

Abstract
Microdissection testicular sperm extraction (micro‐TESE) combined with intracytoplasmic sperm injection is a standard therapeutic option for patients with non‐obstructive azoospermia (NOA). Hormonal treatment has been believed to be ineffective for NOA because of high gonadotropin levels; however, several studies have stimulated spermatogenesis before or after micro‐TESE by using anti‐estrogens, aromatase inhibitors, and gonadotropins. These results remain controversial; however, it is obvious that some of the patients showed a distinct improvement in sperm retrieval by micro‐TESE, and sperm was observed in the ejaculates of a small number of NOA patients. One potential way to improve spermatogenesis is by optimizing the intratesticular testosterone (ITT) levels. ITT has been shown to be increased after hCG‐based hormonal therapy. The androgen receptor that is located on Sertoli cells plays a major role in spermatogenesis, and other hormonal and non‐hormonal factors may also be involved. Before establishing a new hormonal treatment protocol to stimulate spermatogenesis in NOA patients, further basic investigations regarding the pathophysiology of spermatogenic impairment are needed. Gaining a better understanding of this issue will allow us to tailor a specific treatment for each patient.
Keywords: Gonadotropins, Hormonal therapy, Intratesticular testosterone, Non‐obstructive azoospermia, Testicular sperm extraction
Introduction
Although microdissection testicular sperm extraction (micro‐TESE) provides patients with non‐obstructive azoospermia (NOA), which is the most prevalent form of azoospermia, the possibility of fathering a child when used in combination with intracytoplasmic sperm injection (ICSI), NOA remains the most challenging condition in male infertility. The typical success rate of sperm retrieval by micro‐TESE is 40–50 %. Recent studies suggest that nearly 60 % of men with NOA have some level of sperm production in the testes [1]; however, a number of patients remain surgically unmanageable because of maturation arrest (MA) and/or Sertoli cell‐only syndrome. Because of high gonadotropin levels, medical therapies, including hormonal treatments, have been believed to be ineffective at improving spermatogenesis to a degree that sperm is evident in the ejaculate. As such, it is not thought to improve the chances of a successful retrieval by micro‐TESE. Specific and effective gonadotropin treatment is available for men with male hypogonadotropic hypogonadism (MHH), and the induction of spermatogenesis improves patients’ quality of life [2]; however, these patients are uncommon in male infertility clinics.
In this review, we summarize the mechanisms and outcomes of hormonal therapies for NOA, including anti‐estrogens, gonadotropins, and aromatase inhibitors, which are primarily used prior to micro‐TESE, and hCG/follicle‐stimulating hormone (FSH) therapy, which is generally used prior to the second micro‐TESE. Based on the findings from basic and clinical studies, we then hypothesize about mechanisms that may restore spermatogenesis and discuss the future of hormonal treatment for NOA patients.
Rationale for hormonal therapy and intratesticular testosterone
Spermatogenesis is regulated by a complex endocrine, paracrine, and juxtacrine regulatory cross‐talk that involves Sertoli, peritubular, Leydig, and germ cells. Testosterone is one of the most important factors in this network for maintaining spermatogenesis [3]; however, the precise biological mechanisms that support spermatogenesis are poorly understood. To date, the ability of hormonal therapy to increase sperm production in men with NOA has never been demonstrated in a randomized and controlled fashion. One goal of hormonal treatment is optimizing intratesticular testosterone (ITT) levels because many men with NOA have low testosterone and an abnormal testosterone to estradiol ratio (T/E2 ratio). This abnormal ratio can be corrected using anti‐estrogen, hCG, or aromatase inhibitors. Hussein et al. reported the usefulness of clomiphene citrate as the initial treatment prior to micro‐TESE [4], and Reifsnyder et al. also reported a partial effect of testosterone optimization on the micro‐TESE outcome following treatment with clomiphene citrate, hCG, or aromatase inhibitors [5]. Ramasamy et al. reported that, in Klinefelter syndrome, men with a low baseline testosterone level who responded to medical therapy with a resultant testosterone level of greater than 250 ng/dl had a 77 % chance of sperm retrieval versus 55 % for men that did not respond to the therapy initiated to enhance testosterone production [6]. These observations suggest that men who can respond to increased testosterone levels may also display increased levels of the other growth factors secreted from Leydig and Sertoli cells [7].
Sertoli cells are considered to be the target cells for the activity of testosterone and dihydrotestosterone through the androgen receptor (AR) [8]. In humans and rodents, the AR has been reported to localize to the nuclei of Sertoli cells, peritubular myoid cells, Leydig cells, and fibroblasts [9, 10, 11, 12]. Two reports have also described AR immunoexpression in germ cells, primarily in spermatogonia and spermatocytes [13, 14]. Using human testicular biopsy specimens, we confirmed the absence of AR immunoexpression in germ cells and the presence of intense staining in Sertoli cells (Sertoli cell‐specific AR, SCAR) [15]. In rodents, spermiogenesis, the final step of spermatogenesis, is considered to be highly dependent on ITT [16]. On the other hand, high ITT is also necessary for the transition from type A to type B spermatogonia [17]. These findings support the hypothesis that Sertoli cells are most likely mediators of androgen activity in spermatogenesis because of their intimate anatomical and functional interactions with developing germ cells [18, 19]. When testosterone activity is insufficient, spermatogenesis is blocked at meiosis and spermiogenesis, as previously shown using AR knockout mice [20, 21, 22]. Hazra et al. developed transgenic Sertoli cell‐specific AR (TgSCAR) gain‐of‐function mice and reported that the TgSCAR mice demonstrated premature postnatal spermatogenic development, as shown by increased levels of meiotic and post‐meiotic germ cells [23]. These findings demonstrate that SCAR is particularly important for the development of pre‐meiotic spermatocytes. This animal model histologically resembles MA in men with NOA and supports the observation that SCAR expression is lower in early MA compared with late MA [15].
