

Abstract
The Massachusetts General Hospital Radiochemistry Program, in collaboration with Pfizer, has developed unique 11C and 18F-labeling strategies to synthesize isotopologs of lorlatinib (PF-06463922) which is undergoing phase III clinical trial investigations for treatment of non-small-cell lung cancers with specific molecular alterations. A major goal in cancer therapeutics is to measure the concentrations of this drug in the brain metastases of patients with lung cancer, and penetration of the blood–brain barrier is important for optimal therapeutic outcomes. Our recent publication in Nature Communications employed radiolabeled lorlatinib and positron emission tomography (PET) studies in preclinical models including nonhuman primates (NHPs) that demonstrated high brain permeability of this compound. Our future work with radiolabeled lorlatinib will include advanced PET evaluations in rodent tumor models and normal NHPs with the goal of clinical translation.
Keywords: lorlatinib, positron emission tomography, carbon-11, fluorine-18, ALK, ROS1
We have recently published the application of unique radiochemistry strategies to radiolabel the brain penetrant anticancer drug lorlatinib (PF-06463922),1 which is undergoing phase I-III clinical trial investigations for treatment of non-small-cell lung cancers (NSCLC; http://clinicaltrials.gov/ct2/show/NCT01970865). Lorlatinib is a next-generation small molecule macrocyclic inhibitor of both the orphan receptor tyrosine kinase c-ros oncogene 1 (ROS1) and anaplastic lymphoma kinase (ALK) and is the first in class to show significant brain penetration.2 In addition to improvement in potency against both ALK and the various mutants, lorlatinib was designed with improved permeability to cross the blood–brain barrier (BBB), though in vivo data would be critical to confirm this, and to ensure continued investment in the drug’s development. The Pfizer Oncology Team needed to prove that lorlatinib had sufficient central nervous system (CNS) exposure to target brain metastases. Our manuscript titled “Synthesis and preliminary positron emission tomography (PET) imaging of 11C- and 18F-isotopologs of the ROS1/ALK inhibitor lorlatinib in non-human primates” worked toward this goal through a multi-institutional collaboration among chemists with expertise in synthetic organic and medicinal chemistry, as well as radiochemistry and molecular imaging from Harvard Medical School, Massachusetts General Hospital, and Pfizer.1
As novel drug candidates enter clinical testing, it is vital that data are collected to demonstrate proof of mechanism. Fundamentally, there are 3 criteria that need to be measured, termed the “Three Pillars of Survival” by Morgan et al.3 Pillar 1, defined as “exposure at the target site of action,” is a core principle as without exposure one cannot achieve the subsequent 2 pillars, namely, target engagement through binding to the pharmacological target (pillar 2) and downstream expression of pharmacology (pillar 3). While direct measurement of pillar 1 can be difficult to perform, PET offers a way to achieve this if the drug candidate can be readily radiolabeled with an appropriate radionuclide in a manner that causes no structural changes. Overall, the utilization of PET imaging as a tool in drug development has expanded over the past few decades and has proved to be fruitful in confirming target exposure (pillar 1), optimization of drug scheduling, patient selection/stratification, and disease/treatment monitoring.
Our primary aim is to measure the concentrations of lorlatinib in brain tumor lesions of patients with lung cancer and confirm its penetration across the BBB, which is considered to be important for an optimal therapeutic outcome. Toward our goal of assessing the biodistribution and whole-body dosimetry of lorlatinib by PET, we prepared both 11C and 18F isotopologs of lorlatinib (Figure 1). Carbon-11-labeled lorlatinib was routinely prepared with good radiochemical yields, and PET imaging in nonhuman primates confirmed its high BBB permeability (vide infra). Novel radiolabeling strategies including an automated multistep 11C-labeling process and a spirocyclic iodonium ylide (SCIDY)-based radiofluorination were critical to implement the PET imaging studies when traditional labeling methods failed.4,5 In particular, our SCIDY technology has been validated for other human PET imaging studies6 and is used by academic centers as well as the radiopharmaceutical industry to prepare isotopologs of lead drug molecules that were previously challenging to prepare.
