

Abstract
IMPORTANCE
The use of benzodiazepines to control agitation in delirium in the last days of life is controversial.
OBJECTIVE
To compare the effect of lorazepam vs placebo as an adjuvant to haloperidol for persistent agitation in patients with delirium in the setting of advanced cancer.
DESIGN, SETTING, AND PARTICIPANTS
Single-center, double-blind, parallel-group, randomized clinical trial conducted at an acute palliative care unit at MD Anderson Cancer Center, Texas, enrolling 93 patients with advanced cancer and agitated delirium despite scheduled haloperidol from February 11, 2014, to June 30, 2016, with data collection completed in October 2016.
INTERVENTIONS
Lorazepam (3 mg) intravenously (n = 47) or placebo (n = 43) in addition to haloperidol (2 mg) intravenously upon the onset of an agitation episode.
MAIN OUTCOMES AND MEASURES
The primary outcome was change in Richmond Agitation-Sedation Scale (RASS) score (range, −5 [unarousable] to 4 [very agitated or combative]) from baseline to 8 hours after treatment administration. Secondary end points were rescue neuroleptic use, delirium recall, comfort (perceived by caregivers and nurses), communication capacity, delirium severity, adverse effects, discharge outcomes, and overall survival.
RESULTS
Among 90 randomized patients (mean age, 62 years; women, 42 [47%]), 58 (64%) received the study medication and 52 (90%) completed the trial. Lorazepam + haloperidol resulted in a significantly greater reduction of RASS score at 8 hours (−4.1 points) than placebo + haloperidol (−2.3 points) (mean difference, −1.9 points [95% CI, −2.8 to −0.9]; P < .001). The lorazepam + haloperidol group required less median rescue neuroleptics (2.0 mg) than the placebo + haloperidol group (4.0 mg) (median difference, −1.0 mg [95% CI, −2.0 to 0]; P = .009) and was perceived to be more comfortable by both blinded caregivers and nurses (caregivers: 84% for the lorazepam + haloperidol group vs 37% for the placebo + haloperidol group; mean difference, 47% [95% CI, 14% to 73%], P = .007; nurses: 77% for the lorazepam + haloperidol group vs 30% for the placebo + haloperidol group; mean difference, 47% [95% CI, 17% to 71%], P = .005). No significant between-group differences were found in delirium-related distress and survival. The most common adverse effect was hypokinesia (3 patients in the lorazepam + haloperidol group [19%] and 4 patients in the placebo + haloperidol group [27%]).
CONCLUSIONS AND RELEVANCE
In this preliminary trial of hospitalized patients with agitated delirium in the setting of advanced cancer, the addition of lorazepam to haloperidol compared with haloperidol alone resulted in a significantly greater reduction in agitation at 8 hours. Further research is needed to assess generalizability and adverse effects.
TRIAL REGISTRATION
clinicaltrials.gov Identifier: NCT01949662
Delirium was found in approximately 90% of patients in the last days of life in a 2013 systematic review.1 Approximately 50% to 70% of patients with delirium have hyperactive or mixed subtypes, characterized by restlessness, agitation, or aggressive violent behavior. Agitation can be highly distressing to patients, caregivers, and health care professionals, posing a significant safety risk to those involved.2–4
Clinicians caring for patients with agitated delirium at the end of life currently have few evidence-based treatment options.5,6 Neuroleptics and benzodiazepines represent the main pharmacologic choices; however, the use of benzodiazepines in patients with delirium is often debated because, to our knowledge, no randomized trial has ever compared the effect of a benzodiazepine and placebo on any delirium outcomes. The National Comprehensive Cancer Network clinical practice guideline recommended a trial of benzodiazepines in patients whose agitation did not respond adequately to haloperidol.7 However, some clinicians believe that benzodiazepines should be avoided in the management of delirium8 because lorazepam was found to be inferior to haloperidol and chlorpromazine and contributed to excessive adverse effects in a small randomized clinical trial.9 A better understanding of the efficacy and safety of benzodiazepines for agitation may help clinicians to manage this highly distressing syndrome in which few effective treatment options exist. The objective of this randomized clinical trial was to compare the effect of lorazepam vs placebo as an adjuvant to haloperidol on the intensity of agitation in patients with delirium in the setting of advanced cancer.
Methods
Study Design
This was a double-blind, parallel group, placebo-controlled, randomized clinical trial in which patients with hyperactive or mixed delirium were allocated in a 1:1 ratio to receive lorazepam + haloperidol or placebo + haloperidol as treatment for a single episode of restlessness or agitation. The trial protocol and a list of the revisions with justifications are available in Supplement 1. Key protocol changes related to study objectives, eligibility criteria, and statistical analyses are highlighted in eTables 1 to 3 in Supplement 1. The institutional review board at MD Anderson Cancer Center approved this study. Written surrogate consent was obtained from the medical power of attorney or legal representative. The institutional review board did not require caregivers or nurses to sign an informed consent for their involvement. Enrollment occurred from February 11, 2014, to June 30, 2016. Data collection was completed in October 2016.
Eligibility Criteria
Adult patients who were 18 years or older with a diagnosis of advanced cancer at the acute palliative care unit at the University of Texas MD Anderson Cancer Center in Houston, Texas, were eligible for this study if, in the opinion of the attending physician and bedside nurse, they had a diagnosis of delirium by Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) criteria and had a history of agitation with a Richmond Agitation-Sedation Scale (RASS) score of 2 or more over the past 24 hours despite receiving scheduled haloperidol of 1 mg to 8 mg per day. Patients were excluded if they had dementia, use of benzodiazepines or chlorpromazine within the past 48 hours, contraindications to neuroleptics (ie, Parkinson disease, myasthenia gravis, acute narrow-angle glaucoma, seizure disorders, documented corrected QT interval prolongation, or hypersensitivity) or contraindications to benzodiazepines (ie, hypersensitivity). Eligibility criteria revisions during the study are outlined in eTable 2 in Supplement 1.
The acute palliative care unit was selected as the study setting because of the high prevalence of persistent agitated delirium at the end of life and because patients received standardized care for delirium by an experienced interdisciplinary palliative care team consisting of physicians, nurses, and pharmacists. Patients were routinely treated for any potentially reversible causes (eg, opioid neurotoxicity, polypharmacy, infections, hypercalcemia, and other metabolic causes) and provided with nonpharmacologic measures (eg, orientation cues, avoiding unnecessary stimuli, window light, and caregiver education and involvement) and intensive symptom management. Blinded physicians and nurses were involved in the identification of potential patients, administration of study medications and documentation of study outcomes. A do-not-resuscitate order was not required for admission. The bedside nurses conducted shift change sign-out at 7 AM and 7 PM at the bedside to communicate patient care issues to maximize continuity of care and study data collection.
