

Abstract
The present case report describes the infrequent coexistence of squamous cell transformation and the epidermal growth factor receptor (EGFR) T790M mutation as resistance mechanisms to first line treatment with tyrosine kinase inhibitors. The patient was a 44-year-old female, diagnosed with a primitive advanced lung adenocarcinoma with bone metastases. The tumor was positive for the EGFR exon 19 deletion, therefore the patient was treated with afatinib (40 mg/day, orally) and radiotherapy for bone lesions. After 16 months, the patient developed resistance. Cytological examination of the pleural effusion confirmed an adenocarcinoma positive for the EGFR exon 19 deletion and the T790M mutation within exon 20, while a biopsy from the upper left bronchus revealed a keratinizing squamous cell carcinoma positive for the EGFR exon 19 deletion. In addition, the EGFR mutations were concomitantly detected in circulating cell-free tumour DNA. Due to the presence of the T790M mutation, the patient underwent osimertinib therapy (80 mg/day, orally), which resulted in a partial tumour regression at the 2-month follow-up, whereas the squamous lesions were treated with radiotherapy. The adenocarcinoma and squamous carcinoma components may share the same origin, according to the presence of the EGFR exon 19 deletion in both lesions. More accurate characterization of resistance mechanisms may lead to the development of improved treatment regimens.
Keywords: lung adenocarcinoma, epidermal growth factor receptor, tyrosine kinase inhibitor, resistance mechanisms
Introduction
A significant proportion (15–20%) of patients with lung adenocarcinoma harbor epidermal growth factor receptor (EGFR) activating mutations (1) and can benefit from first-line treatment with tyrosine kinase inhibitors (TKI), including gefitinib (2) or erlotinib (3) (first generation TKI) and afatinib (second generation TKI) (4). However, after 12–16 months of TKI treatment almost all patients develop acquired resistance and experience tumour progression (5,6).
The most common resistance mechanism, detectable in ~50% of TKI resistant tumors, is the emergence of a secondary T790M mutation in exon 20 of EGFR (5,7). Other well-known resistance mechanisms, in patients with non-small cell lung cancer treated with EGFR-TKI, include the amplification of MET proto-oncogene tyrosine kinase receptor (MET) (20%) (8), the development of small cell lung cancer transformation (14%) (9,10) and the presence of acquired phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PI3KCA) mutations (5%) (6). Less common resistance mechanisms (30%) include the activation of Insulin-Like Growth Factor-1 Receptor (11), the epithelial to mesenchymal transition (12), and more rarely squamous cell transformation (13–16). Different resistance mechanisms may be observed in the same patient (6) due to intratumor and intrametastatic heterogeneity (17), which strongly influence the patient's response to treatment.
The identification of molecular alterations responsible for acquired TKI resistance is crucial for patient management, as multiple novel treatment strategies are available to overcome this issue (18). For instance, in cases of T790M-mediated resistance, the use of a third-generation TKI, which irreversibly and selectively blocks T790M mutant clones, has been demonstrated to increase the potency of EGFR-TK inhibition (19,20).
A second tumour biopsy is recommended in case of TKI resistance; however, a single biopsy specimen may not mirror all the biological properties of a tumour. Combining re-biopsy analysis with molecular characterization of circulating cell-free tumour (ct) DNA represents a good strategy to describe the molecular landscape of a tumour (21–23). In addition, temporal changes to EGFR activating and resistance mutations in plasma DNA are directly linked to treatment efficacy (24,25).
In the present report the case of a patient who developed two resistance mechanisms in response to first-line afatinib, the T790M mutation and the rare squamous cell transformation, is described. To the best of our knowledge, only a few similar cases have previously been described and they focused on patients with lung adenocarcinoma who were treated with erlotinib and gefitinib (13–15).
