

Abstract
Aims: Many progressive neurological disorders, including Alzheimer's disease (AD), Huntington's disease, and Parkinson's disease (PD), are characterized by accumulation of insoluble protein aggregates. In prospective trials, the cyclooxygenase inhibitor aspirin (acetylsalicylic acid) reduced the risk of AD and PD, as well as cardiovascular events and many late-onset cancers. Considering the role played by protein hyperphosphorylation in aggregation and neurodegenerative diseases, and aspirin's known ability to donate acetyl groups, we asked whether aspirin might reduce both phosphorylation and aggregation by acetylating protein targets.
Results: Aspirin was substantially more effective than salicylate in reducing or delaying aggregation in human neuroblastoma cells grown in vitro, and in Caenorhabditis elegans models of human neurodegenerative diseases in vivo. Aspirin acetylates many proteins, while reducing phosphorylation, suggesting that acetylation may oppose phosphorylation. Surprisingly, acetylated proteins were largely excluded from compact aggregates. Molecular-dynamic simulations indicate that acetylation of amyloid peptide energetically disfavors its association into dimers and octamers, and oligomers that do form are less compact and stable than those comprising unacetylated peptides.
Innovation: Hyperphosphorylation predisposes certain proteins to aggregate (e.g., tau, α-synuclein, and transactive response DNA-binding protein 43 [TDP-43]), and it is a critical pathogenic marker in both cardiovascular and neurodegenerative diseases. We present novel evidence that acetylated proteins are underrepresented in protein aggregates, and that aggregation varies inversely with acetylation propensity after diverse genetic and pharmacologic interventions.
Conclusions: These results are consistent with the hypothesis that aspirin inhibits protein aggregation and the ensuing toxicity of aggregates through its acetyl-donating activity. This mechanism may contribute to the neuro-protective, cardio-protective, and life-prolonging effects of aspirin. Antioxid. Redox Signal. 27, 1383–1396.
Keywords: : aspirin (acetylsalicylic acid), (protein) acetylation, (protein) phosphorylation, (protein) aggregation, neurodegeneration, inflammation
Introduction
Acetylsalicylic acid (ASA or aspirin), a cyclooxygenase inhibitor introduced more than a century ago, is widely used for the prevention of primary and especially secondary cardiovascular events. Several large prospective trials have also demonstrated protective effects against Alzheimer's disease (AD) and Parkinson's disease (PD), as well as a variety of cancers (45). A meta-analysis of primary-prevention trials with aspirin suggested a 7% decrease in all-cause mortality (69); the survival benefits were more pronounced and significant for men with type 2 diabetes (56). A three-site study of genetically heterogeneous male mice demonstrated significant life extension (8%, p < 0.01) by aspirin (70). We and others have found that aspirin and an aspirin pro-drug significantly increase the longevity of wild-type Caenorhabditis elegans (∼21% and 23%, respectively) (5, 76).
Innovation.
Aspirin is a highly effective prospective treatment that is used to reduce the incidence of Alzheimer's and Parkinson's diseases, but the mechanisms by which it confers protection remain controversial. We now show that a well-established property of aspirin, the donation of its acetyl group to diverse proteins, broadly suppresses protein aggregation. This appears to be a general mechanism, since interventions that increase acetylation lower protein aggregation, whereas interference with acetyltransferases elevates it. Although there are individual exceptions, on average, acetylation tends to oppose phosphorylation of proteins, and to reduce aggregation. These principles may guide the design of drugs with improved anti-aggregative properties.
Mechanisms of these protective effects of aspirin remain conjectural, although abatement of reactive oxygen species (ROS), insulin-like signaling, and Jun/mitogen-activated protein kinase (MAPK) signaling appear to contribute to life extension (5, 76). Aspirin possesses a variety of anti-inflammatory, anti-oxidant, and acetylation-mediated properties (3). It is possible that aspirin, a nonselective cyclooxygenase inhibitor, suppresses inflammation by redirecting arachidonate metabolism from biosynthesis of prostaglandins to the formation of lipoxins (27). Aspirin also reduces oxidative damage and dampens ROS-mediated signaling (65). Aspirin-mediated lifespan extension in C. elegans appears to depend in large measure on acetylation (5), requires adenosine monophosphate-activated protein kinase (AMPK), and may involve a dietary-restriction pathway by altering the ATP/AMP ratio (76).
Aspirin lowers the risk of developing solid cancers, including those of the colon, breast, lung, and prostate (31, 35, 45), which has been attributed to its inhibition of NF-κB (67) and Wnt/β-catenin signaling (9, 52), and also to inhibition of platelet aggregation (62). Aspirin acetylates a large number of proteins (3), including numerous mitochondrial proteins (77). Disruption of mitochondrial protein acetylation triggers mitochondrial autophagy, or “mitophagy” (77). A recent study showed that aspirin reduces SOD-1 aggregation in ALS via its acetylation (1), and other work implies that similar effects may apply to many other acetylation targets (5, 80).
Extending our previous report that aspirin reduces age-associated protein aggregation in a C. elegans model of Huntington's disease (5), we now ask whether it is also protective in other models of myotoxic and neurotoxic protein aggregation, including C. elegans strains expressing human tau, transactive response DNA-binding protein 43 (TDP-43), or amyloid beta peptide (Aβ1–42), and in cultured human neuroblastoma cells stably overexpressing the Swedish mutant of amyloid precursor protein (APPSw), as a human-cell model of β-amyloid aggregation.
Results
Protein aggregation and associated traits are ameliorated by aspirin
Protein aggregation increases during normal aging (4, 20, 48) and in neurodegenerative diseases (4, 23, 47, 48). In this study, we examined protein aggregation in C. elegans models expressing Aβ1–42, tau, TDP-43, or Q40::YFP (a transgenic synthetic protein comprising a tract of 40 glutamines [Q40] fused to yellow fluorescent protein [YFP]), and in cultured human neuroblastoma cells overexpressing APPSw.
We first tested the effects of aspirin on behavioral changes associated with aggregate formation in neurons of transgenic worm strains modeling neurodegeneration-associated protein aggregation. C. elegans strains expressing human tau, TDP-43, or Aβ1–42 in neurons show substantial deficits in chemotaxis—movement toward a chemo-attractant (23, 42), here n-butanol—which also decline with normal aging of wild-type worms (21, 58). In each model of neuropathic aggregation, aspirin prevented most of the deficits in responsiveness; typical results are shown in Figure 1, comparing vehicle controls (V) with groups treated with aspirin (ASA).
FIG. 1.
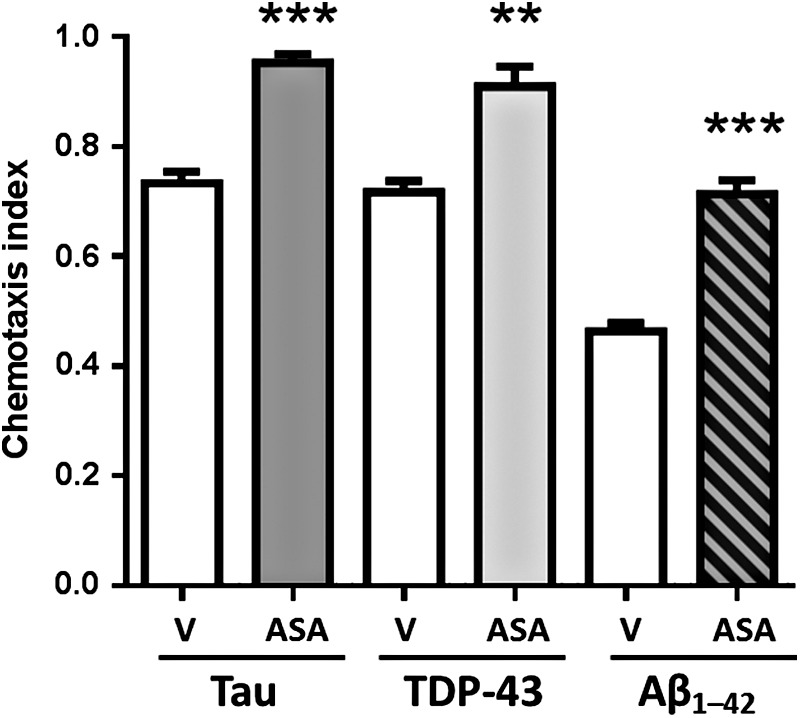
Aspirin treatment significantly protects nematodes from loss of chemotaxis after neuronal expression of human tau, TDP-43, or Aβ1–42 transgenes. Worms of each strain were exposed to aspirin (ASA, 0.5 mM) or vehicle control (V), continuously from the time of hatching. Chemotaxis toward n-butanol was assessed in worms on day 5 posthatch. Significance of control-ASA differences, determined by two-tailed heteroscedastic t-tests: **p ≤ 0.001; ***p ≤ 0.0001. Aβ1–42, amyloid beta peptide (amino acids 1–42).