Study Interventions, Randomization, and Blinding
Web-based simple randomization was used to assign patients to the 2 treatment groups. All enrolled patients immediately initiated a standardized open-label regimen with haloperidol (2 mg) every 4 hours intravenously and another 2 mg every hour as needed for agitation. Because of the fluctuating nature of delirium, we monitored the RASS score of each patient every 2 hours until the score was 2 or more and required rescue medication according to the bedside nurse’s judgment before administering the blinded study medications (lorazepam or placebo). Once the patient met this threshold, a single dose of 3 mg of lorazepam in 25 mL of 0.9% normal saline solution or identically appearing placebo (25 mL of 0.9% normal saline) was infused intravenously over 1.5 minutes. The timing of the primary outcome was 8 hours from when the blinded study medication was administered. Patients in both groups also received 2 mg of haloperidol intravenously immediately afterwards. The RASS score threshold for blinded study medication administration was revised to 1 or more in September 2014 to ensure that patients who had any agitation could proceed to the blinded phase. All patients had at least 2 days of delirium with documentation of agitation before starting the study intervention. The use of other medications and withholding of scheduled haloperidol were permissible as per standard of practice according to the clinical judgment of the attending physician and bedside nurse.
A single dose of study medication was examined instead of repeated dosing because of the very short survival rate among our patient population (ie, hours to days) and the uncertain risks associated with lorazepam in a frail population. We used lorazepam in this study because it has a rapid onset of action (5–20 minutes), a moderate duration of action (hours), a short elimination half-life (12.9 hours), a low risk of accumulation, no major active metabolites, and a predictable bioavailability.10 A 3-mg dose was chosen because a previous study using this dose reported that it provided a physiologic effect lasting at least 8 hours without significant adverse events.11
Research staff conducting the study assessments, bedside nurses, attending physicians, patients, and caregivers were blinded to the allocation of the study medication and study outcomes throughout the entire study. To ensure proper blinding, a separate clinical nurse administered the study medication instead of the bedside nurse who conducted the RASS score assessments. Allocation was concealed by using a secured website that was only accessible to the study pharmacist, who then assigned patients to the study intervention.
Study Outcomes and End Points
Our prespecified primary outcome was the RASS score, a validated 10-point numeric rating scale that ranges from −5 to 4, at 8 hours.12,13 The score definitions were as follows: −5, unarousable; −4, deep sedation; −3, moderate sedation; −2, light sedation; −1, drowsy; 0, alert and calm; 1, restless; 2, agitated; 3, very agitated; 4, combative. This was assessed by the bedside nurse immediately prior to study medication administration and then at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, and 8 hours. Subsequently, RASS scores were documented daily until discharge or death. To determine interrater agreement, the research staff and nurses both rated RASS scores independently at the time of study enrollment.
Secondary outcomes defined a priori included (1) the severity of delirium assessed with the Memorial Delirium Assessment Scale (MDAS; range, 0–30; higher scores indicate greater severity) at baseline, 2, 4, and 8 hours and then daily until discharge, (2) the use of any additional psychotropic agents during the first 8 hours after study medication administration and then daily until discharge, (3) the Edmonton Symptom Assessment System (ESAS; range, 0–10; higher scores indicate greater severity) with proxy ratings provided by family caregivers daily until discharge,14,15 (4) patient comfort perceived by caregivers and bedside nurses daily (5-point Likert scale ranging from “strongly agree” to “strongly disagree”), (5) the recalled frequency of 6 delirium symptoms (ie, disorientation to time, disorientation to place, visual hallucinations, tactile hallucinations, auditory hallucinations, delusional thoughts, and psychomotor agitation) and related distress in the rater recorded by family caregivers and bedside nurses daily until discharge (range 0–4; higher scores indicate greater frequency or distress),2,4 (6) communication capacity perceived by caregivers and bedside nurses was assessed daily, (7) adverse effects related to the use of benzodiazepines and neuroleptics were documented using the Udvalg for Kliniske Undersøgelser assessment (range 0–3; higher scores indicate greater severity),16 (8) duration of stay in acute palliative care unit, and (9) overall survival from the time of study medication administration. Further details of study assessment are available in the eAppendix in Supplement 2. Salivary biomarkers were also collected but results are not reported here. Race/ethnicity data were collected based on patient or family caregiver self-report as mandated by National Cancer Institute using fixed categories.17
Statistical Analysis
The protocol was designed in 2013 to recruit 17 patients per group, which would provide 80% power to detect an effect size of 1.0 in RASS score between groups with an a of .05 using 2-sided t tests. After funding was secured, the sample size was revised to 26 patients per group to detect an effect size of 0.79 (mean difference of 0.5, assuming a within-group SD of 0.63) in September 2015. Enrollment continued until 52 patients received the treatment and completed the first 8 hours of observation.
Baseline characteristics were summarized by descriptive statistics. The prespecified primary outcome, change in RASS score from immediately before blinded study medication administration (time 0) to 8 hours, was compared between study groups by using the Wilcoxon rank sum test. Because of the nature of the study population, many patients died or were discharged before requiring the study medication; thus, a modified intention-to-treat analysis including only patients who started the study interventions was specified a priori. Because the RASS score is a momentary measure, we also conducted post hoc analyses to assess the proportion of patients documented to have any RASS score of 1 or more documented during the first 8 hours.
The change in secondary outcomes before and after medication administration was compared between groups using a 2-tailed Wilcoxon rank sum test for continuous variables and 2-tailed Fisher exact test for categorical variables. The difference in the change of these end points before and after treatment was summarized by mean, median, and proportion along with the associated 95% CIs for parametric, nonparametric continuous, and categorical variables, respectively. The Kaplan-Meier method was used for time-to-event analysis and the log-rank test and univariate Cox regression analysis to compare overall survival between groups. All analyses were 2-sided tests. For our prespecified primary outcome analysis, a 2-sided P value of .05 or less was considered to be statistically significant. We did not adjust for multiple comparisons and all secondary findings are considered to be hypothesis-generating.
The interrater reliability of RASS scores between the bedside nurse and the research nurse were determined at the time of study enrollment using the κ statistic. In post hoc analyses, missing data on the primary outcome were imputed using the multiple imputation method under the assumption of a monotone missing pattern (eTable 1 in Supplement 2). Post hoc worst-case sensitivity analysis was conducted by assuming that the patients who started but did not complete the study intervention had no change in RASS score from baseline at 8 hours. Missing data were not imputed for secondary outcomes.
SAS (SAS Institute), version 9.4, was used for statistical analysis.
Results
Enrollment and Baseline Characteristics
Among the 144 eligible patients, 93 (65%) were enrolled and 90 (63%) were randomized and started on the standardized haloperidol regimen. After a median observation period of 6.4 hours (interquartile range [IQR], 4.4 to 15.7), 58 patients (64%) developed an agitation episode requiring rescue medication and received lorazepam or placebo in conjunction with haloperidol. Fifty-two patients (90%) had at least 8 hours of monitoring (Figure 1). Among the 32 patients (36%) who did not receive the study medication, 27 (84%) did not develop further agitation necessitating intervention until discharge or death after the standardized dose increase of haloperidol, 4 (13%) dropped out, and 1 (3%) was deemed ineligible (Figure 1).
Figure 1. Flow of Patients Through the Study.
