

Abstract
Blocking transforming growth factor (TGF)β1 signal transduction has been a central strategy for scar reduction; however, this approach appears to be minimally effective. Here, we show that fibromodulin (FMOD), a 59-kD small leucine-rich proteoglycan critical for normal collagen fibrillogenesis, significantly reduces scar formation while simultaneously increasing scar strength in both adult rodent models and porcine wounds, which simulate human cutaneous scar repair. Mechanistically, FMOD uncouples pro-migration/contraction cellular signals from pro-fibrotic signaling by selectively enhancing SMAD3-mediated signal transduction, while reducing AP-1-mediated TGFβ1 auto-induction and fibrotic extracellular matrix accumulation. Consequently, FMOD accelerates TGFβ1-responsive adult fibroblast migration, myofibroblast conversion, and function. Furthermore, our findings strongly indicate that, by delicately orchestrating TGFβ1 activities rather than indiscriminately blocking TGFβ1, FMOD elicits fetal-like cellular and molecular phenotypes in adult dermal fibroblasts in vitro and adult cutaneous wounds in vivo, which is a unique response of living system undescribed previously. Taken together, this study illuminates the signal modulating activities of FMOD beyond its structural support functions, and highlights the potential for FMOD-based therapies to be used in cutaneous wound repair.
Introduction
Cutaneous wounds, acquired from surgery or trauma, can cause pathologic scarring with significant functional and psychological sequelae. These pathologies cost the global healthcare system over $8.6 billion and affect 100 million patients annually.1,2 Unfortunately, knowledge of processes regulating dermal wound healing is still incomplete, which leaves patients with suboptimal treatment options. For example, the current standards of care for cutaneous scarring exhibit numerous undesirable side effects:3 Local corticosteroid injections can result in reduced scar strength, pigmentation changes, granulomas, and skin atrophy, while wound irradiation can cause growth inhibition, decreased scar strength, and increased long-term cancer risks.
Although multiple growth factors and cytokines contribute to cutaneous repair, transforming growth factor (TGF)β1 has been recognized as a central modulator of wound healing.4,5 The well-documented inflammatory and pro-fibrotic effects of TGFβ1 have precipitated a considerable body of research and clinical efforts in TGFβ1 signaling blockade to reduce scarring (‘low TGFβ1’ strategies).4,6 However, application of TGFβ1-neutralizing antibodies did not effectively prevent scar formation in adult rat cutaneous wounds.7 Additionally, a synthetic inhibitor peptide of TGFβ failed to reduce fibrosis in a porcine silicone pericapsular fibrotic model.8 Beyond this, in human clinical trials, the knock-down of connective growth factor (CTGF) expression, a known downstream mediator of TGFβ1 bioactivity,9 was inadequate in reducing scar formation.10,11 Even though studies have examined TGFβ1 signaling blockades for possible benefits, it is important to consider off target effects of TGBβ1 blockade. Transgenic mice studies reveal that simple TGFβ1 blockade does not promote ideal wound healing, and instead, TGFβ1-deficient mice die from overwhelming systemic inflammation within 3–4 weeks after their birth.12 When crossed with severe combined immunodeficiency mice, TGFβ1-deficient mice survive but exhibit impaired granulation tissue formation and delayed cutaneous wound repair.13 Finally, inducible fibroblast lineage-specific type II TGFβ receptor-knockout mice demonstrate severely impaired fibroblast migration, myofibroblast development, and myofibroblast function, which leads to deficient granulation tissue formation and defective wound contraction.14 These results indicate that TGFβ1 signaling has multiple roles in the wound healing process, and as such, non-specific TGFβ-blockade is detrimental to cutaneous repair.
Before a particular gestation age, fetal cutaneous wounds can heal scarlessly with apparent restoration of the normal dermal extracellular matrix (ECM) architecture and dermal appendages.15 In rodents, skin wounds begin to scar at approximately day 18.5 of a roughly 20-day gestation period, whereas in humans, cutaneous scarring begins approximately at week 24 of gestation.15 Accumulating data indicate that fetal-type scarless healing is likely inherent to the unique properties of fetal cells, ECM, and growth factor/cytokine profiles that comprise fetal skin, rather than the intrauterine environment.15 Using a rodent fetal wound healing model, we previously reported a rapid decline in fibromodulin (FMOD) expression coinciding with the transition from fetal-type scarless repair to adult-type scarring repair.16,17 FMOD is a 59-kD small leucine-rich proteoglycan (SLRP) with well-described roles in collagen fibrillogenesis.18 FMOD loss- and gain-of-function models have assisted in confirming the importance of FMOD in fetal-type scarless repair, as well as determining TGFβ ligand and receptor levels in fetal and adult skin.16,17 In addition, FMOD deficiency in adult mice has been shown to increase scar size and elicit a severely deficient dermal fibroplasia response, which is characterized by profoundly impaired fibroblast migration and delays in granulation tissue formation.19 Both of these attributes are strikingly similar to those of the inducible, fibroblast lineage-specific TβRII-knockout mice.14
Collectively, these results suggest that both FMOD and TGFβ signal transduction are critical to early dermal fibroblast function during fetal and adult wound repair. We previously described that FMOD deficiency markedly alters the spatiotemporal expression patterns and levels of TGFβ ligands and receptors during adult mouse wound healing.19,20 These findings indicate that FMOD is required for proper TGFβ expression and function during wound repair. In this report, we systematically investigated the interplay between FMOD and TGFβ1 for their roles in the regulation of fibroplasia during adult cutaneous wound repair in preclinical animal models among different species, including mouse, rat and pig.
Materials and methods
Study design
The objective of this study was to assess the effects of FMOD on adult dermal fibroblasts and its therapeutic potential for adult cutaneous wound healing. For all in vitro studies, we used adult rat dermal fibroblasts (RDFs) since dermal fibroblasts are the predominant cell type required for cutaneous wound repair. Primary closure wound models were used in this study to simulate post-surgical wounds, which occur in 55 million elective operations and 25 million traumatic injury operations annually.2 Management of the resulting unwanted scarring requires approximately $3 billion each year.2 To begin, we used mouse and rat cutaneous wounds to test the efficacy of FMOD. Rodent animals were randomly assigned to each experimental group, and the sample size was determined based on previous studies.19–21 Rodents are loose-skinned animals, and as such, their skin can slide and retract over the subcutaneous fascia to produce a large gap initially.22 On the contrary, the pig and human skin is firmly attached to the underlying structure.23,24 Accordingly, a porcine model was then chosen for clinical relevance.23,24 Porcine wounds were randomly treated with phosphate-buffered saline (PBS) control or FMOD among different pigs. Initial porcine wound numbers were determined using power analysis to give α=0.05 and power=0.8, based on preliminary data. All animal surgeries were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals set forth under the institutionally approved protocols provided by the Chancellor’s Animal Research Committee at UCLA (protocol number: 2000-058 and 2008-016). Sutures were removed after stable wound closure based on our preliminary observations (1- (for rodent) or 2 weeks (for pig) post-injury). Early termination of animal experiments was based on either significant morbidity for animals or a loss of body weight more than 10%, in accordance with the approved protocols provided by the Chancellor’s Animal Research Committee at UCLA, although no animals were sacrificed for early termination during this study. The number of experimental replicates is defined in each figure legend. All outliers were included in the analysis, and no data were excluded. Investigators were blinded to treatment groups in all subjective in vivo measurements.