Worms with pan-neuronal expression of human tau [which is hyperphosphorylated and forms aggregates in AD, PD, fronto-temporal dementia (FTD), and amyotrophic lateral sclerosis (ALS) (79)] lost 26% of the normal (wild-type) chemotactic response by day 5 posthatch; however, in identical worms treated with aspirin, only 5% was lost (p < 0.0001), rescuing more than 80% of the deficit. Similarly, a nematode strain with neuronal expression of TDP-43 [observed in aggregates of FTD, PD, and ALS (24, 37)] reduced chemotaxis by 29% at day 5, but only ∼8% in aspirin-treated worms (p = 0.0013), rescuing ∼70% of the deficit. Likewise, chemotaxis was reduced by 65% in worms expressing neuronal Aβ1–42 [characteristic of amyloid plaque in AD and some Lewy body dementias (39)] but only declined by 28% in aspirin-treated worms (p < 0.0001), indicating a 57% rescue.
We next studied worms that express an Aβ1–42 transgene in muscle cells (a model of amyloidopathy similar to inclusion body myositis). This strain (CL4176) develops paralysis within 2 days of induction (42) or in an age-dependent manner in uninduced worms (4). Aspirin treatment reduced paralysis by 66% after Aβ1–42 induction in C. elegans muscle (p ≈ 0.002, Fig. 2A), and it delayed paralysis accompanying age-dependent Aβ1–42 expression by nearly 50% (p < 0.001; Fig. 2B). The decrease in paralysis is not due to lower β-amyloid expression in treated worms, since the intensity of Aβ1–42 signal on Western blots (Fig. 2C) remained quite uniform as paralysis progressed in both ASA-treated and control worms. Amyloid aggregates, stained in situ with thioflavin T, were reduced by an average of 58% (37%, 60%, and 72% in three experiments, each p < 0.005) after aspirin exposure; a typical experiment is illustrated in Figure 2D.
FIG. 2.

Aspirin significantly impedes paralysis in Caenorhabditis elegans adults expressing Aβ1–42 in muscle, by reducing Aβ1–42 aggregation but not its expression. (A) CL4176 worms were maintained from hatch on 0.5-mM aspirin or vehicle (control). Paralysis was assessed 48 h after an upshift to 25°C (at the L3/L4 transition) to induce Aβ1–42 synthesis. (B) Without Aβ1–42 induction, CL4176 worms undergo age-dependent paralysis, presumably due to leaky expression of Aβ1–42 (4). This decline (dashed trace) was delayed by 0.5-mM aspirin (solid trace). Significance was tested by the two-tailed heteroscedastic t-test (A), or the Gehan-Wilcoxon log-rank test (B). (C) Aβ1–42 (monomer and oligomers) was quantified by ImageJ on Western blots by using a primary antibody to Aβ1–42 (AB11132; Abcam). Total signal was not altered in young-adult worms with aspirin treatment (ASA, 0.5 mM) relative to vehicle controls (V) at varying times after induction: 27 h (0% paralysis), 36 h (70%), or 42 h (100%). (D) Amyloid aggregation, measured as spectrally shifted thioflavin-T fluorescence 36 h postinduction, was reduced by 60% in aspirin-treated worms (p < 0.0001).
The most widely studied C. elegans model of age-dependent protein aggregation is the AM141 strain, in which muscle expression of Q40::YFP leads to progressive accrual of fluorescent protein aggregates and, eventually, to paralysis (47, 49). Aspirin treatment reduced the number of Q40::YFP aggregates by >30% (5), and it blocked or delayed the age-dependent increase in abundance and complexity (diversity in size and isoelectric point) of total aggregate proteins (Fig. 3). Each aspirin-treated group contained ∼60% less protein in sarcosyl-insoluble aggregates on adult day 3, and 80% less by day 7 (each p < 0.0001), than untreated controls.
FIG. 3.

Insoluble protein aggregates in C. elegans muscle expressing Q40::YFP, resolved on 2D gels, increase with age but are attenuated by aspirin treatment. Worms were maintained from hatch on 0.5-mM aspirin (+ASA, panels B, D, F) or vehicle (untreated controls, panels A, C, E), and sarcosyl-insoluble aggregates were isolated at days 1, 3, and 5 after the L4/adult molt (∼3.5, 5.5, and 7.5 days posthatch). Panels display the central regions of 2D gels separating aggregate fractions by isoelectric focusing (horizontal) over the pI ranges indicated, followed by polyacrylamide SDS-gel electrophoresis (vertical). 2D, two-dimensional; pI, isoelectric point; SDS, sodium dodecyl sulfate.
Aspirin treatment also reduced aggregate protein complexity as visualized by Sypro Ruby staining after aggregate proteins are separated on two-dimensional (2D) gels (see Fig. 3C–F). Since aspirin was shown to influence the expression of several genes (33, 44), we quantified Q40::GFP protein in AM141 and found no significant change with age in its expression (Fig. 4). This implies that the decrease in aggregate count in aspirin-treated AM141 cannot be explained by decreased steady-state levels of the protein that triggers aggregation, but must instead reflect less formation or stability of aggregates.
FIG. 4.
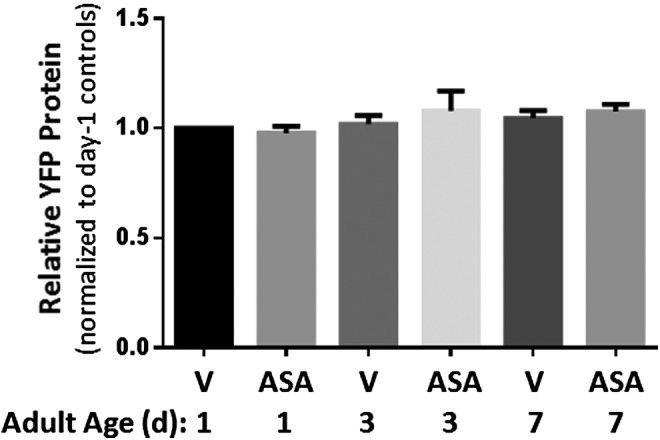
Q40::YFP expression does not change with age or aspirin treatment. AM141 worms (expressing Q40::YFP in muscle) were maintained continuously on 0.5-mM aspirin (ASA) or vehicle (V), from hatch. Worms were lysed at 1, 3, or 7 days of adult age, and total protein was extracted. Proteins separated by SDS-PAGE electrophoresis were blotted and probed with an antibody to GFP, which also recognizes YFP. No significant changes in Q40::YFP expression were observed with aspirin treatment or age.
Aspirin reduces amyloid aggregates in cultured human neural cells expressing APPSw
We next evaluated the effects of aspirin on amyloid accumulation in SH-SY5Y-APPSw, human neuroblastoma cells overexpressing a mutant amyloid precursor protein (APPSw) associated with familial AD. Elevated APPSw leads to substantial accrual of amyloid plaque, visualized by fluorescence microscopy after thioflavin-T staining on days 3 and 7 of culture. Thioflavin-fluorescence intensity in these cells was reduced 67% by aspirin treatment (Fig. 5; p < 0.0001).
FIG. 5.

Aspirin reduces amyloid accumulation in human neuroblastoma cells expressing an aggregation-prone mutation of amyloid precursor protein (APPSw). Amyloid foci were stained with thioflavin T in SH-SY5Y-APPSw cells that were treated for 2 days with (A) vehicle [V], or (B) 0.5-mM aspirin [ASA]. (C) Summary of data, quantified by ImageJ, compiled from three independent experiments. Error bars indicate standard error of the mean, and significance was based on a two-tailed heteroscedastic t-test, in each case treating each experiment as a single data point per group. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
We also examined the effect of aspirin on the quantity of total protein aggregates (large complexes insoluble in 1% sarcosyl) that accumulated in SH-SY5Y-APPSw cells, and we found that aspirin treatment reduced the low-mobility fractions of aggregate proteins, that is, the top 3 gel slices (Fig. 6; each p < 0.03 for combined data from three independent experiments), which are likely to be enriched for highly modified or cross-linked proteins. Proteins in the remainder of the gel were not significantly affected by aspirin treatment.
FIG. 6.

