

Abstract
Background
Cold is a major abiotic stress limiting the production of tropical and subtropical crops in new production areas. Sugarcane (Saccharum spp.) originates from the tropics but is cultivated primarily in the sub-tropics where it frequently encounters cold stress. Besides regulating plant growth, miRNAs play an important role in environmental adaption.
Results
In this study, a total of 412 sugarcane miRNAs, including 261 known and 151 novel miRNAs, were obtained from 4 small RNA libraries through the Illumina sequencing method. Among them, 62 exhibited significant differential expression under cold stress, with 34 being upregulated and 28 being downregulated. The expression of 13 miRNAs and 12 corresponding targets was validated by RT-qPCR, with the majority being consistent with the sequencing data. GO and KEGG analysis indicated that these miRNAs were involved in stress-related biological pathways. To further investigate the involvement of these miRNAs in tolerance to abiotic stresses, sugarcane miR156 was selected for functional analysis. RT-qPCR revealed that miR156 levels increased in sugarcane during cold, salt and drought stress treatments. Nicotiana benthamiana plants transiently overexpressing miR156 exhibited better growth status, lower ROS levels, higher anthocyanin contents as well as the induction of some cold-responsive genes, suggesting its positive role in the plant cold stress response.
Conclusions
This study provides a global view of the association of miRNA expression with the sugarcane response to cold stress. The findings have enriched the present miRNA resource and have made an attempt to verify the involvement of miR156 in plant response to cold stress.
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4231-3) contains supplementary material, which is available to authorized users.
Keywords: Saccharum spp., High-throughput sequencing, miR156, Cold, Transcriptome
Background
Small non-coding RNAs of 18–40 nucleotides (nt) in length, including microRNAs (miRNAs) and small interference RNAs (siRNAs), can regulate gene expression by posttranscriptional mechanisms and epigenetic modifications to influence a number of biological processes [1]. These endogenous 18–25 nt non-coding RNAs [2] can target mRNA cleavage or translational repression by guiding the RNA-induced silencing complex (RISC) to bind to the target mRNA [3]. Primary miRNAs (pri-miRNAs) are transcribed as long precursors in the nucleus, and can be transferred to the cytoplasm to form mature miRNAs after a series of processing steps [4, 5]. The physiological significance of miRNAs in plants has been demonstrated in various biological processes [6, 7]. In order to understand the molecular mechanism of miRNAs in non-model organisms, more miRNAs need to be identified and characterized. With the advantages of identifying large amounts of miRNAs effectively in a short time and at low cost [8], high-throughput sequencing has become the main method for obtaining miRNAs [9, 10].
Cold stress, including chilling (<20 °C) and freezing (< 0 °C), is one important limiting factor that can affect the geographical distribution and planting season of crops, especially for tropical and subtropical crops [11]. Low temperature can suppress plant growth and development by inhibiting metabolic reactions, leading to osmotic, oxidative and other stresses [12]. Plants usually possess a series of strategies to respond to temperature fluctuations, such as the remodeling of cell and tissue structures and gene expression, and metabolism reprogramming [11–13]. Cold-related gene expression can be regulated at the transcriptional, post-transcriptional and post-translational levels [14]. During low temperature stress, C-repeat binding factors (CBFs), which bind to dehydration-responsive elements in gene promoters to activate the downstream cold-responsive (COR) genes, is regulated by the inducer of C-repeat binding factor expression 1 (ICE1). This is known as the ICE-CBF-COR pathway [14, 15]. Furthermore, reports have shown that some miRNAs also take part in the plant response to cold stress and play crucial roles in this process [16, 17]. In sugarcane (Saccharum spp.), the up-regulation of miR319 and down-regulation of its targets, including a Myb transcription factor (GAMyB) and a TCP transcription factor (PCF5), were observed in both cold -tolerant and -sensitive sugarcane varieties under treatment at 4 °C [18]. However, their expression changes were delayed in cold tolerant varieties [18]. Similar results were found in Oryza sativa L., whereby the overexpression of osa-miR319 led to enhanced cold tolerance in transgenic rice seedlings [19]. In addition, some other cold-related miRNAs, such as miR156k from rice [20] and miR394 from Arabidopsis [21], have also been reported.
Sugarcane accounts for ∼80% of the global sugar production and has successfully been developed to be one of the most promising energy biofuels in Brazil [22]. However, sugarcane growth and development is affected by various factors, and due to its tropical origin, domestic varieties are restricted to tropical and subtropical regions [23, 24]. Although low temperature is beneficial for sucrose accumulation in sugarcane [25], it also leads to bud browning, resulting in poor growth [26]. Sugarcane harvest normally begins in winter and continues until spring.
In the present study, a high-throughput small RNA deep sequencing method was used to obtain cold-related miRNAs in sugarcane. The interaction networks of these miRNAs and their targets were investigated based on Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. In addition, the roles of several miRNAs, such as miR156, miR168, miR169 and miR408, in the plant response to abiotic stress, were explored. Our aim was to gain insight into the molecular mechanism of sugarcane cold tolerance and the role of miRNAs in this process.
Methods
Samples and small RNA library preparation
The nodes of ROC22 (relative cold sensitivity) and FN39 (relative cold tolerance) sugarcane cultivars (Saccharum spp. hybrid) were harvested from the field in the Key Laboratory of Sugarcane Biology and Genetic Breeding, Ministry of Agriculture in Fuzhou City, China. The related information of cold tolerance is indicated in Additional file 10: Fig. S1. These nodes were firstly treated with flowing water for 2 days to promote the sprouting of buds and remove impurities, and then they were cultivated in an incubator (Dongqi, Ningbo, China) at 28 °C in the dark and at 65% relative humidity for 4 days for sprouting using the moisture culture method [26]. The cold treatment (4 °C) was performed on corresponding buds from both cultivars for 0 (before the cold treatment), 3, 12, 24 and 48 h. Three biological replications were used for each time point and 6 buds for each sample. The total RNA of all the collected samples was extracted using Trizol™ Reagent (Invitrogen, Carlsbad, CA), qualified by electrophoresis with a 1% agarose gel and quantified using a NanoVue Plus (GE healthcare, Little Chalfont, UK). Usually, gene expression changes occur early upon cold stress, as attested by already published reports [27, 28] and previous results obtained in our lab (unpublished data). For this reason, samples collected at 0 and 3 h of cold stress treatment were assumed to be suitable for target molecular analyses. Therefore, the total RNA (10 μg) of the 0 and 3 h samples of FN39 and ROC22 cultivars was sent to BGI (Shenzhen, China) on dry ice for high-throughput small RNA deep sequencing using the Illumina HiSeq high-throughput sequencing platform.
Healthy sugarcane plantlets exhibiting constant growth status derived from the tissue culture of the ROC22 cultivar were grown under 16 h light/ 8 h dark conditions at 28 °C for 1 week in a incubator. Then, the plantlets were treated with 250 mM NaCl and 25% PEG8000 as the salt and drought stresses, respectively [29]. The samples subjected to salt and drought treatments for 0, 6, 12 and 24 h were collected for RNA extraction. Three biological replications were setup and 5 plantlets were used for each sample.
Bioinformatics analysis
Following size fractionation of the 18–30 nucleotide small RNAs and 5′ adaptor ligation and 3′ adaptor ligation, the small RNAs were reversely transcribed into cDNA, amplified and sequenced. The clean reads were obtained from the raw data from small RNA deep sequencing by removing the low-quality reads and impurities, including the reads without insertions; without 3′ -primer; with 5′-primer contaminants; those with poly A tails as well as small fragment sequences. The clean reads were firstly used to analyze the common/specific sequences and length distributions [30]. The ribosomal RNA (rRNA), small cytoplasmic RNA (scRNA), small nucleolar RNA (snoRNA), small nuclear RNA (snRNA) and transfer RNA (tRNA) were annotated and removed from the clean reads via a BLASTn search on the NCBI GenBank database and Rfam (10.1) database (e = 0.01). The remaining clean reads were aligned with the known miRNAs in miRBase (http://www.mirbase.org/) using the procedure and parameter blastall -p blastn -F F -e 0.01. The detail criterion is that firstly align the clean reads to the miRNA precursor in miRBase with no mismatch. Secondly, the obtained tags align with the mature miRNAs in miRBase with at least 16 nt overlap allowing offsets. Those miRNAs satisfying both criteria will be counted to get their expression, and will be analyzed to get base bias on the first position of the identified miRNAs with certain length, and base bias on each position of all identified miRNAs respectively. The novel miRNAs were predicted using the Mireap software (http://sourceforge.net/projects/mireap/) developed by BGI through considering the biological characteristics of miRNA, such as the secondary structure, the location of mature miRNA on its precursor and the minimum free energy [31]. Specific parameters were set as follows: minimal miRNA sequence length was 18; maximal miRNA sequence length was 25; minimal miRNA reference sequence length was 20; maximal miRNA reference sequence length was 23; maximal copy number of miRNAs on reference was 20; maximal free energy allowed for a miRNA precursor was −18 kcal/mol; maximal space between miRNA and miRNA* was 300, minimal base pairs of miRNA and miRNA* was 16; maximal bulge of miRNA and miRNA* was 4; maximal asymmetry of miRNA/miRNA* duplex was 4; flank sequence length of miRNA precursor was 20 [31].
The comparison analysis between the control and the cold-treated sugarcane was performed according to the following steps. The miRNA expression quantity was normalized using the formula: . Then, the normalized expression quantity was used to calculate the fold change using the formula:.
Mireap software was also used to predict the potential target genes according to the rules as previously reported [32, 33]. In order to identify the functions of potential targets of differentially expressed miRNAs, the number of targets mapping to each gene ontology term in the Gene Ontology database was calculated. Then, the significantly enriched GO terms were obtained using the super geometry inspection method. The GO enrichment analysis can provide clues for the identification of the main biological functions of the potential targets [34]. To further understand the biological functions, KEGG pathway analysis was also conducted, which provides information on the main biochemical pathways and signal transduction pathways the potential targets involved [35]. The GO enrichment analysis was used on predicted target gene candidates of miRNAs. This method firstly map all target gene candidates to GO terms in the database (http://www.geneontology.org/), and calculate gene numbers for each term, and then use hypergeometric test to find significantly enriched GO terms in target gene candidates comparing to the reference gene background. The calculating formula is:, within which N is the number of all genes with GO annotation; n is the number of target gene candidates in N; M is the number of all genes that are annotated to a certain GO term and m is the number of target gene candidates in M. The Bonferroni Correction for the p-value is used to obtain the corrected p-value. GO terms with corrected p-value <0.05 are defined as significantly enriched in target gene candidates. KEGG pathway analysis was also used for the target gene candidates. The calculating formula is the same as that in GO analysis. In this formula, N is the number of all genes with KEGG annotation, n is the number of target gene candidates in N, M is the number of all genes annotated to a certain pathway, and m is the number of target gene candidates in M. Genes with FDR(false discovery rate) <0.05 are considered as significantly enriched in target gene candidates. The KEGG analysis reveals the main pathways which the target gene candidates are involved in.
