

Summary
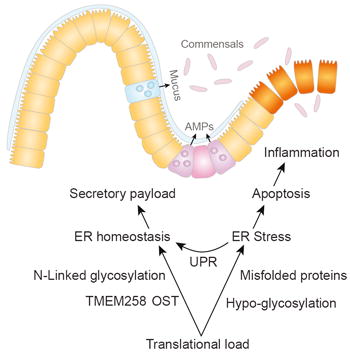
Significant insights into disease pathogenesis have been gleaned from population-level genetic studies; however, many loci associated with complex genetic disease contain numerous genes, and phenotypic associations cannot be assigned unequivocally. In particular, a gene-dense locus on chromosome 11 (61.5-61.65 Mb) has been associated with inflammatory bowel disease, rheumatoid arthritis, and coronary artery disease. Here we identify TMEM258 within this locus as a central regulator of intestinal inflammation. Strikingly, Tmem258 haploinsufficient mice exhibit severe intestinal inflammation in a model of colitis. At the mechanistic level, we demonstrate that TMEM258 is a required component of the oligosaccharyltransferase complex and is essential for N-linked protein glycosylation. Consequently, homozygous deficiency of Tmem258 in colonic organoids results in unresolved endoplasmic reticulum (ER) stress culminating in apoptosis. Collectively, we demonstrate that TMEM258 is a central mediator of ER quality control and intestinal homeostasis.
Introduction
Mucosal homeostasis maintains a delicate balance between host defense against pathogens and tolerance to commensal microorganisms. In the case of inflammatory bowel disease (IBD), this balance is disturbed as a result of genetic and environmental factors that collectively trigger an unrestrained inflammatory response to commensals (Graham and Xavier, 2013; Khor et al., 2011). While the etiology of IBD is incompletely understood, significant insights into its pathophysiology have been ascertained from genome-wide association studies (GWASs). To date, GWASs have implicated more than 200 loci in IBD (Jostins et al., 2012; Liu et al., 2015), yet many of these loci contain multiple genes that are in linkage disequilibrium (LD), and conclusive identification of the IBD risk gene in most loci has been challenging. In a few notable examples, causal variants associated with IBD risk have been identified by GWAS (e.g., NOD2 and ATG16L1) (Huang, 2015). In the vast majority of IBD loci, more targeted approaches are necessary to establish causality. Successful approaches include fine mapping by dense genotyping (Jostins et al., 2012), deep exome sequencing to identify rare coding variants (Beaudoin et al., 2013; Rivas et al., 2011), and integrative functional approaches combining genotyping and metadata such as epigenetics and expression quantitative trait loci (eQTL) (Farh et al., 2015; Okada et al., 2014).
Despite successful approaches to fine mapping causal variants, several complex loci associated with IBD remain unresolved. One such locus on human chromosome 11 (61.5-61.65 Mb) has been associated with IBD and also phenotypes related to lipid metabolism, coronary artery disease (CAD), and rheumatoid arthritis (RA) (Ameur et al., 2012; Hester et al., 2014; Jostins et al., 2012; Liu et al., 2015; Okada et al., 2014). However, it remains unclear whether these disease phenotypes are controlled by a common genetic mechanism, or if these phenotypes are mediated by distinct genes that are in LD within the chromosome 11 locus. In fact, this locus contains several genes comprising a haplotype that arose very recently in human evolution. The derived haplotype D has rapidly become the common allele in modern humans and is associated with more efficient synthesis of long-chain polyunsaturated fatty acids (LC-PUFAs) and increased hepatic expression of the rate-limiting enzymes in this process (FADS1 and FADS2) (Ameur et al., 2012). It has been proposed that haplotype D imparted a selective advantage for early humans with limited dietary intake of LC-PUFA precursors (Ameur et al., 2012), but in combination with a modern high-fat diet, it is associated with increased proinflammatory lipids (arachidonic acids), risk for CAD (Kwak et al., 2011; Lappalainen et al., 2013; Li et al., 2013; Martinelli et al., 2008; Tanaka et al., 2009), and RA (Okada et al., 2014). Whereas haplotype D is associated with a proinflammatory lipid profile and RA, it is not associated with IBD. In contrast, the IBD risk allele tagged by rs174537 is associated with the ancestral haplotype A and is characterized by lower levels of proinflammatory arachidonic acids (Hester et al., 2014; Jostins et al., 2012; Lappalainen et al., 2013; Liu et al., 2015; Tanaka et al., 2009). Furthermore, evidence supporting a contribution of LC-PUFAs to IBD is lacking (Ananthakrishnan et al., 2014; Costea et al., 2014; Guan et al., 2014). Collectively, these observations reveal the limitations of genetics in assigning causality of risk loci to specific genes and highlight the need for a combined functional-genetic approach to link genes with IBD risk. Here, we provide genetic evidence implicating TMEM258 in IBD risk and functionally identify the role for this gene in controlling ER stress responses in the colonic epithelium.
