

Abstract
The epithelial-mesenchymal transition (EMT) is a hallmark of cancer metastasis, and the associated molecular signaling pathways are regarded as therapeutic targets for cancer treatment. Thus, suppressing EMT with a natural chemical compound may be of therapeutic benefit. Eupatolide is a natural chemical compound extracted from the medicinal plant Inula britannica, which is used in Eastern Asia to treat bronchitis, disorders of the digestive system and inflammation. Besides the anti-inflammatory function of eupatolide, the present study found that eupatolide suppressed the migration and invasion of breast cancer cells, which was associated with the downregulation of vimentin in MDA-MB-231 cells and the upregulation of E-cadherin in MCF-7 cells. Treatment with eupatolide also significantly inhibited the migration and invasion of breast cancer cells that had been stimulated with transforming growth factor-β1 (TGF-β1). Eupatolide also suppressed TGF-β1-induced EMT via downregulation of mothers against decapentaplegic homolog 3 (SMAD3) phosphorylation and transcriptional repression of TGF-β receptor 1 (ALK5). In addition to this canonical pathway, the non-canonical protein kinase B (AKT) and extracellular signal-regulated kinase (ERK) pathways were also inhibited by eupatolide treatment. In summary, the results suggest that eupatolide suppresses the migration and invasion of breast cancer cells by blocking the canonical ALK5-SMAD3 signaling pathway and the non-canonical ERK and AKT signaling pathways.
Keywords: eupatolide, epithelial-mesenchymal transition, transforming growth factor-β1, transforming growth factor-β receptor 1, MDA-MB-231 cells, MCF-7 cells
Introduction
Metastasis is the process by which tumor cells migrate from their original site to another host organ by infiltrating the blood stream (1). The first step in metastasis is a change in the cells from an epithelial to a mesenchymal phenotype, termed the epithelial-mesenchymal transition (EMT) (2,3). During EMT, cells lose their basal polarity and their contact with one another, allowing for penetration of the basement membrane (4), via modulation of protein kinase B (AKT) activation (5). The phenotypic transformation includes a transcriptional change in associated genes, including vimentin, N-cadherin, fibronectin, Snail, zinc finger E-box-binding homeobox (Zeb), and Twist (6,7). These genes are involved in the morphological changes and survival of transited tumor cells (8).
Transforming growth factor β (TGF-β) is a multifunctional cytokine that acts as an inhibitor of early-stage cancer, but promotes EMT in late-stage cancer, as it plays a key part in the wound healing process, which requires cell migration (8–11). Upon TGF-β binding to the TGF-β type 2 receptor activin receptor-like kinase 1 (ALK1), the TGF-β type 1 receptor (ALK5) is phosphorylated (10,12), which then results in the phosphorylation of receptor-regulated mothers against decapentaplegic homologs (R-SMADs). R-SMADs include SMAD2 and SMAD3, which transduce extracellular ligand signaling from TGF-β (10,12). The activated R-SMAD combines with the common-mediator SMAD4, which translocates to the nucleus (13), where regulation of the transcriptional target genes involved in EMT occurs (14). There is also a TGF-β-associated non-SMAD signaling pathway that interacts with EMT (15). The pathway is initiated by activation of phosphoinositide-3 kinase (PI3K)-AKT-mechanistic target of rapamycin complex 1 (mTORC1) or extracellular signal-regulated kinase (ERK) signaling, and also leads to cellular changes (14,16).
In Eastern Asia, the Inula britannica plant is used to treat bronchitis, disorders of the digestive system, and inflammation (17,18). A number of biologically active sesquiterpene lactones have been isolated from this plant, of which eupatolide is one (19). Several studies have revealed the biological functions of eupatolide: Eupatolide exerts anti-inflammatory effects by inhibiting nuclear factor κ-light-chain-enhancer of activated B cells activation and nitric oxide and tumor necrosis factor (TNF) production (13,15). In addition, we hypothesized that eupatolide may exert antitumor activity, as eupatolide inhibits platelet-derived growth factor (PDGF)-induced proliferation and migration of vascular smooth muscle cells (VSMCs) through heme oxygenase 1 (HO-1) induction via the reactive oxygen species (ROS)/nuclear factor (erythroid-derived 2)-like 2 (NRF2) pathway (20), and stimulates TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by downregulation of cellular FLICE-like inhibitory protein, an inhibitor of the TRAIL signaling pathway (21).
The aim of the present study was to investigate the effect of eupatolide on proliferation, migration and invasion in breast cancer cells. Breast cancer cells were treated with eupatolide to investigate whether eupatolide inhibited migration and invasion in breast cancer cells. To identify a signaling pathway by which eupatolide affects migration and invasion, western blotting and Matrigel assay were used.