FMOD production
cDNA of a human FMOD transcript (Genbank assessor number: NM_002023) was subcloned into a commercially available vector pSecTag2A (Life Technology, Grand Island, NY, USA) with C-terminal His-tag, and transfected into CHO-K1 cells (ATCC, Manassas, VA, USA).19 After establishing a stable expression clone, the FMOD was produced and purified by a contract research organization, GenScript (Piscataway, NJ, USA). Briefly, a stable human recombinant FMOD-expressing CHO-K1 cell line was cultured in 1 l serum-free Freestyle CHO Expression Medium (Thermo Fisher Scientific, Canoga Park, CA, USA) at 37 °C with 5% CO2 in an Erlenmeyer flask. Cell culture supernatant was harvested on day 10 for purification with HiTrap IMAC HP, 1-ml column (GE Healthcare, Uppsala, Sweden). The fractions from a 100 mm imidazole elution were collected and dialyzed against 20 mm PBS, pH 7.4. After that, the sample with low conductivity was loaded onto HiTrapQ HP 1-ml column (GE Healthcare) for further purification. FMOD was then purified under non-reducing conditions, dialyzed again,25 and then subjected to lyophilization. The purity of the FMOD product is 85%. FMOD is reconstituted in PBS, followed by sterilization through a 0.22-μm filter (Thermo Fisher Scientific) before usage.
Adult rat skin wound model
Adult male Sprague-Dawley (SD) rats (weighing ~300 g) were anesthetized, and the dorsal skin was sterilely prepared. Six full-thickness, 10 mm×3 mm skin ellipses, with the underlying panniculus carnosus muscles, were excised on the dorsum of each animal. Each open wound edge was injected with 25 μl PBS, or 25 μl 0.4 or 2.0 mg ml−1 FMOD in PBS (25 μl×2 edges=50 μl total/wound). For the inhibitor-FMOD combination treatment groups, SMAD3-specific inhibitors (described below) were used with 2.0 mg ml−1 FMOD. Wounds were then marked with permanent dye and closed primarily with 4-0 Nylon using two simple interrupted sutures consistently placed at one-third intervals in each 10-mm length wound. All wounds were separated by at least 2 cm to minimize adjacent wound effects. Sutures were removed 1 week after injury, and wounds were collected 2 weeks after injury. Skin tissues from identical locations of unwounded animals were collected as controls. Wounds were harvested by excising a 4 mm×2 mm full-thickness skin strip, which was divided in two along its short axis.
Adult mouse skin wound model
Three-month old male 129/sv wild-type (WT) and Fmod-null (Fmod−/−) mice were anesthetized, and the dorsal skin was sterilely prepared. Four full-thickness, 10 mm×3 mm skin ellipses, with the underlying panniculus carnosus muscles, were excised on each mouse. Each open wound edge was injected with 25 μl PBS, 25 μl 0.4 mg ml−1 FMOD in PBS, or left untreated (25 μl×2 edges=50 μl total per wound). Wounds were then primarily closed with 5-0 Nylon using two simple interrupted sutures consistently placed at one-third intervals in each 10-mm length wound. All wounds were separated by at least 2 cm to minimize adjacent wound effects. Sutures were removed day 7 post-injury, and wounds were harvested 14 days post-injury (9 separate animals for each genotype; N=18 wounds per condition).19,26
Porcine wound healing model
Primary closure wound porcine models were used in this study, as described previously.24 Briefly, 20-kg female Yorkshire pigs (S&S Farms, Ramona, CA, USA) were sedated with Telazol (Tiletamine and Zolazepam, Fort Dodge Animal Heath, Fort Dodge, IA, USA), a 5 mg kg−1 intramuscular injection. The pigs were then endotracheally intubated, and maintained under a surgical plane of anesthesia with isoflurane at 0.5–2.5% in room air. The flank and back hair was clipped, and the skin was sterilely prepared with three alternating scrubs of povidone iodine solution and alcohol. Since porcine skin is significantly thinker than rodent skin, full-thickness wounds, down to the fascia, were created with a #15 surgical blade by excising a 1.5 cm×0.5 cm ellipse of skin with its long axis running perpendicular to the lines of minimal tension. All wounds were separated by at least 2 cm to minimize adjacent wound effects. Each open wound edge was injected with 100 μl PBS or 2.0 mg ml−1 FMOD in PBS (100 μl×2 edges=200 μl total/wound). Wounds were then marked with permanent dye, and primarily closed with 3-0 Nylon mattress sutures. 24 h later, an additional 50 μl PBS or FMOD was injected into each edge of the wound. A total of 24 wounds were created in each animal. Sutures were removed 2 weeks post-injury, and wounds were harvested at either 4 or 8 weeks post-injury (N=6 wounds from 3 Yorkshire pigs). Unwounded skin tissues, distant from the wounds of each animal, were collected as control samples. Tissues were bisected centrally between the sutures and perpendicular to the long axis of each wound.
Visual appearance evaluation of scarring
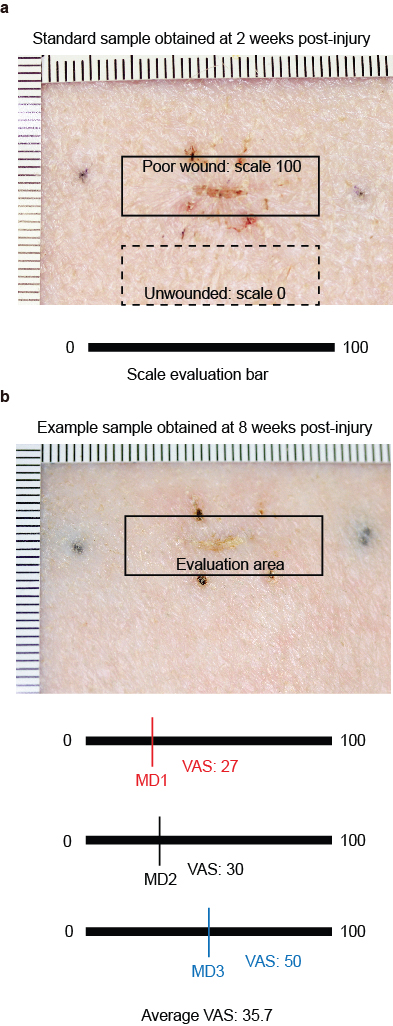
A gross visual assessment of the scar was performed in a randomized, double-blind fashion with an adaptation of Visual Analogue Score (VAS), as described previously.27 Briefly, all scar images were taken by a highly sensitive digital camera (DSLR DS126181, Canon, Tokyo, Japan) and individually presented to 3 experienced medical doctor (MD) assessors for a maximum of 20 s. The assessors placed a mark on a horizontal, 100-mm line to represent the scar quality, with 0 indicating unwounded skin and 100 indicating a poor scar (which represents a raised, hyperpigmented, or red scar with less acceptability and observer comfort; Supplementary Figure 1). When the individual images were displayed, the assessors confirmed and documented a VAS score independently.27
Tensile strength measurement
Tensile strength was determined using an Instron 5565 Universal Testing Machine (Instron, High Wycombe, UK). Using pneumatic grips to avoid specimen slippage, an exact 4 cm×1 cm full-thickness rat or pig skin strip was obtained by meticulous dissection, with the sample being obtained by precisely bisecting the wound. A 1-cm square area of skin was clamped on either side of the wound. The load to failure (breaking strength), measured in Newtons (N), was recorded.
Histological and immunohistological staining
After fixation in 10% neutral buffered formalin (Thermo Fisher Scientific) for 24 h, skin samples were dehydrated, paraffin-embedded, and sectioned at 5-μm increments for either hematoxylin and eosin (H&E) or Masson’s trichrome staining, and 10-μm increments for Picrosirius red (PSR) staining. To ensure a more precise quantification, wounds were bisected centrally, and the total scar area was normalized to varying dermal thickness using the Scar Index, as previously described.19,26 Among these staining methods, PSR coupled polarized light microscopy (PLM) distinguishes clear boundaries between scar tissue and unwounded normal dermis (Supplementary Figure 2). Immunohistochemical staining was performed and analyzed, as previously described.19,20 The primary antibodies used are listed in Supplementary Table 1.