Results
Expression of TMEM258 in Ileal Biopsies Exceeds All Other Genes in the Chromosome 11: 61.5-61.65 Locus
Given the gene-dense landscape of the chromosome 11 (61.5-61.65 Mb) locus and sparse functional annotation of the genes therein, we sought to prioritize candidate causal genes that contribute to IBD pathophysiology. First, we compared the most significant SNPs in the chromosome 11 locus and their respective associations with three phenotypes: CD risk (Jostins et al., 2012), RA risk (Okada et al., 2014), and elevated arachidonic acid in blood samples (Lappalainen et al., 2013). For all three phenotypes, an overlapping set of associated SNPs span coding regions for TMEM258, FADS1, and FADS2 (Figure 1A). By interrogating expression levels of candidate genes in ileum biopsies (Haberman et al., 2014), we found that TMEM258 was expressed at dramatically higher levels than all other neighboring genes in its locus (Figure 1B). Moreover, examination of Tmem258 expression in a published dataset of single-cell transcriptomics (Grun et al., 2016) revealed highest expression in secretory cell types such as goblet and Paneth cells with slightly lower expression in absorptive enterocytes (Figure 1C). Given these findings, we prioritized TMEM258 for deep functional characterization.
Figure 1. Expression of TMEM258 in Ileal Biopsies Exceeds All Other Genes in the Chromosome 11: 61.5-61.65 Locus.
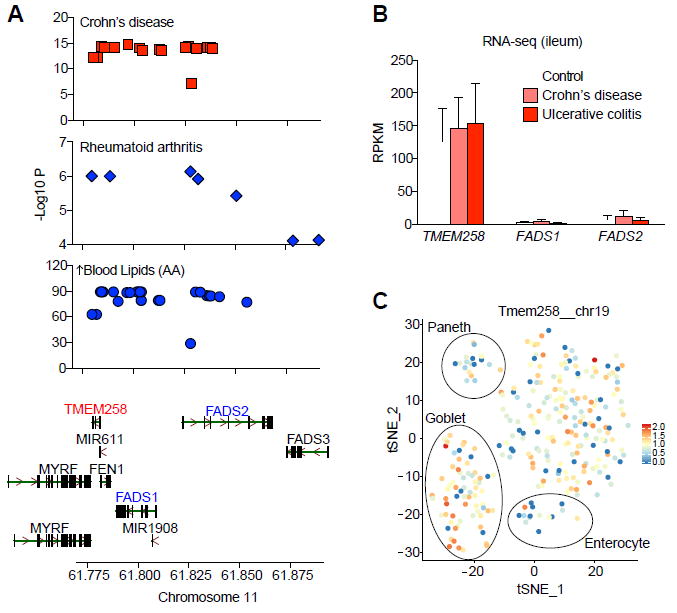
(A) Top SNPs from GWAS (Jostins et al., 2012; Lappalainen et al., 2013; Okada et al., 2014) for each phenotype mapped as chromosomal location versus significance (-Log10 P).
(B) Expression of candidate genes determined by RNA-seq from ileum biopsies of IBD patients and controls (Haberman et al., 2014). RPKM, reads per kilobase per million. UC, ulcerative colitis. CD, crohn’s disease. Ctrl, control. Data represent mean and SD.