Materials and methods
Cell culture, chemicals and antibodies
Human breast cancer MDA-MB-231 and MCF-7 cells were obtained from American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco's modified Eagle's medium (HyClone; GE Healthcare, Chicago, IL, USA) with 10% fetal bovine serum (HyClone; GE Healthcare). Cells were incubated in a humidified atmosphere at 37°C with 5% CO2 (22). Human epidermal growth factor (EGF) and human transforming growth factor-β1 (TGF-β1) were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). The isolation of eupatolide from I. britannica and its chemical structure have been reported previously (23). Antibodies against Snail (cat. no. ab53519), Slug (cat. no. ab27568), SMAD4 (cat. no. ab40759) and phospho-EGFR (cat. no. ab76195) were purchased from Abcam (Cambridge, UK). Antibodies against vimentin (cat. no. 3932), phospho-AKT (Ser473; cat. no. 9271), ERK1/2 (cat. no. 9102), phospho-ERK1/2 (cat. no. 9106), EGF receptor (EGFR; cat. no. 4267), phospho-SMAD3 (Ser423/425; cat no. 9520), SMAD3 (cat no. 9523), phospho-SMAD1/5 (cat. no. 9516), SMAD1 (cat. no. 6944) and SMAD5 (cat. no. 12534) were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). The antibody specific for E-cadherin was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Proliferation assay
The proliferation assay was performed by direct counting. MCF-7 and MDA-MB-231 cells were seeded in 12-well plates at a density of 1×104 cells/well. Once cells were fully attached to the surface of the plate, they were treated with either PBS or 10 µM eupatolide for various durations. After 24, 48 or 72 h of treatment, the proliferating cells were detached using a trypsin-EDTA solution and counted using a Neubauer haemocytometer (Miltrnyi Biotech GmbH, Bergisch Gladbach, Germany). The experiment was performed in triplicate as described previously (24).
Wound healing assay
To assess the effect of eupatolide on the migration of breast cancer cells, MDA-MB-231 and MCF7 cells were seeded in 96-well plates at a density of 3×104 cells/well. The confluent cell monolayer was wounded by scraping with a Wound-Maker provided with the IncuCyte system (Essen Bioscience, Ann Arbor, MI, USA). Cells were then treated with EGF (100 ng/ml) or TGF-β1 (50 ng/ml) in the presence or absence of 10 µM eupatolide for 24 h and control cells were treated with PBS. The kinetics of cell migration were monitored using the IncuCyte system, as described previously (25).
Invasion assay
MCF-7 and MDA-MB-231 cells (2×104 per well) suspended in 100 µl serum-free DMEM were seeded into Transwell inserts (pore size, 8 µm; Costar; Corning Incorporated, NY, USA) coated with Matrigel. The lower chamber was filled with 500 µl DMEM containing 10% FBS. The cells were then treated with EGF (100 ng/ml) or TGF-β1 (50 ng/ml) in the presence or absence of 10 µM eupatolide for 24 h and control cells were treated with PBS. After 24 h, the cells invading the lower chamber were stained with crystal violet and images of the invaded cells in the bottom chamber were imaged. To quantify the results, crystal violet stains were dissolved in 33% acetic acid and then the absorbance at a wavelength of 570 nm was evaluated using the Epoch microplate reader (BioTek Instruments Inc., Winooski, VT, USA). Each experiment was performed three times.
RNA isolation and reverse transcription-polymerase chain reaction (RT-PCR)
MDA-MB-231 cells were seeded and pre-treated with either PBS or 10 µM eupatolide for 30 min, followed by treatment with 50 ng/ml TGF-β1 for 1, 2 or 3 h. Total RNA was isolated using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA), and 4 µg RNA was reverse transcribed using 0.5 µg of random primers, reverse transcriptase, 1.5 mM of dNTP and 5X buffer from RevertAid RT Reverse Transcription kit (Fermentas; Thermo Fisher Scientific, Inc.) at 42°C for 1 h. Once cDNA was synthesized, ALK1, ALK2, ALK3, ALK4 and ALK5 expression levels were amplified using the AccuPower RT-PCR PreMix kit (Bioneer Corporation, Daejeon, Korea) with the following primer sequences: Slug forward, 5′-GGTCAAGAAGCATTTCAAC-3′ and reverse, 5′-GGTAATGTGTGGGTCCGA-3′; Snail forward, 5′-CAGACCCACTCAGATGTCAA-3′ and reverse, 5′-CATAGTTAGTCACACCTCGT-3′; ALK1 forward, 5′-GTGGCAGCAAGTCACAAAGG-3′ and reverse, 5′-TACCAACTTGCCGGACCATC-3′; ALK2 forward, 5′-CTCGGCTCTGTCCAGTTTGT-3′ and reverse, 5′-TCACTGGGGTACTCGGAGAG-3′; ALK3 forward, 5′-TCTCCAAAAGCCCAGCTACG-3′ and reverse, 5′-CCTATGACAACAGGGGGCAG-3′; ALK4 forward, 5′-GTGGTGACGTGGCTGTGAAA-3′ and reverse, 5′-TTTGGAGCAATGTCTATGGT-3′; ALK5 forward, 5′-ATCCCAACAGATGGCAGAG-3′ and reverse, 5′-GGAGAGTTCAGGCAAAGCTG-3′; and GAPDH forward, 5′-GTGTTCCTACCCCCAATGTGT-3′ and reverse, 5′-ATTGTCATACCAGGAAATGAGCTT-3′. GAPDH primers were used as an internal control. PCR was performed for 30 cycles consisting of 30 sec denaturation at 94°C, 30 sec annealing at 56°C and 30 sec extension at 72°C. Each PCR was performed in triplicate. The PCR products were separated by 1% w/v agarose gel electrophoresis, stained with GelRed (Jomar Life Research, Melbourne, Victoria, Australia), and then the band intensities were quantitated by ImageJ software (version 1.51j8; National Institutes of Helath, Bethesda, MD, USA).