Confocal laser scanning microscopy
Following PSR staining, the dermal collagen deposition pattern of the upper dermis was evaluated by confocal microscopy on a Carl Zeiss LSM 510 META laser scanning confocal microscope (Carl Zeiss AG, Oberkochen, Germany), using previously published methods.17,19,26 Collagen organization architecture was assessed by Fractal dimension (FD) and Lacunarity (L) analyses, as previous described,26 since FD and L analyses are more sensitive than traditional methods, such as polarized light microscopy (PLM), X-ray diffraction, laser scattering, and Fourier transform analysis.26
RT2 profiler PCR array analysis of rat wounds
To minimize the contamination of the surrounding unwounded tissue, wound tissues were collected for RNA isolation by manual microdissection from paraffin-embedded tissue sections.28 Total RNA was isolated using RNeasy FFPE Kit (Qiagen, Hilden, Germany). 2.5 μg RNA isolated from the wounds was injected into RT2 First Stand Kit (Qiagen) for reverse transcription. Afterward, real-time PCR was performed in a 96-well rat wound healing RT2 PCR Array (PARN-121A, Qiagen) on a 7300 Real-Time PCR system (Thermo Fisher Scientific), according to the manufacturer’s protocol. For each sample, three arrays were tested. Data analysis was achieved using the manufacturer’s online services.
RDF isolation and maintenance
Adult RDFs were isolated from the dorsal skin of adult male SD rats and maintained in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific) supplemented with penicillin/streptomycin (1% v/v; Thermo Fisher Scientific) and fetal bovine serum (10% v/v; Thermo Fisher Scientific) as previously described.29 RDFs were tested negatively for mycoplasma contamination by the Universal Mycoplasma Detection Kit (ATCC, Manassas, VA, USA; Supplementary Figure 3). RDFs at passage 3 were used for all in vitro tests.
Cell proliferation assay
RDFs were seeded at a density of 2×103 cells per well on 96-well cell culture plates. Since serum is a known source of multiple growth factors, including TGFβ1, serum starvation was carried out to standardize the cell cycle phase and eliminate the influence of growth factor persistence to accurately assess the bioactivities of FMOD. After 16-h serum starvation, cells were treated with 100 μl fresh medium containing FMOD and/or TGFβ1 (Sigma-Aldrich, St Louis, MO, USA). After 48-h incubation, cell proliferation was measured by the Vybrant MTT Cell Proliferation Assay Kit (Thermo Fisher Scientific).
Cell migration assay
For wound scratch assays, RDFs were grown in 6-well tissue culture plates until confluence. After 16-h serum starvation, 1-mm width ‘scratch’ wounds were created by scraping confluent cell monolayers using a sterile pipette. To accurately assess cellular migration, each scratch was examined immediately after scraping under microscope. Only the scratches with a width of 1±0.1 mm were used for further investigation. The wounded monolayer was washed three times with PBS to remove dead cells prior to 20-h incubation in treatment medium: DMEM±2.5 ng ml−1 (100 pm) TGFβ1±12 μg ml−1 (200 nm) FMOD. Concentrations of TGFβ1 and FMOD used were based on previous report.19 Photographs taken immediately after scraping and 20 h later documented cell migration. Migration was quantified by measuring the average wound gaps between the wound edges before and after treatment, using the commercially available software Image-Pro Plus 6.0 (Media Cybernetics, Rockville, MD, USA).
Cell invasion assay
Cell invasion assays were performed in 24-well tissue culture plates using HTS Fluoroblok inserts with 8 μm pore size Fluorescence Blocking PET track-etched membranes (BD Biosciences, Franklin Lakes, NJ, USA). The upper surface of the inserts were coated with 200 μl collagen matrices, rinsed with DMEM, and placed into 24-well tissue culture plates containing 500 μl treatment medium (as described above). After 16-h serum starvation, 2×104 RDFs in 100 μl treatment medium were added to each insert chamber and allowed to invade toward the underside of the membrane for 20 h. Non-invading cells were removed by wiping the upper side of the membrane with a cotton swab. Cells that invaded were fixed and stained with 0.4 mg ml−1 4′,6-diamino-2-phenlindole (DAPI; Sigma-Aldrich) before counting.19
Immunocytochemical staining
RDFs were seeded at a density of 1×104 cells per well on 4-well Lab-TEK II chamber slides (Thermo Fisher Scientific). After 16 h serum starvation, cells were incubated with treatment medium (as described above) for 48 h before immunostaining. Images were obtained using a Leica TCS-SP2-AOBS confocal microscope (Leica Microsystems, Buffalo Grove, IL, USA). The primary antibodies used are listed in Supplementary Table 1.
Flow cytometry
RDFs were harvested 48 h after treatment and filtered through a 40-μm strainer. Cell counts and viability were determined using trypan blue exclusion (Sigma-Aldrich) on a hemocytometer. Single cell suspensions were fixed with 4% formaldehyde, permeabilized with 0.1% Triton X-100 for 15 min, and then blocked with 3% BSA for 30 min before being incubated with α-smooth muscle actin (α-SMA) antibody (Abcam, Cambridge, UK) for 30 min at room temperature. Samples were assayed immediately by LSRFortessa Cytometer (BD Biosciences), and FCS Express 4 software (De Novo Software, Glendale, CA, USA).
Collagen-based cell contraction assay
After 16-h serum starvation, 2×106 RDFs per well were used to establish a 3D collagen matrix in a 24-well plate following the manufacturer’s instructions (Cell Contraction Assay, Cell Biolabs, San Diego, CA, USA). After collagen polymerization, 1.0 ml of treatment medium (as described above) was added atop each collagen gel lattice. Cultures were incubated for 2 days for stress development. Afterwards, collagen gels were gently released from the sides of the culture dishes with a sterile spatula. The collagen gel size changes were measured at 0, 20, 40 and 60 min.
Western blotting
RDFs were seeded at a density of 1×105 cells per dish on 10-cm tissue culture dishes. After 16 h serum starvation, cells were incubated with treatment medium (as described above). Cells were lysed using RIPA buffer (Thermo Fisher Scientific) supplied with Halt Protease and Phosphates Inhibitor Cocktail (Thermo Fisher Scientific) for Western blotting. The antibodies used in this study are also summarized in Supplementary Table 1. Bands on Western blots were quantified using QuantityOne (BioRad Laboratories, Hercules, CA, USA), and values were expressed in relative densitometry units.
Cellular gene expression assay
RDFs were seeded at a density of 1×105 cells per dish on 10-cm tissue culture dishes. After 16 h serum starvation, cells were incubated with treatment medium (as described above). RNAs were extracted using RNeasy Mini Kit (Qiagen) with DNase (Qiagen) treatment, followed by reverse transcription with SuperScript III First-Strand Synthesis System for RT-PCR (Thermo Fisher Scientific). qRT-PCR was performed on a 7300 Real-Time PCR system (Thermo Fisher Scientific) according to the manufacturer’s protocol.30 The primers and probes used in this study are listed in Supplementary Table 2. For each assay, at least three separate sets of qRT-PCR were performed from a different cDNA template. Concomitant glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was performed in separate tubes, as a house-keeping standard since the ΔCT levels of Gapdh did not differ significantly among the treatment conditions (data not shown). Relative gene expression was analyzed with the ΔCT method.19,20,30
Luciferase assays
RDFs were seeded at a density of 1×105 cells per dish on 10-cm tissue culture dishes. After 16 h serum starvation, cells were transiently transfected with the A3-Luc system (that is, CAGA-box reporter; specific for SMAD3 signal transduction)31 or the Cignal AP-1 Reporter (luc) Kit (Qiagen; for AP-1 signal transduction). Luciferase activity was quantified at 2 and 24 h using the Dual Luciferase Assay (Promega, Madison, WI, USA). Values were normalized by Renilla luciferase activity expressed from pRL-TK (Promega).