Expression validation of miRNAs and potential target genes by RT-qPCR
The cold responsive genes, CBF1 (C-repeat binding factor gene), CBF3 and NAC23 (NAM-ATAF-CUC gene), have been reported to be induced by cold stress and were used to verify the efficiency of cold treatment in this study by the reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR) method. In order to validate the presence and expression levels of miRNAs, 13 miRNAs were randomly detected by RT-qPCR method. Total RNA was extracted from the samples of 0, 3, 12, 24 and 48 h cold-treated sugarcane buds using Trizol™ Reagent (Invitrogen, CA), followed by DNase (Promega, USA) treatment to remove the genomic DNA. For the miRNA, the cDNA synthesis was performed according to the instruction of the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, USA). For each miRNA, the specific stem loop primer (Additional file 1: Table S1) was added to the reaction system. The reaction was performed at 16 °C for 30 min, 42 °C for 30 min and 85 °C for 5 s. All cDNA samples were 25-fold diluted and then used as a template in the miRNA RT-qPCR analysis. For the potential target genes, the cDNA synthesis was performed according to the instruction of the PrimeScript™ RT Reagent Kit (TaKaRa, Dalian, China). The reaction procedure included reverse transcription at 37°C for 15 min and the inactivation of reverse transcriptase with heat treatment at 85 °C for 5 s. The expression patterns of miRNAs and potential targets were detected according to the SYBR Green Master (ROX) (Roche, China) manufacturer instruction on a 7500 real time PCR system (Applied Biosystems, USA) (Additional file 2: Table S2). 18S ribosomal RNA (18S rRNA) [26] and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [18] were used as the internal control for miRNA and potential targets, respectively. The expression was calculated using the 2-△△Ct method [36].
All the statistical analysis was conducted using the Statistical Product and Service Solutions (SPSS20.0 version) software. Data was expressed as the mean ± standard error (SE). Significance (p-value <0.05) was calculated using one-way Analysis of Variance (ANOVA) followed by Duncan’s multiple range test.
The transient expression of miRNA in Nicotiana benthamiana
The precursor of miRNA, MIRNA, cloned from sugarcane was sub-cloned with BamH I and Xba I sites into the overexpression vector pCAMBIA 1301, and then transferred into the Agrobacterium tumefaciens strain EHA105. After inoculation in LB medium containing kanamycin (50 μg/mL) and rifampicin (34 μg/mL) twice, the cells of the EHA105 strain were collected and diluted to OD600 = 0.8 with Murashige and Skoog (MS) liquid medium (containing 200 mM acetosyringone). The Agrobacterium-mediated transformation was performed according to a previously described method [37]. The 1 mL syringe was used to infiltrate diluted bacterial suspension into the leaves of tobacco plants at the 6–8-leaf stage. After cultivation for 48 h under 16 h light/ 8 h dark at 23 °C conditions, the injected tobacco plants were treated with 4 °C stress for 12, 24and 48 h in an incubator (Dongqi, Ningbo, China). Then the injected tobacco leaves were collected for RNA extraction and DAB staining. Six biological replications were used.
3, 3′-diaminobenzidine (DAB) solution staining was used to assess H2O2 production [37]. The collected leaves were put into DAB solution (1 mg/mL, pH = 5.8) overnight under dark condition for staining. After boiling in 95% alcohol until the green color faded, the leaves were rinsed in 95% alcohol and photographed.
Anthocyanin quantification was performed according to a previously described method [38]. Briefly, 0.2 g fresh injected tobacco leaves were exacted with MeOH: HCl: H2O (80:5:15) at 4 °C overnight in the dark. After centrifugation at 15,000×g for 5 min, the anthocyanin levels in the extracts were qualified at 530 nm (T6 spectrophotometer, Beijing Purkinje General Instruments).
Results
Data analysis of small RNA libraries
In order to investigate the expression of miRNAs in sugarcane under cold stress, samples were collected at 0, 3, 12, 24 and 48 h over the 4 °C treatment. To validate the cold treatment, the expression of three cold responsive genes (CBF1, CBF3 and NAC23) was examined in these collected samples by RT-qPCR. The results showed that the transcripts of the three genes were all increased compared to those under control condition (Additional file 11: Figure S2), indicating that the cold treatment to sugarcane was effective in this study. In order to identify the miRNAs and other small RNAs involved in cold tolerance, two sugarcane cultivars including FN39 (relative cold tolerance) and ROC22 (relative cold sensitivity) were used to generate four small RNA libraries: F0 (samples of FN39 cultivar without cold treatment), F3 (samples of FN39 cultivar with 3 h cold treatment), R0 (samples of ROC22 cultivar without cold treatment) and R3 (samples of ROC22 cultivar with 3 h cold treatment). These libraries were sequenced by Illumina sequencing, which generated between 20 and 25 million clean reads after filtering out the low quality reads and contaminants, which accounted for 0.78% (F0), 0.97% (F3), 0.78% (R0) and 0.90% (R3) of the raw reads, respectively (Table 1). In all the above libraries, the reads with 24 nt and 21 nt lengths were the most abundant (Fig. 1). The analysis of 18–30 nt miRNA nucleotide bias showed that the first base of 21 nt and 22 nt miRNAs was uracil (U), and the miRNA nucleotide bias at the tenth-eleventh position, usually as the cleavage site, was an adenine (A) in all these libraries.
Table 1.
Small RNA categorization in sugarcane
| Categorization | Unique sRNA (%) | Total sRNA (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| F0 (%) | F3 (%) | R0 (%) | R3 (%) | F0 (%) | F3 (%) | R0 (%) | R3 (%) | |
| miRNA | 67,373 (0.53) | 57,249 (0.56) | 59,960 (0.54) | 58,772 (0.58) | 2,386,352 (9.41) | 1,638,016 (7.89) | 2,635,873 (10.12) | 2,302,177 (9.52) |
| rRNA | 56,395 (0.44) | 80,912 (0.79) | 81,039 (0.73) | 98,210 (0.97) | 729,453 (2.88) | 1,613,788 (7.78) | 1,453,854 (5.58) | 2,451,555 (10.14) |
| repeat | 412 (0) | 345 (0) | 447 (0) | 376 (0) | 957 (0) | 799 (0) | 1019 (0) | 774 (0) |
| snRNA | 4811 (0.04) | 4600 (0.05) | 4096 (0.04) | 4259 (0.04) | 12,452 (0.05) | 12,260 (0.06) | 12,838 (0.05) | 13,164 (0.05) |
| snoRNA | 2152 (0.02) | 2197 (0.02) | 2584 (0.02) | 2396 (0.02) | 4567 (0.02) | 4583 (0.02) | 5840 (0.02) | 5522 (0.02) |
| srpRNA | 0 (0) | 2 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (0) | 0 (0) | 0 (0) |
| tRNA | 9007 (0.07) | 13,069 (0.13) | 13,810 (0.12) | 15,819 (0.16) | 214,125 (0.84) | 17,084,377 (1.94) | 467,304 (1.79) | 696,897 (2.88) |
| unann | 12,602,337 (98.8) | 10,044,731 (98.45) | 10,917,735 (98.54) | 9,907,437 (98.22) | 22,010,746 (86.8) | 17,084,337 (1.94) | 21,461,967 (82.42) | 18,702,763 (77.37) |
| Clean reads | 12,742,487 (100) | 10,203,105 (100) | 11,079,671 (100) | 10,087,269 (100) | 25,358,652 (100) | 20,755,724 (100) | 26,038,695 (100) | 24,172,852 (100) |
| Raw reads | – | – | – | – | 25,556,975 | 20,959,989 | 26,243,698 | 24,392,550 |
The number represents the raw data generated directly from small RNA deep sequencing. F0 and F3 represent the bud sample from sugarcane cultivar FN39 with 0 h and 3 h cold (4 °C) treatment, respectively, while R0 and R3 represent the similar samples from cultivar ROC22, respectively
Fig. 1.
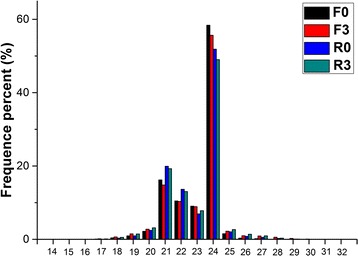
The length distribution of small RNAs in 4 small RNA libraries. F0 and F3 represent the bud samples from sugarcane cultivar FN39 with 0 and 3 h cold (4 °C) treatment, respectively, while R0 and R3 represent the similar samples from cultivar ROC22 with 0 and 3 h cold (4 °C) treatment, respectively
Identification of known miRNAs in sugarcane
To identify the known miRNAs, the clean reads were aligned with known miRNAs in miRBase. There were 219, 207, 210 and 209 known miRNAs identified in the F0, F3, R0 and R3 small RNA libraries, respectively (Additional file 3: Table S3). Compared with F0, F3 had 24 differentially expressed miRNAs (|log2 ratio| ≥ 1, p ≤ 0.05, Fig. 2a, Fig. 3a) including 17 upregulated and seven downregulated (Table 2). Similarly, 15 differentially expressed miRNAs (all upregulated) were identified upon comparison of R3 with R0 (Table 2). Among these differentially expressed miRNAs, there were eight common miRNAs (miR2199, miR5054, miR5059, miR5072, miR5077, miR5152-3p, miR5813 and miR8155) in both cultivars; all of which were upregulated (Fig. 2a). Furthermore, some known differentially expressed miRNAs were identified in either FN39 or ROC22. Among them, 16 known miRNAs, including nine upregulated and seven downregulated miRNAs, were only found in FN39 libraries, accounting for 66.7% (Table 2), including miR157a, miR160a, miR169b, miR6300, miR6196 and miR64785. Conversely, there were seven known differentially expressed miRNAs that existed only in ROC22, accounting for 46.7%, including miR5272, miR863-5p, miR5242, miR5072, miR5221, miR395o-3p and miR1861d.