Western blot analysis
MCF-7 and MDA-MB-231 were treated with either PBS or 10 µM eupatolide and lysed using a buffer containing 150 mM NaCl, 1.0% NP-40, 50 mM Tris-HCl at pH 8.0, 1 mM EDTA, protease inhibitors and phosphatase inhibitors (Roche Applied Science, Penzberg, Germany). The concentration of protein was determined using the BCA protein assay kit (Pierce; Thermo Fisher Scientific, Inc.), the 10 µg of prepared whole-cell lysate samples were separated according to their size on 10% SDS-PAGE, and proteins were transferred onto a 0.45-µm pore-size nitrocellulose membrane for 2 h at room temperature. The membrane was blocked with 3% BSA in TBS-T for 30 min at 25°C and incubated for overnight at 4°C with the 1:1,000 dilution of targeted primary antibody (anti-vimentin, anti-GAPDH, anti-E-cadherin, anti-Snail, anti-Slug, anti-SMAD3, anti-p-SMAD3 S423/425, anti-SMAD1/5, anti-p-SMAD1/5 S423/425, anti-SMAD4, anti-p-ERK1/2 T202/Y204, anti-ERK1/2, anti-p-AKT S473, anti-AKT, anti-SP1, anti-p-SP1 T453 and anti-β-actin antibodies). The horseradish peroxidase-conjugated goat anti-mouse (cat. no. A90-131P) or goat anti-rabbit (cat. no. A120-118P) antibodies (Bethyl Laboratories, Montgomery, TX, USA) at 1:3,000 dilution in 3% BSA in TBS-T were incubated with the membrane at 25°C for 2 h, and then the membrane were washed 3 times for 5 min with TBS-T buffer. The membranes were developed by ECL reagent (GE Healthcare) and bands were detected using the LAS-3000 Image Reader (Fujifilm, Tokyo, Japan). The protein bands were quantitated by ImageJ software (version 1.51j8; National Institutes of Health).
Statistical analysis
Values are presented as the mean ± standard deviation. Multiple comparisons between groups were performed by one-way analysis of variance (ANOVA). P<0.05 was considered to indicate a statistically significant difference. All statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA, USA).
Results
Eupatolide suppresses the migration and invasion of breast cancer cells
To examine whether eupatolide has an anti-proliferative effect on breast cancer cells, estrogen receptor-positive MCF-7 breast cancer cells and estrogen receptor-negative MDA-MB-231 cells were treated with eupatolide for 3 days, with cell numbers measured each day. To determine the statistical significance of changes in cell numbers during the 3 days of treatment, ANOVA was performed. In MDA-MB-231 cells, no significant difference in proliferation rate was observed between those treated with eupatolide or PBS (Fig. 1A); however, MCF-7 cells treated with eupatolide exhibited a significantly decreased cell number compared with the control on the third day of treatment (P<0.01; Fig. 1B).
Figure 1.

Eupatolide suppresses the proliferation of breast cancer cells. (A) MDA-MB-231 and (B) MCF-7 cells were treated with 10 µM eupatolide, and cell numbers were counted at each indicated time point. Experiments were performed in triplicate. **P<0.01. EuTL, eupatolide.
To examine whether eupatolide has an anti-migratory effect on breast cancer cells stimulated with EGF and TGF-β1 (critical growth factors that can induce EMT and metastasis) (26), a confluent monolayer of cells was scratched and then treated with eupatolide for 24 h. As the migration of MCF-7 cells could be affected by the anti-proliferative effect of eupatolide, cells were treated with eupatolide for 24 h only to exclude this influence, and analyzed using the IncuCyte live-cell imaging analyzer. EGF and TGF-β1 treatment greatly increased the migration of cells compared that in the untreated control group, and treatment with eupatolide clearly inhibited the migration induced by EGF and TGF-β1 (Fig. 2A and B). MCF-7 cells migrated more slowly than MDA-MB-231 cells (Fig. 2B); the slow migration of MCF-7 cells was possibly due to their non-metastatic nature (27,28).
Figure 2.

Eupatolide inhibits the migration and invasion of breast cancer cells. (A) MDA-MB-231 and (B) MCF-7 cells were co-treated with either 100 ng/ml of EGF or 50 ng/ml of TGF-β1, as well as 10 µM eupatolide or a control, and the migration of cells was monitored for 24 h using an IncuCyte live-cell imaging system. After 24 h, relative wound density was acquired by IncuCyte. (C and D) MDA-MB-231 cells were seeded into Transwell chambers and stimulated with (C) 100 ng/ml of EGF or (D) 50 ng/ml TGF-β1 independently, or were co-treated with 10 µM eupatolide. The number of invaded cells in the bottom chamber was counted to measure invasion levels. *P<0.05, **P<0.01. EGF, epidermal growth factor; TGF-β1, transforming growth factor-β1; EuTL, eupatolide.
Whether eupatolide also affects the migration of cells under serum-starvation conditions was assessed. MDA-MB-231 and MCF-7 cells arrested at G0 phase were not significantly migrated compared to non-arrested cells. These findings indicated that eupatolide more efficiently inhibited the migration of growth factor-stimulated cells than unstimulated cells. The result was quantified by measuring cell density in the wound area at 2-h intervals for 24 h (Fig. 2A and B). To confirm this effect on migration further, an invasion assay was performed using Transwell plates. Because MCF-7 cells are non-metastatic, only MDA-MB-231 cells were used for the invasion assay. MDA-MB-231 cells were loaded into Transwell chambers and treated with EGF and TGF-β1 as well as eupatolide or control. EGF and TGF-β1 treatment increased the invasion of MDA-MB-231 cells, but co-treatment with eupatolide markedly reduced EGF- and TGF-β1-enhanced invasion (Fig. 2C and D). Eupatolide exhibited an anti-proliferative effect in non-metastatic MCF-7 cells and an anti-migration ability in metastatic MDA-MB-231 cells. However, the anti-migratory effect of eupatolide is unlikely to be caused by the anti-proliferative effect in MCF-7 cells, as the anti-proliferative effect only reached significance on the third day following the treatment with eupatolide.