ELISA
ELISA of matrix metalloproteinase (MMP)2 was performed using the commercially available MMP-2 Quantikine ELISA Assay Kit (R&D Systems, Minneapolis, MN, USA). Briefly, 50 μl of provided diluent solution and 50 μl of conditioned cell culture supernatant were added to each well, and incubated at room temperature for 2 h. Following incubation, the wells were washed four times with 400 μl of provided wash buffer. Subsequently, 200 μl of MMP2 conjugate was added to each well and incubated at room temperature for 2 h before being washed with 200 μl of provided wash buffer four times, and incubated with provided 200 μl of substrate solution at room temperature for 30 min. Afterward, 50 μl of stop solution was added to each well. MMP2 activity was measured using microplate reader (BioTek Instruments, Winooski, VT, USA) at 450 nm, subtracted by readings at 540 nm. Mmp2 concentrations were then calculated by creating a standard curve. All experiments were performed in triplicate.
Inhibitors
SMAD3-specific inhibitors naringenin (4′,5,7-trihydroxyflavanone, which selectively inhibits the expression of SMAD3, but not SMAD2, SMAD4 or SMAD7;32 Sigma-Aldrich; 50 μM) and SIS3 ((2E)-1-(6,7-Dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)-3-(1-methyl-2-phenyl-1H-pyrrolo(2,3–b]pyridin-3-yl)-propenone hydrochloride, CAS 1009104-85-1, which attenuates phosphorylation of SMAD3, but not SMAD2, MAPK, ERK, or PI3-K;33 Calbiochem, San Diego, CA, USA; 5 μM) were used to block SMAD3 signal transduction.
Statistical analysis
All statistical analyses were conducted in consultation with the UCLA Statistical Biomathematical Consulting Clinic. Statistical analysis was computed by OriginPro 8 (Originlab, Northampton, MA, USA). Data were generally presented as mean±s.d., and compared by one-way analysis of variance (ANOVA) and two-sample t-tests. Mann–Whitney and Kruskal–Wallis ANOVA tests were used for non-parametric data. P<0.05 was considered statistical significance.
Data availability
All data generated or analyzed during this study are included in this article (and its Supplementary Information files).
Results
FMOD significantly optimizes adult cutaneous wound healing
Previously, we reported that wounds in the adult Fmod−/− mouse healed with increased scar formation, delayed wound closure, and reduced angiogenesis, which could be partially rescued by exogenous FMOD administration.19,21 Additionally, FMOD administration markedly augmented cutaneous wound vascularity21 and notably decreased scar size in adult WT mice (Supplementary Figure 4). Using an adult rat wound healing model, we demonstrated that exogenous FMOD significantly reduced scar size when compared with PBS control samples as evidenced by PSR staining-coupled PLM (Figure 1a and b). Meanwhile, FMOD-treated adult rat wounds also demonstrated and orderly collagen architecture as documented by PSR staining-coupled confocal laser scanning microscopy (CLSM) (Figure 1a), which was accompanied by increased tensile strength (Figure 1c).
Figure 1.

FMOD reduced scar formation and increased wound tensile strength in adult rat cutaneous wounds. Histology (H&E staining), picrosirius red (PSR) staining-coupled polarized light microscopy (PLM), PSR staining-coupled confocal laser scanning microscopy (CLSM) (a; insert, unwounded skin), and quantitative analyses (b) demonstrated that FMOD significantly reduced the scar size of adult rat skin wounds at day 14 post-injury, while increasing tensile strength in a dose-dependent manner (c). Moreover, SMAD3-specfic inhibitors naringenin and SIS3 eliminated the effects of FMOD on reducing scar formation and increasing wound tensile strength. Scar areas are outlined by blue (H&E) or yellow (PSR-PLM) lines. Scale bar=500 μm (blue) or 25 μm (red). Mann–Whitney test was used for statistical analysis, while mean values are presented. Wound numbers (WN) and animal numbers (AN) are indicated in the figure. *P<0.05, **P<0.01.
Among mammalian skin, porcine skin most closely approximates human skin in anatomic structure, mechanical properties, and wound healing dynamics.23,24 In order to rigorously test the wound healing effects of FMOD treatment in a model that more closely approximates human skin, we excised and primarily closed skin wounds in adult female Yorkshire pigs. FMOD injection at the time of wound closure significantly improved the gross appearance of the pig wounds when assessed at 8 weeks post-injury (Figure 2a and b). Using PSR-PLM, which distinguishes clear boundaries between scar tissue and unwounded dermis, FMOD-treated Yorkshire pig wounds exhibited significantly reduced scar size compared with controls when measured at 4 and 8 weeks post-injury (Figure 2c and d). The dermal collagen architecture was also assessed by PSR-CLSM (Figure 2c), and then quantified FD and L analyses.26 FD provides a measure of how completely an object fills space, which quantifies an object by shape, regularity, lack of smoothness, size, and number of self-similarities, that is, invariance regardless of scale.34 In general, a higher FD value indicates a uniform distribution.34 For example, wounds have lower mean FD values when compared with unwounded skin, corresponding to the uneven and disorganized collagen distribution patterns (Figure 2e). In contrast, L permits an analysis of density, packing, or dispersion through scales.34 Objects with lower L values correspond to a finer texture, while objects with higher L values are more spatially unorganized.35 In this study, we noted that FMOD treatment significantly increased the mean FD values of the wounds, and even approximated FD values of unwounded skin when quantified at 8 weeks post-injury (Figure 2c and e). FMOD treatment also decreased the mean L values to a similar level of the L values of unwounded skin at 8 weeks post-injury, which indicates a notably more organized distribution of uniformly sized collagen fibers in the FMOD-treated wounds (Figure 2c and f). Importantly, the reduced scar size in FMOD-treated wounds did not occur at the expense of diminished tensile strength. In fact, tensile strength was significantly increased in all FMOD-treated groups, relative to controls, at 4 and 8 weeks post-injury (Figure 2g). Overall, these data demonstrate that FMOD application reduced scar size, increased tensile strength, and improved dermal collagen architecture organization in preclinical animal models.
Figure 2.

FMOD reduced scar formation and increased wound tensile strength in adult Yorkshire pig cutaneous wounds. Representative photographs of the gross appearance of adult female Yorkshire pig wounds at 4 and 8 weeks post-injury are shown (a). Compared with PBS-treated wounds, 2.0 mg ml−1 FMOD-treated wounds exhibited improved gross appearance (as evidenced by a decreased Visual Analogue Score; b). Histology of adult female Yorkshire pig wounds at 4 and 8 weeks post-injury are shown (c). Scars are outlined by blue or yellow lines, while dermis layers are outlined by dashed magenta lines in PSR-PLM photographs. Quantitative analyses demonstrated that FMOD significantly reduced scar size (scar index; d). Collagen architecture was evaluated by Fractal dimension (e) and Lacunarity (f) analyses. FMOD treatment also significantly increased wound tensile strength in comparison with PBS control (g). UW, unwounded skin control. Scale bar=500 μm (red), or 25 μm (green). Mann–Whitney test was used for statistical analysis, while mean values are presented. N=24 (b, 4 weeks post-injury), 12 (b, 8 weeks post-injury), or 6 (d-g) wounds from 3 pigs per time point. *P<0.05.