Case report
Written informed consent for the publication of this report was obtained from the patient. In October 2014, a 44-year-old female with an 8 pack/year smoking history presented at the University Hospital of Pisa (Pisa, Italy) with back pain. A few weeks later the patient underwent magnetic resonance imaging of the vertebral column, which revealed a number of osteoblastic bone lesions (S1-3; D2-3 and D7-10 laminas). A computed tomography (CT) scan revealed a left lower lobe mass, and pleural and pericardial effusions (PE). The patient underwent endobronchial ultrasound biopsy and pleural fluid analyses. Histological and cytological samples examination identified an adenocarcinoma, further characterized using cell-block (stained with 10% buffered formalin at room temperature for 24 h) paraffin-embedded sections (thickness, 2 µm) by immunohistochemical staining using the ultraView Universal DAB Detection kit (Ventana Medical Systems. Inc., Tucson, AZ, USA), according to the manufacturer's protocol, with anti-thyroid transcription factor (TTF-1) antibody (mouse monoclonal primary antibody; clone 8G7G3/1; ready-to-use; catalog no. 790-438; Ventana Medical Systems, Inc.) for 44 min at 37°C, which demonstrated a strong positive nuclear stain. An Olympus BX51 light microscope (Olympus Italia Srl; Segrate, Italy) was used for the analysis. The final diagnosis was adenocarcinoma (26), consistent with a lung primary cancer with bone metastases.
An extensive molecular analysis was performed on the PE. Fluorescent In Situ Hybridization (FISH) was performed to evaluate translocations of anaplastic lymphoma kinase (ALK) (using a Vysis ALK Dual-Color Break Apart FISH probe kit; Abbott Laboratories, Abbott Park, IL, USA), proto-oncogene tyrosine-protein kinase ROS (ROS1; using a ROS1 6q22 Break Probe; Kreatech; Leica Microsystems, Ltd., Milton Keynes, UK) and RET proto-oncogene (RET; using a RET 10q11 Break Probe; Kreatech; Leica Microsystems, Ltd.), and to assess the presence of MET amplification (Vysis MET Spectrum Red and CEP7 D7Z1 Spectrum Green; Abbott Laboratories). FISH analysis was performed according to the manufacturers'protocols. All FISH tests were negative: ALK, 4% of neoplastic rearranged cells (cut-off 15%); ROS1, 0% of neoplastic rearranged cells (cut-off 15%); RET, 5% of neoplastic rearranged cells (cut-off 15%); and MET, MET/CEP7=1,1 (cut-off ≥2).
Mutational analysis of KRAS, BRAF, NRAS, PIK3CA, ALK, ERBB2, DDR2, MAP2K1, EGFR, RET was performed using a Sequenom Mass-Array (matrix assisted laser desorption ionization-time of flight mass spectrometry) using the Myriapod Lung Status kit (Diatech Pharmacogenetics SRL, Jesi, Italy) together with the analysis software MASSARRAY® TYPER 4.0 (Diatech Pharmacogenetics, Jesi SRL) according to the manufacturer's protocol (limit of detection: 2.5–5% for EGFR and 2.5–10% for all other genes). The PE demonstrated a deletion in EGFR exon 19 (ex19del).
In November 2014, the patient started treatment with afatinib (40 mg/day, orally). The bone lesions required a radio therapeutic approach due to nerve peduncle compression, therefore the patient received radiation (30 Gy in five fractions on S1-3; 25 Gy in five fractions on D7-10 and 30 Gy in five fractions on D2-3). In addition, the patient was treated with denosumab (120 mg every 28 days, intravenously). After 4 weeks of afatinib, a CT scan revealed a partial response in the lung mass, and a total response for the effusions and bone lesions.
The patient tolerated the therapy well, with mild diarrhea and post-actinic pneumonia, which was treated with antibiotics and anti-inflammatory therapy. Foci of post-actinic pneumonia were observed, primarily on paravetebral and medium lobe sites. At subsequent medical examinations, after 5, 7 and 9 months of treatment, the patient was stable and no EGFR mutations were detected on ctDNA from plasma collected at each visit (Fig. 1A). ctDNA was purified from 4 ml of plasma using a QIAmp Circulating Nucleic Acid kit (Qiagen, Inc., Valencia, CA, USA) and EGFR mutational analysis was performed using an Easy®EGFR Quantitative Real Time PCR kit (Diatech Pharmacogenetics SRL) according to the manufacturer's protocol. The Easy®EGFR quantitative Real Time PCR kit is validated for use on liquid biopsy and its limit of detection ranges from 0.5 to 2%.
Figure 1.