(C) The expression of Tmem258 was overlaid on intestinal single-cell RNAseq profiles (Grun et al., 2016). Enterocyte, goblet, and Paneth cell markers were used to construct a t-SNE plot.
TMEM258 Controls the ER Stress Response
TMEM258 encodes a small 76-amino acid protein containing two predicted transmembrane domains and no additional functional domains that can be readily identified. Nevertheless, TMEM258 is highly conserved from mammals to flies and worms and ubiquitously expressed across human tissues (Figures S1A and S1B). To determine a biological role for TMEM258, we initially sought to define its function by knockout, so as to then evaluate the effects of IBD-associated variants on its function. Thus, we aimed to identify functional pathway-level perturbations after ablating TMEM258 expression. Towards this end, we knocked down TMEM258 in HeLa cells by means of lentiviral transduction with shRNAs and performed transcriptomic analyses by RNA sequencing (RNA-seq). Strikingly, TMEM258 knockdown led to significant upregulation of genes related to glycoprotein modification, chaperone-mediated protein folding, proteostasis, and ER homeostasis (Figures 2A, S1C, and S1D). In light of these findings, we hypothesized that TMEM258 regulates ER stress responses. To test this hypothesis, we compared genes upregulated by TMEM258 knockdown to previously defined ER stress signatures associated with the transcription factors ATF4, XBP1, and ATF6 (Figure 2A). An unbiased gene set enrichment analysis revealed statistically significant overlap between TMEM258 knockdown and the ER stress response induced by tunicamycin, which inhibits protein glycosylation (Figure 2B). In fact, genetic knockdown of TMEM258 induced hypersensitivity to tunicamycin, resulting in cell death (Figure 2C). Our findings indicate that ER-localized TMEM258 (Figure 2D) controls ER homeostasis, and its depletion leads to an unresolved state of pathological ER stress.
Figure 2. Knockdown of TMEM258 Induces ER Stress.
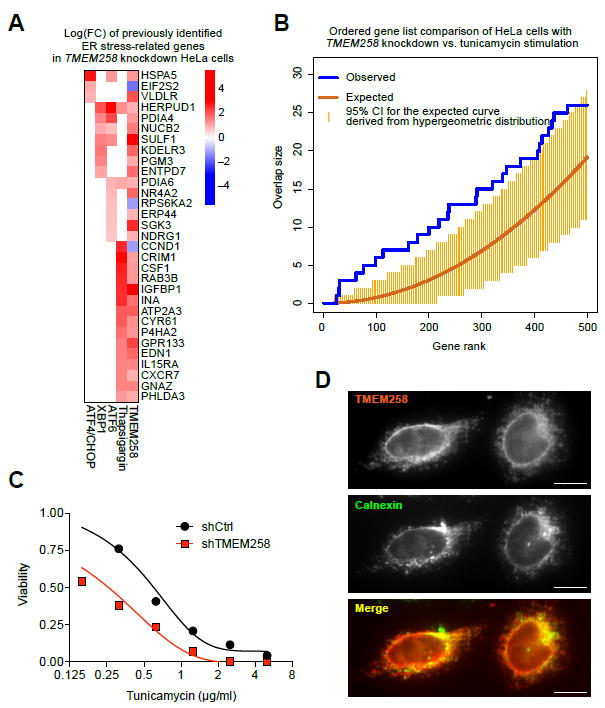
(A) TMEM258 was knocked down in HeLa cells by lentiviral delivery of shRNA. RNA-seq expression profiles were obtained 5 days after transduction and computationally queried against ER stress signatures obtained from GEO datasets (see Experimental Procedures).
(B) Comparison of differentially expressed genes in HeLa cells upon TMEM258 knockdown or tunicamycin treatment to induce ER stress.
(C) Sensitivity of HeLa cells to tunicamycin was determined at day four after drug treatment. Viability was measured by Alamar blue fluorescence.
(D) V5-tagged TMEM258 was ectopically expressed in HeLa cells by lentiviral transduction. Immunofluorescence imaging was performed by staining for TMEM258 with anti-V5 antibody (red) and ER with anti-Calnexin antibody (green). Scale bar, 10 μm.