Although NOA is often accompanied by borderline to low serum testosterone levels and increased serum FSH levels, the serum testosterone level does not always correlate with ITT, and, of course, the serum testosterone and FSH levels do not reflect the focal areas of spermatogenesis that are found during micro‐TESE. ITT is present in high concentrations within the testes, ranging from 100 to 1000‐fold higher than the concentrations found in the circulation, as reported by several laboratories [24, 25, 26]. The ideal concentration of ITT for optimal spermatogenesis in men with NOA, which may be caused by a variety of factors, remains unknown. Although the optimal ITT concentration remains unknown, Shinjo et al. used intratesticular fluid obtained by micro‐TESE to show that the basal ITT was lower in men who responded to hormonal treatment than those from whom spermatozoa could be retrieved at the 2nd micro‐TESE. This indicates that ITT deficiency is one of the causes of NOA and this condition can be improved by hormonal therapy [27]. This is of particular importance in Klinefelter syndrome patients whose serum testosterone levels tend to be lower than common NOA cases. Although baseline serum testosterone levels per se are not predictive of sperm retrieval, the preoperative testosterone serum level after 3 months of medical treatment is predictive of success at sperm retrieval by micro‐TESE in men with Klinefelter syndrome. Ramasamy et al. have shown that men with low baseline testosterone who responded to medical therapy with a resultant testosterone level of greater than 250 ng/dl had a 77 % chance of sperm retrieval vs. 55 % for men that did not respond to therapy [6]. Serum markers that may be able to predict ITT have been described, including insulin‐like factor 3, anti‐mullerian hormone, inhibin B, and 17a‐hydroxyprogesterone, the testosterone precursor. The correlations between these parameters and ITT should be further investigated to enable non‐invasive monitoring of ITT.
Anti‐estrogens
Clomiphene citrate and tamoxifen are non‐steroidal anti‐estrogens and are safe, well‐established medications that can be used empirically to treat idiopathic oligospermia [28] and NOA [4]. Clomiphene citrate and tamoxifen are estrogen receptor modulators that block the negative feedback exerted by estrogen at the hypothalamus and anterior pituitary, thus increasing the serum FSH and luteinizing hormone (LH) levels that then stimulate spermatogenesis and testosterone production. A recent meta‐analysis showed that anti‐estrogens can stimulate the spermatogenesis in NOA patients [29]. Anti‐estrogen treatment is often called an “empirical” therapy; however, the patients’ hormonal parameters should be carefully monitored during this treatment because the responses to increased serum testosterone as well as spermatogenesis and its duration are different in each patient, resulting in controversial results among the publications. Anti‐estrogen therapy is generally safe, but can sometimes be harmful. For example, increased red blood cells may be observed, especially in patients whose serum testosterone is supraphysiologically high (more than 800–900 ng/dl). Hussein et al. treated 42 men with NOA, including cases of MA (43 %) and hypospermatogenesis (57 %), but no cases of Sertoli cell‐only syndrome, with clomiphene citrate at an initial dose of 50 mg on alternating days for 2 weeks, and then serially increasing doses until the testosterone levels reached 600–800 ng/dL [4]. If no effective testosterone increase was observed after this treatment, hCG, recombinant human (rh)FSH, or a combination of the two therapeutics was initiated. Semen analysis was performed at regular intervals, and if azoospermia persisted beyond 6 months of treatment, a testicular biopsy was performed. The results revealed that sperm could be detected in approximately 60 % of the treated by micro‐TESE, while the sperm retrieval rate of the untreated group was 33.6 %. Testicular histology of more than half of the participants in this study showed hypospermatogenesis, indicating that testosterone optimization was effective in post‐meiotic arrest.