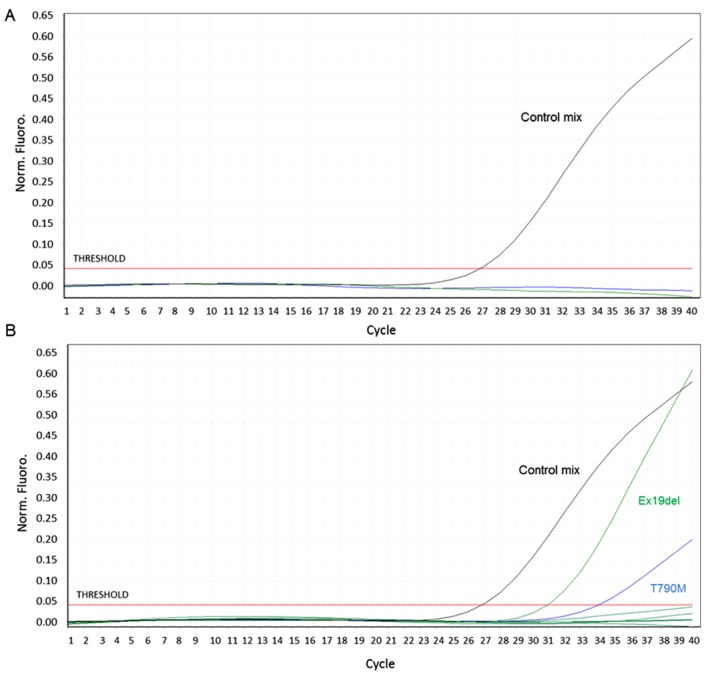
EGFR molecular analysis on ctDNA. Graphs from the quantitative polymerase chain reaction showing EGFR molecular analysis on ctDNA after (A) 9 and (B) 16 months of treatment with afatinib. Black curve: Control mix, including primers and probe for a non-polymorphic EGFR region. The amplification of the control region allows for the evaluation of ctDNA quality and quantity. Green curve: ex19del mix, including primers and probe for 21 types of EGFR deletions. Blue curve: T790M mix, including primers and probe for EGFR T790M mutation. EGFR, epidermal growth factor receptor; ctDNA, circulating cell-free tumour DNA.
In February 2016, a CT scan control detected an almost complete response on the primitive lung mass and bone lesions. The patient carried on the therapy with afatinib and denosumab. Three months later (May 2016), a CT scan demonstrated a large area of atelectasia of the upper left lung lobe partially involving the lower lobe with PE. The cytological examination of PE confirmed an adenocarcinoma with a positive immunohistochemical stain for TTF-1. Cytological samples from pleural effusion were positive for EGFR ex19del and T790M; both mutations were concomitantly detected in ctDNA (Fig. 1B). Broncospic investigation of the upper left bronchus revealed a partial obstruction and infiltration from a whitish neoformation. This lesion was biopsied. The obtained tissue was fixed in 10% buffered formalin (room temperature, 24 h), paraffin-embedded and cut into 5 µm thick sections. The hematoxylin-eosin stain (room temperature, 1 h 26 min) revealed a keratinizing squamous cell carcinoma confirmed by immunohistochemical examination, performed as aforementioned, using a p40 antibody (mouse monoclonal primary antibody; clone BC28; ready-to-use; catalog no. 790-4950; Ventana Medical System, Tucson, AZ, USA) for 40 min at 42°C, the results of which were strongly positive, and for TTF-1 antibody, the result of which was negative. The two lesions harboured the ex19del mutation. The same FISH and mutational tests as those executed on pre-TKI specimen were performed on the post-TKI adenocarcinoma and squamous cell carcinoma samples. Again, all FISH tests gave negative results: ALK, 0% of neoplastic rearranged cells; ROS1, 0% of neoplastic rearranged cells; RET, 0% of neoplastic rearranged cells; and MET, MET/CEP7=1 in the adenocarcinoma. In the squamous cell carcinoma: ALK, 2% of neoplastic rearranged cells; ROS1, 0% of neoplastic rearranged cells; RET, 0% of neoplastic rearranged cells; and MET, MET/CEP7=1. In addition, HER2 amplification (Vysis HER-2/neu SpectrumOrange/CEP17 SpectrumGreen Probes; Abbott Laboratories) was evaluated in post-TKI samples, for which the results were negative: HER2/CEP17=0.9 (cut-off ≥2) in the adenocarcinoma, HER2/CEP17=1 (cut-off ≥2) in the squamous cell carcinoma.
The patient underwent osimertinib (80 mg/day, orally) therapy and radiotherapy for squamous lesions. At a 2-month follow-up T790M positive lesions exhibited a partial regression. Fig. 2 presents the patient's CT-images and histological examinations, and Fig. 3 presents the patient's clinical response.
Figure 2.