Aspirin reduces the abundance of low-mobility proteins in insoluble aggregates from neuroblastoma cells, but it does not prevent exclusion of acetylated proteins. Sarcosyl-insoluble aggregates were isolated from SH-SY5Y-APPSw cells after growth in medium containing 0.5-mM aspirin (ASA) or vehicle alone (V). (A) SyproRuby staining of total protein from SH-SY5Y-APPSw aggregates. (B) Ratios of ASA/control lane intensity in successive gel slices, ±SD (standard deviation) for three independent experiments. Poorly dissolved and low-mobility proteins were 20%–30% less abundant after ASA. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Aspirin but not salicylate reduces aggregation in SY5Y-APPSw cells and AM141 worms
Salicylate treatment confers roughly half of aspirin's protection against age-associated traits in C. elegans (5). We compared aspirin with salicylate for the reduction of aggregates in C. elegans strain AM141, a model of Huntington's disease, and in SH-SY5Y-APPSw neuroblastoma cells as a model of AD. In both C. elegans AM141 and human neuroblastoma cells, the reduction in aggregate levels was greater and more significant for aspirin treatment (ASA) than for salicylate (SA) (Fig. 7A, B).
FIG. 7.

Aspirin is more effective than salicylate in reducing protein aggregation in C. elegans and human-neuroblastoma models of neurodegeneration. (A) C. elegans strain AM141, expressing Q40::YFP in muscle, was exposed from hatch to 0.5-mM aspirin (ASA), 0.5-mM salicylate (SA), or vehicle control (V). YFP-fluorescence images of worms were captured, and aggregates per worm were counted with ImageJ software at day 1 of adulthood. (B) SH-SY5Y-APPSw neuroblastoma cells, expressing an aggregation-prone mutation of amyloid precursor protein (APPSw), were assessed for amyloid deposition after treatments (as in A). Cells were detached by brief digestion in trypsin-EDTA, removing pre-existing extracellular amyloid; they were replated at ∼20% confluence, allowed to attach, and finally grown for 48 h (to 70%–80% confluence) in the presence of drug or solvent. Formalin-fixed cells were stained for amyloid with thioflavin T; nuclei were counterstained with DAPI. Amyloid per cell is shown as the ratio of thioflavin-T to DAPI fluorescence (proportional to cell number). Aspirin reduced both aggregate count per worm (A) and amyloid per cell (B), relative to vehicle controls or salicylic acid. Significance (2-tail heteroscedastic t-tests): *p ≈0.02; **p ≈0.006; ****p < 0.0001. DAPI, 4′,6-diamidino-2-phenylindole; EDTA, ethylene diamine tetraacetic acid; NS, not significant.
Protein acetylation impedes aggregation
Phosphorylation of cytosolic (nonaggregated) proteins was measured by the fluorescence of protein bands that were stained with ProQ Diamond (Life Technologies), a phosphoprotein-specific dye. Aspirin treatment produced a consistent decrease in phosphorylation signal from most protein bands in wild-type C. elegans, extending to greater than fivefold (Fig. 8A) relative to vehicle-only controls (p < 0.001, combining data from three experiments).
FIG. 8.
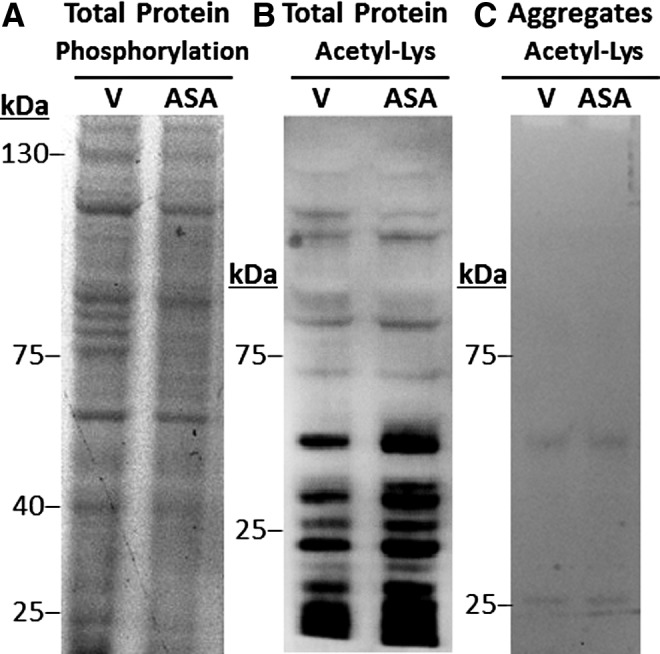
Aspirin reduces phosphorylation but increases acetylation of total protein in wild-type C. elegans adults. Total protein was isolated from worms that were maintained on 0.5-mM aspirin (ASA) or vehicle (V), and it was electrophoresed in one dimension. Phosphoprotein was visualized by staining with ProQ Diamond (A), whereas a subset of acetylated proteins was detected by immunostaining with an antibody to acetyl-lysine (B, C). Aggregate proteins for (C) were isolated as described in the “Materials and Methods” section.
A complementary increase in protein acetylation, visualized with an antibody to acetyl-lysine (Cell Signaling), was demonstrated for the most intensely stained aggregate proteins (Fig. 8B). Increased acetylation was expected after treatment with an acetyl donor, but the observation of a discrete set of medium- to high-mobility proteins targeted for acetylation by aspirin (lower half of Fig. 8B) was unanticipated. In contrast to total protein, very little acetylation was apparent in proteins from sarcosyl-insoluble aggregates, whether worms had been exposed to aspirin or not (Fig. 8C).
We then examined the effect of aspirin on the acetylation of aggregate proteins isolated from C. elegans strain CL4176, which forms amyloid aggregates ∼48 h after the induction of human Aβ1–42 in muscle cells (4). Although levels of total and cytosolic (unaggregated) proteins were not affected by aspirin exposure (Fig. 9A), the majority of protein bands in aggregates (detergent-soluble or -insoluble) were less abundant after aspirin treatment (Fig. 9B). Aggregate phosphoproteins comprise a subset of total aggregate proteins, many of which appear to be preferentially diminished by aspirin exposure (Fig. 9C). In these Aβ-expressing worms, just as in wild-type worms (Fig. 8), very few acetylated proteins were detected in aggregates (Fig. 9D), although one band detected by an antibody to acetyl-lysine (∼34 kDa, mainly in the insoluble fraction) was markedly elevated in aspirin-treated C. elegans (Fig. 9D, bands marked by white <).
FIG. 9.

Aspirin reduces abundance of most aggregate proteins and phosphoproteins, while increasing acetylation of a few proteins, in a nematode model of amyloidopathy. Aggregates from C. elegans strain CL4176, expressing Aβ1–42 in muscle, were isolated and partitioned by solubility in 1% sarcosyl. (A) Total and cytosolic (nonaggregated) protein profiles are unaffected by aspirin treatment. (B) Most aggregate proteins are reduced in quantity by aspirin treatment, especially in sarcosyl-insoluble fractions (examples are marked with “>,” and exceptions are marked with a white “<”). (C) Most phosphoproteins are reduced in aggregates after aspirin. (D) Very few proteins are acetylated in Aβ1–42 aggregates, ±aspirin.
Acetyl modifications were also significantly less abundant (by 30%–45%, p < 0.001) in soluble and insoluble aggregate proteins, relative to total protein, in SH-SY5Y-APPSw cells exposed only to vehicle (Fig. 10A, B). Aspirin treatment reduced those levels by a further twofold or more, relative to controls (Fig. 10B). These results suggest that post-translational acetylation makes proteins less prone to aggregation. Hyperphosphorylation of specific proteins can strongly promote their aggregation, for example, tau and TDP-43 in neurological diseases (2, 53), and increasing evidence indicates cross-talk between phosphorylation and acetylation, with effects on cardiovascular disease and neurological disorders (10, 57, 74).
FIG. 10.

Acetylated proteins are underrepresented in both soluble and insoluble aggregates of human neuroblastoma cells. Total proteins, cytosolic proteins, and sarcosyl-soluble and -insoluble aggregates were isolated from human neuroblastoma cells (SH-SY5YSw) after growth in medium containing 0.5-mM aspirin (ASA) or vehicle only (V). (A) Western blot of proteins from SH-SY5Y-APPSw cells, probed with an antibody to acetyl-lysine. (B) Bars indicate mean ± SD for lane integrals for gels (as in A) from three independent experiments, each normalized to the “total protein” control mean. Aggregate proteins show 35%–80% less acetylation than corresponding total-protein lanes, despite uniform protein loads. Significances, based on two-tailed heteroscedastic t-tests: **p = 0.001, ***p ≤ 0.0006. Asterisks above brackets refer to differences between the bars (groups) connected by each bracket. Asterisks below brackets (just above bars) compare each marked bar with its corresponding (V or ASA) “Total Protein” sample.