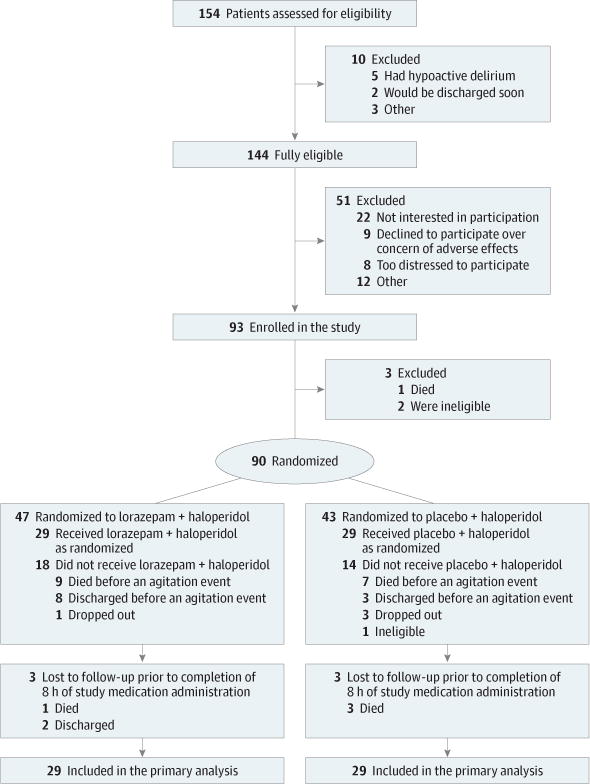
At the time of enrollment, patients were randomized to lorazepam or placebo. All enrolled patients immediately began a standardized regimen with haloperidol 2 mg every 4 hours intravenously and 2 mg every hour as needed for agitation. Because of the fluctuating nature of delirium, the Richmond Agitation-Sedation Scale score of each patient was monitored every 2 hours until the patient’s score was 1 or more and required rescue medication per the judgment of the bedside nurse. Once the dose of haloperidol was increased and standardized, 27 of 90 randomized patients (30%) did not develop further agitation until death or discharge and thus did not require the study medication.
Table 1 shows the baseline characteristics of the 58 patients who received study medication. The mean age was 65 years (range, 30–90), 27 patients (47%) were women, and 44 patients (76%) were white. The median overall survival was 73 hours (95% CI, 49 to 106), with a median follow-up of 164 hours (95% CI, 92 to 195).
Table 1.
Baseline Characteristics of Patients With Advanced Cancer and Agitated Delirium by Study Group
| No. of Patients(%) | ||
|---|---|---|
| Lorazepam + Haloperidol (n = 29) | Placebo + Haloperidol (n = 29) | |
| Age, mean (range), y | 66 (43–90) | 64 (30–88) |
| Women | 11 (37.9) | 16 (55.2) |
| Race/ethnicity | ||
| White | 21 (72.4) | 23 (79.3) |
| Black | 4 (13.8) | 4 (13.8) |
| Hispanic | 2 (6.9) | 0 |
| Other | 2 (6.9) | 2 (6.9) |
| Education | ||
| High school or less | 7 (24.1) | 8 (27.6) |
| Some college | 11 (37.9) | 6 (20.7) |
| Completed college | 10 (34.5) | 14 (48.3) |
| Not available | 1 (3.5) | 1 (3.5) |
| Cancer type | ||
| Breast | 0 | 5 (17.2) |
| Gastrointestinal | 9 (31.0) | 4 (13.8) |
| Genitourinary | 2 (6.9) | 1 (3.4) |
| Gynecological | 2 (6.9) | 2 (6.9) |
| Head and neck | 0 | 1 (3.4) |
| Hematological | 8 (27.6) | 2 (6.9) |
| Respiratory | 4 (13.8) | 10 (34.5) |
| Other | 4 (13.8) | 4 (13.8) |
| Cancer stage | ||
| Metastatic | 20 (69.0) | 26 (89.7) |
| Locally advanced | 1 (3.4) | 0 |
| Recurrent or persistent | 8 (27.5) | 3 (10.3) |
| Karnofsky performance status, %a | ||
| 10 | 7 (24.1) | 5 (17.2) |
| 20 | 15 (51.7) | 12 (41.4) |
| 30 | 4 (13.8) | 10 (34.5) |
| 40 | 3 (10.3) | 2 (6.9) |
| Reason for acute palliative care unit admissionb | ||
| Delirium | 14 (48.3) | 17 (58.6) |
| Pain | 22 (75.9) | 26 (89.7) |
| Dyspnea | 13 (44.8) | 8 (27.6) |
| Other | 13 (44.8) | 10 (34.5) |
| MDAS score, median (IQR)c | 30.0 (23.0–30.0) | 28.0 (19.0–30.0) |
| Medication in prior 48 h | ||
| Haloperidol scheduled | 29 (100) | 29 (100) |
| Haloperidol as needed | 26 (89.7) | 20 (69.0) |
| Chlorpromazine scheduled | 0 | 0 |
| Chlorpromazine as needed | 5 (17.2) | 1 (3.4) |
| Benzodiazepine scheduled | 0 | 0 |
| Benzodiazepine as needed | 1 (3.5) | 1 (3.5) |
| Haloperidol use in prior 24 h | ||
| Scheduled, median (IQR), mg | 3.0 (2.0–5.0) | 3.0 (2.0–5.0) |
| Rescue, median (IQR), mg | 4.0 (2.0–6.0) | 2.0 (0.0–4.0) |
| No. of breakthrough doses, median (IQR) | 2.0 (1.0–3.0) | 2.0 (0.0–3.0) |
| RASS score immediately prior to treatment, mean (SD)d | 1.6 (0.6) | 1.6 (0.6) |
Abbreviation: IQR, interquartile range; MDAS, Memorial Delirium Assessment Scale; RASS, Richmond Agitation-Sedation Scale.
A validated assessment of performance status that ranges from 0% (deceased) to 100% (normal, no complaints).
Patients could have multiple reasons for admission.
A validated 10-item, clinician-rated assessment scale for delirium in patients with cancer18,19 that examines the level of consciousness, disorientation, memory, recall, attention, disorganized thinking, perceptual disturbance, delusions, psychomotor activity, and sleep; assigning a score range for each category of 0 to 3, for a total score range of 0 to 30; a total score of 13 or higher indicates delirium.
A validated 10-point numeric rating scale that ranges from −5 (unarousable) to 4 (very agitated and combative); a score of 0 indicates that a patient is alert and calm.
Agitation and RASS Score
The mean RASS score prior to medication administration was 1.6 points (SD, 0.6) in both groups. Lorazepam + haloperidol was associated with a significantly greater reduction of RASS score at 8 hours than placebo + haloperidol (−4.1 points for the lorazepam + haloperidol group vs −2.3 points for the placebo + haloperidol group; mean difference, −1.9 points [95% CI, −2.8 to −0.9]; P < .001) (Figure 2A and Table 2). As shown in Figure 2A, patients in the lorazepam + haloperidol group had a significant within-group reduction in RASS score within the first 30 minutes of treatment administration and this effect was maintained at 8 hours (mean change, −3.6 points at 30 minutes and −4.1 points at 8 hours). A smaller decrease in RASS score was also observed in the placebo + haloperidol group at 30 minutes and at 8 hours (mean change, −1.6 points at 30 minutes and −2.3 points at 8 hours). The κ for RASS score assessment at the time of study enrollment between research staff and nurse was 0.79 (P < .001).
Figure 2. Change in Richmond Agitation-Sedation Scale (RASS) Over the First 8 Hours After Treatment.