CT images and histological examinations of the patient. (A) CT image of patient at the time of diagnosis; (B and C) CT images of patient at the time of progression. (D) Pre-afatinib treatment of original adenocarcinoma in pleural effusion (hematoxylin and eosin, magnification, ×200); (E) TTF-1 positive stain. (F) Post-afatinib treatment adenocarcinoma in pleural effusion (hematoxylin and eosin). (G) TTF-1 positive stain. (H) Post-afatinib squamous cell transformation in core needle biopsy of the upper left bronchus (hematoxylin and eosin); (I) p40 positive stain. Scale bar, 100 µm. Magnification for histochemical staining images, ×200. CT, computed tomography; TTF-1, thyroid transcription factor-1.
Figure 3.
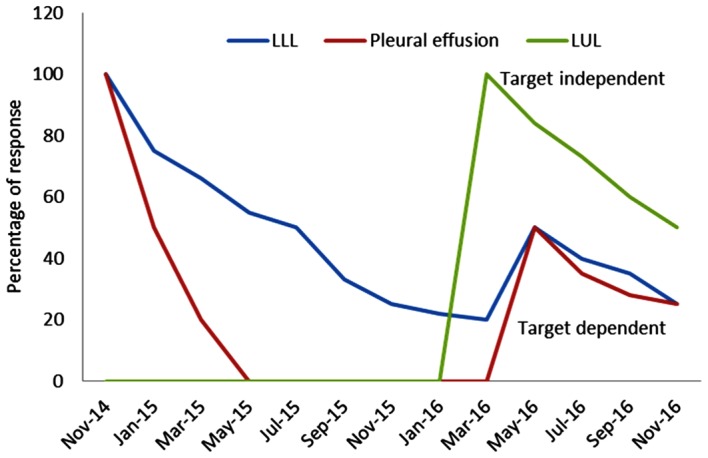
Diagram illustrating the longitudinal and clinical evaluation of the patient. Target dependent, lesions responding to afatinib treatment (epidermal growth factor receptor positive) (LLL and pleural effusion); target independent, lesions resistant to afatinib treatment [LUL (squamous cell carcinoma)]. LLL, left lower lobe; LUL, left upper lobe.
Discussion
Patients with EGFR mutant lung adenocarcinoma treated with TKI typically develop resistance within 1 year of treatment (5). The existence of different acquired TKI resistance mechanisms together with tumour heterogeneity constitutes a major challenge for clinical practice (6).
The present report describes the case of a patient with lung adenocarcinoma treated with afatinib who developed the T790M mutation and squamous cell transformation. To date, only a few cases of squamous cell transformation, with (13,15) and without (14) concomitant T790M, have been reported in response to erlotinib and gefitinib, and no similar cases have been reported in response to afatinib (27). Longo et al (16) recently reported a case of lung cell adenocarcinoma positive for the EGFR exon 21 L858R mutation, who, following TKI treatment, developed squamous cell carcinoma change together with an EGFR exon 20 S768I secondary mutation.
In our case, histological transformation may have been a consequence of TKI treatment or it could have been enhanced by radiotherapy, as reported in other types of cancer, including prostate cancer (28).
All the histological evaluations have been performed on specimens obtained by needle biopsy, and although there were different morphological and immunohistochemical characteristics in pre and post-TKI lesions, the presence of the squamous cell carcinoma prior to EGFR-TKI therapy in form of an adenosquamous carcinoma cannot be excluded. However, lung adenocarcinoma exhibits a different molecular landscape compared with squamous cell carcinoma, for instance EGFR mutations are present in 10–40% of cases of adenocarcinoma, but rarely in squamous cell carcinoma (29). The presence of ex19del in both lesions in the present study suggests that the adenocarcinoma and squamous carcinoma components share the same clonal origin and a mixed tumour is unlikely on the basis of the different location of the two lesions. Furthermore, all the tumour lesions were extensively characterized from a molecular point of view and the only difference was the presence of the T790M mutation, which was detected only in the post-TKI adenocarcinoma specimen.
The reported case highlights the role of intra-tumour heterogeneity, defined as the presence within the same tumour of distinct cellular populations with specific phenotypic features, in response to TKI treatment, which selects clones with intrinsic or acquired resistance that drive disease progression (30–32). However, a complete characterization of the mechanisms of response and resistance is essential to provide patients with the greatest clinical benefit and several studies and case reports confirm this issue (5,6,9,10,13–16,18).