Aggregation of amyloid peptide Aβ1–42 is predicted to be opposed by acetylation
Amyloid β1–42 is a polypeptide that is believed to initiate the formation of toxic amyloid plaque in affected cerebral cortex regions of patients with AD (25). Acetylation of lysine-624 in APP modulates its cleavage to favor release of Aβ1–42 (34). We performed simulations to assess the possibility that aspirin-mediated acetylation of the same lysine (numbered as Lys28 in Aβ1–42) might also inhibit oligomerization of amyloid peptide and subsequent formation of insoluble aggregates. Acetylation at lysine-28 markedly reduced the propensity of Aβ1–42 to form tetrameric or octameric complexes (Fig. 11), as predicted by several computer-modeling approaches. First, oligomers modeled with docking software (HEX 6.1) were used as inputs for molecular-dynamic simulations in GROningen MAchine for Chemical Simulation (GROMACS), a versatile modeling package that reproduces observed protein-interaction properties with remarkable accuracy (4, 67). The simulations comprise 20-ns, three-dimensional random walks for every atom of each molecule in the simulation, constrained by all linked and nearby atoms in a fully solvated aqueous environment, and by realistic limits to free energy increase of the entire ensemble.
FIG. 11.

Acetylation reduces compactness and stability of Aβ1–42 oligomers. Molecular docking models and molecular-dynamic simulations indicate that lysine acetylation in Aβ1–42 (“Kac”) destabilizes its assembly into tetramers and octamers. Optimized docking models are shown for: (A) native Aβ1–42 tetramer, (B) acetylated Aβ1–42 tetramer, (C) native Aβ1–42 octamer, and (D) acetylated Aβ1–42 octamer. Molecular-dynamic simulations in GROMACS indicate increased dispersion (radius of gyration) for acetylated tetramer (E) and octamer (F), and decreased hydrogen bonding for acetylated octamer (G), relative to their unmodified forms. (E–G) Properties of lysine-acetylated oligomers (Kac) are shown in red, and those of unmodified Aβ oligomers are indicated in black.
Typical “snapshots” of structures (Fig. 11A–D) indicate that tetrameric and octameric complexes (Fig. 11A, C) of native Aβ1–42 are much more compact than those of monoacetylated [Kac28] peptide (Fig. 11B, D). In agreement with these predicted structures, the radius of gyration (Rg), a measure of structural dispersion, gradually declined for native Aβ1–42 oligomers during GROMACS simulations (black tracings in Fig. 11E, F), indicating condensation into more compact complexes (40, 73); whereas oligomers of the corresponding acetylated [Kac28] Aβ1–42 peptide (red tracings) displayed larger radii of gyration that also fluctuated more widely, and overall did not decline with time.
This indicates that the acetylated Aβ1–42 peptide fails to coalesce into the stable complexes formed by native Aβ1–42 (Fig. 11E, F). This difference in behavior can be attributed, in part, to the decline in hydrogen bonds during the course of the simulation (an average loss of 10–15 bonds per octamer) for Kac28, versus a small increase over time for native Aβ1–42 octamers (Fig. 11G). For proteins in general, there is a linear correlation between complex stability and the number of hydrogen bonds formed (54).
Protein aggregation and associated traits correlate inversely with overall protein acetylation
Diverse inhibitors of protein deacetylases and acetyl transferases, including both drugs and RNA-interference constructs targeting specific genes, were used to perturb the average protein acetylation state either upward or downward. To reduce acetylations, C. elegans adults were fed with bacteria that express RNA interference (RNAi) constructs targeting histone/protein acetyltransferase genes nath-5 and nat-6. To increase acetylations, adult worms were exposed to either (i) bacteria expressing RNAi constructs that individually target deacetylase genes hat-1, nceh-1, or sin-3; (ii) CTPB (N-(4-chloro-3-trifluoromethyl-phenyl)-2-ethoxy-benzamide; abcam), an activator of histone/protein acetyltransferases; or (iii) PDI, a cocktail of protein deacetylase inhibitors (sc-362323; SantaCruz Biotech.). After each treatment, or exposure to control bacteria that carry an empty vector, AM141 adults (expressing Q40::YFP in muscle) were assessed for aggregate counts, and CL4176 adults (expressing human Aβ1–42 in muscle) were tested for motility/paralysis.
The results (Fig. 12) indicate a perfect inverse concordance between the expected effects of treatments on protein acetylation and the observed levels of aggregate formation in strain AM141 (p < 0.02; Fig. 12A) or of its consequence, paralysis, measured in strain CL4176 (p < 0.01; Fig. 12B). All but one of these intervention effects were individually significant at p < 0.01 to p < 0.0005. RNAi knockdown of age-1, which encodes the catalytic subunit of class-I phosphatidylinositol 3-kinase and was reported to be a potent positive modulator of protein aggregation (49), is included in Figure 12A for comparison.
FIG. 12.

Interventions that elevate protein acetylation reduce aggregation, and vice versa. (A) In a C. elegans model of Huntington-like Q40 aggregation, the number of foci was reduced by RNAi knockdown of genes encoding protein deacetylases (sin-3, nceh-1) or by an enhancer of acetylation activity (CTPB); each of these interventions is expected to increase protein acetylation. Conversely, aggregate count was increased by RNAi targeting n-acetyltransferase genes (nat-6, nat-25), and knockdowns were expected to reduce acetylation. Significance was determined by two-tailed heteroscedastic t-tests: #p ≈0.07, *p ≤ 0.01, **p < 0.003, and ***p < 0.0005. (B) Fraction paralyzed after inhibition of acetyl transferase or deacetylase genes by RNAi or by chemical activators of acetyltransferases or inhibitors of deacetylases. Significance was determined by two-tailed heteroscedastic t-tests: *p < 0.01, **p < 0.002, and ***p < 0.0005. RNAi, RNA interference.
Discussion
Aspirin is a widely used, nonsteroidal anti-inflammatory compound that exhibits diverse biological activities and reduces or delays the onset of age-associated diseases, including cardiovascular disease, AD, ALS, PD, and a variety of cancers (1, 5, 8, 18, 59). Here, we present in vivo results for the nematode C. elegans (employing several models of neuropathic aggregation), and in vitro data for human SH-SY5Y-APPSw neuroblastoma cells overexpressing APPSw and forming amyloid plaque. Through these parallel studies, we sought to identify additional mechanisms by which aspirin may contribute to neuroprotection.
Although it is unlikely to be the sole mechanism, here we show that protein acetylation by aspirin contributes to its ability to delay or prevent neurotoxic protein aggregation. These effects are not limited to aspirin, but they are produced by modulation of protein acetylation with drugs and RNAi constructs that target acetyltransferases or deacetylases.
Among the diverse post-translational modifications of proteins, acetylation has been chiefly implicated in chromatin remodeling (66). Phosphorylation is an even more common protein modification that is instrumental in cell signaling via kinase cascades that either activate or (more rarely) inactivate their targets (30). Previous reports had indicated that aspirin alters phosphorylation of diverse key cellular proteins, including insulin receptor substrate-1, Bcl-2, MAPK, integrin receptors, and leukocyte-specific protein-1 (7, 16, 38, 46, 55, 60, 63, 71). We observed a decrease in the phosphorylation levels of several signaling proteins in human endothelial cells, after aspirin treatment (data not shown, manuscript in preparation). An inverse correlation between target-site acetylation and phosphorylation (implied by the earlier references and Fig. 8) could arise from local steric hindrance and/or allosteric effects. However, specific instances may also reflect cross-talk between sites on interacting proteins; as an example, acetylation of Sip2, the regulatory subunit of yeast SNF-1/Sip2 complex, reduces phosphorylation of Sch9 (an Akt ortholog), slowing growth and increasing longevity (41). Such complex interactions must have evolved in a piecemeal fashion, which would not explain the observed inverse correlation. An alternative rationale is that competitive suppression between phosphorylation and acetylation of the same protein may have evolved frequently and independently to create positive-feedback loops, each with two alternative stable states [e.g., see (72)].
Chromatin remodeling and the regulation of transcription are largely accomplished via addition and removal of site-specific methyl and acetyl groups on histone tails (32). These changes can have surprisingly “concerted” functional consequences; for example, knockout of histone deacetylase-2 in mice improves memory and accelerates learning (50). Nevertheless, many such studies have assumed, rather than demonstrated, that histones are the relevant deacetylation target—despite documentation of many other protein-deacetylase substrates (6, 26).