Eupatolide suppresses EMT by blocking the expression of Snail and Slug
Since eupatolide inhibited the migration of breast cancer cells, the expression of genes associated with EMT was assessed. Since vimentin maintains the cytoskeletal architecture and tissue integrity, it is involved in migration of cells. Therefore, MDA-MB-231 cells were treated with eupatolide for 24, 48 or 72 h at a concentration of 10 µM to examine the effect on vimentin expression. Eupatolide treatment markedly inhibited vimentin expression in a time-dependent manner (Fig. 3A). The adherens junction protein E-cadherin contributes to the maintenance of the epithelial barrier function by homotypic interactions (29). Since MCF-7 cells express much higher levels of E-cadherin than MDA-MB-231 cells (30), MCF-7 cells were treated with eupatolide. Treatment with eupatolide increased E-cadherin expression in a time-dependent manner (Fig. 3B), indicating that eupatolide-mediated E-cadherin expression contributed to the suppression of cell migration. Subsequently, whether eupatolide could inhibit the expression level of transcription factors known to regulate EMT-associated gene expression was assessed. MDA-MB-231 and MCF-7 cells were treated with eupatolide and the expression levels of Snail and Slug were measured by western blotting. The expression levels of the two proteins were decreased in both types of cell in a dose-dependent manner (Fig. 3C). As Snail and Slug are unstable proteins (7), the mRNA levels of Snail and Slug were also examined using RT-qPCR in duplicate. No significant difference was observed between the eupatolide-treated and control groups; thus, treatment with eupatolide did not affect Snail and Slug mRNA expression in these cells (Fig. 3D), indicating that eupatolide contributes to the destabilization of Snail and Slug proteins.
Figure 3.

Eupatolide inhibits epithelial-mesenchymal transition through the downregulation of Snail and Slug expression. (A) MDA-MB-231 and (B) MCF-7 cells were treated with 10 µM eupatolide for the indicated times, and the expression of vimentin and E-cadherin were examined by western blotting. (C) MDA-MB-231 and MCF-7 cells were treated at the indicated concentrations of eupatolide for 24 h, and the expression levels of Snail and Slug were examined by western blotting. (D) Levels of Snail and Slug mRNA were examined using reverse transcription-polymerase chain reaction performed in duplicate; no significant differences were observed. EuTL, eupatolide.
Eupatolide blocks TGF-β1-induced SMAD3 activation
To investigate which signaling pathway was targeted by eupatolide, the SMAD signaling pathway was examined, as TGF-β1-mediated SMAD3 activation increases Snail expression (31). MDA-MB-231 cells were treated with TGF-β1 in the presence of eupatolide and then examined by western blotting to determine the level of phospho-SMAD3 and phospho-SMAD1/5. The levels of phospho-SMAD3, which were increased by TGF-β1 treatment, were decreased by eupatolide treatment in a time-dependent manner (Fig. 4A). However, SMAD1/5 phosphorylation was not affected by eupatolide treatment. Certain non-SMAD-dependent signaling pathways are also induced by TGF-β in EMT. TGF-β induces the phosphorylation of tyrosine residues on both type-I (ALK5) and type-II (ALK1) TGF-β receptors and/or on SHC adaptor protein 1 (32). The phosphorylated tyrosine residues recruit growth factor receptor-bound protein 2/son of sevenless homolog (GRB2/SOS) to activate ERK through RAS proto-oncogene, GTPase (RAS), Raf-1 proto-oncogene, serine/threonine kinase (RAF) and their downstream mitogen-activated protein kinase (MAPK) cascades. ERK then regulates target gene transcription through downstream transcription factors, in conjunction with SMADs, to control EMT (33). To investigate whether eupatolide affects these signaling pathways, levels of phospho-ERK were examined. Pre-treatment with eupatolide 30 min prior to TGF-β1 treatment inhibited ERK phosphorylation at 1 h after treatment (Fig. 4B), indicating that eupatolide also inhibited the TGF-β1-mediated non-SMAD pathway. The PI3K/AKT pathway is another non-SMAD pathway that contributes to TGF-β-induced EMT (15). Thus, whether eupatolide affected the AKT signaling pathway was investigated. Contrary to what was expected, TGF-β1 inhibited the basal level of AKT phosphorylation at the S473 residue, which was recovered by incubation with eupatolide (Fig. 4B).
Figure 4.