Aromatase inhibitors
Aromatase is present primarily in the adipose tissue, liver, testes, skin, and brain where it converts testosterone and other androgens to estradiol. Steroidal (testolactone) and non‐steroidal (anastrozole, letrozole) aromatase inhibitors have been used to stimulate spermatogenesis in men with NOA. Aromatase inhibitors inhibit the conversion of androgens to estrogens, thereby increasing serum testosterone and, presumably, the ITT levels. Furthermore, feedback inhibition of the pituitary and hypothalamus is decreased due to decreased serum estrogen levels, resulting greater gonadotropin release [30]. In oligospermic men, aromatase inhibitors increase the T/E2 ratio to normal and have been suggested to improve semen parameters significantly [30, 31]. Fertile men have a mean T/E2 ratio of 14.5, whereas men with NOA have a T/E2 ratio of 6.9 [31], and men with Klinefelter syndrome have a T/E2 ratio of 4.4 [30]. Therefore, it appears that at least a small subset of male infertility patients, including those with NOA, who have a decreased T/E2 ratio may benefit from treatment with an aromatase inhibitor (anastrozole 1 mg daily, testolactone 100–200 mg daily, or other aromatase inhibitors) [30]. Candidates for aromatase inhibition have usually been identified as men with serum T/E2 ratios of <10 [31]. The report by Schiff et al. regarding their experience using pretreatment with testolactone, hCG, and anastrozole in men with non‐mosaic Klinefelter syndrome prior to TESE/ICSI shows that the hormonal therapies had favorable results [32]. However, in a more recent update of their data, this group identified no significant impact of the pretreatment on a successful outcome [6]. Evidence regarding the use of aromatase inhibitors for controlled ovarian stimulation is accumulating; however, the optimal dose to stimulate spermatogenesis remains unknown. Additional basic and clinical studies are needed to establish the dose and timing of aromatase inhibitor medication.
Gonadotropins
Cao et al. reported an NOA case that was treated with hCG only in which testicular sperm was found by TESE following hormonal treatment [33]. As shown in retrospective clinical studies, previous attempts to improve spermatogenesis in men with NOA via treatment with either rhFSH alone or in combination with hCG have been partially successful [34, 35]. Selman et al. reported that a man with Y chromosome microdeletions who received rhFSH and hCG before a successful ICSI procedure was able to produce ejaculated sperm that numbered in the few thousands [34]. In their later study, 49 men with NOA were treated with rhFSH. They were initially treated with 75 IU of rhFSH on alternate days for 2 months, followed by 150 IU for 2 months, and then an additional 2,000 IU of hCG for 2 months, three times a week. All men remained azoospermic at the end of treatment; however, while none had mature sperm in the pre‐treatment testicular biopsy, sperm were found in the biopsies of 22 % men after treatment [34]. Efesoy et al. reported sperm in the ejaculate of 2 of 11 azoospermic men with MA who were treated with rhFSH [35]. Similarly, Ramasamy et al. reported improved outcomes of primary TESE following gonadotropin therapy in men with NOA and Klinefelter syndrome [6]. These studies included a variety of NOA patients. By selecting the candidates more strictly and treating the patients with more specificity, gonadotropin therapies may be very effective at stimulating spermatogenesis in men with NOA.
Dysfunction of gonadotropin per se is very rare, but should not be discounted because the efficacy of gonadotropin treatment is extremely favorable. LH‐inactivating mutations include the following the genotypic abnormalities: a missense mutation that impaired receptor binding [36]; a point mutation that disrupted a vital cystine knot motif and abrogated the heterodimerization of LH [37]; a nine base pair (9‐bp) deletion in exon 2 that impaired the cystine knot folding motif [38]; a G to C substitution in intron 2 that disrupted splicing, generating an abnormality in the processing of the LH β‐subunit mRNA [39]; and an IVS2 + 1G → C substitution that prevented the proper assembly of the mutant LH β‐subunit with the LH α‐subunit and the secretion of the heterodimer [40]. These cases were diagnosed as due to hypogonadism, not infertility, but spermatogenesis as well as hypogonadism treatment was also completed after hCG‐based hormonal therapy. In contrast to these distinct loss‐of‐function mutations, variant LH β has also been reported and may result in a fertile eunuch [41].
The human FSH β gene is highly conserved, and amino acid changing mutations are extremely rare [42]. To date, several inactivating mutations in the FSH β gene have been found that resulted in delayed puberty and isolated FSH deficiency in 3 women and 2 men. A 2‐bp deletion/premature STOP codon in exon 3 of the FSH β gene resulted in an absence of bioactivity [43, 44]. Two missense mutations in exon 3 also ablated bioactivity [45, 46]. Although no information regarding treatment with rhFSH was noted, these male patients are considered to be good responders with regard to the restoration of their spermatogenesis. These mutations are very rare, but should be considered in the management of NOA, especially when standard treatments fail. A number of inactivating gonadotropin receptors have been reported (reviewed by Themmen [47]); however, the ability of gonadotropins to restore spermatogenesis in these men is doubtful. FSH receptor gene polymorphisms have been studied as potential risk factors for spermatogenetic failure and may result in primary testicular failure, but there have been no publications regarding the involvement of these mutations in NOA. Selice et al. evaluated the ability of FSH treatment to restore sperm production in men with polymorphisms in the FSH receptor and found that only subjects with at least one serine in position 680 showed a statistically significant increase in semen parameters.