Recent studies have shown that Transcription Factor EB (E-box), which is involved in regulating lysosomal functions when deacetylated, promotes Aβ1–42 clearance from microglia (6). In a Drosophila model of Huntington's disease, “histone deacetylase inhibitors” were found to relieve neuronal degeneration induced by polyglutamate expansion (68). Acetylation was also shown to protect enzymes from inactivation arising from denaturation and aggregation in vitro (64).
In this study, we investigated whether the protection conferred by aspirin against neurodegenerative diseases (and perhaps other age-related pathologies) might reflect a lower propensity of acetylated proteins to become enmeshed in aggregates. Our rationale was that age-progressive neurodegenerative diseases, including AD, PD, and Huntington's disease, are accompanied by the gradual accumulation of neuronal or peri-neuronal protein aggregates or “inclusion bodies,” in many cases shown to be neurotoxic. For each neuropathy, characteristic aggregate constituents provide diagnostic markers. In some instances, for example, tau and TDP-43, hyperphosphorylation strongly promotes inclusion in aggregates, and we reasoned that acetylation may oppose de novo protein phosphorylations, thereby reducing aggregation. Although a recent study showed that acetylation of a specific residue can promote aggregation of hyperphosphorylated TDP-43 (17), this appears to be an unusual exception.
The simple hypothesis that phosphorylation most commonly favors aggregation, and is usually opposed by acetylation, is consistent with our observation of increased acetylation and decreased phosphorylation of total protein in aspirin-treated C. elegans (Figs. 8 and 9). Aspirin at 0.5 mM, within the range of human therapeutic doses, reduced the levels of aggregated proteins (Figs. 3–7) and rescued many aggregation-associated phenotypes, including behavioral traits, in four nematode models of diverse aggregation-mediated human diseases (Figs. 1 and 2). We also demonstrated in silico, through docking simulations and molecular-dynamic modeling, that a single acetylation can markedly destabilize oligomerization of Aβ1–42, the precursor of amyloid formation in AD.
Previous studies (chiefly from the Calabrese group) had shown that acetylcarnitine, an acetyl donor delivered from peroxisomes to mitochondria, responds to oxidative stress by altering the expression of diverse heat shock proteins and the NRF-2 transcription factor (5, 15, 19). Acetylcarnitine directly or indirectly regulates multiple genes that are involved in caloric restriction and longevity pathways, including sirtuin/mTOR signaling (5, 13–15). Acetylcarnitine-mediated acetylation may also post-translationally modulate the functions of proteins that are involved in oxidative stress response and longevity.
Proteasomal degradation is a key mechanism of proteostasis, and it accounts for removal of most unfolded proteins in eukaryotes (36). Although polyubiquitin-tagged proteins are cleared chiefly by 26S proteasomes (28), 20S proteasomes to which the proteasome activator PA200 has bound can degrade peptides and unstructured proteins that lack ubiquitin tags (22). This mechanism, which is critical in the clearance of histones during DNA repair and spermatogenesis, is ubiquitin-independent but requires acetylation of histones (61).
In principle, these diverse responses could be mediated by different activities of aspirin. Our data implicate acetylation as a key mechanism, based on the novel observation that protein aggregation and associated paralysis are reduced whenever deacetylase activities are inhibited (whether by pharmacologic or epigenetic means), but are elevated by inhibition of acetyltransferases (Fig. 12). From this, we infer that protein acetylation may be a useful biomarker of resistance to aggregation, and that acetyl donors may be a class of drug interventions that deserves further exploration. We conclude that general protein acetylation by aspirin is both necessary and sufficient to disrupt protein aggregation and to reduce the sequelae of toxicity and behavioral impairment—either by preventing the entry of acetylated proteins into aggregates or by promoting the degradation of aggregation-prone proteins via 20S proteasomes.
The opposing effects of protein phosphorylation and acetylation, with respect to aggregation, suggest a mechanism based on electrostatic interactions. Phosphorylated sites can form electrovalent (ionic) bonds with basic residues of other proteins, and might thus promote their co-aggregation, whereas acetylation is a more neutral modification (serving chiefly to neutralize the positive charges of lysines) that does not usually increase the aggregation propensity of proteins. Although a simplistic model (“phosphorylation promotes aggregation, acetylation opposes it”) provides a superficially adequate explanation of our results, it is unlikely to be the entire story. None of our data would exclude, for example, the possibility that a few critical phosphorylases are negatively regulated by acetylation, and/or key phosphatases are activated by acetylation.
Similarly, a relatively small repertoire of protein acetylases (conceivably just one, with multiple targets) may be negatively regulated by phosphorylation, and/or deacetylases might be positively regulated by phosphate additions. Indeed, any one or any combination of the interactions mentioned earlier could account for the drug and knockdown effects we have observed. It is clear, however, that aspirin acetylates many protein targets and reduces phosphorylation overall, and that acetylation of some subset of targets is responsible for reducing protein aggregation.
The results presented here highlight a general mechanistic target for interventions to reduce protein aggregation (Figs. 2, 3, 5–7, 10, and 12) and, therefore, rescue behavioral deficits arising from aggregation, for example, failure of chemotaxis in nematode models of neuronal aggregation seeded by tau, TDP-43, and Aβ1–42 (Fig. 1). Remarkably, aspirin delayed by almost 1.5-fold the age-dependent paralysis attributed to “leaky” expression of Aβ1–42 in muscle of uninduced worms (Fig. 2B). Taken together, these data establish a causal link between protein acetylation and a substantial reduction or delay of protein aggregation and its behavioral sequelae, and they thus define a novel therapeutic strategy to combat neurodegenerative diseases.
Materials and Methods
C. elegans strains
Wild-type Bristol N2, AM141 (unc-54/Q40::YFP), CL4176 (smg-1ts [myo-3/Aβ1–42/long 3′-untranslated region (UTR)]), and CL2355 (smg-1ts [snb-1/Aβ1–42/long 3′-UTR]) were obtained from the Caenorhabditis Genetics Center (CGC). Strains with CK10 expressing transgenic tau in neurons (Paex-3/Tau), and CK405 expressing TDP43 (Psnb-1/TDP-43) in neurons were kind gifts from Brian Kramer (37). All strains were maintained in 10-cm Petri dishes, on the surface of agar containing nematode growth medium (NGM) overlaid with Escherichia coli (OP50) at 20°C. Strain CL4176 was upshifted to 25°C at the L3/L4 transition to induce neuronal synthesis of Aβ1–42. Synchronized, well-fed worms were lysed on day 3 posthatch (day 1 of adulthood) to release eggs, placed on NGM agar seeded with E. coli OP50 and brought to 1.0-mM aspirin (Sigma, St. Louis, MO), or equilibrated with vehicle as a control.
Chemotaxis assay
Chemotaxis was assessed in transgenic strains expressing tau, TDP-43, or Aβ1–42 in all neurons, as previously described (4) Briefly, synchronized worms from hatch were grown on control (empty feeding vector) bacteria with or without aspirin. The aspirin stock solution was 0.5 M in 95% (v/v) ethanol, and it was diluted 1:1000 in NGM. Expression of Aβ1–42 was induced by an upshift to 25°C, as described earlier. Worms at day 5 posthatch were collected after multiple washes to remove bacteria, and they were assayed in 100-mm NGM-agar plates without bacteria. Worms (≥50) were placed near centers of plates spotted on one edge with ∼5 μl n-butanol (as chemo-attractant) mixed with sodium azide (0.34% w/v, final), and on the opposite edge with S buffer containing only 0.34% sodium azide. Assay plates were incubated at 20°C, and the chemotaxis index (CI) was scored every 30 min, expressed as CI = [(number of worms near attractant) − (number of worms near control)]/(total worms per plate) (21). Each experiment was performed at least three times.
Cell growth and harvesting
SH-SY5Y-APPSw (a gift from Dr. Steve Barger, University of Arkansas for Medical Sciences, Little Rock AR) is a human neuroblastoma cell line that was originally obtained from a 4-year-old girl, expressing a mutant amyloid precursor protein (APPSw) associated with familial AD, introduced as a stably integrated transgene. Cells were grown in DMEM containing 10% (v/v) fetal bovine serum at 37°C, containing 1-μM aspirin or ethanol vehicle (control). Cells were trypsinized before confluence and rinsed in buffer for replating or for harvesting.
Isolation of aggregated proteins
Cultured neuronal cells or C. elegans adults were collected, drained of excess liquid, flash frozen in liquid nitrogen, pulverized in a dry-ice-cooled mortar, and suspended in buffer with nonionic detergent (20-mM Hepes pH 7.4, 300-mM NaCl, 2-mM MgCl2, 1% [v/v] NP40, and protease/phosphatase inhibitors [CalBiochem]) at 0°C (11, 12, 49). Cuticle or cell debris was removed by brief centrifugation (5 min at 3000 rpm). After removal of cytosolic proteins that dissolved in NP40 (18 min centrifugation at 13,000 g), insoluble protein pellets were brought to pH 7.4 with 0.1-M Hepes buffer containing ionic detergent (1% sarcosyl [v/v] final concentration) and 5-mM ethylene diamine tetraacetic acid (EDTA), and they were centrifuged for 30 min at 100,000 g. The pellet and supernatant are the detergent-insoluble and -soluble fractions, respectively (37).