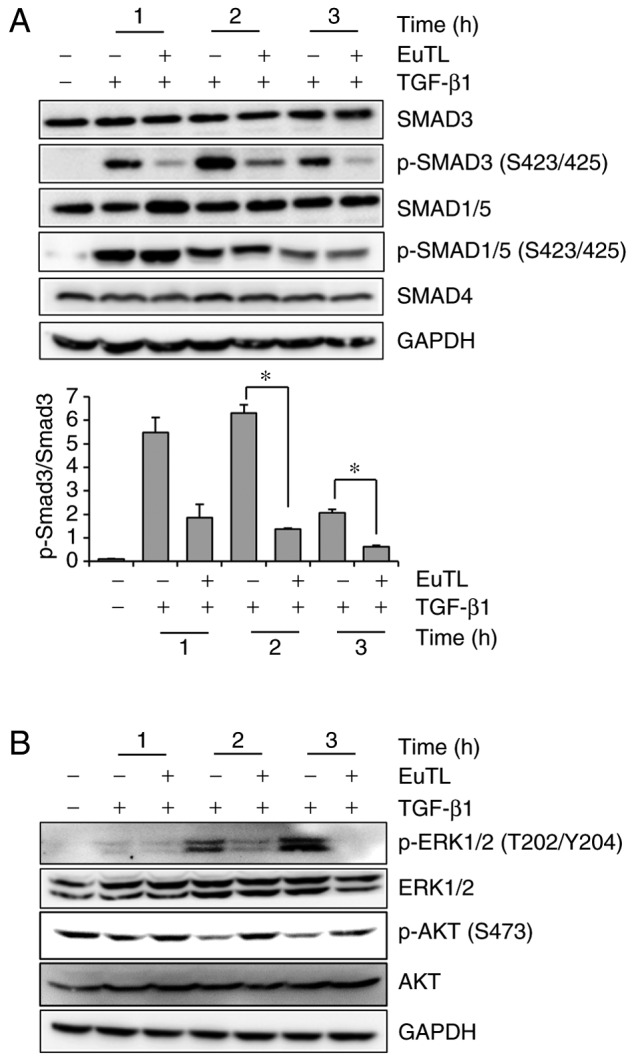
Eupatolide inhibits TGF-β1-induced SMAD3 activation. (A and B) MDA-MB-231 cells were pre-treated for 30 min with 10 µM eupatolide or control and additionally incubated in the presence or absence of 50 ng/ml TGF-β1 for the indicated time. Cells were lysed, and phosphorylation levels of (A) SMADs and (B) ERK1/2 and AKT were measured by western blot analysis. *P<0.05. SMAD3, mothers against decapentaplegic homolog 3; TGF-β1, transforming growth factor-β1; EuTL, eupatolide; p-, phosphorylated; ERK1/2, extracellular signal-regulated kinase 1/2; AKT, protein kinase B.
Eupatolide represses TGF-β1-induced ALK5 expression
Since SMAD3 activation is mediated by the heterodimerization of ALK5 and ALK1, the transcript levels of ALK1, ALK2, ALK3, ALK4, and ALK5 were examined following eupatolide treatment. TGF-β1 treatment increased the level of ALK5 transcription, which was decreased upon co-treatment with eupatolide in a time-dependent manner. However, co-treatment with eupatolide and TGF-β1 did not affect the levels of ALK1, ALK2, ALK3 and ALK4 transcription (Fig. 5A). As TGF-β is a potent enhancer of cell migration and invasion, and ALK5 is the critical receptor component of the TGF-β receptor heterodimer, the inhibition of ALK5 transcription by eupatolide is potentially a critical mechanism for the suppression of the TGF-β1 signaling pathway (34). The Sp1 transcription factor (SP1) is known to increase ALK5 expression. To assess whether SP1 activation was blocked by eupatolide treatment, cells were treated with TGF-β1 and eupatolide, and the levels of SP1 and of phospho-SP1 were examined. TGF-β1-activated SP1 phosphorylation was not affected by eupatolide treatment (Fig. 5B). We therefore hypothesize that eupatolide inhibits TGF-β1 signaling pathway in two ways: By inhibiting ALK5 transcription and TGF-β1-activated ERK expression, leading to Snail and Slug degradation (Fig. 5C). Thus, eupatolide could be used as an inhibitor of the TGF-β1 signaling pathway, leading to the suppression of EMT.
Figure 5.

Eupatolide inhibits TGF-β1-induced ALK5 expression. MDA-MB-231 cells were pre-treated for 30 min with 10 µM eupatolide and additionally incubated in the presence or absence of 50 ng/ml TGF-β1 for the indicated times. (A) The transcript levels of ALK1, ALK2, ALK3, ALK4 and ALK5 were measured using reverse transcription-polymerase chain reaction. (B) SP1 and p-SP1 levels were measured by western blotting. (C) Schematic diagram showing the TGF-β signaling pathway inhibited by eupatolide. TGF-β1, transforming growth factor-β1; EuTL, eupatolide; p-SP1, phosphorylated transcription factor Sp1; ALK5, activin receptor-like kinase 5.
Discussion
The majority of carcinomas are of epithelial origin and capable of metastasizing to other organs (35). For this process, epithelial tumor cells acquire mesenchymal characteristics and lose epithelial characteristics in the process of EMT. This is required for cells to translocate to other organs, mediated via an increase in the migration and invasion of tumor cells. As cancer cells lose epithelial characteristics during EMT, it is worthwhile examining whether eupatolide affects the epithelial phenotype of cancer cells. Since eupatolide treatment inhibits the migration and invasion of breast cancer cells by increasing E-cadherin expression and decreasing expression of vimentin, we hypothesize that treatment of eupatolide inhibits the metastasis of cancer cells by enhancement of the epithelial phenotype.