FMOD markedly alters gene expression during wound healing
To further understand the molecular basis underlying FMOD’s wound healing enhancing effects, a RT2 profiler PCR array was utilized to screen for FMOD-responsive genes during the adult rat wound healing period (Supplementary Figure 5). We previously observed an inverse correlation between Fmod and Tgfβ1 levels during fetal wound healing in rodents. High Fmod levels correlated with decreased Tgfβ1 expression and scarless repair, while low Fmod levels correlated with increased Tgfβ1 expression and scar formation.17 Corresponding to our fetal rodent data, FMOD application to adult rats reduced early (days 1, 3, and 7) Tgfβ1 transcripts (Figure 3a). Not surprisingly, FMOD treatment also decreased expression of TGFβ1-responsive genes that encode fibrotic collagen, specifically collagen (Col)1α1 and Col1α2 (days 3 and 7; Figure 3b and c), and Col3α1 (day 3; Figure 3d). These collagen genes contribute to excessive ECM production and scar formation at early time points post-injury, and are subsequently upregulated during the remodeling phase of wound healing (day 14). Somewhat unexpectedly, FMOD application elevated another subset of TGFβ1-responsive genes implicated in ECM remodeling, fibroblast migration/invasion, and myofibroblast differentiation/contraction;4,9,36 all of which are events that occur before wound closure, typically by post-injury day 5. These genes include matrix metalloproteinase 2 (Mmp2; Figure 3e), Ctgf (Figure 3f), and Actα2 (encoding α-Sma; Figure 3g). After complete wound re-epithelialization, these genes were noticeably downregulated in the FMOD-treated groups. Consequently, FMOD application appears to be associated with both the early upregulation of TGFβ1-responsive genes that promote cellular migration and myofibroblastic conversion, and the early downregulation of TGFβ1-responsive genes that promote fibrotic ECM deposition and scarring (Figure 3h).
Figure 3.

FMOD administration altered the expression of TGFβ1-related genes during adult rat cutaneous wound healing. Expression of Tgfβ1 (a), Col1α1 (b), Col1α2 (c), Col3α1 (d), Mmp2 (e), Ctgf (f), and Actα2 (g) was compared between PBS (control) and 2.0 mg ml−1 FMOD-treated adult rat cutaneous wounds. Clustergram plot further indicated that FMOD diversely modulated the expression of TGFβ1-related genes during adult rat cutaneous wound healing (h). A two-sample t-test was used for statistical analysis. Data were normalized to unwounded skin (dashed lines), and are shown as mean±the standard deviation. N=3 wounds from 3 animals pooled for each sample (9 wounds from 9 animals in total). *P<0.05.
FMOD modulates the behavior of adult dermal fibroblasts
As fibroblasts are the predominant cell type required for dermal repair, we tested FMOD’s effects on adult RDFs. First, up to 200 nm FMOD alone did not affect RDF proliferation; however, FMOD significantly stimulated RDF proliferation in the presence of TGFβ1 (Supplementary Figure 6). Second, while FMOD alone did not increase cell motility, FMOD+TGFβ1 significantly promoted RDF migration and invasion (Figure 4a and b) and markedly enhanced myofibroblast differentiation and contractility, as shown by α-Sma staining (Figure 4c and d). Quantitative analysis of 3D collagen gel contraction studies confirmed an increased contractility of FMOD+TGFβ1-induced myofibroblasts, relative to contractility induced by TGFβ1 alone (Figure 4e). In contrast, FMOD alone temporarily decreased RDF Col3α1 expression in the absence of exogenous TGFβ1 administration (Supplementary Figure 7a) and significantly inhibited the ability of TGFβ1 to stimulate RDF Col1α1 and Col1α2 transcription (Supplementary Figures 7b and c). Additionally, FMOD temporarily enhanced RDF Mmp2 expression, regardless the existence of TGFβ1 (Supplementary Figures 7d and e). Overall, FMOD-treated adult RDFs exhibited increased migration, and myofibroblast differentiation and contractility, with less fibrotic ECM production. These findings paralleled the reduced scar formation and accelerated wound closure seen in FMOD-treated adult rat wounds in vivo (Figure 1).
Figure 4.

FMOD enhanced TGFβ1-induced rat dermal fibroblast (RDF) migration and invasion, and myofibroblast conversion and contraction. FMOD+TGFβ1 significantly promoted TGFβ1-mediated RDF migration (a) and invasion through collagen matrices (b). Flow cytometry tests revealed that FMOD significantly enhanced TGFβ1-induced rat dermal fibroblast-myofibroblast conversion (c), which corresponds with immunofluorescent staining (d). Interestingly, after 48 h treatment, α-Sma+ myofibroblasts formed stress fibers (yellow arrows) in the FMOD+TGFβ1 group. Additionally, FMOD+TGFβ1 significantly stimulated TGFβ1-mediated myofibroblast contraction in collagen gel (e). Dosages: TGFβ1 (100 pm) and FMOD (200 nM). Scale bar=100 μm (a, b), or 50 μm (d). Two-sample t-test was used for statistical analysis, and data are shown as mean±the standard deviation N=4 (a, b), 3 (c), or 6 (e). *P<0.05, **P<0.01.
Consistent with our in vivo findings (Figure 3a) and previous studies,17,19,20 FMOD strongly suppressed Tgfβ1 expression and auto-induction in adult RDFs (Figure 5a). FMOD also significantly repressed TGFβ1-induced expression of plasminogen activator inhibitor (Pai)1, a well-known pro-fibrotic molecule that contributes to excessive accumulation of collagen,37 in adult RDFs (Figure 5b). The downregulation of Tgfβ1 and Pai1 in FMOD-treated RDFs could be attributed to FMOD significantly decreasing the AP-1-mediated TGFβ1 signal transduction (Figure 5c), which has been reported to control TGFβ1 auto-induction and PAI1 expression.37,38 In contrast, FMOD treatment promoted rapid Smad2 phosphorylation/activation in adult RDFs (Figure 5d), which correlates with Mmp2 expression. Since SMAD2, but not SMAD3, has been reported to regulate MMP2 expression,39 these findings suggest that FMOD-induced phosphorylation of SMAD2 may be responsible for the transient upregulation of Mmp2. Notably, FMOD stimulation of Smad2 signaling occurred concomitantly with FMOD repression of AP-1 activation. Understanding that AP-1 promotes TGFβ1 auto-induction,38 while SMAD2 regulates MMP2 expression,39 may explain the seemingly paradoxical coupling of decreased TGFβ1 expression while upregulating TGFβ1 downstream target MMP2 in earlier human dermal fibroblasts studies after adenovirus-mediated FMOD overexpression.40
Figure 5.

FMOD delicately orchestrated Tgfβ1 signal transduction in adult RDFs. Western blotting and qRT-PCR revealed that FMOD reduced Tgfβ1 expression and Tgfβ1 auto-induction (a), and inhibited TGFβ1-dependent Pai1 transcription in adult RDFs (b). Using an AP-1-luciferase reporter system, we further revealed that FMOD significantly downregulated AP-1-mediated non-canonical TGFβ1 signal transduction (which predominately regulates TGFβ1 auto-induction and Pai1 expression) up to 24 h post-treatment (c). Meanwhile, FMOD stimulated adult RDF Smad2 phosphorylation/activation when measured over a short amount of time (2 h) in the presence or absence of TGFβ1, but did not have an obvious effect on Smad2 expression (d). Although FMOD alone downregulated initial Smad3 expression up to 24 h post-treatment, when combined with TGFβ1, FMOD did not affect Smad3 expression in the 48-h experiment period, but it significantly prolonged TGFβ1-responsive Smad3 phosphorylation/activation (e), which was confirmed using a CAGA-box (specific for SMAD3 signal transduction) Luciferase reporter system (f). Dosages: TGFβ1 (100 pM) and FMOD (200 nM). A two-sample t-test was used for statistical analysis. Data were normalized to untreated RDFs at time 0 (dashed lines) and shown as mean±the standard deviation. N=3 (a, b, d, e) or 5 (c, f). *P<0.05; a blue star indicates the significance in comparison with FMOD administration, and a magenta star indicates the significance in comparison with TGFβ1 administration.