In the present context, a single tumour biopsy, limited by the presence of geographic heterogeneity, may be inadequate to detect all cancer gene mutations, explaining the lack of a direct correlation between molecular alteration and clinical efficacy of treatment (23). The liquid biopsy and analysis of ctDNA furthers understanding of intra-tumour heterogeneity, since it detects contributions from multiple tumour sites. In the present case report, as soon as the patient experienced clinical progression, activating and resistance mutations became detectable on ctDNA, supporting its value in agreement with previously published data (24,25).
However, according to current knowledge and reported cases, neither liquid biopsy nor solid biopsy on their own can suffice for the monitoring of cancer therapy. In spite of the non-invasiveness of liquid biopsy and its high informative value, resistance mechanisms, including phenotypic changes, cannot be evaluated without a histopathological analysis of tumour tissues; for this reason tissue biopsy should be performed whenever possible.
In conclusion, the present case report underlines the complementarity of tumour re-biopsies and analysis of ctDNA in order to have a more complete view of temporal evolution and molecular diversity of TKI-resistant disease, thus improving therapeutic regimens.
References
- 1.Ileana EE, Wistuba II, Izzo JG. From uniplex to multiplex molecular profiling in advanced non-small cell lung carcinoma. Cancer J. 2015;21:413–424. doi: 10.1097/PPO.0000000000000150. [DOI] [PubMed] [Google Scholar]
- 2.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 3.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 4.Yang JC, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, Zhou C, Hu CP, O'Byrne K, Feng J, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16:141–151. doi: 10.1016/S1470-2045(14)71173-8. [DOI] [PubMed] [Google Scholar]
- 5.Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, Riely GJ. Analysis of tumour specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris M, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 9.Furugen M, Uechi K, Hirai J, Aoyama H, Saio M, Yoshimi N, Kinjo T, Miyagi K, Haranaga S, Higa F, et al. An autopsy case of two distinct, acquired drug resistance mechanisms in epidermal growth factor receptor-mutant lung adenocarcinoma: Small cell carcinoma transformation and epidermal growth factor receptor T790M mutation. Intern Med. 2015;54:2491–2496. doi: 10.2169/internalmedicine.54.5481. [DOI] [PubMed] [Google Scholar]
- 10.Alì G, Bruno R, Giordano M, Prediletto I, Marconi L, Zupo S, Fedeli F, Ribechini A, Chella A, Fontanini G. Small-cell lung cancer transformation and the T790M mutation: A case report of two acquired mechanisms of TKI resistance detected in a tumor rebiopsy and plasma sample of EGFR-mutant lung adenocarcinoma. Oncol Lett. 2016;12:4009–4012. doi: 10.3892/ol.2016.5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peled N, Wynes MW, Ikeda N, Ohira T, Yoshida K, Qian J, Ilouze M, Brenner R, Kato Y, Mascaux C, Hirsch FR. Insulin-like growth factor-1 receptor (IGF-1R) as a biomarker for resistance to the tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. Cell Oncol (Dordr) 2013;36:277–288. doi: 10.1007/s13402-013-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, Iwata KK, Gibson N, Haley JD. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65:9455–9462. doi: 10.1158/0008-5472.CAN-05-1058. [DOI] [PubMed] [Google Scholar]
- 13.Scher KS, Saldivar JS, Fishbein M, Marchevsky A, Reckamp KL. EGFR-mutated lung cancer with T790M-acquired resistance in the brain and histologic transformation in the lung. J Natl Compr Canc Netw. 2013;11:1040–1044. doi: 10.6004/jnccn.2013.0126. [DOI] [PubMed] [Google Scholar]
- 14.Levin PA, Mayer M, Hoskin S, Sailors J, Oliver DH, Gerber DE. Histologic transformation from adenocarcinoma to squamous cell carcinoma as a mechanism of resistance to EGFR inhibition. J Thorac Oncol. 2015;10:e86–e88. doi: 10.1097/JTO.0000000000000571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jukna A, Montanari G, Mengoli MC, Cavazza A, Covi M, Barbieri F, Bertolini F, Rossi G. Squamous cell carcinoma ‘Transformation’ concurrent with secondary T790M mutation in resistant EGFR-mutated adenocarcinomas. J Thorac Oncol. 2016;11:e49–e51. doi: 10.1016/j.jtho.2015.12.096. [DOI] [PubMed] [Google Scholar]