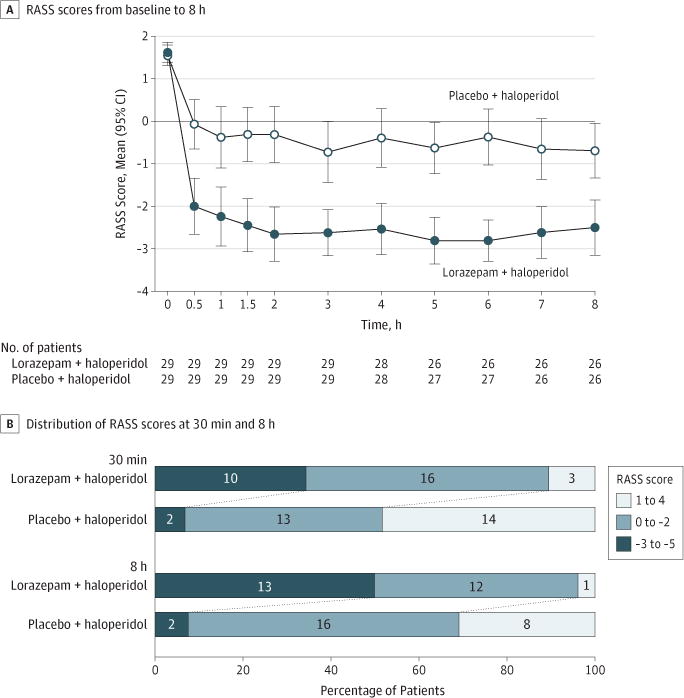
A, Time 0 indicates immediately before treatment administration. Error bars indicate 95% CIs. Both treatments were associated with significant reduction in the mean RASS score within the first 30 minutes of treatment. RASS score remained relatively stable for both groups over the 8-hour observation period. Lorazepam + haloperidol was associated with a significantly greater reduction in RASS score than placebo + haloperidol at 8 hours (P < .001, 2-sided Wilcoxon rank sum test). B, A larger proportion of patients had hyperactivity (RASS score, 1 to 4) in the placebo + haloperidol group at both 30 minutes and 8 hours (P = .001 for both time points). In contrast, a larger proportion of patients had sedation in the lorazepam group (RASS score, −3 to −5). The 2-sided Fisher exact test was used to compare the 3 categories of RASS scores between groups.
Table 2.
Study Outcomes Among Patients With Advanced Cancer and Agitated Delirium Receiving Lorazepam + Haloperidol vs Placebo + Haloperidol
| Lorazepam + Haloperidol Group(n = 29) | Placebo + Haloperidol Group (n = 29) | Difference Between Groups (95% CI) [Range] | P Valuea | |||
|---|---|---|---|---|---|---|
| No. of Patientsb | Mean (95% CI) | No. of Patientsb | Mean (95% CI) | |||
| Primary Outcomes | ||||||
| Change in RASS score (baseline to 8 h), pointsc | 26 | −4.1 (−4.8 to −3.4) | 26 | −2.3 (−2.9 to −1.6) | −1.9 (−2.8 to −0.9) | <.001 |
| Absolute RASS score at 8 h, pointsc | 26 | −2.5 (−3.2 to −1.9) | 26 | −0.7 (−1.3 to −0.1) | 1.8 (−2.7 to −0.9) | <.001 |
| Secondary Outcomes | ||||||
| Change in RASS score from baseline to 30 minc | 29 | −3.6 (−4.3 to −2.9) | 29 | −1.6 (−2.2 to −1.0) | −2.0 (−2.9 to −1.1) | <.001 |
| RASS score ≥1 within 8 h, No. of observations (%)c | 29 | 8 (28) | 29 | 22 (76) | % (95% CI), −48 (−71 to −26) | <.001 |
| Neuroleptic use within 8 hd | ||||||
| HEDD, median (IQR) [range], mg | ||||||
| Scheduled | 29 | 2.0 (2.0 to 4.0) [0.0 to 6.0] | 29 | 2.0 (2.0 to 4.0) [0.0 to 5.0] | 0 (0 to 0) [−5 to 6] | .68 |
| Rescue | 29 | 2.0 (2.0 to 2.0) [0.0 to 8.0] | 29 | 4.0 (2.0 to 5.0) [0.0 to 19.0] | −1.0 (−2.0 to 0) [−19.0 to 8.0] | .009 |
| No. of rescue doses | 29 | 1.0 (1.0 to 1.0) [0.0 to 4.0] | 29 | 2.0 (1.0 to 2.0) [0.0 to 7.0] | −0.5 (−1 to 0) [−7.0 to 4.0] | .008 |
| Total | 29 | 6.0 (4.0 to 6.0) [2.0 to 14.0] | 29 | 6.0 (4.0 to 8.0) [2.0 to 21.0] | −1.0 (−2 to 0) [−19.0 to 12.0] | .03 |
| Chlorpromazine, No. of doses (%) | 29 | 2 (6.9) | 29 | 4 (13.8) | −0.1 (−0.3 to 0.2) | .67 |
| Change in MDAS score (baseline to 8 h)e | 25 | 2.5 (0.7 to 4.4) | 25 | 0.4 (−2.2 to 3.0) | 2.1 (−1.0 to 5.2) | .18 |
| Change in respiratory rate/min (baseline to 8 h)e | 21 | −1.5 (−2.7 to −0.3) | 25 | −0.5 (−2.6 to 1.6) | −1.0 (−3.4 to 1.4) | .80 |
| Change in ESAS score (baseline to day 1), mean (SD)f | ||||||
| Pain | 18 | −2.4 (−3.7 to −1.0) | 7 | −1.7 (−5.6 to 2.1) | −0.7 (−3.6 to 2.2) | .67 |
| Fatigue | 11 | 0.1 (−1.2 to 1.3) | 6 | −1.8 (−5.2 to 1.5) | 1.9 (−0.7 to 4.5) | .23 |
| Nausea | 13 | −0.7 (−2.8 to 1.4) | 6 | −2.7 (−6.7 to 1.4) | 2.0 (−1.7 to 5.7) | .49 |
| Depression | 15 | −1.4 (−3.6 to 0.8) | 6 | 0.2 (−2.8 to 3.2) | −1.6 (−5.3 to 2.2) | .56 |
| Anxiety | 18 | −3.4 (−5.3 to −1.6) | 7 | −2.1 (−6.5 to 2.2) | −1.3 (−5.0 to 2.4) | .55 |
| Drowsiness | 17 | 1.9 (0.2 to 3.7) | 7 | −2.0 (−4.9 to 0.9) | 3.9 (0.8 to 7.1) | .03 |
| Shortness of breath | 18 | −1.0 (−2.1 to 0.1) | 7 | −0.4 (−4.6 to 3.7) | −0.6 (−3.3 to 2.2) | .41 |
| Appetite | 15 | 0.6 (−0.3 to 1.5) | 7 | 2.1 (−0.9 to 5.1) | −1.5 (−3.6 to 0.6) | .26 |
| Sleep | 18 | −2.9 (−4.8 to −1.0) | 7 | −2.4 (−6.0 to 1.1) | −0.5 (−4.0 to 3.1) | .74 |
| Feeling of well-being | 14 | −2.3 (−4.2 to −0.4) | 6 | −1.5 (−4.9 to 1.9) | −0.8 (−4.2 to 2.6) | .51 |
| Improvement in comfort on day 1, No. of patients (%)g | % (95% CI) | |||||
| Assessed by caregiver | 19 | 16 (84) | 19 | 7 (37) | 47 (14 to 73) | .007 |
| Assessed by nurse | 22 | 17 (77) | 20 | 6 (30) | 47 (17 to 71) | .005 |
| UKU adverse effects, No. of patients with increased level on day 3 vs baseline (%)h | ||||||
| Dystonia | 16 | 0 | 15 | 0 | ||
| Rigidity | 16 | 0 | 15 | 0 | ||
| Hypokinesia or akinesia | 16 | 3 (18.8) | 15 | 4 (26.7) | −8 (−40 to 28) | .68 |
| Hyperkinesia | 16 | 1 (6.3) | 15 | 2 (13.3) | −7 (−40 to 28) | .60 |
| Tremor | 16 | 0 | 15 | 0 | ||
| Akathisia | 16 | 3 (18.8) | 15 | 1 (6.7) | 12 (−22 to 45) | .60 |
| Epilepticseizure | 16 | 0 | 15 | 0 | ||
| Paresthesia | 16 | 0 | 15 | 0 | ||
| Discharged alive from the acute palliative care unit, No. of patients (%) | 29 | 9 (31) | 29 | 7 (24.1) | 6.9 (−20 to 33) | .77 |
| Duration of acute palliative care unit stay, median (IQR),d | 29 | 6 (4 to 9) | 29 | 6 (3 to 8) | Difference, 1(−1 to 3)i | .35 |
| Overall survival from treatment administration, median (95% CI), hj | 29 | 68 (49 to 130) | 29 | 73 (38 to 106) | HR, 1.2 (0.7 to 2.2) | .56 |
Abbreviations: ESAS, Edmonton Symptom Assessment System; HEDD, haloperidol equivalent daily dose; HR, hazard ratio; IQR, interquartile range; MDAS, Memorial Delirium Assessment Scale; RASS, Richmond Agitation-Sedation Scale; UKU, Udvalg for Kliniske Undersøgelser.