With respect to Smad3, FMOD alone slightly inhibited its expression in RDFs, while FMOD+TGFβ1 combination significantly prolonged Smad3 phosphorylation/activation without altering its gene or protein expression (Figure 5e). Using a luciferase-reporter system specific for SMAD3 signal transduction (that is, CAGA-box reporter), we confirmed that FMOD prolonged TGFβ1-induced Smad3 signaling in RDFs in vitro (Figure 5f). Similarly, increased phosphorylated SMAD3 was detected in FMOD-treated adult rat wounds at day 3 post-injury (Supplementary Figure 8). FMOD-mediated increase in Smad3 signal transduction in FMOD+TGFβ1-treated RDFs could explain the elevated expression of Smad3-dependent molecules Ctgf and α-Sma (Figure 6), which are known to significantly augment migration and contraction.9,36 Furthermore, two specific inhibitors of SMAD3-signal transduction, naringenin and SIS3, were used to attenuate Smad3-signaling in adult RDFs. Both inhibitors diminished the positive effects of FMOD on Ctgf and α-Sma expression, migration, invasion, and contraction of RDFs (Supplementary Figures 9 and 10). Importantly, co-administration of specific inhibitors of SMAD3-signal transduction with FMOD completely eliminates the ability of FMOD to reduce scar size and increase tensile strength in adult rat wounds (Figure 1). Comprehensively, these results confirmed that FMOD’s pro-migratory and pro-contractility effects that reduce scar size and increase scar strength require intact TGFβ1-responsive SMAD3 signaling activation.
Figure 6.

FMOD enhanced TGFβ1-responsive Ctgf and α-Sma expression in adult RDFs. In the presence of TGFβ1, FMOD significantly increased expression of Ctgf (a) and a-Sma (b) of RDFs. Dosages: TGFβ1 (100 pM) and FMOD (200 nm). A two-sample t-test was used for statistical analysis. Data were normalized to untreated RDFs at time 0 (dashed lines) and shown as mean±the standard deviation. N=3. *P<0.05; a blue star indicates the significance in comparison with FMOD administration, and a magenta star indicates the significance in comparison with TGFβ1 administration.
Discussion
In this study, we demonstrate that intradermal recombinant human FMOD injection significantly reduced scar size by 50–70% in adult animal wounds of loose-skin rodent and fixed-skin porcine models.41 Importantly, FMOD-mediated improvements in wound healing are accompanied by a marked increase in wound tensile strength, which represents a potential significant improvement over the current standards of care. For example, corticosteroids, used in the current standard of care, have been found to impair wound healing and decrease scar strength.42,43 We believe our finding provides the first successful case demonstrating feasibility in shifting the paradigm of reducing scar formation from multifarious therapies involving cytokine-growth factor-steroid cocktails and laser treatments to a simple, biological approach using native ECM molecule(s).
Our current study elucidates the critical function of FMOD in regulating wound healing. Much to our surprise, FMOD selectively augments early pro-migratory and pro-contractile canonical TGFβ1 signaling to decrease scar size and increase wound tissue tensile strength, while simultaneously repressing TGFβ-stimulated fibrotic gene expression to decrease scar tissue formation (Figure 7 and Supplementary Video 1). For the first time, we can predictably uncouple the beneficial pro-migration/contraction effects of TGFβ1 that promote faster and stronger wound closure from the pro-fibrotic effects of TGFβ1 that promote excessive scarring. Future studies will further examine how FMOD delicately orchestrates TGFβ signal transduction, such as uncoupling the canonical and non-canonical TGFβ1 signaling pathways.
Figure 7.

FMOD delicately orchestrated TGFβ1-signaling during adult cutaneous wound healing. Rather than indiscriminately blocking TGFβ1 signals, FMOD selectively regulated TGFβ1 bioactivities in a signal transduction pathway-dependent manner.
Importantly, we also demonstrate the critical role that the FMOD-TGFβ1-CTGF nexus plays in rapid cell migration and myofibroblast function. TGFβ1-CTGF signal transduction, which has been thought of as a culprit contributing to excessive scar formation, can now be considered as part of the solution when in the presence of FMOD. This paradigm-shifting concept fundamentally challenges the current drug development focus on ‘low TGFβ1/CTGF’ and/or ‘high TGFβ3’ targets for minimizing human cutaneous scarring. Indeed, our data show that increased TGFβ1-mediated CTGF signaling in the early period after skin injury is actually beneficial for: (1) rapid fibroblast migration (2), rapid fibroblast to myofibroblast conversion (3), increased myofibroblast contractility to promote wound closure, and (4) increased wound tensile strength. Publically available data appear to validate our hypothesis that indiscriminate blockade of TGFβ1 and its downstream mediator CTGF, especially at early stage of wound healing, is not optimal for human scar reduction. TGFβ1-neutralizing antibodies and peptides have failed to reduce scar formation in preclinical animal studies.7,8 Moreover, clinical trials appear to be halted for PF-0673871/EXC001, an antisense oligonucleotide product targeting CTGF,10 while recent Phase 2 clinical trial results from RXI Pharmaceuticals show that application of RXI-109—another RNA interference (RNAi) product targeting CTGF—starting 2 weeks post-surgery leads to better scar reduction scores than an application immediately after injury.11 Interestingly, our present work that underscores the importance of early fibroblast migration, and myofibroblast conversion and contraction, for optimal repair—and FMOD’s role in these processes—may also explain the lack of efficacy observed in Phase 3 clinical trials for Juvista (Avotermin, recombinant human TGFβ3 by Renovo, Manchester, UK).44 Although TGFβ3 has been widely described as a potent anti-fibrotic molecule4 initially identified through scarless fetal wound healing studies, a lesser known effect of TGFβ3 is its well-documented inhibition of adult dermal fibroblast proliferation and migration.45,46 In agreement with this effect, we found TGFβ3 levels to be dramatically elevated in adult Fmod−/− mouse wounds that exhibited severely delayed fibroblast migration and granulation tissue formation, which ultimately healed with increased scar size.19 Moreover, we showed that TGFβ3 addition markedly inhibited fibroblast migration, and that FMOD could directly reverse this effect in vitro and in vivo to restore fibroblast migration and reduce scar size.19
Recent discoveries highlight how SLRP functions extend well beyond their originally described roles in ECM structural support.18,47 SLRPs are now widely recognized as key regulators of intracellular signal cascades that govern multiple processes, such as angiogenesis and cell fate determination.21,25,30,48,49 Our previous studies identified that FMOD alone can reprogram somatic cells to a multipotent state under a serum-free situation in vitro.25,30 We also found that FMOD could restore the fetal-type scarless healing with hair follicle regeneration in late-gestation fetal wounds, which normally heal but form a scar.16,17 In the current study, we observed that FMOD treatment results in adult fibroblasts and wounds bearing several striking similarities to the attributes of fetal fibroblasts and fetal wounds: first, like fetal wounds,15 FMOD treatment to adults wounds causes reduced and more transient TGFβ1 expression; second, like fetal wounds,50 FMOD treatment induces high levels of SMAD2 and SMAD3 phosphorylation, and low levels of several fibrosis-associated targets; third, like fetal fibroblasts,51 FMOD treatment results in a more migratory and contractile phenotype; fourth, like fetal fibroblasts, FMOD treatment exhibits higher TGFβ1-stimulated CTGF expression levels for increased myofibroblast differentiation and contraction;52 and lastly, much like fetal wounds,53 the markedly reduced Actα2 expression at days 7 and 14 post-injury (Figure 3g) in the FMOD-treated groups suggests that FMOD treatment results in more rapid myofibroblast clearance from the wound. Taken together, FMOD administration in adult wound models elicits a similar phenotype to fetal wounds at the molecular, cellular, and gross morphological levels. This unique response of living systems to FMOD, an extracellular SLRP molecule involved in collagen fibrillogenesis,18 has not been previously described. Considering that the expression of FMOD significantly decreased in senescent fibroblasts,54,55 the fetal-like phenotypes induced by FMOD in adult dermal fibroblasts in vitro and adult cutaneous wounds in vivo, as demonstrated in the current study, aptly qualifies FMOD as a matricellular regulator with diverse functions beyond its structural support roles, and may open a new avenue to develop regenerative medicines that ‘turn back the clock’ for more youthful living.