See also Figure S1.
TMEM258 Is a Component of the Oligosaccharyltransferase Complex and Is Required for N-linked Protein Glycosylation
To gain deeper insight into the role of TMEM258 in controlling ER stress responses, we devised a proteomic approach to identify interaction partners by ectopic expression of TMEM258 in two cell lineages, BV-2 macrophages and BW5147 T lymphocytes (Figure S2 and Table S1). Strikingly, we detected nearly identical interaction partners for TMEM258 in both cell lines, despite their distinctly specialized lineages (Figure 3A). Moreover, we identified all known subunits of the OST complex as TMEM258 interactors (Figures 3A and 3B). In this context, the OST is strictly required for N-linked protein glycosylation, wherein it transfers a 14-sugar oligosaccharide from dolichol to asparagine residues on nascently translated proteins. Thus, N-linked glycosylation facilitates protein folding and maintains ER quality control. Consistent with this notion, we found that TMEM258 silencing dramatically reduced glycoprotein surface content, as determined by reactivity to concanavalin A, a lectin that specifically binds to α-D-mannosyl and α-D-glucosyl moieties (Figure 3C).
Figure 3. TMEM258 Interacts with the OST Complex and Regulates Protein N-linked Glycosylation.
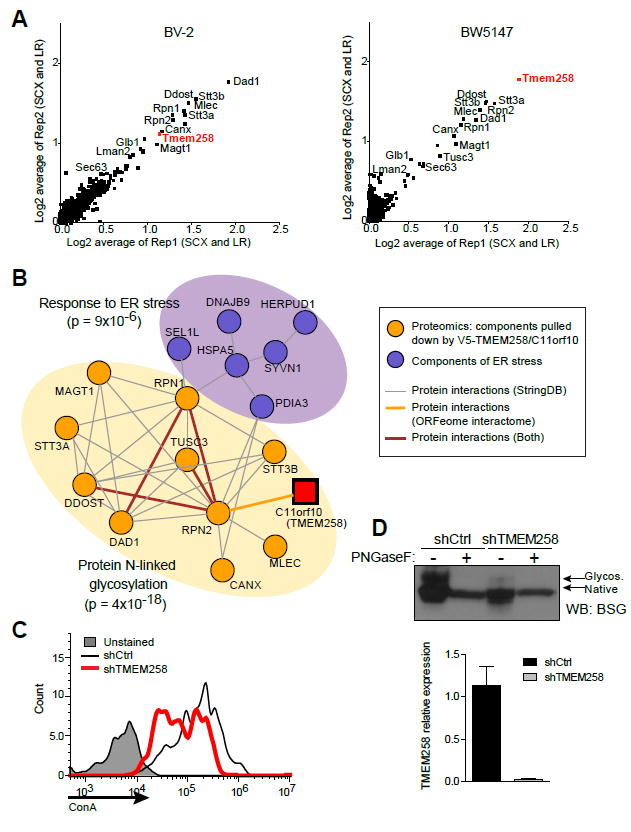
(A) TMEM258 interaction partners were identified in BV-2 cells (left) and BW5147 cells (right) expressing V5-tagged TMEM258. High-performance liquid chromatography-mass spectrometry was employed to detect proteins after immunoprecipitation with anti-V5 antibody.
(B) Network analysis integrating TMEM258 interaction partners with the OST complex and ER stress response.
(C) TMEM258 was knocked down in HeLa cells, and glycoproteins were detected by surface staining with FITC-labeled concanavalin A (ConA) followed by FACS.
(D) Measurement of N-linked glycosylation on the prototypical glycoprotein basigen (BSG) was monitored by western blot. Where indicated, samples were deglycosylated with PNGaseF. Knockdown of TMEM258 in HeLa cells was verified by qPCR. shCtrl, control shRNA. Data represent mean and SD.
See also Figure S2.