Salvage hCG/FSH therapy
In men with NOA, the gonadotropin receptors are usually down‐regulated due to the high levels of endogenous gonadotropins; this is one reason why gonadotropin treatment is believed to be ineffective at restoring spermatogenesis. This down regulation in turn allows a “resetting” of the FSH and LH receptors and “resting” of the Leydig and Sertoli cells, whose functions then improves [48]. We have reviewed the data regarding repeated TESE in men with NOA who had no sperm retrieved during the first TESE [49] (Fig. 1). In spite of their high gonadotropin levels, the serum testosterone levels of all of the participants were statistically increased after hCG treatment, and half of the men showed decreased endogenous gonadotropin secretion due to the negative feedback regulation exerted by the increased serum testosterone. Half of the patients did not show any gonadotropin decrease because the extent of serum testosterone increase was mild an/or the responsiveness to testosterone and estradiol in the hypothalamus and related central nervous system was weak. This condition is very unique for the intratesticular environment because there is an “FSH resetting” during which the Sertoli cells are resting. On the other hand, the ITT level is extremely high [27] because of the supraphysiologically stimulated Leydig cells. The precise mechanisms responsible for inducing spermatogenesis are under investigation, and histological data have shown that men with hypospermatogenesis or late MA are likely to respond to hormonal treatment [49], thereby indicating that spermiogenesis, a step requiring high ITT levels, can be stimulated by hormonal therapy [50]. In the current study, 43 NOA men from whom no sperm could be obtained by micro‐TESE received 5,000 IU of hCG three times a week for 3 months. The men whose FSH declined significantly received an additional 150 IU of recombinant FSH three times a week for 2 months, while the other men continued to receive hCG until the repeat TESE. The second TESE was successful in 19 % of men who received this salvage hormonal therapy (Fig. 2). In our preliminary report, the number of men who exhibited hypospermatogenesis and late MA was relatively high compared to Sertoli cell‐only (SCO) syndrome and early MA [49]. A multi‐institutional prospective study, including SCO and early MA, to validate this protocol is on‐going, and the results will be reported in the near future. Suzuki et al. reported an 87 % sperm retrieval rate for the second micro‐TESE (Annual Meeting of Japan Society for Reproductive Medicine, 2013).
Figure 1.
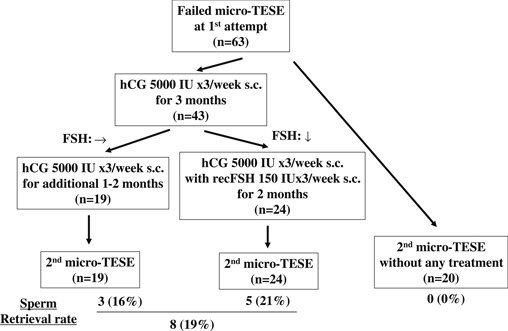
Salvage hormonal treatment protocol, patient selection, and the accumulated outcome
Figure 2.
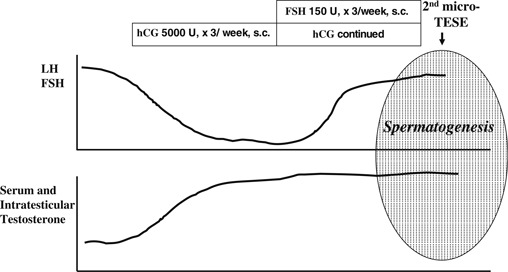
Hormonal changes during salvage hormonal therapy
Proposed mechanisms of hormonal therapy for NOA
The first step in spermatogenesis is spermatogonial proliferation. This step can be evaluated by detecting the proliferating cell nuclear antigen (PCNA) expression in humans [50]. PCNA is expressed in the nucleus during all phases of the cell cycle, with a maximum expression in the S‐ and G2‐phases. In the normal seminiferous epithelium, PCNA is expressed in the nuclei of mitotic spermatogonia primary spermatocytes. Given the observation that a higher expression level of spermatogonial PCNA is associated with increased germ cell maturation [50, 51] and a higher sperm output [52], the increased expression of PCNA may have positive effects on spermatogenesis. Previous studies have shown that up to 40–50 % of the spermatogonia were positive for PCNA in the testes of men with normal spermatogenic function; however, the PCNA expression was decreased in the NOA group [27, 50, 51, 52, 53, 54]. Spermatogonial PCNA expression in men has been shown to be gonadotropin‐independent [53, 54], whereas we showed that the spermatogonial expression of PCNA increased significantly after hCG‐based hormonal therapy [27]. In agreement with previously reported findings that FSH stimulates DNA synthesis in rat spermatogonia and spermatocytes in vitro [55], the stimulatory effect of rhFSH on spermatogonial PCNA expression was also observed in our series [27]. Continuous overnight monitoring of gonadotropin secretion showed that the decreased relative gonadotropin pulse amplitude observed in patients with NOA was caused by high basal gonadotropin levels [49]. This decrease indicates that the stimulation of Leydig and Sertoli cells by gonadotropin is paradoxically weak because of the down‐regulation of receptors [56]. Therefore, the exogenous administration of rhFSH is likely to stimulate Sertoli cells, even under hypergonadotropic conditions.
In the absence of testosterone or AR, spermatogenesis does not proceed beyond the meiosis stage [20, 21, 22]. Taken together with our result of salvage hormonal therapy, elevation of ITT may play a partial, but important role in the steps of spermiogenesis [27, 49], suggesting that the temporal expression and localization of AR are also important. AR in human testes is localized to the Sertoli cells, but not germ cells, indicating that ITT acts indirectly through Sertoli cells. Testosterone also plays a role in the final maturation of Sertoli cells [57], and both testosterone and FSH have additive effects on SCAR expression [58]. Rather than FSH stimulation, SCAR expression has been shown to be primarily dependent on the ITT level [59]. Furthermore, there was no significant association between ITT and SCAR expression in our study [15]. The exogenous administration of rhFSH had a positive effect on SCAR expression in NOA, as determined using human testicular samples obtained at micro‐TESE [15, 27]. In our experience, the basal ITT and AR expression levels had no predictive value for sperm recovery [15, 27]; however, the ITT after medical treatment is predictive of successful sperm retrieval by micro‐TESE.