The change in study outcomes was compared before and after medication administration between groups using 2-tailed Wilcoxon Rank Sum test for continuous variables and 2-tailed Fisher exact test for categorical variables. All secondary outcomes should be considered hypothesis-generating.
Patients with data available for each analysis are shown. The number of patients with missing data varied because of attrition (eg, death), the specific timing of study assessments (eg, day 1 vs day 3), and the availability of caregivers and bedside nurses.
RASS is a validated 10-point numeric rating scale that was assessed by the bedside nurse immediately prior to study medication administration and then at 0.5 h, 1 h, 1.5 h, 2 h, 3 h, 4 h, 5 h, 6 h, 7 h, and 8 h. (score range, −5 [unarousable] to 4 [very agitated and combative]; 0 indicates that a patient is alert and calm).
The total dose of neuroleptics during the first 8 h was calculated based on the concept of HEDD, in which 8 mg of parenteral haloperidol is equivalent to 100 mg of parenteral chlorpromazine.20 This concept has been used in multiple studies to examine neuroleptic use.21,22 The 95% CIs for the median between-group difference were estimated by the Hodges-Lehmann method of location shift for 2-sample data.
Avalidated 10-item, clinician-rated assessment scale for delirium in patients with cancer18,19 that examines the level of consciousness, disorientation, memory, recall, attention, disorganized thinking, perceptual disturbance, delusions, psychomotor activity, and sleep (each item score range, 0–3; total score range, 0–30; a score of13 or higher indicates delirium). This assessment was conducted by the bedside nurse or research coordinator at the time of enrollment; time of study medication administration; at 2 h, 4 h, 8 h, and 24 h after study medication administration; and then daily until discharge.
The ESAS is a symptom battery that has been validated and widely used in different clinical settings, including the acute palliative care unit.14,15,23 Because patients were delirious, caregivers were asked to provide their proxy rating of ESAS daily. It assessed the symptom intensity of 10 symptoms (pain, fatigue, nausea, depression, anxiety, drowsiness, shortness of breath, appetite, sleep, and feeling of well-being) over the past 24 h. Each symptom was assessed using an 11-point numeric rating scale (range, 0–10; a higher score indicates worse symptoms).
Assessed by asking the following question independently: “In my opinion, the patient was more comfortable after the study medication.” The responses ranged from “strongly agree,” “agree,” “neutral,” “disagree,” and “strongly disagree.”
In this study, “strongly agree” and “agree” were combined for analysis.
Eight neurologic symptoms were documented using the UKU adverse effects rating scale at baseline and day 3 (dystonia, rigidity, hypokinesia or akinesia, hyperkinesia, tremor, akathisia, epileptic seizures, paresthesias). Each item was assigned a score from 0 (absent) to 3 (most severe) based on symptom severity of the last 3 d.16
Estimated by the Hodges-Lehmann method of location shift for 2-sample data.
Overall survival was calculated from time of study medication administration to death or last follow-up.
In post hoc analyses, multiple imputation with 20 iterations and worst-case sensitivity analysis were both consistent with the primary findings (multiple imputation: −4.0 for the lorazepam + haloperidol group vs −2.4 for the placebo + haloperidol group; mean difference, −1.6 [95% CI, −2.6 to −0.6], P = .001; worst-case sensitivity analysis: −3.7 for the lorazepam + haloperidol group vs −2.0 for the placebo + haloperidol group; mean difference, −1.7 [95% CI, −2.7 to −0.7], P = .003). The proportion of patients who developed a RASS score of 1 or more anytime during the first 8 hours was significantly lower in the lorazepam + haloperidol group than the placebo + haloperidol group (28% in the lorazepam + haloperidol group vs 76% in the placebo + haloperidol group; absolute risk reduction, 48% [95% CI, 26% to 71%]; P < .001).
Figure 2B shows that a larger proportion of patients in the placebo + haloperidol group had hyperactivity (RASS score, 1 to 4) at both 30 minutes and 8 hours. When data were plotted for individuals, there were consistent and rapid decreases in RASS score among patients who received lorazepam + haloperidol, and a variable response with placebo + haloperidol (eFigure 1 in Supplement 2). Daily RASS scores documented after the initial 8-hour period showed few patients had a RASS score of −3 or less in either study group (eFigure 2 in Supplement 2).
Secondary Outcomes
Patients in the lorazepam + haloperidol group required significantly lower doses of rescue neuroleptics, total neuroleptics, and a fewer number of rescue neuroleptics during the first 8 hours (median haloperidol equivalent daily dose of rescue neuroleptics: 2.0 mg in the lorazepam + haloperidol group vs 4.0 mg in the placebo + haloperidol group; median difference, −1.0 mg [95% CI, −2.0 to 0], P = .009; median total neuroleptics: 6.0 mg in the lorazepam + haloperidol group vs 6.0 mg in the placebo + haloperidol group; median difference, −1.0 mg [95% CI, −2.0 to 0], P = .03; median No. of rescue neuroleptic doses: 1.0 in the lorazepam + haloperidol group vs 2.0 in the placebo + haloperidol group; median difference, −0.5 [95% CI, −1.0 to 0], P = .008) (Table 2). Moreover, patients in the lorazepam + haloperidol group were perceived to be in greater comfort after study medication administration by both caregivers and nurses (caregivers: 84% in the lorazepam + haloperidol group vs 37% in the placebo + haloperidol group; mean difference, 47% [95% CI, 14% to 73%], P = .007; nurses: 77% in the lorazepam + haloperidol group vs 30% in the placebo + haloperidol group; mean difference, 47% [95% CI, 17% to 71%], P = .005).