To further define the role for TMEM258 in N-linked glycosylation, we monitored maturation of the prototypical glycoprotein basigen (BSG). In control knockdown experiments, nascently translated BSG was N-glycosylated in the ER and trafficked to the Golgi apparatus for further glycosylation. These glycosylation events were reflected as an upshift in gel mobility, which was reversed by treatment with the endoglycosidase PNGaseF (Figure 3D). Strikingly, BSG gel mobility in TMEM258 knockdown samples was equivalent to PNGaseF-treated samples from control knockdown, indicating that TMEM258 is required for N-linked glycosylation (Figure 3D). Collectively, these findings demonstrate that TMEM258 depletion results in defective N-linked glycosylation, profound ER stress (Figure 2), and perturbed ER homeostasis. Thus, N-linked glycosylation is tightly regulated to maintain ER homeostasis, and TMEM258 expression levels are a significant factor in governing the efficiency of glycosylation.
Tmem258 Controls Intestinal Inflammation in Mice
To determine the role of TMEM258 in intestinal inflammation, we developed a knockout mouse model (Figures S3A and S3B) with the aim of eliciting ER stress by perturbing N-linked glycosylation. Whereas haploinsufficient Tmem258+/- mice were viable and fertile, we did not obtain any homozygous Tmem258-/- mice from multiple breeding pairs, nor did we detect any signs of fetal resorption in timed matings. Given that complete deficiency of Tmem258 is early embryonic lethal, we reasoned that it functions in a fundamental biological process that may be revealed in haploinsufficient Tmem258+/- mice. Initially, we confirmed that Tmem258+/- mice were generally healthy, development of immune cell subsets was unperturbed, and intestinal barrier function was intact (Figures S3C-S3F, S4). However, after induction of intestinal inflammation by oral administration of 2.5% dextran sodium sulfate (DSS), Tmem258+/- mice exhibited accelerated weight loss and increased mortality relative to WT mice (Figure 4A). Additionally, overall health scores and blinded histopathological scoring indicated exacerbated disease in Tmem258+/- mice (Figures 4B, 4C, and S5). While both groups of mice showed evidence of crypt ablation and inflammatory infiltrates, pathological severity was significantly elevated in Tmem258+/- mice (Figures 4C, S5B, and S5C), and the epithelial healing response was diminished in Tmem258+/- mice (Figures 4B, 4C, and S5B). Consistent with increased histopathological metrics of inflammation, Tmem258+/- mice exhibited a pronounced inflammatory gene expression signature, as determined by RNA-seq of colonic tissue (Figure 4D and Table S2).
Figure 4. Tmem258 Haploinsufficiency Exacerbates DSS Colitis.
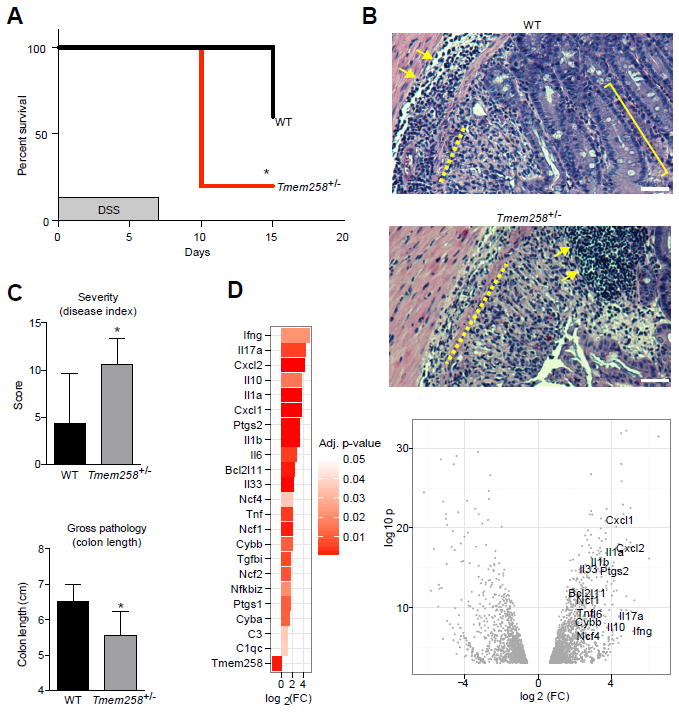
(A) Mice were administered 2.5% DSS in drinking water for 7 days. Mortality was monitored daily. *p < 0.05 Gehan-Breslow-Wilcoxon test (survival). N = 5-6 mice per genotype for 2 independent experiments.