Conclusion
Variable histological patterns in different tubules, even in the same individual, may explain the poor correlation of TESE results with histological patterns. Pathological information at micro‐TESE is extremely important to counsel the patients who need further therapy and to also establish a new hormonal treatment (Table 1). Micro‐TESE can only “seek” sperm, but not “make” sperm. One of the reasons why we call hormonal treatment for NOA empirical, but not specific, is a lack of understanding of the endocrine regulation that induces spermatogenesis. To reveal the pathophysiology of NOA, clinical trials to establish useful hormonal treatment regimens for NOA patients should use strict patient selection criteria.
Table 1.
Pathological findings based on our current protocol for men where micro‐TESE failed to retrieve sperm
| Pathological findings | Treatment options |
|---|---|
| Hypospermatogenesis | (1) Redo micro‐TESE, (2) Clomiphene or hCG therapy prior to 2nd micro‐TESE |
| Late maturation arrest | (1) Clomiphene or hCG therapy prior to 2nd micro‐TESE |
| Early maturation arrest | (1) Developing an FSH‐based hormonal therapy prior to 2nd micro‐TESE |
| Sertoli cell only | (1) Not recommend further treatment, (2) Redo micro‐TESE with agreement of extremely low SRR |
Micro‐TESE microdissection testicular sperm extraction, SRR sperm retrieval rate
Conflict of interest
Koji Shiraishi declares that he has no conflict of interest.
Human rights statements and informed consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national), and with the Helsinki Declaration of 1964 and its later amendments. Informed consent was obtained from all patients for being included in the study.
Animal studies
No animal experiments were used in this study.
References
- 1. Schlegel PN. Nonobstructive azoospermia: a revolutionary surgical approach and results. Semin Reprod Med, 2009, 27, 165–170 10.1055/s‐0029‐1202305 [DOI] [PubMed] [Google Scholar]
- 2. Shiraishi K, Oka S, Matsuyama H. Assessment of quality of life during gonadotrophin treatment for male hypogonadotrophic hypogonadism. Clin Endocrinol (Oxf), 2014, 81 (2) 259–265 10.1111/cen.12435 [DOI] [PubMed] [Google Scholar]
- 3. McLachlan RI. The endocrine control of spermatogenesis. Baillieres Best Pract Res Clin Endocrinol Metab, 2000, 14, 345–362 10.1053/beem.2000.0084 [DOI] [PubMed] [Google Scholar]
- 4. Hussein A, Ozgok Y, Ross L, Rao P, Niederberger C. Optimization of spermatogenesis‐regulating hormones in patients with non‐obstructive azoospermia and its impact on sperm retrieval: a multicentre study. BJU Int, 2013, 111, 110–114 10.1111/j.1464‐410X.2012.11485.x [DOI] [PubMed] [Google Scholar]
- 5. Reifsnyder JE, Ramasamy R, Husseini J, Schlegel PN. Role of optimizing testosterone before microdissection testicular sperm extraction in men with non‐obstructive azoospermia. J Urol, 2012, 188, 532–536 10.1016/j.juro.2012.04.002 [DOI] [PubMed] [Google Scholar]
- 6. Ramasamy R, Ricci JA, Palermo GD, Gosden LV, Rosenwaks Z, Schlegel PN. Successful fertility treatment for Klinefelter's syndrome. J Urol, 2009, 182, 1108–1113 10.1016/j.juro.2009.05.019 [DOI] [PubMed] [Google Scholar]
- 7. Shiraishi K, Matsuyama H. Local expression of epidermal growth factor‐like factors in human testis and its role in spermatogenesis. J Androl, 2012, 33, 66–73 10.2164/jandrol.110.011981 [DOI] [PubMed] [Google Scholar]
- 8. Silva FR, Leite LD, Wassermann GF. Rapid signal transduction in Sertoli cells. Eur J Endocrinol, 2002, 147, 425–433 10.1530/eje.0.1470425 [DOI] [PubMed] [Google Scholar]
- 9. Takeda H, Chodak G, Mutchnik S, Nakamoto T, Chang C. Immunohistochemical localization of androgen receptors with mono‐ and polyclonal antibodies to androgen receptor. J Endocrinol, 1990, 126 (1) 17–25 10.1677/joe.0.1260017 [DOI] [PubMed] [Google Scholar]
- 10. Ruizeveld Winter JA, Trapman J, Vermey M, Mulder E, Zegers ND, Kwast TH. Androgen receptor expression in human tissues: an immunohistochemical study. J Histochem Cytochem, 1991, 39, 927–936 10.1177/39.7.1865110 [DOI] [PubMed] [Google Scholar]