Fig. 2.
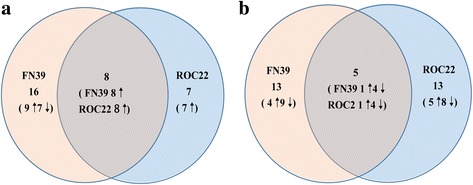
Distribution of differentially expressed miRNAs in FN39 and ROC22 cultivars. a) Known miRNAs; b) novel miRNAs
Fig. 3.
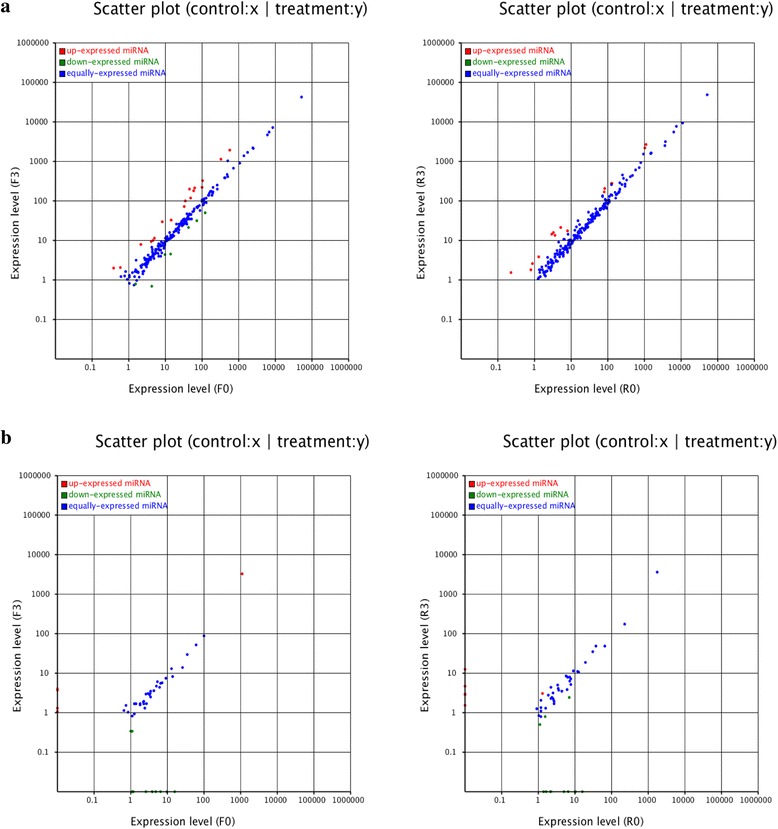
Scatter plots of differentially expressed miRNAs in 4 small RNA libraries. a) Known miRNAs; b) novel miRNAs. Green: significantly downregulated miRNAs; red: significantly upregulated miRNAs; blue: differentially expressed miRNAs (not significant). F0 and F3 represent the bud samples from sugarcane cultivar FN39 with 0 and 3 h cold (4 °C) treatment, respectively, while R0 and R3 represent the similar samples from cultivar ROC22 with 0 and 3 h cold (4 °C) treatment, respectively
Table 2.
Known miRNAs with significantly differential expression in sugarcane buds following cold treatment
| miRNA | sequence | F0-std | F3-std | R0-std | R3-std | Fold change | |
|---|---|---|---|---|---|---|---|
| F0-F3 | R0-R3 | ||||||
| miR1310 | GAGGCATCGGGGGCGCAACGCC | 8.32 | 29.24 | 15.78 | 30.98 | 1.81** | 0.97 |
| miR1520d | ATCAGAACTGGTACGGACAA | 574.08 | 1917.98 | 1131.16 | 2620.83 | −1.74** | 1.21 |
| miR157a | TTGACAGAAGATAGAGAGCAC | 4.65 | 9.68 | 4.34 | 4.26 | 1.06** | −0.02 |
| miR2199 | TGATAACTTGACGGATCGC | 59.42 | 179.46 | 129.61 | 272.82 | 1.59** | 1.07** |
| miR2916 | GGGGCTCGAAGACGATCAGAT | 46.30 | 198.06 | 128.65 | 255.78 | −2.10** | 0.99 |
| miR5054 | TCCCCACGGACGGCGCCA | 50.00 | 117.65 | 81.84 | 164.77 | 1.23** | 1.00** |
| miR5059 | CGTTCCTGGGCAGCAACACCA | 35.92 | 97.80 | 84.02 | 201.88 | 1.44** | 1.26** |
| miR5072 | GTTCCCCAGCGGAGTCGCCA | 2.16 | 7.70 | 5.34 | 20.97 | 1.82** | 1.97** |
| miR5077 | TTCACGTCGGGTTCACCA | 104.90 | 321.31 | 104.90 | 321.30 | 1.62** | 1.62** |
| miR5152-3p | AGTCCTGCTATACCCACCA | 0.59 | 2.02 | 1.30 | 3.76 | 1.77** | 1.52** |
| miR5575 | TGGATTTTGGAATGTTTTGGT | 0.39 | 1.98 | 3.03 | 2.23 | 2.32** | −0.44 |
| miR5671 | CATGGTGGTGACGGGTGAC | 32.73 | 70.96 | 72.74 | 126.88 | 1.12** | 0.80 |
| miR5813 | ACAGCAGGACGGTGGTCATGGA | 63.41 | 206.30 | 132.57 | 269.48 | 1.70** | 1.02** |
| miR6196 | GGGACGAGAAAGATGGGAGGA | 5.12 | 11.12 | 17.93 | 21.15 | 1.12 | 1.09** |
| miR6300 | GTCGTTGTAGTATAGTGGTG | 101.38 | 217.53 | 216.10 | 284.36 | 1.10** | 0.40 |
| miR6478 | CCGACCTTAGCTCAGTTGGTA | 14.70 | 32.52 | 30.53 | 51.46 | 1.14** | 0.75 |
| miR8155 | CGTAACCTGGCTCCGATACCA | 4.14 | 9.20 | 8.10 | 16.96 | 1.15** | 1.06** |
| miR894 | GTTTCACGTCGGGTTCACCA | 334.04 | 1108.12 | 1040.41 | 2131.64 | 1.72** | 1.03 |
| miR160a | TGCCTGGCTCCCTGTATGCCA | 13.92 | 4.43 | 4.64 | 8.89 | −1.65** | 0.94 |
| miR169b | CAGCCAAGGATGACTTGCCGG | 43.54 | 20.76 | 38.28 | 39.38 | −1.06** | 0.04 |
| miR169e-3p | GGCAGTCTCCTTGGCTAGC | 73.19 | 31.02 | 14.24 | 21.84 | −1.24** | 0.62 |
| miR397-5p | TTGACTGCAGCGTTGATGAGC | 9.82 | 12.34 | 2.30 | 3.64 | 1.12** | 0.66 |
| miR528-5p | TGGAAGGGGCATGCAGAGGAG | 123.62 | 49.19 | 303.86 | 262.15 | −1.32** | −0.21 |
| miR5532 | ATGGAAATTGATGACAAAGGGG | 1.58 | 0.77 | 1.46 | 1.20 | −1.03 * | −0.28 |
| miR1861d | TGCGTCATCAGGCAGGAACTGG | – | – | 3.30 | 15.34 | – | 2.22** |
| miR395o-3p | TGAAGTGTTTGGGTGAACTC | 2.60 | 1.64 | 0.88 | 2.56 | −0.66 | 1.54** |
| miR5221 | AACGAGATGGTGTTTTACTT | – | – | 3.00 | 14.23 | – | 2.24** |
| miR5242 | TATTTAGAACAGGCGATGTCA | 1.26 | 1.49 | 3.68 | 13.24 | 0.24 | 1.84** |
| miR5272f | GAATTGATTTGTGTTGGATAAATT | 3.27 | 2.64 | 0.80 | 1.78 | −0.30 | 1.14** |
| miR394a | TTGGCATTCTGTCCACCTCC | 4.26 | 7.78 | 0.88 | 0.74 | 1.03** | −0.96 |
| miR863-5p | TTATAGTCTTGTGGATCAAAT | – | – | 0.23 | 1.53 | – | 2.73** |
F0 and F3 represent the bud sample from sugarcane cultivar FN39 with 0 h and 3 h cold (4 °C) treatment, respectively, while R0 and R3 represent the similar samples from cultivar ROC22, respectively. ** and * indicate a significant difference at p-value <0.01 and p-value <0.05, respectively. “---” indicates no expression
Identification of novel miRNAs in sugarcane
A total of 151, 109, 140 and 132 novel miRNAs were discovered from the F0, F3, R0 and R3 libraries respectively using the Mireap software (Additional file 3: Table S3). There were 18 (five upregulated and 13 downregulated miRNAs) and 18 (six upregulated and 12 downregulated miRNAs) novel miRNAs expressed differentially in FN39 and ROC22 respectively after cold treatment for 3 h (|log2 ratio| ≥ 1, p ≤ 0.05, Fig. 2b, Fig. 3b, Table 3). Compared with known miRNAs, the novel miRNAs exhibited greater differential expression (a 10-fold up-regulation of novel_mir_117 in R3 and 11-fold down-regulation of novel_mir_72 in F 3). A proportion of novel miRNAs also showed cultivar-specificity, being expressed only in FN39 (13 novel, four upregulated and nine downregulated) or in ROC22 (13 novel, 5 upregulated and 8 downregulated).
Table 3.