Figure 1.

Synthesis of (A) 11C- and (B) 18F-labeled isotopologs of lorlatinib.
Imaging of 11C-radiolabeled lorlatinib in nonhuman primates provided unequivocal in vivo confirmation of the desired BBB permeability with rapid brain uptake (Figure 2). Peak-measured brain concentrations were locally high, with the cerebellum exceeding a standardized uptake value of 2 at approximately 10 minutes postinjection. Regional uptake exhibited modest heterogeneity but was generally concordant with expected ALK distribution, with highest radioactivity concentrations in the cerebellum, frontal cortex, and thalamus; intermediate levels in other cortical gray matter; and lowest values in white matter. These PET imaging results support that lorlatinib crosses the BBB at sufficient concentrations to be potentially effective against brain metastases, per pillar 1.
Figure 2.
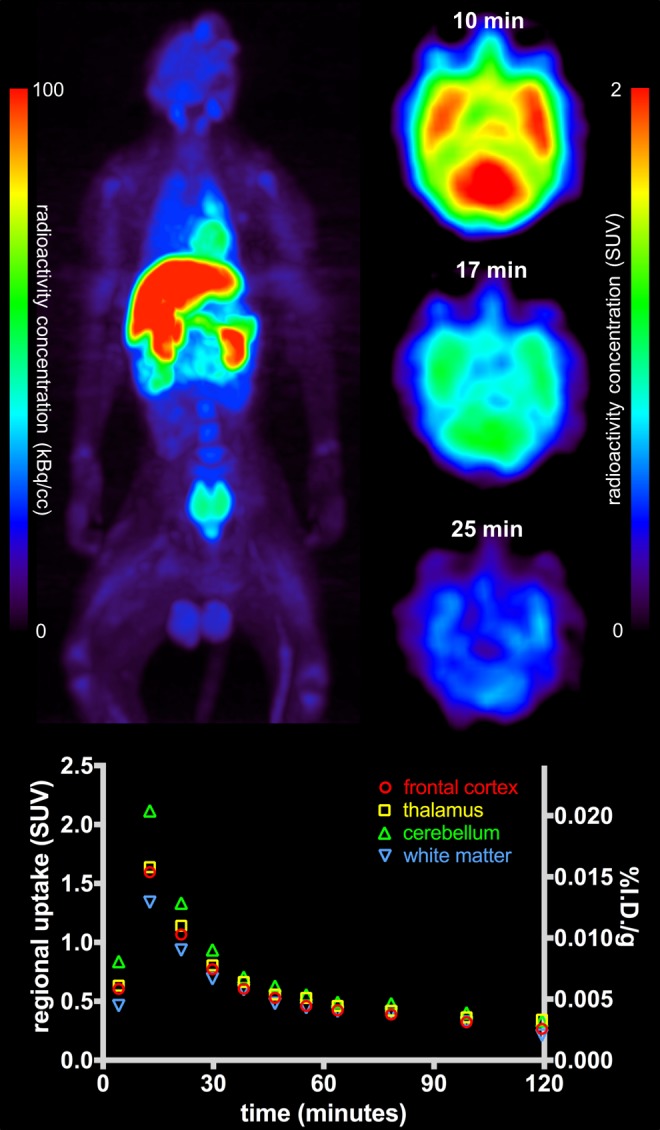
Top panels depict the representative PET imaging of [11C]lorlatinib in rhesus macaque, including a time course from 10 to 25 minutes postinjection of the radiotracer. The corresponding regional time-activity curves (below) show the rapid uptake followed by fast washout of the radiotracer from normal brain tissues.