However, administration of FMOD was not able to fully stimulate a fetal-type scarless healing with hair follicle regeneration or significantly increase the type III collagen content in adult wound models, which incorporate the complexity of the wound healing process and illustrates that other molecules, in addition to FMOD and TGFβ, are also important for scarring. For instance, recent human studies suggest that single-nucleotide polymorphisms in several loci or genes are also associated with hypertrophic scar and keloid formation in different populations.56–59 Therefore, further investigation is required to entirely understand the nature of wound healing.
In summary, we systematically demonstrated that FMOD administration potently reduced scar size, improved scar appearance, and increased scar strength in both an adult rodent model and a porcine model to simulate human wound healing. With pig skin being recognized by the US FDA as the closest animal equivalent to human skin,23,24 there is high translational potential and applicability of our current findings to human patients. To examine whether FMOD’s significant effects on cell migration and contraction can also translate to new therapies for clinical conditions associated with inadequate fibroplasia, such as poor healing from illness, medications or chronic wounds, the preclinical efficacy of FMOD should be validated in splinting excisional animal models.22,60 In addition, in order to apply FMOD clinically, Good Manufacturing Practice (GMP)-compliant high-purity FMOD production should be established, and the safety of FMOD should be fully assessed in advance of clinical trials.
Acknowledgments
Fmod−/− animals were kindly provided by Dr Ake Oldberg (Lund University, Sweden). We thank Miss Ching-Yun Hsu from the School of Dentistry, UCLA for her assistance with FMOD production, and Dr Matthew Schibler (California NanoSystems Institute Advanced Light Microscopy/Spectroscopy Shared Resource Facility, University of California Los Angeles, Los Angeles, CA, USA) for assisting on CLSM analysis. This study was supported by the Plastic Surgery Foundation (2013 National Endowment for Plastic Surgery 269698), NIH-NIAMS (R43AR064126, R44AR064126, and R43AR063558), and NIH-NIDCR (R44DE024692). Aaron James has a T32 training fellowship (5T32DE007296-14). CLSM was performed at the Center for NanoScience Institute Advanced Light Microscopy/Spectroscopy Shared Resource Facility at UCLA. Flow cytometry was conducted with the support of UCLA Broad Stem Cell Research Center Flow Cytometry Core Resource. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Supplementary Information accompanies the paper on the Signal Transduction and Targeted Therapy website (http://www.nature.com/sigtrans)
ZZ, KT and CS are inventors on fibromodulin-related patents assigned to UCLA. ZZ, KT and CS are founders of Scarless Laboratories Inc., which sublicenses fibromodulin-related patents from the UC Regents, who also hold equity in the company. ZZ and CS are also officers of Scarless Laboratories, Inc.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Research, B. Markets for Advanced Wound Management Technologies. BCC Research, Markets for Advanced Wound Management Technologies 2014.
- Sund B, Arrow AK. New Developments in Wound Care. Clinica reports, 2000. [Google Scholar]
- Gauglitz GG, Korting HC, Pavicic T, Ruzicka T, Jeschke MG. Hypertrophic scarring and keloids: pathomechanisms and current and emerging treatment strategies. Mol Med 2011; 17: 113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Kane S, Ferguson MW. Transforming growth factor βs and wound healing. Int J Biochem Cell Biol 1997; 29: 63–78. [DOI] [PubMed] [Google Scholar]
- Ikushima H, Miyazono K. TGF-beta signal transduction spreading to a wider field: a broad variety of mechanisms for context-dependent effects of TGF-beta. Cell Tissue Res 2012; 347: 37–49. [DOI] [PubMed] [Google Scholar]
- Bayat A, Bock O, Mrowietz U, Ollier WER, Ferguson MWJ. Genetic susceptibility to keloid disease and hypertrophic scarring: Transforming growth factor beta 1 common polymorphisms and plasma levels. Plastic Reconstruct Surg 2003; 111: 535–543. [DOI] [PubMed] [Google Scholar]
- Shah M, Foreman DM, Ferguson MW. Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. J Cell Sci 1995; 108: 985–1002. [DOI] [PubMed] [Google Scholar]
- San-Martin A, Dotor J, Martinez F, Hontanilla B. Effect of the inhibitor peptide of the transforming growth factor beta (p144(A (R))) in a new silicone pericapsular fibrotic model in pigs. Aesthet Plast Surg 2010; 34: 430–437. [DOI] [PubMed] [Google Scholar]
- Grotendorst GR. Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev 1997; 8: 171–179. [DOI] [PubMed] [Google Scholar]
- PF-06473871, EXC 001/Pfizer, Isis. Available at: http://sigma.larvol.com/product.php?e1=3966&tab=ott&sourcepg=TZP.
- Galiano R. Overivew of Phase 2 Clinical Trial Results of RXI-109 Treatment to Reduce the Formation of Hypertrophic Dermal Scars.
- Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA 1993; 90: 770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe MJ, Doetschman T, Greenhalgh DG. Delayed wound healing in immunodeficient TGF-beta 1 knockout mice. J Invest Dermatol 2000; 115: 3–11. [DOI] [PubMed] [Google Scholar]
- Denton CP, Khan K, Hoyles RK, Shiwen X, Leoni P, Chen Y et al. Inducible lineage-specific deletion of TbetaRII in fibroblasts defines a pivotal regulatory role during adult skin wound healing. J Invest Dermatol 2009; 129: 194–204. [DOI] [PubMed] [Google Scholar]
- Larson BJ, Longaker MT, Lorenz HP. Scarless fetal wound healing: a basic science review. Plast Reconstr Surg 2010; 126: 1172–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soo C, Hu FY, Zhang X, Wang Y, Beanes SR, Lorenz HP et al. Differential expression of fibromodulin, a transforming growth factor-beta modulator, in fetal skin development and scarless repair. Am J Pathol 2000; 157: 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Zhang X, Dang C, Beanes S, Chang GX, Chen Y et al. Fibromodulin is essential for fetal-type scarless cutanous wound healing. Am J Pathol 2016; 186: 2824–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iozzo RV, Goldoni S, Berendsen AD, Young MF. Small Leucine-Rich Proteoglycans: The Extracellular Matrix: an Overview, (ed. Mecham RP197–231 Springer: Berlin Heidelberg, 2011. [Google Scholar]
- Zheng Z, Nguyen C, Zhang X, Khorasani H, Wang JZ, Zara JN et al. Delayed wound closure in fibromodulin-deficient mice is associated with increased TGF-beta3 signaling. J Invest Dermatol 2011; 131: 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Lee KS, Zhang X, Nguyen C, Hsu C, Wang JZ et al. Fibromodulin-deficiency alters temporospatial expression patterns of transforming growth factor-β ligands and receptors during adult mouse skin wound healing. PLoS One 2014; 9: e90817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Jian J, Velasco O, Hsu CY, Zhang K, Levin A et al. Fibromodulin enhances angiogenesis during cutaneous wound healing. Plastic Reconstruct Surg Glob Open 2014; 2: e275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson JM, Yu F, Opalenik SR. Splinting strategies to overcome confounding wound contraction in experimental animal models. Adv Wound Care 2013; 2: 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan TP, Eaglstein WH, Davis SC, Mertz P. The pig as a model for human wound healing. Wound Repair Regen 2001; 9: 66–76. [DOI] [PubMed] [Google Scholar]
- Gurtner GC, Dauskardt RH, Wong VW, Bhatt KA, Wu K, Vial IN et al. Improving cutaneous scar formation by controlling the mechanical environment: large animal and phase I studies. Ann Surg 1993; 217(4): 391–396; 2011; 254: 217–225. [DOI] [PubMed] [Google Scholar]