Novel miRNAs with significant differential expression in sugarcane buds following cold treatment
| miRNA | sequence | F0-std | F3-std | R0-std | R3-std | Fold change | |
|---|---|---|---|---|---|---|---|
| F3-F0 | R3-R0 | ||||||
| novel_mir_107 | AGGGATGGTATGCACTGAAGC | 0.01 | 3.71 | 0.01 | 4.55 | 8.54** | 8.83** |
| novel_mir_117 | AAGAGGAAGAGAGAGAGGGTG | 0.01 | 0.01 | 0.01 | 12.33 | – | 10.27** |
| novel_mir_134 | GAGGCAAGGAGGTGGAATAGAC | 0.01 | 3.90 | – | – | 8.61** | – |
| novel_mir_136 | AAGCAAGTTGGGGTAGGCTAA | 0.01 | 1.30 | – | – | 7.02** | – |
| novel_mir_142 | TGAAACGGTACGGCTGATAAGTT | 0.01 | 1.06 | – | – | 6.73** | – |
| novel_mir_21 | TAAATATATAACATCAGGACC | 1.10 | 0.01 | – | – | −6.79** | – |
| novel_mir_34 | CGTACCACCGTCCGGGACTAA | 1.14 | 0.01 | – | – | −6.84** | – |
| novel_mir_37 | TCCAATGCATACCGTCCCTAA | 3.94 | 0.01 | 5.03 | 0.01 | −8.62** | −8.97** |
| novel_mir_41 | CCGAGAGGTAGGGCCGGTCGAA | 6.55 | 0.01 | 12.83 | 10.34 | −9.35** | – |
| novel_mir_54 | TCCAAATTATAAGACGTTTTG | 1.03 | 0.34 | 1.57 | 0.79 | −1.60** | −1.00 * |
| novel_mir_57 | GGGCTGGTTCGGCTGGTGGAA | 1.10 | 0.01 | – | – | −6.79** | – |
| novel_mir_68 | ATAAGACGTTTTGGCTTTTCT | 3.90 | 0.01 | – | – | −8.61** | – |
| novel_mir_72 | AAGAGGAAGAGAGAGAGGGTGA | 16.09 | 0.01 | 16.17 | 0.01 | −10.65** | −10.66** |
| novel_mir_74 | TCTTGGATTTGCATTGGATGCC | 1.18 | 0.01 | – | – | −6.89** | – |
| novel_mir_79 | CGGCCGACGCGCCGCGGCGGGC | 2.64 | 0.01 | 2.19 | 0.01 | −8.05** | −7.77** |
| novel_mir_80 | TGGTTGTTGGCTGGCCATGGCTG | 5.44 | 5.93 | 6.57 | 0.01 | – | −9.36** |
| novel_mir_81 | GCTAGAGGCAGCAACTGCATA | 9.94 | 0.01 | – | – | −9.96** | – |
| novel_mir_89 | CAATCGTGGACCAACTAGGCT | 4.85 | 0.01 | 3.53 | 4.01 | −8.92** | 0.18 |
| novel_mir_91 | CCTGCGTCGCACGGATTCGT | 1114.45 | 3231.35 | – | – | 1.54** | – |
| novel_mir_97 | GTCGTCGCCGTCGTCGTCGTC | 1.10 | 0.34 | 1.19 | 0.79 | −1.71** | −0.60 |
| novel_mir_1 | GGAATGTTGTCTGGTCGGAGA | 9.31 | 7.37 | 10.48 | 0.01 | −0.34 | −10.03** |
| novel_mir_130 | GGCATGGGAACATGTAGGAAGG | – | – | 0.01 | 2.81 | – | 8.14** |
| novel_mir_171 | TTTGAATAAGACGAGTGGTCA | – | – | 10.33 | 0.01 | – | −10.01** |
| novel_mir_183 | TAAGCTGGCTGATGCTGTTTT | – | – | 1.65 | 0.01 | – | −7.37** |
| novel_mir_216 | TCGAGGCCGAACGGATAAGTC | – | – | 0.01 | 1.53 | – | 7.26** |
| novel_mir_228 | TTTTTGGTGATTGATGACAAC | – | – | 0.01 | 2.90 | – | 8.18** |
| novel_mir_28 | AGGTCATGCTGTAGTTTCATC | – | – | 1.30 | 3.02 | – | 1.21** |
| novel_mir_3 | GGCTGAGCATGCCGCCGTCGAG | 0.75 | 1.49 | 7.18 | 2.40 | 1.00 | −1.58** |
| novel_mir_58 | CGTCGCCGTCGTCGTCGTCGTC | 2.29 | 1.64 | 1.11 | 0.50 | −0.48 | −1.16 * |
| novel_mir_64 | AGGGGTTGTGCGGCGGTGCCC | – | – | 1.42 | 0.01 | – | −7.15** |
| novel_mir_67 | AAAAACTTTGTACTAAAGCATA | – | – | 2.27 | 0.01 | – | −7.82** |
F0 and F3 represent the bud sample from sugarcane cultivar FN39 with 0 h and 3 h cold treatment, respectively; R0 and R3 represent the bud sample from sugarcane cultivar ROC22 with 0 h and 3 h, respectively. ** and * indicate a significant differences at p-value <0.01 and p-value <0.05, respectively. “---” indicates no expression
Functional analysis of target genes of differential expressed miRNAs
A total of 2805 target genes for 202 known expressed miRNAs were predicted using the Mireap software from the sugarcane EST database and the sugarcane unigene database (Additional file 4: Table S4). In order to provide an insight into their biological functions, GO and KEGG analyses of differentially expressed miRNAs were performed.
GO analysis of the predicted targets of differentially expressed miRNAs includes three ontologies: biological process, cellular component and molecular function (Additional file 5: Table S5). On the biological process, the result of the GO analysis indicated that the targets of the differentially expressed known and novel miRNAs were mostly involved in cellular and metabolic processes (Fig. 4). On the cellular component, the predicted targets of differentially expressed known miRNAs were mainly associated with cell and cell part, while for the novel miRNAs, their targets were mainly related with the macromolecular complex (Fig. 4). The molecular function of these targets was mainly related to the binding and catalytic activity (Fig. 4).
Fig. 4.
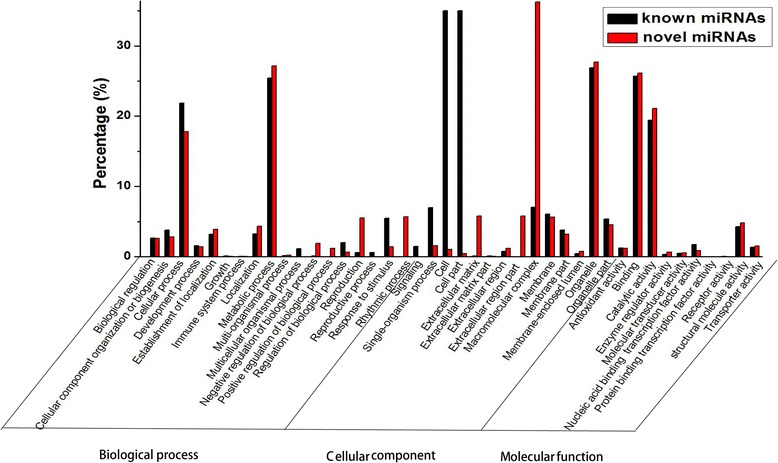
GO analysis of the predicted target genes for known and novel differentially expressed miRNAs
KEGG analysis of target genes was performed (Additional files 6, 7, 8 and 9: Table S6-S9). In our study, six pathways related to cold stress were analyzed, including plant hormone signal transduction, ABC transporters, peroxisomes, phosphatidylinositol signaling systems, ubiquitin mediated proteolysis and calcium signal pathways (Fig. 5). There were a number of miRNAs that are associated with the regulation of hormone signal pathways (Fig. 6). These miRNAs could affect plants in a number of ways, including cell enlargement, stomatal closure, stem growth, germination, cell division, shoot initiation, fruit ripening and senescence through regulating their target genes.
Fig. 5.
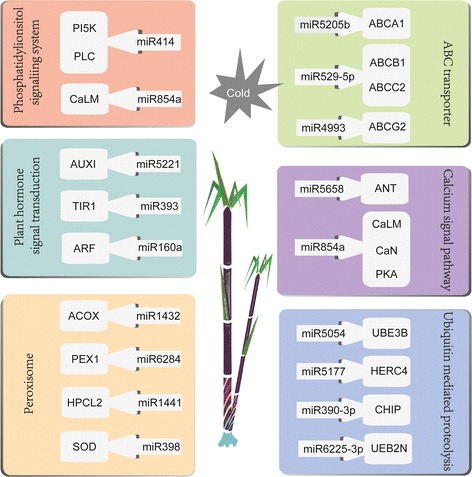
Predicted regulation networks between miRNAs and their target genes involved in six biological pathways in the sugarcane response to cold stress
Fig. 6.