- 11. Iwamura M, Abrahamsson P‐A, Benning M, Cockett AT, Sant'Agnese PA. Androgen receptor immunostaining and its tissue distribution in formalin fixed, paraffin‐embedded sections after microwave treatment. J Histochem Cytochem, 1994, 42, 783–788 10.1177/42.6.8189040 [DOI] [PubMed] [Google Scholar]
- 12. Roijen JH, Assen S, Kwast TH, Rooij DG, Boersma WJA, Vreeburg JTM, Weber RFA. Androgen receptor immunoexpression in the testes of subfertile men. J Androl, 1995, 16, 510–516 [PubMed] [Google Scholar]
- 13. Kimura N, Mizokami A, Oonuma T, Sasano H, Nagura H. Immunocytochemical localization of androgen receptor with polyclonal antibody in paraffin‐embedded human tissues. J Hisiochem Cytochem, 1993, 41, 671–678 10.1177/41.5.8468448 [DOI] [PubMed] [Google Scholar]
- 14. Guillaume E, Pineau C, Evrard B, Dupaix A, Moertz E, Sanchez JC, Hochstrasser DF et al. Cellular distribution of translationally controlled tumor protein in rat and human testes. Proteomics, 2001, 1, 880–890 10.1002/1615‐9861(200107)1:7<880::AID‐PROT880>3.0.CO;2‐2 [DOI] [PubMed] [Google Scholar]
- 15.Kato Y, Shiraishi K, Matsuyama H. Expression of testicular androgen receptor in non‐obstructive azoospermia and its change after hormonal therapy. Andrology 2014, in press. [DOI] [PubMed]
- 16. Singh J, Handelsman DJ. Neonatal administration of FSH increases Sertoli cell numbers and spermatogenesis in gonadotropin‐deficient (hpg) mice. J Endocrinol, 1996, 151, 37–48 10.1677/joe.0.1510037 [DOI] [PubMed] [Google Scholar]
- 17. McLachlan RI, O'Donnell L, Meacham SJ, Stanton PG, Krester DM, Pratis K et al. Identification of specific sites of hormonal regulation in spermatogenesis in rats, monkeys, and man. Recent Prog Horm Res, 2002, 57, 149–179 10.1210/rp.57.1.149 [DOI] [PubMed] [Google Scholar]
- 18. Griswold MD Griswold MD. Perspective on the function of Sertoli cells. Sertoli cell biology, 2005. San Diego: Elsevier Science; 15–18 [Google Scholar]
- 19. O'shaughnessy PJ, Verhoeven G, Gerndt K, Monteiro A, Abel MH. Direct action through the Sertoli cells is essential for androgen stimulation of spermatogenesis. Endocrinology, 2010, 151, 2343–2348 10.1210/en.2009‐13332871953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chang C, Chen YT, Yeh SD, Xu Q, Wang RS, Guillou F et al. Infertility with defective spermatogenesis and hypotestosteronemia in male mice lacking the androgen receptor in Sertoli cells. Proc Natl Acad Sci USA, 2004, 101, 6876–6881 10.1073/pnas.0307306101406435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gendt K, Swinnen JV, Saunders PT, Schoonjans L, Dewerchin M, Devos A, Tan K, Atanassova N et al. A Sertoli cell‐selective knockout of the androgen receptor causes spermatogenic arrest in meiosis. Proc Natl Acad Sci USA, 2004, 101, 1327–1332 10.1073/pnas.0308114100337052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holdcraft RW, Braun RE. Androgen receptor function is required in Sertoli cells for the terminal differentiation of haploid spermatids. Development, 2004, 131, 459–467 10.1242/dev.00957 [DOI] [PubMed] [Google Scholar]
- 23. Hazra R, Corcoran L, Robson M, McTavish KJ, Upton D, Handelsman DJ, Allan CM. Temporal role of Sertoli cell androgen receptor expression in spermatogenic development. Mol Endocrinol, 2013, 27, 12–24 10.1210/me.2012‐1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jarow JP, Chen H, Rosner TW, Trentacoste S, Zirkin BR. Assessment of the androgen environment within the human testis: minimally invasive method to obtain intratesticular fluid. J Androl, 2001, 22, 640–645 [PubMed] [Google Scholar]
- 25. Coviello AD, Matsumoto AM, Bremner WJ, Herbst KL, Amory JK, Anawalt BD et al. Low‐dose human chorionic gonadotropin maintains intratesticular testosterone in normal men with testosterone‐induced gonadotropin suppression. J Clin Endocrinol Metab, 2005, 90, 595–602 10.1210/jc.2004‐1857 [DOI] [PubMed] [Google Scholar]
- 26. Matthiesson KL, McLachlan RI, O'Donnell L, Frydenberg M, Robertoson DM, Stanton PG et al. The relative roles of follicle‐stimulating hormone and luteinizing hormone in maintaining spermatogonial maturation and spermiation in normal men. J Clin Endocrinol Metab, 2006, 91, 3962–3969 10.1210/jc.2006‐1145 [DOI] [PubMed] [Google Scholar]