Fractions were individually equilibrated in Microcon spin columns with Laemmli loading buffer containing 2% (w/v) sodium dodecyl sulfate (SDS) and 50-mM freshly diluted dithiothreitol (DTT), heated for 2 min at 100°C to dissolve proteins, and resolved by polyacrylamide gel electrophoresis.
2D gel display
Aggregate proteins were dissolved in 8-M urea, 2-M thiourea, 2% v/v CHAPS, and ampholytes for isoelectric focusing to span the desired pH range (e.g., pH 3–10). The second dimension is a 4%–20% acrylamide gradient gel (Invitrogen). Gels were incubated with SYPRO Ruby (ThermoFisher) to stain total protein.
Paralysis assay
We initiated a synchronous CL4176 cohort by lysing gravid hermaphrodites in alkaline sodium hypochlorite; eggs were transferred onto 60-mm culture plates that were seeded with bacteria with or without aspirin. Muscle expression of Aβ1–42 was induced by upshifting worms from 20°C to 25°C at the L3–L4 transition, and resultant paralysis (i.e., movement of the head, but not the body, in response to touch) was scored at 36 h post-upshift (4). Each experiment was performed at least three times.
Visualization of aggregate reporters
Q40::YFP aggregates were counted, and fluorescence images were captured on an Olympus DP71 camera that was mounted on an Olympus BX51 fluorescence microscope. Incident light was filtered to 490 ± 20 nm and emitted light to 535 ± 30 nm. Protein aggregates were quantified from digital images using NIH ImageJ software. SH-SY5Y-APPSw cells expressing mutant amyloid precursor protein were plated and incubated for 24 h at 37°C to allow attachment and proliferation. Aspirin in ethanol, or the same amount of ethanol for controls, was added in minimal volume (final concentration of ethanol, v/v, 0.1%–0.3%), and 0.8 mg/ml thioflavin-T was added 24 h later. Fluorescence intensity was scored 15 min afterward. The excitation peak for thioflavin-T shifted to 450 nm, and the emission maximum shifted to 482 nm on amyloid binding (78).
Quantitation of thioflavin-T stained SY5Y-APPSw cells
SY5Y-APPSw cells were seeded at a density of 7000 cells/well in 96-well plates and cultured for 16 h before adding aspirin or salicylate. After growth for a further 1–2 days, cells were fixed in 4% paraformaldehyde (v/v), rinsed, and stained with thioflavin T (0.1%, w/v) for 10 min in the dark. After four washes in phosphate-buffered saline (PBS), cells were stained in DAPI (4′,6-diamidino-2-phenylindole), 10 min at 0.4 μg/ml, and washed 3 × in PBS. Fluorescence of Aβ amyloid stained by thioflavin-T was measured on a SpectraMax Plate Reader (440 nm excitation, 482 nm emission) and divided by the number of nuclei assessed by ImageJ analysis of DAPI-stained images (358 nm excitation, 461 nm emission). Data were combined for at least three independent experiments.
Affinity purification, protein fractionation, and Western blotting
Cells or age-synchronized worms were pelleted, drained, and flash frozen in liquid nitrogen. Proteins were extracted as described earlier, suspended in Laemmli loading buffer (see Isolation of aggregated proteins), and heated for 5 min at 100°C to dissolve proteins before gel electrophoresis in 1% SDS, 4%–12% polyacrylamide gradient gels (Life Technologies, Grand Island, NY).
Phosphoproteins were imaged by fluorescence after incubation with phosphoprotein-specific stain (Pro-Q Diamond; Life Technologies). Total protein was then visualized with SYPRO Ruby (Life Technologies). In each case, digital images (BioRad Digital Imager) were quantified with QuantityOne software (BioRad, Hercules, CA). Gels were also blotted onto nylon membranes, which were then probed with rabbit antibody to acetyl-lysine (Cell Signaling, Danvers, MA), followed by incubation with HRP-coupled murine secondary antibody against rabbit IgG (Sigma). Finally, filters were developed by using an HRP-imaging kit (BioRad).
Modeling of Aβ1–42
We simulated the structure of amyloid peptide Aβ1–42 and its acetylated form and assessed aggregation propensity and ability to form several distinct oligomers, by using GROMACS molecular-dynamic modeling.
Molecular docking simulations of Aβ1–42 oligomers
For docking simulation studies, the NMR-resolved structure of Aβ1–41 (1IYT) was retrieved from the Protein data base. We used Modeller 9.13 (University of California, San Francisco, CA) to build Aβ1–42 from the Aβ1–41 peptide. After energy minimization, Aβ1–42 (native) monomer was used for further studies. Vienna-PTM 2.0 server was used to build acetylated Lys28 (Kac28) Aβ1–42 (43). To make oligomers of Aβ1–42 (WT and Kac28), docking studies were performed by using Hex-8.0 with default parameters, with the sole exception that OPLS minimization was run in Hex-8.0 after each docking to derive the energy-minimized complex (http://hex.loria.fr/hex.php). Explicit molecular-dynamic simulations were performed on tetrameric and octameric complexes of Aβ1–42, both native and acetylated, at lysine 28 (Kac28).
Molecular-dynamic simulations
To check the dynamic behavior and aggregation propensity of both native Aβ1–42, and Aβ1–42 acetylated on lysine-28 (Kac28), explicit molecular-dynamic simulations were performed by using GROMACS 5.0 (73). Both tetramers and octamers of each Aβ1–42 were immersed in a cubic box of 0.7-nm sides, filled with SPC16 solvent, and finally neutralized by using NaCl for counterions. A modified GROMOS 54A7 force field was used for all simulations involving post-translational modifications (73). Each system was then energy minimized by using the steepest descent algorithm for ∼2000 steps and equilibrated with number of particles, volume, and temperature (NVT) and number of particles, pressure, and temperature (NPT) protocols for 200 ps before the actual run.
Molecular-dynamic simulations were performed by using the leap-frog integrator for ∼20 ns. Collection and analysis of parameters were as previously described (4). Output parameters such as radius of gyration, root-mean-square deviation, and hydrogen bonds were calculated within the GROMACS package. All structures were visualized by using Visual Molecular Dynamics (VMD) (29, 73).
Statistical analyses
All statistical analyses were performed using GraphPad Prism 6. Results are expressed as mean ± standard deviation, unless otherwise indicated. Differences between groups were generally evaluated for significance by two-tailed heteroscedastic t-tests, appropriate to samples of unequal or undetermined variance. In survival comparisons (Fig. 2B), inter-group differences were evaluated by the Gehan-Wilcoxon log-rank test. P values are given without adjustment for multiple comparisons, to enable the reader to make appropriate allowance for multiple endpoints.