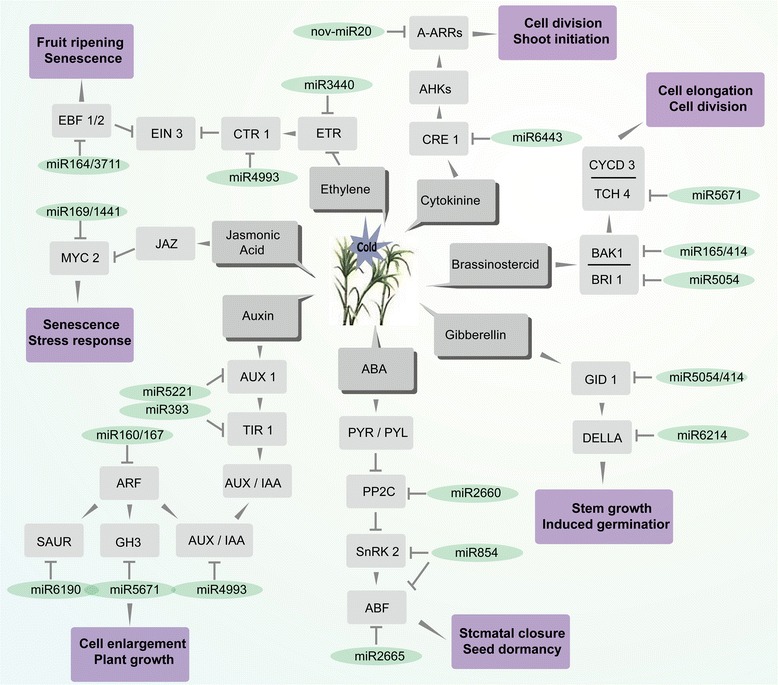
The involvement of miRNAs and their predicted targets into plant hormone signaling pathways in sugarcane response to cold stress. The miR3440-ETR, miR4993-CTR1, miR164/3711-EBF1/2 were involved in the ethylene signal pathway which is related to the fruit ripening senescence. miR6443-CRE, nov-miR20-A-ARRs were involved in the cytokinine signal pathway which is related to the cell division shoot initiation. miR5054-BRI1, miR165/414-BAK1, miR5671-TCH4 are involved in the brassinostercid signal pathway which is related to cell elongation and cell division. miR5054/414-GID1, miR6214-DELLA are involved in the gibberellin signal pathway which is related to stem ABF growth and induced germination. miR2660-PP2C, miR854-SnRK2/ABF, miR2665-ABF are involved in ABA signal pathway which is related to stomatal closure and seed dormancy. miR5221-AUX1, miR393-TIR1, miR160/167-ARF, miR6190-SAUR, miR5671-GH3, miR4993-AUX/IAA are involved in auxin signal pathway which is related to cell enlargement and plant growth. miR169/1441-MYC2 are involved in jasmonic acid signal pathway which is related to senescence and stress response
Expression analysis of miRNAs and their targets by RT-qPCR
RT-qPCR was used to detect the expression levels of miRNAs and their targets. A total of 13 miRNAs and 12 corresponding targets including four transcription factors and eight protein coding genes, were tested in the samples of the two sugarcane cultivars collected at different time points under cold stress (Fig. 7). In total, there were 11 miRNAs, sharing a similar expression profile with those in the deep sequencing data when detected by RT-qPCR. As shown in Fig. 8, the miRNA expression patterns (up- or down-regulation) as measured by quantitative analysis and the sequencing results of the miRNAs were largely consistent, except miR394 and miR5177. From the expression profiles detected by RT-qPCR, there were several patterns among these 13 miRNAs. The transcripts of miR160, miR167 and miR319 were all induced by cold treatment in FN39 and ROC22. In contrast, the expressions of miR169, miR5177 and miR5564 were all suppressed. MiR168 was induced at the early stage, but was inhibited afterwards. The other six miRNAs showed different expression patterns in FN39 and ROC22. MiR156 showed a trend of increase firstly, then decrease in FN39, while it was downregulated in ROC22. The expression levels of miR397 and miR398 showed first decrease then increase in FN39, while the opposite trend was observed in ROC22. In FN39, the expression levels of miR393, miR394 and miR408 were all induced by the cold stress, while in ROC22, the expression levels of miR393 and miR408 were first increased then decreased, and the miR394 was inhibited all the time. The different expression patterns of miRNAs in relative cold tolerance FN39 and relative cold sensitivity ROC22 may indicate their important roles in plant response to cold stress. Most of the above 13 miRNAs exhibited a negative correlation with their predicted targets in both cultivars (FN39 and ROC22) with the exception of miR168 and miR394 (Fig. 7). The inaccuracy of target prediction or regulatory mechanism other than miRNAs modulating the expression of targets in sugarcane may explain this result.
Fig. 7.
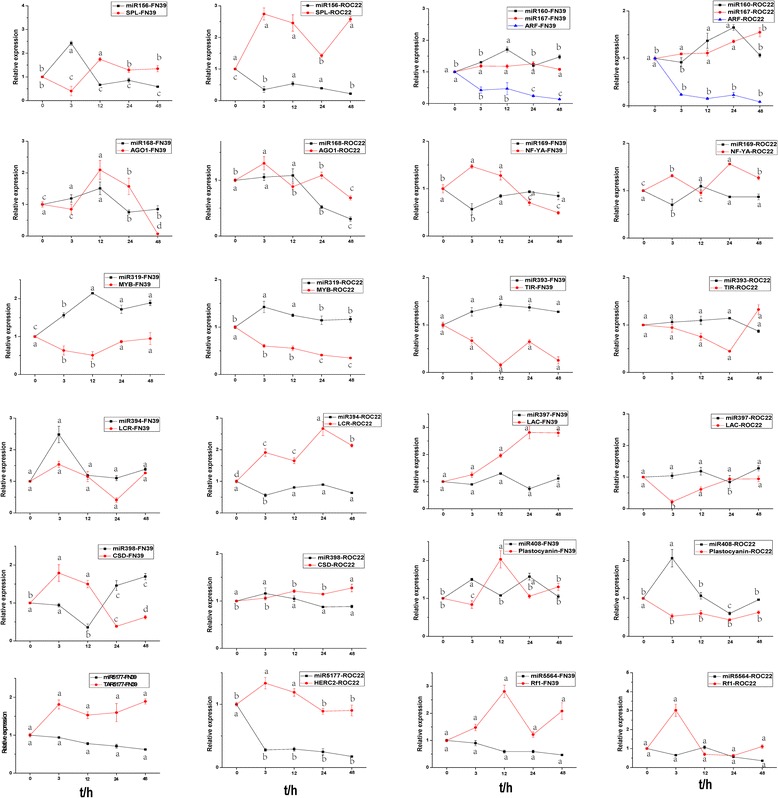
The expression profiles of 13 miRNAs and their targets in the two sugarcane cultivars FN39 and ROC22. The relative expression levels of miRNAs and their predicted target genes were normalized to the 18S rRNA and GAPDH expression levels, respectively. Each bar represented as means of three replicates (n = 3) ± standard error. Lowercase letters in each graph attest significant differences within the same gene/miRNA over the treatment period, as determined by Duncan’s new multiple range test (p-value <0.05)
Fig. 8.
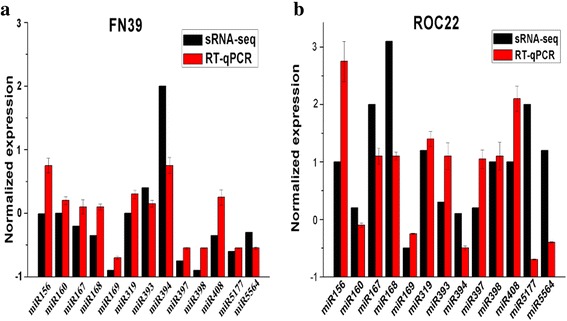
qRT-PCR validation of 13 randomly selected miRNAs identified by small RNA sequencing. a) miRNAs expressed in FN39. b) miRNAs expressed in ROC22. Sugarcane buds of FN39 and ROC22 treated with cold stress for 48 h were used as qRT-PCR samples. The data of qRT-PCR were normalized to the 18S rRNA expression level and represented as means of three replicates (n = 3) ± standard error
Transient expression analysis of miR156 in tobacco
Homologues of miR156, miR168, miR169 and miR408 have been implicated in abiotic stresses [39, 40]. In addition to cold stress (Fig. 7), the expression profiles of these four miRNAs were detected under drought and salt stresses using stem loop RT-qPCR (Fig. 9). Compared with the other three miRNAs, miR156 showed the largest differential expression, with 4-fold and 3-fold up-regulation respectively under drought and salt stresses. The other three miRNAs exhibited smaller differences compared to the control. This suggests a role for miR156 in plant response to abiotic stress in sugarcane.
Fig. 9.
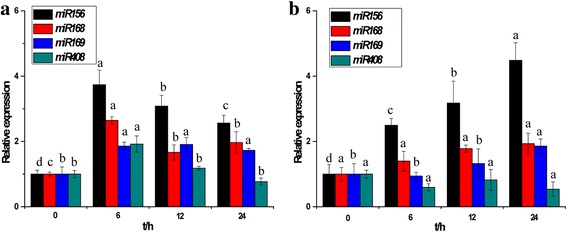
The expression of miR156, miR168, miR169 and miR408 in sugarcane under salt (a) and drought (b) stresses. The four miRNAs showed different responses to the salt and drought treatments. Compared with the other three miRNAs, miR156 showed the largest differential expression. The other three miRNAs exhibited smaller differences compared to the control. 18S rRNA was used as the internal control gene. Each bar represented as means of three replicates (n = 3) ± standard error. Lowercase letters in each graph attest significant differences within the same gene/miRNA over the treatment period, as determined by Duncan’s new multiple range test (p-value <0.05)
MIR156 was transiently overexpressed in tobacco leaves using Agrobacterium expressing pCAMBIA1301-MIR156. Significant levels (p-value <0.05) of the MIR156 transcript were detected (Fig. 10a), but not in the control. After 24 h cold treatment, the tobacco plants showed a visual phenotype. Compared with the control, the leaves overexpressing MIR156 exhibited better growth status (Fig. 10b, c). A number of assays were performed on the tobacco leaves. The extent of H2O2 accumulation under cold stress was assayed using the DAB staining method. A deeper staining was produced in mock leaves, indicating lower H2O2 accumulation in leaves overexpressing MIR156 (Fig. 10d). Three members of the early-responsive dehydration (ERD) gene family, ERD10B, ERD10C and ERD10D, which are downstream genes in the ICE-CBF-COR pathway [41], and the LEA gene, whose encoded protein protects cells from water stress [42], were upregulated in pCAMBIA1301-MIR156 overexpressing tobacco leaves after 12 h cold treatment (Fig. 10e). Additionally, under cold stress, anthocyanin levels in leaves overexpressing pCAMBIA1301-MIR156 were higher than those in the control (Fig. 10f). These results suggest that tobacco leaves overexpressing MIR156 have enhanced cold tolerance. In addition, the transient expression of the other three miRNAs, miR168, miR169 and miR408, produced no obvious phenotype or difference in reactive oxygen species (ROS).
Fig. 10.

