che X, Fang W, Gu L, Yan Y, Qian J, Ni Z: Identification of mannose-binding lectin as a mechanism in progressive immunoglobulin A nephropathy. Int J Clin Exp Pathol 8: 1889–1899, 2015 [PMC free article] [PubMed] [Google Scholar]
- 17.Pirulli D, Boniotto M, Vatta L, Crovella S, Spano A, Morgutti M, Zezlina S, Bertola L, Roccatello D, Scolari F, Peruzzi L, Savoldi S, Amoroso A: Polymorphisms in the promoter region and at codon 54 of the MBL2 gene are not associated with IgA nephropathy. Nephrol Dial Transplant 16: 759–764, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Santos IK, Costa CH, Krieger H, Feitosa MF, Zurakowski D, Fardin B, Gomes RB, Weiner DL, Harn DA, Ezekowitz RA, Epstein JE: Mannan-binding lectin enhances susceptibility to visceral leishmaniasis. Infect Immun 69: 5212–5215, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Floege J, Feehally J: The mucosa-kidney axis in IgA nephropathy. Nat Rev Nephrol 12: 147–156, 2016 [DOI] [PubMed] [Google Scholar]
- 20.Thiel S, Holmskov U, Hviid L, Laursen SB, Jensenius JC: The concentration of the C-type lectin, mannan-binding protein, in human plasma increases during an acute phase response. Clin Exp Immunol 90: 31–35, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turner MW: The lectin pathway of complement activation. Res Immunol 147: 110–115, 1996 [DOI] [PubMed] [Google Scholar]
- 22.Roos A, Rastaldi MP, Calvaresi N, Oortwijn BD, Schlagwein N, van Gijlswijk-Janssen DJ, Stahl GL, Matsushita M, Fujita T, van Kooten C, Daha MR: Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J Am Soc Nephrol 17: 1724–1734, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Miller SA, Dykes DD, Polesky HF: A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16: 1215, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Feldman HI, Kusek JW, Eqqers P, Van Lente F, Greene T, Coresh J; CKD-CPI (Chronic Kidney Disease Epidemiology Collaboration) : A new equation to estimate glomerularfiltration rate. Ann Intern Med 150: 604–612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberts IS, Cook HT, Troyanov S, Alpers CE, Amore A, Barratt J, Berthoux F, Bonsib S, Bruijn JA, Cattran DC, Coppo R, D’Agati V, D’Amico G, Emancipator S, Emma F, Feehally J, Ferrario F, Fervenza FC, Florquin S, Fogo A, Geddes CC, Groene HJ, Haas M, Herzenberg AM, Hill PA, Hogg RJ, Hsu SI, Jennette JC, Joh K, Julian BA, Kawamura T, Lai FM, Li LS, Li PK, Liu ZH, Mackinnon B, Mezzano S, Schena FP, Tomino Y, Walker PD, Wang H, Weening JJ, Yoshikawa N, Zhang H; Working Group of the International IgA Nephropathy Network and the Renal Pathology Society : The Oxford classification of IgA nephropathy: Pathology definitions, correlations, and reproducibility. Kidney Int 76: 546–556, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Trimarchi H, Barratt J, Cattran DC, Cook HT, Coppo R, Haas M, Liu ZH, Roberts IS, Yuzawa Y, Zhang H, Feehally J; IgAN Classification Working Group of the International IgA Nephropathy Network and the Renal Pathology Society, Conference Participants : Oxford classification of IgA nephropathy 2016: An update from the IgA nephropathy classification working group. Kidney Int 91: 1014–1021, 2017 [DOI] [PubMed] [Google Scholar]
- 27.Gera M, Slezak JM, Rule AD, Larson TS, Stegall MD, Cosio FG: Assessment of changes in kidney allograft function using creatinine-based estimates of glomerular filtration rate. Am J Transplant 7: 880–887, 2007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.