Eupatolide is a sesquiterpene lactone that can be isolated from traditional herbal extracts, and which exhibits various biological functions, including anti-inflammatory, anti-proliferative and anti-migratory activities (19,20,36). Since eupatolide inhibits the PDGF-mediated proliferation and migration of vascular smooth muscle cells through HO-1 induction via the ROS-NRF2 pathway (20), it is conceivable that eupatolide may inhibit the migration of breast cancer cells regardless of their estrogen receptor expression status. In a previous study, it was shown that eupatolide sensitizes breast cancer cells to TRAIL-induced apoptosis (19); it is therefore possible that the eupatolide-mediated inhibition of migration in cancer cells could be caused by the suppression of cell proliferation. The present study indicates that eupatolide inhibited the proliferation of MCF-7 cells on day 3 after treatment, although this did not occur in MDA-MB-231 cells. As MCF-7 cells are non-metastatic, whereas MDA-MB-231 cells are, it is likely that the inhibition of migration in metastatic cells is not caused by the inhibition of proliferation. To prove this hypothesis, various non-metastatic and metastatic breast cancer cells require examination.
The TGF-β ligand superfamily is classified into three subgroups, TGF-βs, activins and bone morphogenetic proteins (33,37), and there are five type-II receptors and seven different type-I receptors, termed ALK1-ALK7 (38). Binding of each TGF-β ligand induces type II-type I receptor heterodimerization and the combination of type II-type I receptor is dependent on a binding ligand type. After the receptor heterodimerization, constitutively active type II receptor phosphorylates and activates the type I receptor. The specificity and versatility of TGF-β signaling depend on which SMADs are activated (38). Receptor complexes containing ALK1, ALK2, ALK3 and ALK6 phosphorylate SMAD1, SMAD5 and SMAD8 (39), whereas receptor complexes containing ALK4, ALK5, and ALK7 phosphorylate SMAD2 and SMAD3 (40). In the present study, eupatolide suppressed SMAD3 phosphorylation but did not affect SMAD1/5 phosphorylation. Thus, ALK4, ALK5 and ALK7 may be upstream kinases, rather than ALK1, ALK2, ALK3 and ALK6. Transcriptional analysis revealed that eupatolide specifically suppressed ALK5 transcription, even though a transcription factor responsible for downregulation of ALK5 transcription was not identified.
TGF-β antisense oligonucleotides have been used widely in preclinical studies for cancer therapy (41). Treatment with AP-12009, an antisense oligonucleotide against TGF-β2, resulted in a significant increase in survival time (42) another antisense oligonucleotide targeted against TGF-β1, AP-11014, is being developed for treatment of human non-small cell lung, colorectal and prostate cancer (43). In addition to these relatively large TGF-β inhibitors, developing small molecular TGF-β inhibitors that are selectively targeted to inhibit ALK5 is of interest, as they could inhibit all TGF-β isoforms (44), unlike, for instance, antisense oligonucleotides that only target a specific TGF-β isoform. In addition to the SMAD signal transduction pathway, TGF-β also activates other intracellular signaling pathways, including the MAPK, PI3K/AKT, and Rho-like GTPase pathways (45). TGF-β phosphorylates its receptors on tyrosine residues, leading to the recruitment of GRB2/SOS to activate ERK through RAS proto-oncogene/Raf-1 proto-oncogene (RAS/RAF) cascades (46,47). Activated ERK then stimulates TGF-β-target gene transcription in conjunction with SMADs, also stabilizing Snail and Slug to control EMT (48). In the present study, eupatolide suppressed ERK activation, which could contribute to the destabilization of Snail and Slug. Therefore, eupatolide is a promising candidate small anti-TGF-β inhibitor, as it is able to inhibit TGF-β-mediated Snail and Slug stabilization via ERK and ALK5 transcription.
The PI3K/AKT pathway is another non-SMAD pathway that contributes to TGF-β-induced EMT (33). In numerous other TGF-β-induced responses, the activation of PI3K or AKT antagonizes TGF-β-induced apoptosis and growth inhibition via a physical interaction between AKT and SMAD3 (37,38), which is independent of AKT phosphorylation. The interaction between AKT and SMAD3 prevents ALK5-mediated phosphorylation and the nuclear localization of SMAD3, which leads to the inhibition of SMAD3-mediated transcription (31,32). It means that PI3K/AKT can negatively regulate TGF-β-induced SMADs signaling pathways (49). The present study found that TGF-β inhibited the basal level of AKT phosphorylation rather than activating AKT, whereas treatment with eupatolide increased AKT phosphorylation. Thus, the eupatolide-mediated increase in AKT phosphorylation may enhance the degree of interaction between AKT and SMAD3, which contributes to the inhibition of TGF-β signal transduction. On the other hand, AKT activation is known to inhibit glycogen synthase kinase-3β (GSK-3β), leading to the stabilization of Snail and Slug. However, treatment with eupatolide destabilized Snail and Slug, with a corresponding activation of AKT. This discrepancy can be explained as follows: AKT binds directly to unphosphorylated SMAD3 regardless of AKT phosphorylation, leading to the prevention of SMAD3 activation. Thus, regardless of the eupatolide-mediated increase in AKT phosphorylation, if phosphorylated AKT and SMAD3 interaction is enhanced, AKT phosphorylation can contribute to the inhibition of the TGF-β signaling pathway. Conversely, Snail destabilization with AKT activation upon eupatolide treatment may be explained if GSK-3β inactivation upon eupatolide treatment can be affected more by ERK inhibition than AKT activation.