- 27. Shinjo E, Shiraishib K, Matsuyama H. The effect of human chorionic gonadotropin‐based hormonal therapy on intratesticular testosterone levels and spermatogonial DNA synthesis in men with non‐obstructive azoospermia. Andrology, 2013, 1, 929–935 10.1111/j.2047‐2927.2013.00141.x [DOI] [PubMed] [Google Scholar]
- 28. Sigman M, Vance ML. Medical treatment of idiopathic infertility. Urol Clin North Am, 1987, 14, 459–469 [PubMed] [Google Scholar]
- 29. Chua ME, Escusa KG, Luna S, Tapia LC, Dofitas B, Morales M. Revisiting oestrogen antagonists (clomiphene or tamoxifen) as medical empiric therapy for idiopathic male infertility: a meta‐analysis. Andrology, 2013, 1, 749–757 10.1111/j.2047‐2927.2013.00107.x [DOI] [PubMed] [Google Scholar]
- 30. Raman JD, Schlegel PN. Aromatase inhibitors for male infertility. J Urol, 2002, 167, 624–629 10.1016/S0022‐5347(01)69099‐2 [DOI] [PubMed] [Google Scholar]
- 31. Pavlovich CP, King P, Goldstein M, Schlegel PN. Evidence of a treatable endocrinopathy in infertile men. J Urol, 2001, 165, 837–841 10.1016/S0022‐5347(05)66540‐8 [PubMed] [Google Scholar]
- 32. Schiff JD, Palermo GD, Veeck LL, Goldstein M, Rosenwaks Z, Schlegel PN. Success of testicular sperm extraction [corrected] and intracytoplasmic sperm injection in men with Klinefelter syndrome. J Clin Endocrinol Metab, 2005, 90, 6263–6267 10.1210/jc.2004‐2322 [DOI] [PubMed] [Google Scholar]
- 33. Cao YX, Zhang ZG. A successful pregnancy outcome using testicular sperm from an infertile male pretreated with hCG. Arch Androl, 2007, 53, 1–3 10.1080/01485010600889076 [DOI] [PubMed] [Google Scholar]
- 34. Selman HA, Cipollone G, Stuppia L, Santo M, Sterzik K, El‐Danasouri I. Gonadotropin treatment of an azoospermic patient with a Ychromosome microdeletion. Fertil Steril, 2004, 82, 218–219 10.1016/j.fertnstert.2003.11.055 [DOI] [PubMed] [Google Scholar]
- 35. Efesoy O, Cayan S, Akbay E. The efficacy of recombinant human follicle stimulating hormone in the treatment of various types of male‐factor infertility at a single university hospital. J Androl, 2009, 30, 679–684 10.2164/jandrol.108.007278 [DOI] [PubMed] [Google Scholar]
- 36. Weiss J, Axelrod L, Whitcomb RW, Harris PE, Crowley WF, Jameson JL. Hypogonadism caused by a single amino acid substitution in the β subunit of luteinizing hormone. N Engl J Med, 1992, 326, 179–183 10.1056/NEJM199201163260306 [DOI] [PubMed] [Google Scholar]
- 37. Valdes‐Socin H, Salvi R, Daly AF, Valdes‐Socin H, Salvi R, Daly AF et al. Hypogonadism in a patient with a mutation in the luteinizing hormone β subunit gene. N Engl J Med, 2004, 351, 2619–2625 10.1056/NEJMoa040326 [DOI] [PubMed] [Google Scholar]
- 38. Achard C, Courtillot C, Lahuna O, Meéduri G, Soufir JC et al. Normal spermatogenesis in a man with mutant luteinizing hormone. N Engl J Med, 2009, 361, 1856–1863 10.1056/NEJMoa0805792 [DOI] [PubMed] [Google Scholar]
- 39. Lofrano‐Porto A, Barra GB, Giacomini LA, Nascimento PP, Latronico AC, Casulari LA et al. Luteinizing hormone β mutation and hypogonadism in men and women. N Engl J Med, 2007, 357, 897–904 10.1056/NEJMoa071999 [DOI] [PubMed] [Google Scholar]
- 40. Basciani S, Watanabe M, Mariani S, Paseri M, Persichetti A, Fiore D et al. Hypogonadism in a patient with two novel mutations of the lutteinizing hormone β‐subunit gene expressed in a compound heterozygous form. J Clin Endocrinol Metab, 2012, 97, 3031–3038 10.1210/jc.2012‐1986 [DOI] [PubMed] [Google Scholar]
- 41. Shiraishi K, Naito K. Fertile eunuch syndrome with the mutations (Trp8Arg and Ile15Thr) in the beta subunit of luteinizing hormone. Endocr J, 2003, 50, 733–737 10.1507/endocrj.50.733 [DOI] [PubMed] [Google Scholar]
- 42. Lamminen T, Jokinen P, Jiang M et al. Human FSHβsubunit gene is highly conserved. Mol Hum Reprod, 2005, 11, 601–605 10.1093/molehr/gah198 [DOI] [PubMed] [Google Scholar]
- 43. Matthews CH, Borgato S, Beck‐Peccoz P, Adams M, Tone Y, Gambino G et al. Primary amenorrhoea and infertility due to a mutation in the b‐subunit of follicle‐stimulating hormone. Nat Genet, 1993, 5, 83–86 10.1038/ng0993‐83 [DOI] [PubMed] [Google Scholar]
- 44. Phillip M, Arbelle JE, Segev Y, Parvari R. Male hypogonadism due to a mutation in the gene for the b‐subunit of follicle stimulating hormone. N Engl J Med, 1998, 338, 1729–1732 10.1056/NEJM199806113382404 [DOI] [PubMed] [Google Scholar]