Abbreviations Used
- 2D
two-dimensional
- Aβ1–42
amyloid beta peptide (amino acids 1–42)
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- AMPK
adenosine monophosphate-activated protein kinase
- APPSw
amyloid precursor protein (Sw: Swedish mutation, favoring AD and aggregation)
- ASA
acetylsalicylic acid
- CI
chemotaxis index
- DAPI
4′,6-diamidino-2-phenylindole
- DTT
dithiothreitol, a reducing agent
- EDTA
ethylene diamine tetraacetic acid, a chelating agent
- FTD
fronto-temporal dementia
- GROMACS
GROningen MAchine for Chemical Simulation, a command-line software package for molecular-dynamic simulations for >108 particles (www.gromacs.org)
- NGM
nematode growth medium
- PD
Parkinson's disease
- Q40::YFP
a transgenic synthetic protein comprising a tract of 40 glutamines (Q40) fused to YFP (yellow fluorescent protein)
- Rg
radius of gyration
- RNAi
RNA interference (procedure to “knock down” transcript levels in targeted genes)
- ROS
reactive oxygen species
- SD
standard deviation
- SDS
sodium dodecyl sulfate, a strong ionic detergent
- TDP-43
transactive response DNA-binding protein 43
- UTR
untranslated region
Acknowledgments
The authors thank Brian Kraemer (University of Washington and Puget Sound Veterans Healthcare Center, Seattle, WA) for generously providing C. elegans strains CK10 and CK405, and W. Sue T. Griffin for a careful reading of this article. This work was supported by grants (Merit Review and Senior Research Career Scientist Award) from the Dept. of Veteran Affairs to R.J.S.R., and by Program Project grant 2P01AG012411-17A1 (W.S.T. Griffin) from the National Institute on Aging (NIA/NIH). Additional support for research reported herein was provided by an Institutional Development Award (IDeA grant P20 GM109005) from the National Institute of General Medical Sciences (NIGMS/NIH). Support of High-Performance Computing (HPC) at the University of Arkansas at Little Rock Computational Research Center (CRC) was provided by the U.S. National Science Foundation (grants CRI CNS‐0855248, EPS‐0701890, EPS‐0918970, and MRI CNS‐0619069).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Abdolvahabi A, Shi Y, Rhodes NR, et al. Arresting amyloid with Coulomb's Law: acetylation of ALS-linked SOD1 by aspirin impedes aggregation. Biophys J 108: 1199–1212, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alavi Naini SM. and Soussi-Yanicostas N. Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid Med Cell Longev 2015: 151979, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alfonso LF, Srivenugopal KS, and Bhat GJ. Does aspirin acetylate multiple cellular proteins? (Review). Mol Med Rep 2:533–537, 2009 [DOI] [PubMed] [Google Scholar]
- 4.Ayyadevara S, Balasubramaniam M, Gao Y, et al. Proteins in aggregates functionally impact multiple neurodegenerative disease models by forming proteasome-blocking complexes. Aging Cell 14: 35–48, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayyadevara S, Bharill P, Dandapat A, et al. Aspirin inhibits oxidant stress, reduces age-associated functional declines, and extends lifespan of Caenorhabditis elegans. Antioxid Redox Signal 18: 481–490, 2013 [DOI] [PubMed] [Google Scholar]
- 6.Bao J, Zheng L, Zhang Q, et al. Deacetylation of TFEB promotes fibrillar Abeta degradation by upregulating lysosomal biogenesis in microglia. Protein Cell 7: 417–433, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barreiro GC, Prattali RR, Caliseo CT, et al. Aspirin inhibits serine phosphorylation of IRS-1 in muscle and adipose tissue of septic rats. Biochem Biophys Res Commun 320: 992–997, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Bartolucci AA, Tendera M, and Howard G. Meta-analysis of multiple primary prevention trials of cardiovascular events using aspirin. Am J Cardiol 107: 1796–1801, 2011 [DOI] [PubMed] [Google Scholar]
- 9.Bos CL, Kodach LL, van den Brink GR, et al. Effect of aspirin on the Wnt/beta-catenin pathway is mediated via protein phosphatase 2A. Oncogene 25: 6447–6456, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Bryson BD. and White FM. Quantitative profiling of lysine acetylation reveals dynamic crosstalk between receptor tyrosine kinases and lysine acetylation. PLoS One 10: e0126242, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Byrum S, Smart SK, Larson S, and Tackett AJ. Analysis of stable and transient protein-protein interactions. Methods Mol Biol 833: 143–152, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byrum SD, Taverna SD, and Tackett AJ. Quantitative analysis of histone exchange for transcriptionally active chromatin. J Clin Bioinforma 1: 17, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calabrese V, Giuffrida Stella AM, Calvani M, and Butterfield DA. Acetylcarnitine and cellular stress response: roles in nutritional redox homeostasis and regulation of longevity genes. J Nutr Biochem 17: 73–88, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Calabrese V, Ravagna A, Colombrita C, et al. Acetylcarnitine induces heme oxygenase in rat astrocytes and protects against oxidative stress: involvement of the transcription factor Nrf2. J Neurosci Res 79: 509–521, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Calabrese V, Scapagnini G, Davinelli S, et al. Sex hormonal regulation and hormesis in aging and longevity: role of vitagenes. J Cell Commun Signal 35: 369–384, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi BH, Chakraborty G, Baek K, and Yoon HS. Aspirin-induced Bcl-2 translocation and its phosphorylation in the nucleus trigger apoptosis in breast cancer cells. Exp Mol Med 45: e47, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen TJ, Hwang AW, Restrepo CR, et al. An acetylation switch controls TDP-43 function and aggregation propensity. Nat Commun 6: 5845, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cook NR, Lee IM, Zhang SM, et al. Alternate-day, low-dose aspirin and cancer risk: long-term observational follow-up of a randomized trial. Ann Intern Med 159: 77–85, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cornelius C, Perrotta R, Graziano A, et al. Stress responses, vitagenes and hormesis as critical determinants in aging and longevity: mitochondria as a “chi”. Immun Ageing 10: 15, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.David DC, Ollikainen N, Trinidad JC, et al. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol 8: e1000450, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dosanjh LE, Brown MK, Rao G, et al. Behavioral phenotyping of a transgenic Caenorhabditis elegans expressing neuronal amyloid-beta. J Alzheimers Dis 19: 681–690, 2010 [DOI] [PubMed] [Google Scholar]
- 22.Erales J. and Coffino P. Ubiquitin-independent proteasomal degradation. Biochim Biophys Acta 1843: 216–221, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fatouros C, Pir GJ, Biernat J, et al. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum Mol Genet 21: 3587–3603, 2012 [DOI] [PubMed] [Google Scholar]
- 24.Ferrari R, Forabosco P, Vandrovcova J, et al. Frontotemporal dementia: insights into the biological underpinnings of disease through gene co-expression network analysis. Mol Neurodegener 11: 21, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finder VH, Vodopivec I, Nitsch RM, and Glockshuber R. The recombinant amyloid-beta peptide Abeta1–42 aggregates faster and is more neurotoxic than synthetic Abeta1–42. J Mol Biol 396: 9–18, 2010 [DOI] [PubMed] [Google Scholar]
- 26.Gao S, Li H, Feng X, et al. Alpha-enolase plays a catalytically independent role in doxorubicin-induced cardiomyocyte apoptosis and mitochondrial dysfunction. J Mol Cell Cardiol 79C: 92–103, 2014 [DOI] [PubMed] [Google Scholar]
- 27.Gilroy DW. The role of aspirin-triggered lipoxins in the mechanism of action of aspirin. Prostaglandins Leukot Essent Fatty Acids 73: 203–210, 2005 [DOI] [PubMed] [Google Scholar]
- 28.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature 426: 895–899, 2003 [DOI] [PubMed] [Google Scholar]
- 29.Humphrey W, Dalke A, and Schulten K. VMD: visual molecular dynamics. J Mol Graph 14: 33–38, 1996 [DOI] [PubMed] [Google Scholar]
- 30.Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 80: 225–236, 1995 [DOI] [PubMed] [Google Scholar]
- 31.Jacobs EJ, Newton CC, Gapstur SM, and Thun MJ. Daily aspirin use and cancer mortality in a large US cohort. J Natl Cancer Inst 104: 1208–1217, 2012 [DOI] [PubMed] [Google Scholar]
- 32.Jaenisch R. and Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33 Suppl: 245–254, 2003 [DOI] [PubMed] [Google Scholar]
- 33.Jiang R, Wei L, Zhu M, et al. Aspirin inhibits LPS-induced expression of PI3K/Akt, ERK, NF-kappaB, CX3CL1, and MMPs in human bronchial epithelial cells. Inflammation 39: 643–650, 2016 [DOI] [PubMed] [Google Scholar]
- 34.Kukar TL, Ladd T, Robertson P, et al. Lysine 624 of the amyloid precursor protein (APP) is a critical determinant of amyloid beta peptide length: support for a sequential model of gamma-secretase intramembrane proteolysis and regulation by the amyloid beta precursor protein (APP) juxtamembrane region. J Biol Chem 286: 39804–39812, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ladabaum U. and Sowers M. Potential population-wide impact of aspirin on colon cancer mortality. Gastroenterology 115: 514–515, 1998 [DOI] [PubMed] [Google Scholar]