- 16.Longo L, Mengoli MC, Bertolini F, Bettelli S, Manfredini S, Rossi G. Synchronous occurrence of squamous cell carcinoma ‘transformation’ and EGFR exon 20 S768I mutation as a novel mechanism of resistance in EGFR-mutated lung adenocarcinoma. Lung Cancer. 2017;103:24–26. doi: 10.1016/j.lungcan.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 17.Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C. Translational implications of tumor heterogeneity. Clin Cancer Res. 2015;21:1258–1266. doi: 10.1158/1078-0432.CCR-14-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piotrowska Z, Sequist LV. Epidermal growth factor receptor-mutant lung cancer: New drugs, New resistance mechanisms, and future treatment options. Cancer J. 2015;21:371–347. doi: 10.1097/PPO.0000000000000147. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Song Z, Jin Y, Tang Z, Kang J, Ma X. Novel selective and potent EGFR inhibitor that overcomes T790M-Mediated resistance in non-small cell lung cancer. Molecules. 2016;21 doi: 10.3390/molecules21111462. pii: E1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pantel K, Diaz LA, Jr, Polyak K. Tracking tumor resistance using ‘liquid biopsies’. Nat Med. 2013;19:676–677. doi: 10.1038/nm.3233. [DOI] [PubMed] [Google Scholar]
- 22.Imamura F, Uchida J, Kukita Y, Kumagai T, Nishino K, Inoue T, Kimura M, Oba S, Kato K. Monitoring of treatment responses and clonal evolution of tumor cells by circulating tumor DNA of heterogeneous mutant EGFR genes in lung cancer. Lung Cancer. 2016;94:68–73. doi: 10.1016/j.lungcan.2016.01.023. [DOI] [PubMed] [Google Scholar]
- 23.Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, Kurtz DM, Stehr H, Scherer F, Karlovich CA, et al. Circulating tumor DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. doi: 10.1038/ncomms13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chia PL, Do H, Morey A, Mitchell P, Dobrovic A, John T. Temporal changes of EGFR mutations and T790M levels in tumor and plasma DNA following AZD9291 treatment. Lung Cancer. 2016;98:29–32. doi: 10.1016/j.lungcan.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Uchida J, Imamura F, Kukita Y, Oba S, Kumagai T, Nishino K, Inoue T, Kimura M, Kato K. Dynamics of circulating tumor DNA represented by the activating and resistant mutations in epidermal growth factor receptor tyrosine kinase inhibitor treatment. Cancer Sci. 2016;107:353–358. doi: 10.1111/cas.12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG. Introduction to the 2015 World Health Organization Classification of tumors of the lung, Pleura, Thymus and heart. J Thorac Oncol. 2015;10:1240–1242. doi: 10.1097/JTO.0000000000000630. [DOI] [PubMed] [Google Scholar]
- 27.Wu SG, Liu YN, Tsai MF, Chang YL, Yu CJ, Yang PC, Yang JC, Wen YF, Shih JY. The mechanism of acquired resistance to irreversible EGFR tyrosine kinase inhibitor-afatinib in lung adenocarcinoma patients. Oncotarget. 2016;7:12404–12413. doi: 10.18632/oncotarget.7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller VA, Reuter V, Scher HI. Primary squamous cell carcinoma of the prostate after radiation seed implantation for adenocarcinoma. Urology. 1995;46:111–113. doi: 10.1016/S0090-4295(99)80174-5. [DOI] [PubMed] [Google Scholar]
- 29.Devarakonda S, Morgensztern D, Govindan R. Genomic alterations in lung adenocarcinoma. Lancet Oncol. 2015;16:e342–e351. doi: 10.1016/S1470-2045(15)00077-7. [DOI] [PubMed] [Google Scholar]
- 30.Yu JY, Yu SF, Wang SH, Bai H, Zhao J, An TT, Duan JC, Wang J. Clinical outcomes of EGFR-TKI treatment and genetic heterogeneity in lung adenocarcinoma patients with EGFR mutations on exons 19 and 21. Chin J Cancer. 2016;35:30. doi: 10.1186/s40880-016-0086-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hata A, Yoshioka H, Fujita S, Kunimasa K, Kaji R, Imai Y, Tomii K, Iwasaku M, Nishiyama A, Ishida T, Katakami N. Complex mutations in the epidermal growth factor receptor gene in non-small cell lung cancer. J Thorac Oncol. 2010;5:1524–1528. doi: 10.1097/JTO.0b013e3181e8b3c5. [DOI] [PubMed] [Google Scholar]
- 32.Morgillo F, Corte CM Della, Fasano M, Ciardiello F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open. 2016;1:e000060. doi: 10.1136/esmoopen-2016-000060. [DOI] [PMC free article] [PubMed] [Google Scholar]