- Li CS, Yang P, Ting K, Aghaloo T, Lee S, Zhang Y et al. Fibromodulin reprogrammed cells: A novel cell source for bone regeneration. Biomaterials 83: 194–2062016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorasani H, Zheng Z, Nguyen C, Zara J, Zhang X, Wang J et al. A quantitative approach to scar analysis. Am J Pathol 2011; 178: 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JA, Bond JS, Mason T, Ludlow A, Cridland P, O’Kane S et al. Visual analogue scale scoring and ranking: a suitable and sensitive method for assessing scar quality? Plast Reconstr Surg 2006; 118: 909–918. [DOI] [PubMed] [Google Scholar]
- Erickson HS, Gillespie JW, Emmert-Buck MR. Tissue microdissection. Methods Mol Biol 2008; 424: 433–448. [DOI] [PubMed] [Google Scholar]
- Freshney RI. Culture of Animal Cells: A Manual of Basic Technique. AR Liss: New York, 1987. [Google Scholar]
- Zheng Z, Jian J, Zhang X, Zara JN, Yin W, Chiang M et al. Reprogramming of human fibroblats into multipotent cells with a single ECM proteoglycan, fibromodulin. Biomaterials 2012; 33: 5821–5831. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGF beta/Smad signaling by oncogenic Ras. Genes & Development 1999; 13: 804–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang W, Hu H, Tang N, Zhang C, Liang W et al. Smad3 specific inhibitor, naringenin, decreases the expression of extracellular matrix induced by TGF-beta1 in cultured rat hepatic stellate cells. Pharm Res 2006; 23: 82–89. [DOI] [PubMed] [Google Scholar]
- Jinnin M, Ihn H, Tamaki K. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-beta1-induced extracellular matrix expression. Mol Pharmacol 2006; 69: 597–607. [DOI] [PubMed] [Google Scholar]
- Smith Jr TG, Lange GD, Marks WB. Fractal methods and results in cellular morphology--dimensions, lacunarity and multifractals. Journal of Neuroscience Methods 1996; 69: 123–136. [DOI] [PubMed] [Google Scholar]
- Ling EJY, Servio P, Kietzig AM. Fractal and lacunarity analyses: quantitative characterization of hierarchical surface topographies. Microsc Microanal 2016; 22: 168–177. [DOI] [PubMed] [Google Scholar]
- Evans R, Tian Y, Steadman R, Phillips A. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp Cell Res. 2003; 282: 90–100. [DOI] [PubMed] [Google Scholar]
- Loskutoff DJ, Quigley JP. PAI-1, fibrosis, and the elusive provisional fibrin matrix. J Clin Invest 2000; 106: 1441–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Angel P, Lafyatis R, Hattori K, Kim KY, Sporn MB et al. Autoinduction of transforming growth factor-beta-1 is mediated by the Ap-1 complex. Mol Cell Biol 1990; 10: 1492–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piek E, Ju WJ, Heyer J, Escalante-Alcalde D, Stewart CL, Weinstein M et al. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem 2001; 276: 19945–19953. [DOI] [PubMed] [Google Scholar]
- Stoff A, Rivera AA, Mathis JM, Moore ST, Banerjee NS, Everts M et al. Effect of adenoviral mediated overexpression of fibromodulin on human dermal fibroblasts and scar formation in full-thickness incisional wounds. J Mol Med 2007; 85: 481–496. [DOI] [PubMed] [Google Scholar]
- Swindle MM, Makin A, Herron AJ, Clubb FJ, Frazier KS. Swine as models in biomedical research and toxicology testing. Vet Pathol 2012; 49: 344–356. [DOI] [PubMed] [Google Scholar]
- Bayat A, McGrouther DA, Ferguson MW. Skin scarring. BMJ 2003; 326: 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CW, Ries WR. Nonoperative techniques for scar management and revision. Facial Plastic Surg 2001; 17: 283–288. [DOI] [PubMed] [Google Scholar]
- Longaker MT, Rohrich RJ, Greenberg L, Furnas H, Wald R, Bansal V et al. A randomized controlled trial of the embrace advanced scar therapy device to reduce incisional scar formation. Plastic Reconstruct Surg 2014; 134: 536–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay B, Fan J, Guan S, Li Y, Chen M, Woodley DT et al. A ‘traffic control’ role for TGFβ3: orchestrating dermal and epidermal cell motility during wound healing. The Journal of Cell Biology 2006; 172: 1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han A, Bandyopadhyay B, Jayaprakash P, Lua I, Sahu D, Chen M et al. The anti-motility signaling mechanism of TGFβ3 that controls cell traffic during skin wound healing. Biol Open 2012; 1: 1169–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merline R, Schaefer RM, Schaefer L. The matricellular functions of small leucine-rich proteoglycans (SLRPs). J Cell Commun Signal 2009; 3: 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi Y, Ehirchiou D, Kilts TM, Inkson CA, Embree MC, Sonoyama W et al. Identification of tendon stem/progenitor cells and the role of the extracellular matrix in their niche. Nat Med 2007; 13: 1219–1227. [DOI] [PubMed] [Google Scholar]
- Jian J, Zheng Z, Zhang K, Rackohn TM, Hsu C, Levin A et al. Fibromodulin promoted in vitro and in vivo angiogenesis. Biochem Biophys Res Commun 2013; 436: 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walraven M, Beelen RHJ, Ulrich MMW. Transforming growth factor-beta (TGF-beta) signaling in healthy human fetal skin: A descriptive study. J Dermatol Sci 2015; 78: 117–124. [DOI] [PubMed] [Google Scholar]
- Sandulache VC, Parekh A, Dohar JE, Hebda PA. Fetal dermal fibroblasts retain a hyperactive migratory and contractile phenotype under 2-and 3-dimensional constraints compared to normal adult fibroblasts. Tissue Eng 2007; 13: 2791–2801. [DOI] [PubMed] [Google Scholar]
- Colwell AS, Krummel TM, Longaker MT, Lorenz HP. Fetal and adult fibroblasts have similar TGF-beta-mediated, Smad-dependent signaling pathways. Plastic And Reconstructive Surgery 2006; 117: 2277–2283. [DOI] [PubMed] [Google Scholar]
- Cass DL, Sylvester KG, Yang EY, Crombleholme TM, Adzick NS. Myofibroblast persistence in fetal sheep wounds is associated with scar formation. J Pediatr Surg 1997; 32: 1017–1021, discussion 1021-1012. [DOI] [PubMed] [Google Scholar]
- Faraonio R, Pane F, Intrieri M, Russo T, Cimino F. In vitro acquired cellular senescence and aging-specific phenotype can be distinguished on the basis of specific mRNA expression. Cell Death Differ 2002; 9: 862–864. [DOI] [PubMed] [Google Scholar]
- Bevilacqua MA, Iovine B, Zambrano N, D'Ambrosio C, Scaloni A, Russo T et al. Fibromodulin gene transcription is induced by ultraviolet irradiation, and its regulation is impaired in senescent human fibroblasts. J Biol Chem 2005; 280: 31809–31817. [DOI] [PubMed] [Google Scholar]
- Nakashima M, Chung S, Takahashi A, Kamatani N, Kawaguchi T, Tsunoda T et al. A genome-wide association study identifies four susceptibility loci for keloid in the Japanese population. Nat Genet 2010; 42: 768–771. [DOI] [PubMed] [Google Scholar]
- Thompson CM, Hocking AM, Honari S, Muffley LA, Ga M, Gibran NS. Genetic risk factors for hypertrophic scar development. J Burn Care Res 2013; 34: 477–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa R, Watanabe A, Than Naing B, Sasaki M, Fujita A, Akaishi S et al. Associations between keloid severity and single-nucleotide polymorphisms: importance of rs8032158 as a biomarker of keloid severity. J Invest Dermatol 2014; 134: 2041–2043. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Liu SL, Xie J, Ding MQ, Lu MZ, Zhang LF et al. NEDD4 single nucleotide polymorphism rs2271289 is associated with keloids in Chinese Han population. Am J Transl Res 2016; 8: 544–555. [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ge J, Tredget EE, Wu Y. The mouse excisional wound splinting model, including applications for stem cell transplantation. Nat Protoc 2013; 8: 302–309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this article (and its Supplementary Information files).