(B and C) At day 8, mice were assigned an overall health score (severity), colon lengths were measured, and tissue sections were obtained. Histopathological scoring (HistoPath) was based on severity of inflammation, depth of inflammation, and crypt damage. Epithelial proliferation in response to injury was scored independently. Arrows, inflammation; bracket, epithelial proliferation; dashed line, crypt damage. Data represent mean and SD of histopathological scores. *p < 0.05, Students’ t test. Scale bar, 25 μm.
(D) Colon tissue was obtained at day 8 and transcriptomic profiles were measured by RNA-seq. Differentially expressed inflammatory genes in Tmem258+/- mice are displayed.
See also Figures S3-S5.
Tmem258 Limits ER Stress and Prevents Apoptosis in Colonic Epithelial Cells
In the context of intestinal pathology, we hypothesized that TMEM258 plays a central role in secretory cell types that have a large translational load, such as Paneth cells and goblet cells, which secrete large quantities of antimicrobial peptides and mucins respectively. In unmanipulated mice, Tmem258 haploinsufficiency did not induce dramatic ER stress in the colonic epithelium, but after induction of colitis with DSS, we observed clear evidence of elevated ER stress in Tmem258+/- mice relative to WT controls. Specifically, Tmem258 haploinsufficiency led to increased frequencies of epithelial cells that stained positive for BiP/GRP78 (HSPA5) (Figures 5A and S6), a major transcriptional target of the unfolded protein response that functions as a chaperone and regulator of ER stress sensors such as IRE1, PERK, and ATF6. Moreover, we detected a greater frequency of apoptotic epithelial cells, as assessed by positive staining for active caspase 3, in Tmem258+/- mice compared to controls. Consistent with the notion that TMEM258 performs a protective function in the context of ER stress-mediated apoptosis, we observed massive crypt ablation and disorganized dispersion of Ki67-positive proliferating cells in Tmem258+/- mice (Figure 5A).
Figure 5. TMEM258 Controls ER Stress and Apoptosis in Colonic Epithelium.
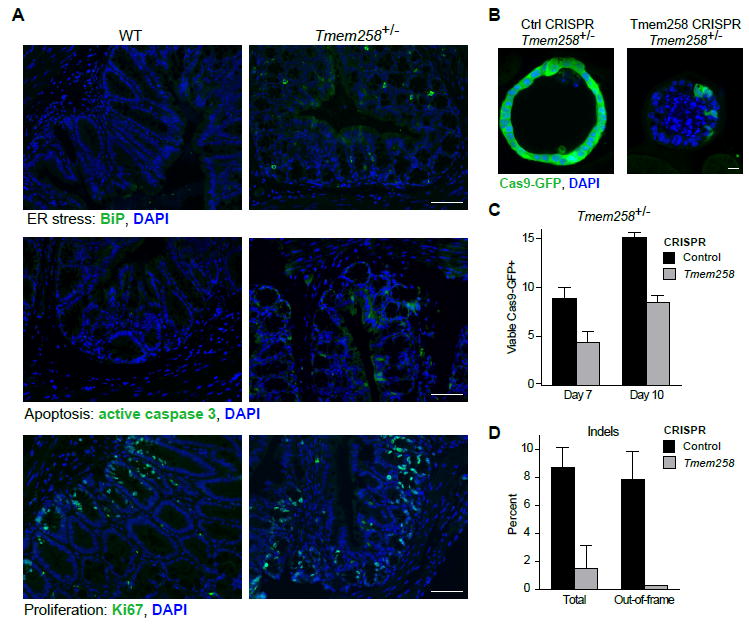
(A) Mice were administered 2.5% DSS in drinking water for 7 days. Colon tissue was obtained at day 8 and processed for immunofluorescence imaging to detect the indicated markers. Scale bar, 25 μm.