The ESAS showed no statistically significant difference between the 2 study groups, except for greater level of drowsiness as rated by caregivers (1.9 in the lorazepam + haloperidol group vs −2.0 in the placebo + haloperidol group; mean difference, 3.9 [95% CI, 0.8 to 7.1]; P = .03) (Table 2). During the first 8 hours after study medication administration, MDAS score and respiratory rate did not differ between study groups and remained stable over time (MDAS score: 2.5 points in the lorazepam + haloperidol group points vs 0.4 points in the placebo + haloperidol group; mean difference, 2.1 points [95% CI, −1.0 to 5.2], P = .18; respiratory rate: −1.5 in the lorazepam + haloperidol group vs −0.5 in the placebo + haloperidol group; mean difference, −1.0 [95% CI, −3.4 to 1.4], P = .80) (Table 2). We did not identify any significant difference in other secondary measures, including delirium recall and related distress and communication capacity (eTable 2 in Supplement 2). The most common adverse effects were hypokinesia and akathisia (hypokinesia: 3 patients [19%] in the lorazepam + haloperidol group and 4 patients [27%] in the placebo + haloperidol group; akathisia: 3 patients [19%] in the lorazepam + haloperidol group and 1 patient [7%] in the placebo + haloperidol group). One patient (3%) in the lorazepam + haloperidol group and 3 patients (10%) in the placebo + haloperidol group died within 8 hours of study medication administration. No significant differences were found in discharge outcomes and overall survival (Table 2 and eFigure 3 in Supplement 2).
Discussion
In this preliminary trial of hospitalized patients with delirium in the setting of advanced cancer, the addition of lorazepam to haloperidol compared with haloperidol alone resulted in a significantly greater reduction in agitation at 8 hours. Patients in the lorazepam + haloperidol group required fewer rescue medications, were perceived to be more comfortable by both caregivers and nurses blinded to treatment assignment, and had no difference in adverse events, respiratory depression, or survival. Taken together, this study supports the judicious use of single-dose lorazepam + haloperidol for patients with persistent agitated delirium after a trial of scheduled haloperidol.
Agitation in the setting of delirium is a common manifestation of the dying process and a management challenge. Trials on agitated delirium are logistically complex because they require surrogate decision makers to enroll their family members during the final days of life when emotional stress levels may be high.
Although haloperidol has been considered the standard therapy for delirium management,7 a recent study raised questions about its safety and effectiveness.24,25 The current study provides some insights into the efficacy of intravenous haloperidol. In the control group, a single 2-mg dose of haloperidol alone resulted in a rapid decrease in agitation level; however, its effect was highly variable and nonsustained. Thus, the control group highlights the need to identify better options to manage persistent agitation.26,27
The use of benzodiazepine for delirium is controversial. A 2009 Cochrane review28 commented on the lack of placebocontrolled randomized clinical trials and concluded that benzodiazepines could not be recommended for delirium that was not related to alcohol withdrawal, and indeed benzodiazepines may precipitate delirium.9,29–31 Instead of benzodiazepine alone, this study tested the combination strategy to take advantage of the different mechanism of action of benzodiazepine and the neuroleptic haloperidol.26
Agitation in the setting of delirium is distressing for patients, their caregivers, and clinicians. The RASS score enables evaluation of the effect of alternative pharmacologic interventions to treat agitation from multiple perspectives. However, the desirable RASS score among patients with agitated delirium is ill defined and is likely to depend on how much caregivers and patients value alertness in the context of the dying process. Patients and their caregivers wish to avoid both agitation (ie, RASS score, ≥1) and excessive sedation (ie, RASS score, ≤−3). In this study, the lorazepam + haloperidol group not only had fewer patients with a RASS score of 1 or more anytime during the first 8 hours, but also required fewer doses of rescue medications, supporting the hypothesis that lorazepam + haloperidol could effectively control agitation. The number needed to treat based on this metric was 2.1 (95% CI, 1.4 to 3.9). Whether a RASS score of 0 to −2 might be considered a more desirable outcome than a RASS score of −3 to −5 is uncertain and is likely to vary among patients, their caregivers, and clinicians. In this trial, the mean RASS score was approximately 0 in the placebo + haloperidol group and was below −2 in the lorazepam + haloperidol group, suggesting a trade-off between more-effective treatment of agitation and higher levels of sedation (Figure 2). Yet, patients in the lorazepam + haloperidol group were perceived to be more comfortable, suggesting that caregivers and nurses valued lack of agitation over the risk of greater sedation. More research is needed to define the optimal RASS score range in the context of terminal delirium. Further research should also examine various pharmacologic combinations and dosing to minimize oversedation while achieving optimal control of agitation.27,35,36,37
In contrast to a majority of clinical trials on delirium that focused on reducing the overall delirium severity or a composite of symptoms,9,24 the primary goal of this study was to control a specific symptom of delirium–agitation–because it causes high levels of distress among patients and caregivers.4 The study findings support the therapeutic role of lorazepam when given in combination with haloperidol as a single-dose rescue to patients with refractory agitation despite scheduled haloperidol. The use of lorazepam in other combinations, populations, and indications needs to be thoroughly investigated in future clinical studies. Currently, patients with severe refractory agitated delirium often require hospitalization for control of this highly distressing syndrome. However, many patients and caregivers prefer to die at home with support from home hospice. Both lorazepam and haloperidol are available as oral medications including a rapid sublingual form of lorazepam. Further research is needed to examine if these treatment options are feasible and effective for patients with agitated delirium in the home setting.
There were no significant between-group differences in multiple exploratory outcomes, such as delirium severity, delirium-related distress, and communication capacity. However, this study was not powered to examine these secondary outcomes. Specifically, there was no significant worsening of agitation, respiratory rate, or other adverse effects. A single dose of lorazepam was not associated with a shortened survival consistent with nonrandomized observational studies examining the effect of continuous benzodiazepine infusion on survival.32,33
Limitations
This study has several limitations. First, this was a singlecenter study conducted at a tertiary care cancer center. Although the mortality rate of this acute palliative care unit is similar to other US centers34 and a majority of patients who were eligible enrolled onto this study, the study findings may not be generalizable to other settings (eg, patients earlier in the disease trajectory or those treated at home) and the external validity needs to be further assessed. Second, only a single dose of study medication was administered as rescue. Future studies will need to assess the effects of repeated dosing. Third, a single lorazepam dose of 3 mg might be too high for some patients, especially those with severe liver failure who cannot metabolize lorazepam. Further studies are needed to examine different doses. Fourth, several secondary outcomes in this study, such as the delirium recall questionnaire, require further validation. Fifth, this study had a small sample size and thus wide CIs in many measures. It was not powered to examine the multiple secondary outcomes and thus the secondary findings should be considered as exploratory.
Conclusions
In this preliminary trial of hospitalized patients with agitated delirium in the setting of advanced cancer, the addition of lorazepam to haloperidol compared with haloperidol alone resulted in a significantly greater reduction in agitation at 8 hours. Further research is needed to assess generalizability and adverse effects.
Supplementary Material
Key Points.