As traditional herbal extracts have been used to treat cancer in East Asia, it is necessary to provide scientific evidence that these traditional anticancer therapies are effective. The present study reveals a partial molecular mechanism by which eupatolide inhibits the migration and invasion of breast tumor cells. However, one limitation of this study is that effect of eupatolide on the inhibition of cell migration was only assessed in vitro, owing to a lack of available eupatolide for the conduction of an in vivo experiment. If similar results are observed in an animal model, the development of eupatolide-derived chemicals to prevent cancer metastasis may be worthwhile.
Acknowledgements
The present study was supported by grants from the National Research Foundation of Korea grant funded by the Korean government (no. NRF-2016R1A2B2011683) and the Science Research Center of the National Research Foundation funded by the Korean government (no. 2016R1A5A1011974).
References
- 1.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalluri R, Weinberg RA. The basics of epithelial-mesenchytnal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19:1438–1449. doi: 10.1038/nm.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin YC, Lee YC, Li LH, Cheng CJ, Yang RB. Tumor suppressor SCUBE2 inhibits breast-cancer cell migration and invasion through the reversal of epithelial-mesenchymal transition. J Cell Sci. 2014;127:85–100. doi: 10.1242/jcs.132779. [DOI] [PubMed] [Google Scholar]
- 7.Wu Y, Zhou BP. Snail: More than EMT. Cell Adh Migr. 2010;4:199–203. doi: 10.4161/cam.4.2.10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marcucci F, Stassi G, De Maria R. Epithelial-mesenchymal transition: A new target in anticancer drug discovery. Nat Rev Drug Discov. 2016;15:311–325. doi: 10.1038/nrd.2015.13. [DOI] [PubMed] [Google Scholar]
- 9.Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFβ in cancer. FEBS Lett. 2012;586:1959–1970. doi: 10.1016/j.febslet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 10.Wakefield LM, Roberts AB. TGF-beta signaling: Positive and negative effects on tumorigenesis. Curr Opin Genet Dev. 2002;12:22–29. doi: 10.1016/S0959-437X(01)00259-3. [DOI] [PubMed] [Google Scholar]
- 11.Kubiczkova L, Sedlarikova L, Hajek R, Sevcikova S. TGF-β - an excellent servant but a bad master. J Transl Med. 2012;10:183. doi: 10.1186/1479-5876-10-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunha SI, Pietras K. ALK1 as an emerging target for antiangiogenic therapy of cancer. Blood. 2011;117:6999–7006. doi: 10.1182/blood-2011-01-330142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitman M, Raftery L. TGFbeta signaling at the summit. Development. 2005;132:4205–4210. doi: 10.1242/dev.02023. [DOI] [PubMed] [Google Scholar]
- 14.Curado F, Spuul P, Egaña I, Rottiers P, Daubon T, Veillat V, Duhamel P, Leclercq A, Gontier E, Génot E. ALK5 and ALK1 play antagonistic roles in transforming growth factor β-induced podosome formation in aortic endothelial cells. Mol Cell Biol. 2014;34:4389–4403. doi: 10.1128/MCB.01026-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schniewind B, Groth S, Müerköster S Sebens, Sipos B, Schäfer H, Kalthoff H, Fändrich F, Ungefroren H. Dissecting the role of TGF-beta type I receptor/ALK5 in pancreatic ductal adenocarcinoma: Smad activation is crucial for both the tumor suppressive and prometastatic function. Oncogene. 2007;26:4850–4862. doi: 10.1038/sj.onc.1210272. [DOI] [PubMed] [Google Scholar]
- 17.Khan AL, Hussain J, Hamayun M, Gilani SA, Ahmad S, Rehman G, Kim YH, Kang SM, Lee IJ. Secondary metabolites from Inula britannica L. and their biological activities. Molecules. 2010;15:1562–1577. doi: 10.3390/molecules15031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee YH, Lee NK, Paik HD. Antimicrobial characterization of inula britannica against helicobacter pylori on gastric condition. J Microbiol Biotechnol. 2016;26:1011–1017. doi: 10.4014/jmb.1510.10001. [DOI] [PubMed] [Google Scholar]
- 19.Lee J, Hwangbo C, Lee JJ, Seo J, Lee JH. The sesquiterpene lactone eupatolide sensitizes breast cancer cells to TRAIL through down-regulation of c-FLIP expression. Oncol Rep. 2010;23:229–237. [PubMed] [Google Scholar]
- 20.Kim N, Hwangbo C, Lee S, Lee JH. Eupatolide inhibits PDGF-induced proliferation and migration of aortic smooth muscle cells through ROS-dependent heme oxygenase-1 induction. Phytother Res. 2013;27:1700–1707. doi: 10.1002/ptr.4924. [DOI] [PubMed] [Google Scholar]
- 21.Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW. Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–383. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- 22.Lee S, Jeong AL, Park JS, Han S, Jang CY, Kim KI, Kim Y, Park JH, Lim JS, Lee MS, Yang Y. IK-guided PP2A suppresses Aurora B activity in the interphase of tumor cells. Cell Mol Life Sci. 2016;73:3375–3386. doi: 10.1007/s00018-016-2162-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin HZ, Lee D, Lee JH, Lee K, Hong YS, Choung DH, Kim YH, Lee JJ. New sesquiterpene dimers from Inula britannica inhibit NF-kappaB activation and NO and TNF-alpha production in LPS-stimulated RAW264.7 cells. Planta Med. 2006;72:40–45. doi: 10.1055/s-2005-873189. [DOI] [PubMed] [Google Scholar]