Functional analysis of miR156 in tobacco infiltrated by Agrobacterium tumefaciens strain EHA105. a) The transcript analysis of MIR156 in infiltrated tobacco leaves; b) and c) the phenotype observed in infiltrated tobacco leaves overexpressing pCAMBIA1301 and pCAMBIA1301-MIR156 with 4 °C treatment for 24 h; d) DAB staining of tobacco leaves with 4 °C treatment for 24 h; e) expression analysis of cold response genes in infiltrated tobacco leaves with 4 °C treatment for 12 h. f) quantification of anthocyanin in tobacco leaves with 4 °C treatment for 24 h. A data point represents the mean ± SE (n = 3) (a, e, f). Lowercase letters in each graph attest significant differences within the same gene/miRNA over the treatment period, as determined by Duncan’s new multiple range test (p-value <0.05)
Discussion
Small RNA analysis of sugarcane response to cold stress
Cold stress is a major limiting factor in the distribution, yield and quality of crops [11]. Although the cold signaling pathway has been well studied [11, 14], the role of small RNAs in low temperature sensing in plants remains unclear. In recent years, several researchers have found a critical role of miRNA in abiotic stress [43, 44]. Cold-related miRNAs in plants have been identified using various methods. Zhou et al. (2008) [16] found 19 upregulated miRNA genes of 11 miRNA families in Arabidopsis thaliana under cold stress using a computational, transcriptome-based approach. In Brachypodium distachyon, 27 conserved and 129 predicted miRNAs involved in the cold stress response were identified by high-throughput sequencing and whole-genome-wide data mining [17]. In Poncirus trifoliate (L.) Raf., a total of 107 conserved and five potentially novel miRNAs were characterized before and after cold treatment through deep sequencing [45]. In this study, a total of 412 sugarcane miRNAs, including 261 known and 151 predicted novel miRNAs, were discovered in two cultivars exhibiting differences in cold tolerance by small RNA deep sequencing and bioinformatics analysis, enriching the present miRNA resources in sugarcane. This is far more than previously identified in other species, except for the possibility of the parameters used were different, it is most probably related to the larger genome and complex genetic background of sugarcane.
These small RNAs obtained in this study were divided into several classes. Due to the limited knowledge of sugarcane genome information, the unannotated small RNAs occupied the majority, which was consistent with the halophyte Halostachys caspica [30] and Morus alba L. [46]. According to a previous report, the 21 nt and 24 nt length sRNA sequences are generated by different DCL proteins [6]. Small RNAs with 20–22 nucleotides are generated by DCL1. Longer small RNAs, 23–25 nucleotides in length, are processed by DCL3. The DCL2 and DCL4 generate the 22 and 24 nucleotides respectively [47]. Usually, the length of miRNAs corresponds to 21 nt or 22 nt, while the siRNAs are distributed in the 24 nt [6]. The 24 nt and 21 nt small RNA length distribution in sugarcane was consistent with the phenomenon in Panicum virgatum [48], Fragaria ananassa [49], rice [50] and H. caspica [30], which verified our data.
Among the total 412 identified miRNAs, 62 miRNAs exhibited significant expression differences (|log2 ratio| ≥ 1, p ≤ 0.05) between the cold-treated and the control samples. Differentially expressed miRNAs were further validated by RT-qPCR. According to previous reports, miR160/167 and miR393 are involved in the auxin response by targeting of auxin response factors (ARF) and transport inhibitor response (TIR) genes, respectively [51, 52]. In this study, miR167 and miR393 were upregulated in both cultivars, while miR160 was upregulated in FN39 (relative cold tolerance cultivar) and downregulated in ROC22 (relative cold sensitivity cultivar). In Lycopersicon esculentum Mill studies using stem-loop RT-qPCR, miR167 and miR393 increased at 4 °C at early time points (0, 1, 4 and 16 h) [53]. In 2-week-old Arabidopsis seedlings, miR393 was strongly induced by cold, NaCl, dehydration and ABA treatments [54]. The up-regulation of miR167 was found in Triticum aestivum, suggesting that miR167 and the trans-acting short-interfering RNA-auxin response factor (tasiRNA-ARF) have a role in regulating the auxin signaling pathway and the developmental response to cold stress [55]. Under unstressed conditions, low levels of these miRNAs may be sufficient for the fine-tuning of their targets. Conversely, under stress conditions, upregulated miRNAs can reduce ARF and TIR transcript levels (Fig. 7), suppressing ARF-mediated auxin responsive gene expression, leading to plant growth and development attenuation and eventually enhancing plant stress tolerance.
There was up-regulation of miR168 and miR408 under cold stress in both the cold tolerant and less cold tolerant sugarcane cultivars. Liu et al. (2008) [39] identified 14 stress-inducible miRNAs using microarray data in Arabidopsis, and found miR168 to be responsive to cold stress. The increased expression of miR408 could lead to the improved tolerance of Arabidopsis to cold and oxidative stresses and the enhancement of cellular antioxidant capacity, manifested by the lower level of ROS and induction of genes related to anti-oxidative function [56]. MiR319 showed upregulated expression in both cultivars in this study, which was consistent with previous research which identified its positive role in the response of sugarcane to cold stress [18]. Some miRNAs exhibited differential expression in the two cultivars: miR156, miR160 and miR394 were upregulated in the cold tolerant FN39, but downregulated in the cold sensitive ROC22, while miR397 and miR398 were downregulated in FN39 and upregulated in ROC22. The opposite expression changes of these miRNAs in these cultivars may be a key factor leading to difference in cold tolerance. MiR156 is a conserved miRNA in many plant species and its function in plant growth and development has been studied during events such as lateral root development [57] and floral transition [58]. MiR156 also has a role in plant response to abiotic stress, such as drought and salt stresses [59].
As the function of miRNAs is performed by targeting of mRNAs, the target genes were predicted with certain described criteria. Several targets of conserved miRNAs in this study, such as the SQUAMOSA promoter-binding protein-like gene (SPL, target of miR156) and nuclear factor Y gene (NF-Y, target of miR169), have been reported in some other plant species, indicating the miRNAs and its targets may have a similar role in different plant species [60]. In our study, a total of 4002 targets were predicted for the 259 miRNAs, showing that some miRNAs have more than one target, which is consistent with earlier reports in Glycine max seeds [61] and trifoliate orange [45]. Another reason for the large amount of predicted targets may be the lack of sufficient sugarcane genome sequences and annotation information, leading to inaccuracies in prediction. Among the targets, transcription factors were included and involved in the response of sugarcane to cold stress, such as the ARF, TIR, MYB and NF-Y. Transcription factors control a series of downstream genes by binding cis-elements in promoter regions [62]. It is conceivable that miRNAs regulating the transcription factors will exert a more extensive influence on gene expression. However, the function of miRNAs and their targets needs to be validated through further experiments.
Functional analysis of miR156 in tobacco under abiotic stress
Several miRNAs, including miR156, miR168, miR169 and miR408, which have been studied in plant response to abiotic stresses [39, 40], were selected for further functional investigation. The highly conserved miR156 family has been reported as a key factor in the regulation of plant growth and development by targeting the SPL genes. In Gossypium hirsutum L., the miR156 was upregulated under 0.25% NaCl or 2.5% PEG stresses in roots [63]. In Zea mays L., miR156 showed a modest up-regulation in two inbred lines under both salt and drought stresses, but also a strong higher expression in the hybrid lines based on northern blot detection. SPL, a target of miR156, showed opposite expression patterns under abiotic stresses, indicating the role of miR156 as a negative regulator in maize during this process [64]. A similar positive role of miR156 in sugarcane was shown in our RT-qPCR results, with a 3.7-fold and 4.5-fold up-regulation under salt and drought stresses, respectively.
The role of miR156 in cold stress has been shown in rice [20]. Transgenic rice with overexpressed OsmiR156k exhibited a lower survival rate, proline content, chlorophyll II, and down-regulation of cold-sensitive genes under cold stress, indicating decreased cold tolerance. They concluded that miR156k worked as a negative regulator of plant tolerance to cold stress [20]. In our work, miR156 showed a positive role in the response of tobacco to cold stress, supported by better growth status and lower ROS accumulation. Additionally, 4 cold-related genes (ERD10B, ERD10C, ERD10D and LEA genes) all showed higher expression levels in tobacco leaves when MIR156 was overexpressed than those in the control. This may be due to the fact that miR156 exerts various affects in different plant species.
Anthocyanin level has been shown to be responsive to many stresses including cold, salt, UV-B and biotic stresses, and protect plants from damage by scavenging the free radicals [65, 66]. Gou et al. (2011) [38] concluded that increased miR156 could promote the accumulation of anthocyanins, while reduced miR156 would be conducive to flavonol accumulation in Arabidopsis. During this process, SPL9, one of the miR156 targets, inhibits the expression of anthocyanin synthesis genes by interacting with the production of the anthocyanin pigments1 (PAP1) component of the MYB-BHLH-WD40 complex, resulting in the interference with the regulation of anthocyanin accumulation [38]. Due to the fact that the greater accumulation of anthocyanins in miR156 transiently overexpressed tobacco leaves was observed, it is possible that the overexpression of sugarcane miR156 leads to the accumulation of anthocyanins to prevent the over production and accumulation of ROS in tobacco leaves. This deduction was consistent with the observation of growth status and DAB staining, in which the MIR156 overexpressing tobaccos displayed better growth status and less ROS accumulation. Considering the better growth status, lower ROS level, higher anthocyanins level with the higher expression levels of four cold-related genes in MIR156 overexpressing tobaccos than those in control, it indicated that the miR156 plays a positive role in plant response to cold stress.
Conclusions
In this study, small RNA sequencing of sugarcane with cold treatment and comprehensive analysis of the cold-responsive miRNAs and their targets were performed. The results showed that miRNAs played an important role in sugarcane under cold stress by regulating several biological processes, including plant hormone signal transduction, ABC transporter function, peroxisomes, phosphatidylinositol signaling systems, ubiquitin mediated proteolysis and calcium signaling pathways. In addition, the role of miR156 in plant response to cold stress was also studied. These findings provide the valuable information for further functional characterization of miRNAs in sugarcane under cold stress.
Additional files
The stem loop primers of miRNAs in sugarcane (DOCX 14 kb)
The real time PCR primers of miRNAs and target gene in sugarcane (DOCX 15 kb)
The known and novel miRNAs in 4 small RNA libraries (F0, F3, R0 and R3) (XLSX 30 kb)
Prediction of target genes of the known and novel miRNAs (XLSX 208 kb)
GO analysis of the predicted targets of known and novel miRNAs (XLSX 243 kb)
KEGG analysis of the predicted targets of known miRNAs in F3/F0 (XLSX 12 kb)
KEGG analysis of the predicted targets of known miRNAs in R3/R0 (XLSX 11 kb)
KEGG analysis of the predicted targets of novel miRNAs in F3/F0 (XLSX 15 kb)
KEGG analysis of the predicted targets of novel miRNAs in R3/R0 (XLSX 12 kb)
The stem longitudinal section of sugarcane FN39 (relative cold tolerance) and ROC22 (relative cold sensitivity) in plant tip part after cold stress, indicating that more serious damage was observed in ROC22 in its stem tissues and growing point (TIFF 31806 kb)
Validation of the cold-stress treatment in sugarcane. The expression patterns of three cold responsive genes, CBF1, CBF3 and NAC23, were detected by RT-qPCR in sugarcane FN39 (A) and ROC22 (B) cultivars. The relative expression at 0 h is equal to 1. GAPDH was used as internal control. Error bars show the range of duplicate analysis of sample in RT-qPCR. Lower case letters in each graph attest significant differences within the same gene/miRNA over the treatment period, as determined by Duncan’s new multiple range test (p-value <0.05) (TIFF 2258 kb)
Acknowledgements
We are grateful to Dr. Andrew C. Allan (New Zealand Institute for Plant and Food Research, Sandringham, Auckland, New Zealand) for his assistance in editing the paper.