Question
Is the combination of lorazepam + haloperidol superior to placebo + haloperidol in the treatment of persistent agitation in patients with delirium and advanced cancer?
Findings
In this randomized trial of 58 patients, the addition of lorazepam to haloperidol compared with haloperidol alone resulted in a significantly greater reduction in agitation at 8 hours (−4.1 vs −2.3 points on the 10-point Richmond Agitation Sedation Scale).
Meaning
The addition of lorazepam to haloperidol may provide superior control of agitation in patients with persistent delirium.
Acknowledgments
Funding/Support: This study was supported by grant R21CA186000-01A1 from the National Cancer Institute (Drs Hui, Bruera, Hess, and Breitbart); a Mentored Research Scholar Grant in Applied and Clinical Research (MRSG-14-1418-01-CCE) from the American Cancer Society (Dr Hui) and the Andrew Sabin Family Fellowship Award (Dr Hui) from the Andrew Sabin Family Foundation; grant P30CA016672 from the National Institutes of Health CancerCenter(Drs Diba and Hess and Ms Liu); and grant R01CA200867 from the National Institutes of Health (Dr Delgado-Guay).
Role of the Funder/Sponsor: The funding sources were not involved in the design and conduct of the study; the collection, management, analysis, and interpretation of the data; the preparation, review, and approval of the manuscript, and the decision to submit for publication.
Footnotes
Author Contributions: Dr Hui had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Concept and design: Hui, Wilson, Dibaj, Nguyen, de la Cruz, Walker, Zhukovsky, Delgado Guay, Hall, Hess, Breitbart, Bruera.
Acquisition, analysis, or interpretation of data: Dibaj, Nguyen, de la Cruz, Zhukovsky, Delgado Guay, Vidal, Epner, Reddy, Tanco, Williams, Hall, Liu, Hess, Amin.
Drafting of the manuscript: Dibaj, Nguyen, Delgado Guay, Hall.
Critical revision of the manuscript for important intellectual content: Hui, Wilson, Dibaj, de la Cruz, Walker, Zhukovsky, Delgado Guay, Vidal, Epner, Reddy, Tanco, Williams, Liu, Hess, Amin, Breitbart, Bruera.
Statistical analysis: Dibaj, Liu, Hess.
Administrative, technical, or material support: Hui, Wilson, Nguyen, Walker, Williams, Hall, Amin, Breitbart, Bruera.
Supervision: Hui, de la Cruz, Reddy, Tanco, Bruera.
Additional Contributions: We thank all the nurses, patients, and caregivers who participated in this study. We also thank Kristy W. Rofheart, MLA, and Swati Bansal, MPH (both from MD Anderson Cancer Center), for assistance with data collection. They received a salary for study coordination. We also thank Duck-Hee Kang, PhD (University of Texas Health Science Center), for her support in the design and conduction of this study. She did not receive compensation for her contribution.
Conflict of Interest Disclosures: All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest and none were reported.
Previous Presentation: This study was presented as an oral abstract at the 2017 American Society of Clinical Oncology Annual Meeting; June 2–6, 2017; Chicago, Illinois.
Contributor Information
David Hui, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Susan Frisbee-Hume, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Annie Wilson, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Seyedeh S. Dibaj, Department of Biostatistics, MD Anderson Cancer Center, Houston, Texas.
Thuc Nguyen, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Maxine De La Cruz, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Paul Walker, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Donna S. Zhukovsky, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Marvin Delgado-Guay, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Marieberta Vidal, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Daniel Epner, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Akhila Reddy, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Kimerson Tanco, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Janet Williams, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Stacy Hall, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
Diane Liu, Department of Biostatistics, MD Anderson Cancer Center, Houston, Texas.
Kenneth Hess, Department of Biostatistics, MD Anderson Cancer Center, Houston, Texas.
Sapna Amin, Department of Investigational Pharmacy, MD Anderson Cancer Center, Houston, Texas.
William Breitbart, Department of Psychiatry and Behavioral Sciences, Memorial Sloan Kettering Cancer Center, New York, New York.
Eduardo Bruera, Department of Palliative Care, Rehabilitation and Integrative Medicine, MD Anderson Cancer Center, Houston, Texas.
References
- 1.Hosie A, Davidson PM, Agar M, Sanderson CR, Phillips J. Delirium prevalence, incidence, and implications for screening in specialist palliative care inpatient settings: a systematic review. Palliat Med. 2013;27(6):486–498. doi: 10.1177/0269216312457214. [DOI] [PubMed] [Google Scholar]
- 2.Breitbart W, Gibson C, Tremblay A. The delirium experience: delirium recall and delirium-related distress in hospitalized patients with cancer, their spouses/caregivers, and their nurses. Psychosomatics. 2002;43(3):183–194. doi: 10.1176/appi.psy.43.3.183. [DOI] [PubMed] [Google Scholar]
- 3.Breitbart W, Alici Y. Agitation and delirium at the end of life: “we couldn’t manage him”. JAMA. 2008;300(24):2898–2910. doi: 10.1001/jama.2008.885. [DOI] [PubMed] [Google Scholar]
- 4.Bruera E, Bush SH, Willey J, et al. Impact of delirium and recall on the level of distress in patients with advanced cancer and their family caregivers. Cancer. 2009;115(9):2004–2012. doi: 10.1002/cncr.24215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kehl KA. Treatment of terminal restlessness: a review of the evidence. J Pain Palliat Care Pharmacother. 2004;18(1):5–30. [PubMed] [Google Scholar]
- 6.Hui D, De La Cruz M, Bruera E. Palliative care for delirium in patients in the last weeks of life: the final frontier. J Palliat Care. 2014;30(4):259–264. [PubMed] [Google Scholar]
- 7.Levy MH, Smith T, Alvarez-Perez A, et al. NCCN clinical practice guidelines in oncology: palliative care. doi: 10.6004/jnccn.2009.0031. https://www.nccn.org/professionals/physician_gls/f_guidelines.asp. Accessed April 19, 2017. [DOI] [PubMed]
- 8.Aggarwal R. Delirium and benzodiazepines. Curr Psychiatr. 2011;10(10):4. http://www.mdedge.com/currentpsychiatry/article/64502/schizophrenia-other-psychotic-disorders/delirium-and- benzodiazepines. Accessed August 11, 2017. [Google Scholar]
- 9.Breitbart W, Marotta R, Platt MM, et al. A double-blind trial of haloperidol, chlorpromazine, and lorazepam in the treatment of delirium in hospitalized AIDS patients. Am J Psychiatry. 1996;153(2):231–237. doi: 10.1176/ajp.153.2.231. [DOI] [PubMed] [Google Scholar]
- 10.de Wit M, Best AM, Epstein SK, Greenblatt DJ. Lorazepam concentrations, pharmacokinetics and pharmacodynamics in a cohort of mechanically ventilated ICU patients. Int J Clin Pharmacol Ther. 2006;44(10):466–473. doi: 10.5414/cpp44466. [DOI] [PubMed] [Google Scholar]
- 11.Greenblatt DJ, Ehrenberg BL, Gunderman J, et al. Kinetic and dynamic study of intravenous lorazepam: comparison with intravenous diazepam. J Pharmacol Exp Ther. 1989;250(1):134–140. [PubMed] [Google Scholar]
- 12.Sessler CN, Gosnell MS, Grap MJ, et al. The Richmond Agitation-Sedation Scale: validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med. 2002;166(10):1338–1344. doi: 10.1164/rccm.2107138. [DOI] [PubMed] [Google Scholar]
- 13.Ely EW, Truman B, Shintani A, et al. Monitoring sedation status over time in ICU patients: reliability and validity of the Richmond Agitation-Sedation Scale (RASS) JAMA. 2003;289(22):2983–2991. doi: 10.1001/jama.289.22.2983. [DOI] [PubMed] [Google Scholar]
- 14.Bruera E, Kuehn N, Miller MJ, Selmser P, Macmillan K. The Edmonton Symptom Assessment System (ESAS): a simple method for the assessment of palliative care patients. J Palliat Care. 1991;7(2):6–9. [PubMed] [Google Scholar]
- 15.Hui D, Bruera E. The Edmonton Symptom Assessment System 25 years later: past, present, and future developments. J Pain Symptom Manage. 2017;53(3):630–643. doi: 10.1016/j.jpainsymman.2016.10.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lingjaerde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K. The UKU side effect rating scale: a new comprehensive rating scale for psychotropic drugs and a cross-sectional study of side effects in neuroleptic-treated patients. Acta Psychiatr Scand Suppl. 1987;334(suppl):1–100. doi: 10.1111/j.1600-0447.1987.tb10566.x. [DOI] [PubMed] [Google Scholar]
- 17.National Institutes of Health. NIH Policy on reporting race and ethnicity data: subjects in clinical research. https://grants.nih.gov/grants/guide/notice-files/NOT-OD-01-053.html. Accessed July 26, 2017.