In support of pillar 2, we sought to establish target engagement through binding to the pharmacological target. Herein, we assessed tumor uptake of [11C]lorlatinib in mice bearing subcutaneous human H3122 (EML4-ALK positive) xenografts by PET-CT imaging in conjunction with blocking studies. These studies showed that the tumor uptake reached its plateau in approximately 30 to 60 minutes after injection of [11C]lorlatinib (>2% ID/g) and co-injection with unlabeled lorlatinib resulted in significant decrease in the tumor uptake (<0.4% ID/g), thereby providing support of pillar 2.
The existence or progression of the cancer into the CNS is associated with poorer prognoses for patients with NSCLC and constitutes a true unmet medical need. In this age of increasing costs and time constraints of clinical trials, herein the case of lorlatinib as with many other studies, a rigorously designed PET imaging study has proven to be an invaluable early clinical development tool in drug discovery and development. By confirming pillar 1 and supporting pillar 2 through in vivo PET imaging studies, this symbiotic academic–industrial partnership has not only led to 2 novel PET radiotracers for an unprecedented imaging target but also inspired the development of new carbon-11 and fluorine-18 labeling methodologies. We anticipate translating radiolabeled lorlatinib for PET clinical research studies in the near future. Given the excitement around potent and selective kinase inhibitors in drug development and related diagnostic radioimaging agents for kinase targets,7–10 we anticipate multiple first-in-human studies around unexplored kinase targets within the CNS.
Footnotes
Authors’ Note: N.V. thanks Pfizer and the National Institute on Ageing of the NIH for funding (R01AG054473). S.H.L. is a recipient of an NIH career development award (DA038000) and an Early Career Award in Chemistry of Drug Abuse and Addiction (ECHEM, DA043507) from the National Institute on Drug Abuse.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Collier TL, Normandin MD, Stephenson NA, et al. Synthesis and preliminary PET imaging of 11C and 18F isotopologues of the ROS1/ALK inhibitor lorlatinib. Nat Commun. 2017;8:15761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnson TW, Richardson PF, Bailey S, et al. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem. 2014;57(11):4720–4744. [DOI] [PubMed] [Google Scholar]
- 3. Morgan P, Van Der Graaf PH, Arrowsmith J, et al. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving phase II survival. Drug Discov Today. 2012;17(9-10):419–424. [DOI] [PubMed] [Google Scholar]
- 4. Rotstein BH, Stephenson NA, Vasdev N, Liang SH. Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics. Nat Commun. 2014;5:4365. [DOI] [PubMed] [Google Scholar]
- 5. Rotstein BH, Wang L, Liu RY, et al. Mechanistic studies and radiofluorination of structurally diverse pharmaceuticals with spirocyclic iodonium(III) ylides. Chem Sci. 2016;7(7):4407–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stephenson NA, Holland JP, Kassenbrock A, et al. Iodonium ylide–mediated radiofluorination of 18F-FPEB and validation for human use. J Nucl Med. 2015;56(3):489–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bernard-Gauthier V, Bailey JJ, Mossine AV, et al. A kinome-wide selective radiolabeled TrkB/C inhibitor for in vitro and in vivo neuroimaging: synthesis, preclinical evaluation and first-in-human. J Med Chem. 2017;60(16):6897–6910. [DOI] [PubMed] [Google Scholar]
- 8. Hicks JW, VanBrocklin HF, Wilson AA, Houle S, Vasdev N. Radiolabeled small molecule protein kinase inhibitors for imaging with PET or SPECT. Molecules. 2010;15(11):8260–8278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Holland JP, Cumming P, Vasdev N. PET of signal transduction pathways in cancer. J. Nucl Med. 2012;53(9):1333–1336. [DOI] [PubMed] [Google Scholar]
- 10. Holland JP, Cumming P, Vasdev N. PET radiopharmaceuticals for probing enzymes in the brain. Am J Nucl Med Mol Imaging. 2013;3(3):194–216. [PMC free article] [PubMed] [Google Scholar]