(B and C) Complete knockout of Tmem258 was achieved by CRISPR targeting in colonic organoids derived from Tmem258+/- mice bred to stop-floxed Cas9-GFP mice. Simultaneous delivery of Cre recombinase and sgRNA targeting Tmem258 (or control) was achieved by lentiviral transduction. Viable Cas9-GFP cells were identified by immunofluorescence staining with anti-GFP antibody (B). Scale bar, 25 μm. In addition, percentages of GFP-positive cells were quantified by FACS (C). Data represent mean + SD.
(D) Sequencing and validation of insertion/deletion mutations in Tmem258 CRISPR organoids. Data represent mean and SD derived from 2 independent sgRNAs for exonic regions and 2 independent sgRNAs for intronic regions of Tmem258.
See also Figure S6.
To precisely define the impact of Tmem258 deficiency in colonic epithelial cells, we sought to generate homozygous null organoid cultures. We hypothesized that complete deficiency of Tmem258 would result in unresolved ER stress and rapid apoptosis. Thus, we devised a CRISPR-based approach to target the remaining Tmem258 allele in heterozygous mice and used an inducible Cas9-GFP reporter to monitor loss of GFP-positive cells as a measure of cell death in targeted cells. Accordingly, we bred Tmem258+/- mice with stop-floxed Cas9-2A-GFP knockin mice and isolated colonic crypts to derive spheroid cultures in vitro. We then introduced sgRNAs targeting Tmem258 exonic (or intronic) sequences by transduction with lentiviral particles co-encoding Cre recombinase. Following Cre-mediated deletion of the stop codon upstream of Cas9-2A-GFP, inducible expression of Cas9 and GFP marks cells that have received a sgRNA. Using this approach, we found that sgRNAs targeting Tmem258 exons (Tmem258 CRISPR) had fewer viable Cas9-GFP-positive cells compared to control sgRNAs targeting noncoding intronic regions of Tmem258 (Ctrl CRISPR) (Figures 5B and 5C). Importantly, this observation was consistent across two independent Tmem258 sgRNAs and two independent control sgRNAs. Moreover, lentiviral titers were similar between all sgRNAs, suggesting that Tmem258 CRISPR induces cell death in organoids. Morphologically, Tmem258 CRISPR organoids contained scattered Cas9-GFP-positive cells within small spheroids that encapsulated numerous dead cells within the lumen (Figure 5B). In contrast, control CRISPR organoids appeared healthier and larger with contiguous Cas9-GFP-positive cells, suggestive of active proliferation (Figure 5B and 5C). Among Cas9-GFP-positive cells, insertion/deletion mutations were observed with both Tmem258 and control CRISPR, but more in-frame mutations were detected in Tmem258 CRISPR (Figure 5D). This observation suggests that out-of-frame alleles are negatively selected, consistent with our finding that Tmem258 deletion increases ER stress and cell death in colonic organoids (Figures 5B and 5C).