- 18.Breitbart W, Rosenfeld B, Roth A, Smith MJ, Cohen K, Passik S. The Memorial Delirium Assessment Scale. J Pain Symptom Manage. 1997;13(3):128–13. doi: 10.1016/s0885-3924(96)00316-8. [DOI] [PubMed] [Google Scholar]
- 19.Fadul N, Kaur G, Zhang T, Palmer JL, Bruera E. Evaluation of the Memorial Delirium Assessment Scale (MDAS) for the screening of delirium by means of simulated cases by palliative care health professionals. Support Care Cancer. 2007;15(11):1271–1276. doi: 10.1007/s00520-007-0247-6. [DOI] [PubMed] [Google Scholar]
- 20.Rijcken CA, Monster TB, Brouwers JR, de Jong-van den Berg LT. Chlorpromazine equivalents versus defined daily doses: how to compare antipsychotic drug doses? J Clin Psychopharmacol. 2003;23(6):657–659. doi: 10.1097/01.jcp.0000096247.29231.3a. [DOI] [PubMed] [Google Scholar]
- 21.Hui D, Bush SH, Gallo LE, Palmer JL, Yennurajalingam S, Bruera E. Neuroleptic dose in the management of delirium in patients with advanced cancer. J Pain Symptom Manage. 2010;39(2):186–196. doi: 10.1016/j.jpainsymman.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 22.Hui D, Reddy A, Palla S, Bruera E. Neuroleptic prescription pattern for delirium in patients with advanced cancer. J Palliat Care. 2011;27(2):141–14. [PubMed] [Google Scholar]
- 23.Hui D, dos Santos R, Chisholm GB, Bruera E. Symptom expression in the last seven days of life among cancer patients admitted to acute palliative care units. J Pain Symptom Manage. 2015;50(4):488–494. doi: 10.1016/j.jpainsymman.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agar MR, Lawlor PG, Quinn S, et al. Efficacy of oral risperidone, haloperidol, or placebo for symptoms of delirium among patients in palliative care: a randomized clinical trial. JAMA Intern Med. 2017;177(1):34–42. doi: 10.1001/jamainternmed.2016.7491. [DOI] [PubMed] [Google Scholar]
- 25.Hui D, Valentine A, Bruera E. Neuroleptics for delirium: more research is needed. JAMA Intern Med. 2017;177(7):1052–1053. doi: 10.1001/jamainternmed.2017.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Menza MA, Murray GB, Holmes VF, Rafuls WA. Controlled study of extrapyramidal reactions in the management of delirious, medically ill patients: intravenous haloperidol versus intravenous haloperidol plus benzodiazepines. Heart Lung. 1988;17(3):238–241. [PubMed] [Google Scholar]
- 27.Hui D, Dev R, Bruera E. Neuroleptics in the management of delirium in patients with advanced cancer. Curr Opin Support Palliat Care. 2016;10(4):316–323. doi: 10.1097/SPC.0000000000000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lonergan E, Luxenberg J, Areosa Sastre A. Benzodiazepines for delirium. Cochrane Database Syst Rev. 2009;(4):CD006379. doi: 10.1002/14651858.CD006379.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marcantonio ER, Juarez G, Goldman L, et al. The relationship of postoperative delirium with psychoactive medications. JAMA. 1994;272(19):1518–1522. [PubMed] [Google Scholar]
- 30.Pandharipande P, Shintani A, Peterson J, et al. Lorazepam is an independent risk factor for transitioning to delirium in intensive care unit patients. Anesthesiology. 2006;104(1):21–26. doi: 10.1097/00000542-200601000-00005. [DOI] [PubMed] [Google Scholar]
- 31.Zaal IJ, Devlin JW, Hazelbag M, et al. Benzodiazepine-associated delirium in critically ill adults. Intensive Care Med. 2015;41(12):2130–213. doi: 10.1007/s00134-015-4063-z. [DOI] [PubMed] [Google Scholar]
- 32.Maeda I, Morita T, Yamaguchi T, et al. Effect of continuous deep sedation on survival in patients with advanced cancer (J-Proval): a propensity score-weighted analysis of a prospective cohort study. Lancet Oncol. 2016;17(1):115–122. doi: 10.1016/S1470-2045(15)00401-5. [DOI] [PubMed] [Google Scholar]
- 33.Maltoni M, Pittureri C, Scarpi E, et al. Palliative sedation therapy does not hasten death: results from a prospective multicenter study. Ann Oncol. 2009;20(7):1163–1169. doi: 10.1093/annonc/mdp048. [DOI] [PubMed] [Google Scholar]
- 34.Hui D, Elsayem A, De la Cruz M, et al. Availability and integration of palliative care at US cancer centers. JAMA. 2010;303(11):1054–1061. doi: 10.1001/jama.2010.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin SH, Hui D, Chisholm G, et al. Frequency and outcome of neuroleptic rotation in the management of delirium in patients with advanced cancer. Cancer Res Treat. 2015;47(3):339–405. doi: 10.4143/crt.2013.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pandharipande PP, Pun BT, Herr DL, et al. Effect of sedation with dexmedetomidine vs lorazepam on acute brain dysfunction in mechanically ventilated patients: the MENDS randomized controlled trial. JAMA. 2007;298(22):2644–2653. doi: 10.1001/jama.298.22.2644. [DOI] [PubMed] [Google Scholar]
- 37.Reade MC, Eastwood GM, Bellomo R, et al. DahLIA Investigators; Australian and New Zealand Intensive Care Society Clinical Trials Group Effect of dexmedetomidine added to standard care on ventilator-free time in patients with agitated delirium: a randomized clinical trial. JAMA. 2016;315(14):1460–1468. doi: 10.1001/jama.2016.2707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.