Funding
This work was funded by the earmarked fund for the Modern Agriculture Technology of China (CARS-17), the 948 Program on the Introduction of International Advanced Agricultural Science and Technique of the Department of Agriculture (2014-S18) and the Scientific Research Foundation of the Graduate School of Fujian Agriculture and Forestry University (324-1122yb025).
Availability of data and materials
The data supporting the conclusions of this article are within the paper and its additional files. All sequencing reads are deposited in the National Center for Biotechnology Information under the BioProject number PRJNA388815 with the Sequence Read Archive (SRA) study accession SRP108561.
Abbreviations
- 18S rRNA
18S ribosomal RNA
- ARF
auxin response factor gene
- CBF
C-repeat binding factor gene
- DAB
3, 3′-diaminobenzidine
- F0
samples of FN39 cultivar without cold treatment
- F3
samples of FN39 cultivar with 3 h cold treatment
- GAMyB
Myb transcription factor
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- ICE1
inducer of C-repeat binding factor expression 1 gene
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- NF-Y
nuclear factor Y gene
- PAP1
production of the anthocyanin pigments1
- PCF5
TCP transcription factor gene
- R0
samples of ROC22 cultivar without cold treatment
- R3
samples of ROC22 cultivar with 3 h cold treatment
- RISC
RNA-induced silencing complex
- ROS
reactive oxygen species
- rRNA
ribosomal RNA
- RT-qPCR
reverse transcription quantitative real-time polymerase chain reaction
- scRNA
small cytoplasmic RNA
- snoRNA
small nucleolar RNA
- snRNA
small nuclear RNA
- SPL
SQUAMOSA promoter-binding protein-like gene
- tasiRNA-ARF
trans-acting short-interfering RNA-auxin response factor
- TIR
transport inhibitor response gene
- tRNA
transfer RNA
Authors’ contributions
YTY, LPX and YXQ conceived, designed and initiated the project. YTY prepared materials. YTY, XZ, YCS, JKZ and ZTW performed experiments and contributed to data analysis and validation. YTY drafted the manuscript. LPX and YXQ helped to revise the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable. This is to confirm that no specific permits were needed for the described experiments, and this study did not involve any endangered or protected species.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4231-3) contains supplementary material, which is available to authorized users.
Contributor Information
Yuting Yang, Email: yytjiayou@126.com.
Xu Zhang, Email: fjnlzhangxu@163.com.
Yachun Su, Email: syc2009mail@163.com.
Jiake Zou, Email: zjk930103@163.com.
Zhoutao Wang, Email: wzt1417@sina.com.
Liping Xu, Email: xlpmail@126.com.
Youxiong Que, Email: queyouxiong@126.com.
References
- 1.Li AL, Mao L. mRNA and small RNA transcriptomes reveal insights into dynamic homoeolog regulation of allopolyploid heterosis in nascent hexaploid wheat. Plant Cell. 2014;26:1878–1900. doi: 10.1105/tpc.114.124388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurihara Y, Watanabe Y. Arabidopsis Micro-RNA biogenesis through dicer-like 1 protein functions. PNAS. 2004;34:12753–12758. doi: 10.1073/pnas.0403115101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brodersen P, Sakvarelidzeachard L, Bruunrasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O. Widespread translational inhibition by plant miRNAs and siRNAs. Science. 2008;320:1185–1190. doi: 10.1126/science.1159151. [DOI] [PubMed] [Google Scholar]
- 4.Unver T, Budak H. Conserved microRNAs and their targets in model grass species Brachypodium distachyon. Planta. 2009;230:659–669. doi: 10.1007/s00425-009-0974-7. [DOI] [PubMed] [Google Scholar]
- 5.Kumar R. Role of microRNAs in biotic and abiotic stress responses in crop plants. Appl Biochem Biotech. 2014;174:93–115. doi: 10.1007/s12010-014-0914-2. [DOI] [PubMed] [Google Scholar]
- 6.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 7.Jin HL. Endogenous small RNAs and antibacterial immunity in plants. FEBS Lett. 2008;582:2679–2684. doi: 10.1016/j.febslet.2008.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, et al. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One. 2007;e219:2. doi: 10.1371/journal.pone.0000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song C, Chen W, Zhang C, Korir NK, Yu H, Ma Z, et al. Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange ( Citrus trifoliata) BMC Genomics. 2010;11:431. doi: 10.1186/1471-2164-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge AJ, Shangguan LF, Zhang X, Dong QH, Han J, Liu H, et al. Deep sequencing discovery of novel and conserved microRNAs in strawberry (Fragaria×ananassa) Physiol Plantarum. 2013;148:387–396. doi: 10.1111/j.1399-3054.2012.01713.x. [DOI] [PubMed] [Google Scholar]
- 11.Chinnusamy V, Zhu JH, Zhu JK. Cold stress regulation of gene expression in plants. Trend. Plant Sci. 2007;12:444–451. doi: 10.1016/j.tplants.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 12.Thomashow MF. Plant cold acclimation: freezing tolerance genes and regulatory mechanisms. Annu Rev Plant Biol. 1999;50:571–599. doi: 10.1146/annurev.arplant.50.1.571. [DOI] [PubMed] [Google Scholar]
- 13.Viswanathan C, Zhu JK. Molecular genetic analysis of cold-regulated gene transcription. Philos T R Soc B. 2002;357:877–886. doi: 10.1098/rstb.2002.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi YT, Ding YL, Yang SH. Cold signal transduction and its interplay with phytohormones during cold acclimation. Plant Cell Physiol. 2015;56:7–15. doi: 10.1093/pcp/pcu115. [DOI] [PubMed] [Google Scholar]
- 15.Solanke AU, Sharma AK. Signal transduction during cold stress in plants. Physiol Mol Biol Pla. 2008;14:69–79. doi: 10.1007/s12298-008-0006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou X, Wang G, Sutoh K, Zhu JK, Zhang W. Identification of cold-inducible microRNAs in plants by transcriptome analysis. BBA - Gene Regul Mech. 1779;2008:780–788. doi: 10.1016/j.bbagrm.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Zhang JY, YY X, Huan Q, Chong K. Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genomics. 2009;10:449. doi: 10.1186/1471-2164-10-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thiebaut F, Rojas CA, Almeida KL, Grativol C, Domiciano CC, Lamb CRC, et al. Regulation of miR319 during cold stress in sugarcane. Plant Cell Environ. 2012;35:502–512. doi: 10.1111/j.1365-3040.2011.02430.x. [DOI] [PubMed] [Google Scholar]
- 19.Yang CH, Li DY, Mao DH, Liu X, Ji CJ, Li XB, et al. Overexpression of microRNA319 impacts leaf morphogenesis and leads to enhanced cold tolerance in rice (Oryza sativa L) Plant Cell Environ. 2013;36:2207–2218. doi: 10.1111/pce.12130. [DOI] [PubMed] [Google Scholar]
- 20.Cui N, Sun XL, Sun MZ, Jia BW, Duanmu HZ, Lv DK, et al. Overexpression of OsmiR156k leads to reduced tolerance to cold stress in rice ( Oryza Sativa) Mol Breeding. 2015;35:1–11. doi: 10.1007/s11032-015-0402-6. [DOI] [Google Scholar]
- 21.Song JB, Gao S, Wang Y, Li BW, Zhang YL, Yang ZM. miR394 and its target gene LCR are involved in cold stress response in Arabidopsis. Plant Gene. 2016;5:56–64. doi: 10.1016/j.plgene.2015.12.001. [DOI] [Google Scholar]
- 22.Que YX, LP X, QB W, Liu YF, Ling H, Liu YH, et al. Genome sequencing of Sporisorium scitamineum provides insights into the pathogenic mechanisms of sugarcane smut. BMC Genomics. 2014;16:1–20. doi: 10.1186/1471-2164-15-996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li YR, Yang LT. Sugarcane agriculture and sugar industry in China. Sugar Tech. 2015;17:1–8. doi: 10.1007/s12355-014-0342-1. [DOI] [Google Scholar]
- 24.Zhang BQ, Yang LT, Li YR. Physiological and biochemical characteristics related to cold resistance in sugarcane. Sugar Tech. 2015;17:49–58. doi: 10.1007/s12355-014-0340-3. [DOI] [Google Scholar]
- 25.Ebrahim MK, Zingsheim O, El-Shourbagy MN, Moore PH, Komor E. Growth and sugar storage in sugarcane grown at temperatures below and above optimum. J Plant Physiol. 1998;153:593–602. doi: 10.1016/S0176-1617(98)80209-5. [DOI] [Google Scholar]
- 26.Yang YT, Zhang X, Chen Y, Guo JL, Ling H, Gao SW, et al. Selection of reference genes for normalization of microRNA expression by RT-qPCR in sugarcane buds under cold stress. Front Plant Sci. 2016;7:1–10. doi: 10.3389/fpls.2016.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medina J, Catalá R, Salinas J. The CBFs: three Arabidopsis transcription factors to cold acclimate. Plant Sci. 2011;1:3–11. doi: 10.1016/j.plantsci.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 28.Zhao CZ, Lang ZB, Zhu JK. Cold responsive gene transcription becomes more complex. Trends Plant Sci. 2015;8:466–468. doi: 10.1016/j.tplants.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang YT, ZW F, YC S, Zhang X, Li G, Guo JL, et al. A cytosolic glucose-6-phosphate dehydrogenase gene, ScG6PDH, plays a positive role in response to various abiotic stresses in sugarcane. Sci Rep. 2014;4:70–90. doi: 10.1038/srep07090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang RR, Zeng YL, Yi XY, Zhao LJ, Zhang YF. Small RNA deep sequencing reveals the important role of microRNAs in the halophyte Halostachys caspica. Plant Biotech J. 2015;13:395–408. doi: 10.1111/pbi.12337. [DOI] [PubMed] [Google Scholar]