- 45. Layman LC, Lee EJ, Peak DB, Namnoum AB, Vu KV, Lingen BL et al. Delayed puberty and hypogonadism caused by mutations in the follicle‐stimulating hormone b‐subunit gene. N Engl J Med, 1997, 337, 607–611 10.1056/NEJM199708283370905 [DOI] [PubMed] [Google Scholar]
- 46. Lindstedt G, Nyström E, Matthews C, Ernest I, Janson PO, Chatterjee K. Follitropin (FSH) deficiency in an infertile male due to FSHb gene mutation. A syndrome of normal puberty and virilization but underdeveloped testicles with azoospermia, low FSH but high lutropin and normal serum testosterone concentrations. Clin Chem Lab Med, 1998, 36, 663–665 10.1515/CCLM.1998.118 [DOI] [PubMed] [Google Scholar]
- 47. Themmen AP, Huhtaniemi IT. Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary‐gonadal function. Endocr Rev, 2000, 21, 551–583 10.1210/edrv.21.5.0409 [DOI] [PubMed] [Google Scholar]
- 48. Foresta C, Bettella A, Spolaore D, Merico M, Rossato M, Ferlin A. Suppression of the high endogenous levels of plasma FSH in infertile men are associated with improved Sertoli cell function as reflected by elevated levels of plasma inhibin B. Hum Reprod, 2004, 19, 1431–1437 10.1093/humrep/deh255 [DOI] [PubMed] [Google Scholar]
- 49. Shiraishi K, Ohmi C, Shimabukuro T, Matsuyama H. Human chorionic gonadotrophin treatment prior to microdissection testicular sperm extraction in non‐obstructive azoospermia. Hum Reprod, 2012, 27, 331–339 10.1093/humrep/der404 [DOI] [PubMed] [Google Scholar]
- 50. Steger K, Aleithe I, Behre H, Bergmann M. The proliferation of spermatogonia in normal and pathological human seminiferous epithelium: an immunohistochemical study using monoclonal antibodies against Ki‐67 protein and proliferating cell nuclear antigen. Mol Hum Reprod, 1998, 4, 227–233 10.1093/molehr/4.3.227 [DOI] [PubMed] [Google Scholar]
- 51. Bar‐Shira Maymon B, Yogev L, Yavetz H, Lifschitz‐Mercer B, Schreiber L, Kleiman S et al. Spermatogonial proliferation patterns in men with azoospermia of different etiologies. Fertil Steril, 2003, 80, 1175–1180 10.1016/S0015‐0282(03)02161‐7 [DOI] [PubMed] [Google Scholar]
- 52. Salama N, Tsuji M, Tamura M, Kagawa S. Prolifelating cell nuclear antigen in testes of infertile men with varicocele. Scand J Urol Nephrol, 2003, 37, 48–52 10.1080/00365590310008695 [DOI] [PubMed] [Google Scholar]
- 53. Takagi S, Itoh N, Kimura M, Sasao T, Tsukamoto T. Spermatogonial proliferation and apoptosis in hypospermatogenesis associated with nonobstructive azoospermia. Fertil Steril, 2001, 76, 901–907 10.1016/S0015‐0282(01)02732‐7 [DOI] [PubMed] [Google Scholar]
- 54. Ruwanpura SM, McLachlan R, Stanton PG, Meacham SJ. Follicle‐stimulating hormone affects spermatogonial survival by regulating intrinsic apoptotic pathway in adults rats. Biol Reprod, 2008, 78, 705–713 10.1095/biolreprod.107.065912 [DOI] [PubMed] [Google Scholar]
- 55. Henriksén K, Kangasniemi M, Parvinen M, Kaipa A, Hakovirta H. In vitro, follicle‐stimulating hormone prevents apoptosis and stimulates deoxyribonucleic acid synthesis in the rat seminiferous epithelium in a stage‐specific fashion. Endocrinology, 1996, 137 (5) 2141–2149 [DOI] [PubMed] [Google Scholar]
- 56. Freeman DA, Ascoli M. Desensitization of steroidogenesis in cultured Leydig tumor cells: role of cholesterol. Proc Natl Acad Sci USA, 1982, 79, 7796–7800 10.1073/pnas.79.24.7796347435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sharpe RM, McKinnell C, Kivlin C, Fisher JS. Proliferation and functional maturation of Sertoli cells, and their relevance to disorders of testis function in adulthood. Reproduction, 2003, 125, 769–784 10.1530/rep.0.1250769 [DOI] [PubMed] [Google Scholar]
- 58. Verhoeven G, Willems A, Denolet E, Swinnen JV, Gendt K. Androgens and spermatogenesis: lessons from transgenic mouse models. Philos Trans R Soc Lond B Biol Sci, 2010, 365, 1537–1556 10.1098/rstb.2009.01172871915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hill CM, Anway MD, Zirkin BR, Brown TR. Intratesticular androgen levels, androgen receptor localization, and androgen receptor expression in adult rat Sertoli cells. Biol Reprod, 2004, 71, 1348–1358 10.1095/biolreprod.104.029249 [DOI] [PubMed] [Google Scholar]