- 24.Lee S, Han S, Jeong AL, Park JS, Yang Y. Depletion of IK causes mitotic arrest through aberrant regulation of mitotic kinases and phosphatases. FEBS Lett. 2014;588:2844–2850. doi: 10.1016/j.febslet.2014.06.046. [DOI] [PubMed] [Google Scholar]
- 25.Sinha C, Ren A, Arora K, Moon CS, Yarlagadda S, Zhang W, Cheepala SB, Schuetz JD, Naren AP. Multi-drug resistance protein 4 (MRP4)-mediated regulation of fibroblast cell migration reflects a dichotomous role of intracellular cyclic nucleotides. J Biol Chem. 2013;288:3786–3794. doi: 10.1074/jbc.M112.435925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, Qi L, Liang Z, Song W, Liu Y, Wang Y, Sun B, Zhang B, Cao W. Transforming growth factor-β1 induces EMT by the transactivation of epidermal growth factor signaling through HA/CD44 in lung and breast cancer cells. Int J Mol Med. 2015;36:113–122. doi: 10.3892/ijmm.2015.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guttilla IK, Phoenix KN, Hong X, Tirnauer JS, Claffey KP, White BA. Prolonged mammosphere culture of MCF-7 cells induces an EMT and repression of the estrogen receptor by microRNAs. Breast Cancer Res Treat. 2012;132:75–85. doi: 10.1007/s10549-011-1534-y. [DOI] [PubMed] [Google Scholar]
- 28.Vergara D, Valente CM, Tinelli A, Siciliano C, Lorusso V, Acierno R, Giovinazzo G, Santino A, Storelli C, Maffia M. Resveratrol inhibits the epidermal growth factor-induced epithelial mesenchymal transition in MCF-7 cells. Cancer Lett. 2011;310:1–8. doi: 10.1016/j.canlet.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 29.van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008;65:3756–3788. doi: 10.1007/s00018-008-8281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chao YL, Shepard CR, Wells A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer. 2010;9:179. doi: 10.1186/1476-4598-9-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kocic J, Bugarski D, Santibanez JF. SMAD3 is essential for transforming growth factor-β1-induced urokinase type plasminogen activator expression and migration in transformed keratinocytes. Eur J Cancer. 2012;48:1550–1557. doi: 10.1016/j.ejca.2011.06.043. [DOI] [PubMed] [Google Scholar]
- 32.Pannu J, Nakerakanti S, Smith E, ten Dijke P, Trojanowska M. Transforming growth factor-beta receptor type I-dependent fibrogenic gene program is mediated via activation of Smad1 and ERK1/2 pathways. J Biol Chem. 2007;282:10405–10413. doi: 10.1074/jbc.M611742200. [DOI] [PubMed] [Google Scholar]
- 33.Wrighton KH, Lin X, Feng XH. Phospho-control of TGF-beta superfamily signaling. Cell Res. 2009;19:8–20. doi: 10.1038/cr.2008.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cantelli G, Crosas-Molist E, Georgouli M, Sanz-Moreno V. TGFBeta-induced transcription in cancer. Semin Cancer Biol. 2017;42:60–69. doi: 10.1016/j.semcancer.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev Oncog. 2013;18:43–73. doi: 10.1615/CritRevOncog.v18.i1-2.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee J, Tae N, Lee JJ, Kim T, Lee JH. Eupatolide inhibits lipopolysaccharide-induced COX-2 and iNOS expression in RAW264.7 cells by inducing proteasomal degradation of TRAF6. Eur J Pharmacol. 2010;636:173–180. doi: 10.1016/j.ejphar.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 37.Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 38.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 39.Daly AC, Randall RA, Hill CS. Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol Cell Biol. 2008;28:6889–6902. doi: 10.1128/MCB.01192-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell. 2005;16:1987–2002. doi: 10.1091/mbc.E04-08-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Connolly EC, Freimuth J, Akhurst RJ. Complexities of TGF-β targeted cancer therapy. Int J Biol Sci. 2012;8:964–978. doi: 10.7150/ijbs.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schlingensiepen KH, Schlingensiepen R, Steinbrecher A, Hau P, Bogdahn U, Fischer-Blass B, Jachimczak P. Targeted tumor therapy with the TGF-beta 2 antisense compound AP 12009. Cytokine Growth Factor Rev. 2006;17:129–139. doi: 10.1016/j.cytogfr.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 43.Akhurst RJ. Large- and small-molecule inhibitors of transforming growth factor-beta signaling. Curr Opin Investig Drugs. 2006;7:513–521. [PubMed] [Google Scholar]
- 44.Ling LE, Lee WC. Tgf-beta type I receptor (Alk5) kinase inhibitors in oncology. Curr Pharm Biotechnol. 2011;12:2190–2202. doi: 10.2174/138920111798808257. [DOI] [PubMed] [Google Scholar]
- 45.Smith AL, Robin TP, Ford HL. Molecular pathways: Targeting the TGF-β pathway for cancer therapy. Clin Cancer Res. 2012;18:4514–4521. doi: 10.1158/1078-0432.CCR-11-3224. [DOI] [PubMed] [Google Scholar]
- 46.Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, Nicoletti I, Grignani F, Pawson T, Pelicci PG. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell. 1992;70:93–104. doi: 10.1016/0092-8674(92)90536-L. [DOI] [PubMed] [Google Scholar]
- 47.Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM, Derynck R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007;26:3957–3967. doi: 10.1038/sj.emboj.7601818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 49.Guo X, Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009;19:71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]