- 36.Lecker SH, Goldberg AL, and Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 17: 1807–1819, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Liachko NF, Guthrie CR, and Kraemer BC. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J Neurosci 30: 16208–16219, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu H, Ouyang P, Liu ZH, et al. [Aspirin inhibits proliferation and expression of p44/42 MAPK phosphorylation in vascular endothelial cells]. Di Yi Jun Yi Da Xue Xue Bao 24: 1013–1015, 2004 [PubMed] [Google Scholar]
- 39.Lleo A, Cavedo E, Parnetti L, et al. Cerebrospinal fluid biomarkers in trials for Alzheimer and Parkinson diseases. Nat Rev Neurol 11: 41–55, 2015 [DOI] [PubMed] [Google Scholar]
- 40.Lobanov MI, Bogatyreva NS, and Galzitskaia OV. [Radius of gyration is indicator of compactness of protein structure]. Mol Biol (Mosk) 42: 701–706, 2008 [PubMed] [Google Scholar]
- 41.Lu JY, Lin YY, Sheu JC, et al. Acetylation of yeast AMPK controls intrinsic aging independently of caloric restriction. Cell 146: 969–979, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lublin AL. and Link CD. Alzheimer's disease drug discovery: in vivo screening using Caenorhabditis elegans as a model for beta-amyloid peptide-induced toxicity. Drug Discov Today Technol 10: e115–e119, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Margreitter C, Petrov D, and Zagrovic B. Vienna-PTM web server: a toolkit for MD simulations of protein post-translational modifications. Nucleic Acids Res 41: W422–W426, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin G, Wex T, Treiber G, et al. Low-dose aspirin reduces the gene expression of gastrokine-1 in the antral mucosa of healthy subjects. Aliment Pharmacol Ther 28: 782–788, 2008 [DOI] [PubMed] [Google Scholar]
- 45.Mills EJ, Wu P, Alberton M, et al. Low-dose aspirin and cancer mortality: a meta-analysis of randomized trials. Am J Med 125: 560–567, 2012 [DOI] [PubMed] [Google Scholar]
- 46.Minakami K, Watanabe Y, Miyahara M, et al. Effect of indomethacin and aspirin on the TNF-alpha-induced priming and protein tyrosyl phosphorylation of human neutrophils. Physiol Chem Phys Med NMR 25: 55–67, 1993 [PubMed] [Google Scholar]
- 47.Morimoto RI. Stress, aging, and neurodegenerative disease. N Engl J Med 355: 2254–2255, 2006 [DOI] [PubMed] [Google Scholar]
- 48.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev 22: 1427–1438, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morley JF, Brignull HR, Weyers JJ, and Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A 99: 10417–10422, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morris MJ, Mahgoub M, Na ES, et al. Loss of histone deacetylase 2 improves working memory and accelerates extinction learning. J Neurosci 33: 6401–6411, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Narayan PJ, Lill C, Faull R, et al. Increased acetyl and total histone levels in post-mortem Alzheimer's disease brain. Neurobiol Dis 74: 281–294, 2015 [DOI] [PubMed] [Google Scholar]
- 52.Nath N, Vassell R, Chattopadhyay M, et al. Nitro-aspirin inhibits MCF-7 breast cancer cell growth: effects on COX-2 expression and Wnt/beta-catenin/TCF-4 signaling. Biochem Pharmacol 78: 1298–1304, 2009 [DOI] [PubMed] [Google Scholar]
- 53.Neumann M. Molecular neuropathology of TDP-43 proteinopathies. Int J Mol Sci 10: 232–246, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nick TU, Lee W, Kossmann S, et al. Hydrogen bond network between amino acid radical intermediates on the proton-coupled electron transfer pathway of E. coli alpha2 ribonucleotide reductase. J Am Chem Soc 137: 289–298, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohira T, Bannenberg G, Arita M, et al. A stable aspirin-triggered lipoxin A4 analog blocks phosphorylation of leukocyte-specific protein 1 in human neutrophils. J Immunol 173: 2091–2098, 2004 [DOI] [PubMed] [Google Scholar]
- 56.Ong G, Davis TM, and Davis WA. Aspirin is associated with reduced cardiovascular and all-cause mortality in type 2 diabetes in a primary prevention setting: the Fremantle Diabetes study. Diabetes Care 33: 317–321, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parker BL, Shepherd NE, Trefely S, et al. Structural basis for phosphorylation and lysine acetylation cross-talk in a kinase motif associated with myocardial ischemia and cardioprotection. J Biol Chem 289: 25890–25906, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Partridge L. Some highlights of research on aging with invertebrates, 2009. Aging Cell 8: 509–513, 2009 [DOI] [PubMed] [Google Scholar]
- 59.Pomponi MF, Gambassi G, Pomponi M, and Masullo C. Alzheimer's Disease: fatty acids we eat may be linked to a specific protection via low-dose aspirin. Aging Dis 1: 37–59, 2010 [PMC free article] [PubMed] [Google Scholar]
- 60.Prattali RR, Barreiro GC, Caliseo CT, et al. Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in growth hormone treated animals. FEBS Lett 579: 3152–3158, 2005 [DOI] [PubMed] [Google Scholar]
- 61.Qian MX, Pang Y, Liu CH, et al. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 153: 1012–1024, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Santilli F, Boccatonda A, and Davi G. Aspirin, platelets, and cancer: the point of view of the internist. Eur J Intern Med 34: 11–20, 2016 [DOI] [PubMed] [Google Scholar]
- 63.Santos MT, Moscardo A, Valles J, et al. Participation of tyrosine phosphorylation in cytoskeletal reorganization, alpha(IIb)beta(3) integrin receptor activation, and aspirin-insensitive mechanisms of thrombin-stimulated human platelets. Circulation 102: 1924–1930, 2000 [DOI] [PubMed] [Google Scholar]
- 64.Shaw BF, Schneider GF, Bilgicer B, et al. Lysine acetylation can generate highly charged enzymes with increased resistance toward irreversible inactivation. Protein Sci 17: 1446–1455, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shi X, Ding M, Dong Z, et al. Antioxidant properties of aspirin: characterization of the ability of aspirin to inhibit silica-induced lipid peroxidation, DNA damage, NF-kappaB activation, and TNF-alpha production. Mol Cell Biochem 199: 93–102, 1999 [DOI] [PubMed] [Google Scholar]
- 66.Skiniotis G, Moazed D, and Walz T. Acetylated histone tail peptides induce structural rearrangements in the RSC chromatin remodeling complex. J Biol Chem 282: 20804–20808, 2007 [DOI] [PubMed] [Google Scholar]
- 67.Stark LA, Reid K, Sansom OJ, et al. Aspirin activates the NF-kappaB signalling pathway and induces apoptosis in intestinal neoplasia in two in vivo models of human colorectal cancer. Carcinogenesis 28: 968–976, 2007 [DOI] [PubMed] [Google Scholar]
- 68.Steffan JS, Bodai L, Pallos J, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413: 739–743, 2001 [DOI] [PubMed] [Google Scholar]
- 69.Stegeman I, Bossuyt PM, Yu T, et al. Aspirin for primary prevention of cardiovascular disease and cancer. A benefit and harm analysis. PLoS One 10: e0127194, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Strong R, Miller RA, Astle CM, et al. Nordihydroguaiaretic acid and aspirin increase lifespan of genetically heterogeneous male mice. Aging Cell 7: 641–650, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tanaka T, Kurose A, Halicka HD, et al. Nitrogen oxide-releasing aspirin induces histone H2AX phosphorylation, ATM activation and apoptosis preferentially in S-phase cells: involvement of reactive oxygen species. Cell Cycle 5: 1669–1674, 2006 [DOI] [PubMed] [Google Scholar]
- 72.Tazearslan C, Ayyadevara S, Bharill P, and Shmookler Reis RJ. Positive feedback between transcriptional and kinase suppression in nematodes with extraordinary longevity and stress resistance. PLoS Genet 5: e1000452, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van der Spoel D, Lindahl E, Hess B, et al. GROMACS: fast, flexible, and free. J Comput Chem 26: 1701–1718, 2005 [DOI] [PubMed] [Google Scholar]
- 74.van Noort V, Seebacher J, Bader S, et al. Cross-talk between phosphorylation and lysine acetylation in a genome-reduced bacterium. Mol Syst Biol 8: 571, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vlad SC, Miller DR, Kowall NW, and Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70: 1672–1677, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wan QL, Zheng SQ, Wu GS, and Luo HR. Aspirin extends the lifespan of Caenorhabditis elegans via AMPK and DAF-16/FOXO in dietary restriction pathway. Exp Gerontol 48: 499–506, 2013 [DOI] [PubMed] [Google Scholar]
- 77.Webster BR, Scott I, Han K, et al. Restricted mitochondrial protein acetylation initiates mitochondrial autophagy. J Cell Sci 126: 4843–4849, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wolfe LS, Calabrese MF, Nath A, et al. Protein-induced photophysical changes to the amyloid indicator dye thioflavin T. Proc Natl Acad Sci U S A 107: 16863–16868, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wolfe MS. Tau mutations in neurodegenerative diseases. J Biol Chem 284: 6021–6025, 2009 [DOI] [PubMed] [Google Scholar]
- 80.Yue KT, Davis JW, and Aldridge EG. Effect of aspirin and benadryl on human platelet oxidative phosphorylation and aggregation. Thromb Diath Haemorrh 30: 577–585, 1973 [PubMed] [Google Scholar]