- 31.YB L, Yang LT, Qi YP, Li Y, Li Z, Chen YB, et al. Identification of boron-deficiency-responsive microRNAs in Citrus sinensis roots by Illumina sequencing. BMC Plant Biol. 2014;14:1–16. doi: 10.1186/1471-2229-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allen E, Xie Z, Gustafson AM, Carrington JC. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005;121:207–221. doi: 10.1016/j.cell.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D. Specific effects of microRNAs on the plant transcriptome. Dev Cell. 2005;8:517–527. doi: 10.1016/j.devcel.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 34.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Eppig, gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:480–484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 37.Du SC, Hwang IS, Hwang BK. Requirement of the cytosolic interaction between pathogenesis-related protein10 and leucine-rich repeat protein1 for cell death and defense signaling in pepper. Plant Cell. 2012;24:1675–1690. doi: 10.1105/tpc.111.092759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gou JY, Felippes FF, Liu CJ, Weigel D, Wang JW. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell. 2011;23:1512–1522. doi: 10.1105/tpc.111.084525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu HH, Tian X, Li YJ, CA W, Zheng CC. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA. 2008;14:836–843. doi: 10.1261/rna.895308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding D, Zhang LF, Wang H, Liu ZJ, Zhang ZX, Zheng YL. Differential expression of miRNAs in response to salt stress in maize roots. Ann Bot. 2009;103:29–38. doi: 10.1093/aob/mcn205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kasuga M, Miura S, Shinozak K, Yamaguchi-Shinozaki K. A Combination of the Arabidopsis DREB1A gene and stress-inducible rd29A promoter improved drought- and low-temperature stress tolerance in tobacco by gene transfer. Plant Cell Physiol. 2004;45:346–350. doi: 10.1093/pcp/pch037. [DOI] [PubMed] [Google Scholar]
- 42.Wang W, Vinocur B, Altman A. Plant responses to drought, salinity and extreme temperatures: towards genetic engineering for stress tolerance. Planta. 2003;218:1–14. doi: 10.1007/s00425-003-1105-5. [DOI] [PubMed] [Google Scholar]
- 43.Khraiwesh B, Zhu JK, Zhu JH. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. BBA - Gene Regul Mec. 1819;2012:137–148. doi: 10.1016/j.bbagrm.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang BH. MicroRNA: a new target for improving plant tolerance to abiotic stress. J Exp Bot. 2015;66:1740–1761. doi: 10.1093/jxb/erv013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang XN, Li X, Liu JH. Identification of conserved and novel cold-responsive microRNAs in trifoliate orange (Poncirus trifoliata (L.) Raf.) using high-throughput sequencing. Plant Mol Biol Rep. 2013;32:328–341. doi: 10.1007/s11105-013-0649-1. [DOI] [Google Scholar]
- 46.Jia L, Zhang DY, Qi XW, Ma B, Xiang ZH, He NJ. Identification of the conserved and novel miRNAs in mulberry by high-throughput sequencing. PLoS One. 2014;9:e104409. doi: 10.1371/journal.pone.0104409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borges F, Martienssen RA. The expanding world of small RNAs in plants. Nat Rev Mol Cell Bio. 2015;12:727. doi: 10.1038/nrm4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie F, Jr CNS, Taki FA, He Q, Liu H, Zhang B. High-throughput deep sequencing shows that microRNAs play important roles in switchgrass responses to drought and salinity stress. Plant Biotech J. 2014;12:354–366. doi: 10.1111/pbi.12142. [DOI] [PubMed] [Google Scholar]
- 49.XB X, Yin LL, Ying QC, Song HM, Xue DW, Lai TF, et al. High-throughput sequencing and degradome analysis identify miRNAs and their targets involved in fruit senescence of Fragaria ananassa. PLoS One. 2013;8:e70959. doi: 10.1371/journal.pone.0070959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li QW, Long FY, Tai W. Deep sequencing on genome-wide scale reveals the unique composition and expression patterns of microRNAs in developing pollen of Oryza sativa. Genome Biol. 2011;12:1–16. doi: 10.1186/gb-2011-12-4-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glazińska P, Wojciechowski W, Wilmowicz E, Zienkiewicz A, Frankowski K, Kopcewicz J. The involvement of InMIR167 in the regulation of expression of its target gene InARF8, and their participation in the vegetative and generative development of Ipomoea nil plants. J Plant Physiol. 2013;171:225–234. doi: 10.1016/j.jplph.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 52.Chen ZH, Bao ML, Sun YZ, Yang YJ, Xu XH, Wang JH, et al. Regulation of auxin response by miR393-targeted transport inhibitor response protein 1 is involved in normal development in Arabidopsis. Plant Mol Biol. 2011;77:619–629. doi: 10.1007/s11103-011-9838-1. [DOI] [PubMed] [Google Scholar]
- 53.Koc I, Filiz E, Tombuloglu H. Assessment of miRNA expression profile and differential expression pattern of target genes in cold-tolerant and cold-sensitive tomato cultivars. Biotechnol Biotech E. 2015;29:851–860. doi: 10.1080/13102818.2015.1061447. [DOI] [Google Scholar]
- 54.Sunkar R, Zhu JK. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004;16:2001–2019. doi: 10.1105/tpc.104.022830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang ZH, Zhang LP, CG X, Yuan SH, Zhang FT, Zheng YL, et al. Uncovering small RNA-mediated responses to cold stress in a wheat thermosensitive genic male-sterile line by deep sequencing. Plant Physiol. 2012;159:721–738. doi: 10.1104/pp.112.196048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma C, Burd S, Lers A. miR408 is involved in abiotic stress responses in Arabidopsis. Plant J. 2015;84:167–187. doi: 10.1111/tpj.12999. [DOI] [PubMed] [Google Scholar]
- 57.Yu N, Niu QW, Ng KH, Chua NH. The role of miR156/SPLs modules in Arabidopsis lateral root development. Plant J. 2015;83:673–685. doi: 10.1111/tpj.12919. [DOI] [PubMed] [Google Scholar]
- 58.Yu S, Galvão VC, Zhang YC, Horrer D, Zhang TQ, Hao YH, et al. Gibberellin regulates the Arabidopsis floral transition through miR156-targeted SQUAMOSA PROMOTER BINDING–LIKE transcription factors. Plant Cell. 2012;24:3320–3332. doi: 10.1105/tpc.112.101014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cui LG, Shan JX, Shi M, Gao JP, Lin HX. The miR156-SPL9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014;80:1108–1117. doi: 10.1111/tpj.12712. [DOI] [PubMed] [Google Scholar]
- 60.Ni ZY, Hu Z, Jiang QY, Zhang H. GmNFYA3, a target gene of miR169, is a positive regulator of plant tolerance to drought stress. Plant Mol Biol. 2013;82:113–129. doi: 10.1007/s11103-013-0040-5. [DOI] [PubMed] [Google Scholar]
- 61.Song QX, Liu YF, XY H, Zhang WK, Ma B, Chen SY, et al. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 2011;11:1–16. doi: 10.1186/1471-2229-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Singh KB, Foley RC, Oñate-Sánchez L. Transcription factors in plant defense and stress responses. Curr Opin Plant Biol. 2002;5:430–436. doi: 10.1016/S1369-5266(02)00289-3. [DOI] [PubMed] [Google Scholar]
- 63.Wang M, Wang QL, Zhang BH. Response of miRNAs and their targets to salt and drought stresses in cotton (Gossypium hirsutum L) Gene. 2013;530:26–32. doi: 10.1016/j.gene.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 64.Kong Y, Elling AA, Chen B, Deng X. Differential expression of microRNAs in maize inbred and hybrid lines during salt and drought stress. Am J Plant Sci. 2010;1:69–76. doi: 10.4236/ajps.2010.12009. [DOI] [Google Scholar]
- 65.Chalker-Scott L. Environmental significance of anthocyanins in plant stress responses. Photochem Photobiol. 1999;70:1–9. doi: 10.1111/j.1751-1097.1999.tb01944.x. [DOI] [Google Scholar]
- 66.Oh JE, Kim YH, Kim JH, Kwon YR, Lee HJ. Enhanced level of anthocyanin leads to increased salt tolerance in Arabidopsis PAP1-D plants upon sucrose treatment. J Korean Soc Appl Bi. 2011;54:79–88. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The stem loop primers of miRNAs in sugarcane (DOCX 14 kb)
The real time PCR primers of miRNAs and target gene in sugarcane (DOCX 15 kb)
The known and novel miRNAs in 4 small RNA libraries (F0, F3, R0 and R3) (XLSX 30 kb)
Prediction of target genes of the known and novel miRNAs (XLSX 208 kb)
GO analysis of the predicted targets of known and novel miRNAs (XLSX 243 kb)
KEGG analysis of the predicted targets of known miRNAs in F3/F0 (XLSX 12 kb)
KEGG analysis of the predicted targets of known miRNAs in R3/R0 (XLSX 11 kb)
KEGG analysis of the predicted targets of novel miRNAs in F3/F0 (XLSX 15 kb)
KEGG analysis of the predicted targets of novel miRNAs in R3/R0 (XLSX 12 kb)
The stem longitudinal section of sugarcane FN39 (relative cold tolerance) and ROC22 (relative cold sensitivity) in plant tip part after cold stress, indicating that more serious damage was observed in ROC22 in its stem tissues and growing point (TIFF 31806 kb)
Validation of the cold-stress treatment in sugarcane. The expression patterns of three cold responsive genes, CBF1, CBF3 and NAC23, were detected by RT-qPCR in sugarcane FN39 (A) and ROC22 (B) cultivars. The relative expression at 0 h is equal to 1. GAPDH was used as internal control. Error bars show the range of duplicate analysis of sample in RT-qPCR. Lower case letters in each graph attest significant differences within the same gene/miRNA over the treatment period, as determined by Duncan’s new multiple range test (p-value <0.05) (TIFF 2258 kb)
Data Availability Statement
The data supporting the conclusions of this article are within the paper and its additional files. All sequencing reads are deposited in the National Center for Biotechnology Information under the BioProject number PRJNA388815 with the Sequence Read Archive (SRA) study accession SRP108561.