Increased Expression of TMEM258 Associates with IBD Risk and Impaired Secretion of Glycoproteins
To determine how genetic variation associated with IBD risk impacts TMEM258 function, we fine-mapped eQTL signals within the chromosome 11 (61.5-61.65) locus to determine the impact of the IBD risk allele for rs174537 on gene expression. Using the Geuvadis dataset (Tintle et al., 2015), which is comprised of matched genome sequences and transcriptomes from immortalized lymphoblasts, we found a strong eQTL effect for rs174538 on TMEM258, wherein the minor allele was associated with increased expression. Importantly, rs174538 is in LD with the IBD risk SNP rs174537 (r2 0.86, D’ 0.98), and the minor alleles for both SNPs correspond to IBD risk and increased expression of TMEM258 (Figure 6A). Although we also observed eQTL effects for rs174538 on other genes in the locus, these residual effects regressed-out after adjusting for additional eQTLs in the locus. Thus, the dominant IBD risk SNP (rs174537) significantly increases expression of TMEM258, whereas the dominant RA SNP decreases expression of FADS1 (data not shown). Given these findings, we hypothesized that TMEM258 expression above or below optimal levels may alter the stoichiometry of the OST complex and impair its catalytic assembly or activity. To identify the OST subunits with which TMEM258 interacts, we used V5-tagged TMEM258 as bait in co-immunoprecipitation experiments. Accordingly, we found that TMEM258 specifically interacts with RPN1, and co-expression of both proteins elevates their steady state levels (Figure 6B). Given that RPN1 was previously shown to interact with the lectin MLEC and inhibit secretion of glycoproteins (Qin et al., 2012), we sought to determine the impact of elevated expression of TMEM258 on protein secretion. Toward this end, we transfected HEK293T cells with TMEM258 (or GFP control) and monitored secretion of the glycoprotein alpha 1 anti-trypsin. Strikingly, elevated expression of TMEM258 resulted in impaired glycosylation and secretion of this prototypical secretory protein (Figure 6C). Taken together with evidence from human IBD genetics, our results suggest that perturbation of TMEM258 expression profoundly impacts protein glycosylation and ER stress responses. Moreover, intestinal homeostasis is particularly susceptible to perturbation by pathological ER stress resulting from impaired protein glycosylation.
Figure 6. Increased Expression of TMEM258 Associates with IBD Risk and Impaired Secretion of Glycoproteins.
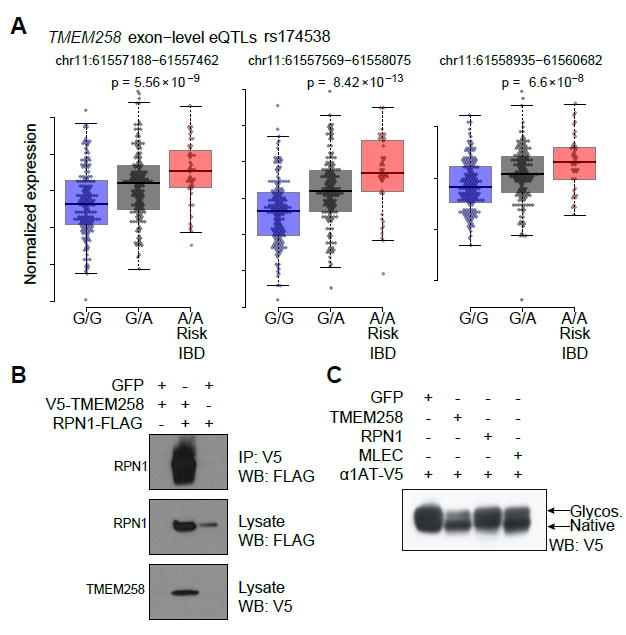
(A) Exon-level eQTLs for rs174538 (LD with rs174537 r2 0.86, D’ 0.98). Data were derived from lymphoblasts in the Geuvadis study (Tintle et al., 2015). Data represent mean and SD.
(B) Coimmunoprecipitation of TMEM258 and the OST subunit RPN1 after ectopic expression in HEK293T cells.
(C) Ectopic expression of TMEM258 in HE293T cells leads to impaired secretion and glycosylation of alpha-1 antitrypsin.
Discussion
The chromosome 11 (61.5-61.65) locus contains five protein-coding genes and has been associated with three distinct phenotypes: IBD (Jostins et al., 2012), RA (Okada et al., 2014), and inflammatory lipid synthesis (Lappalainen et al., 2013). Due to a relatively recent positive selection event acting on the locus, extensive LD spans the region. One feasible explanation for this positive selection is that a derived FADS genotype, associated with efficient lipid synthesis, provided a selective advantage to early humans (Ameur et al., 2012). While overwhelming evidence supports the notion that lipid phenotypes are governed by genetic variants that impact FADS function, positive selection for FADS genotypes may have passively selected all genetic variants in LD with FADS, and more importantly, all phenotypes conferred by those variants. Indeed, the derived haplotype at this locus is more common than the ancestral haplotype, and is associated with increased levels of serum phospholipids and arachidonic acid (Ameur et al., 2012; Hester et al., 2014). While the impact of FADS1 and FADS2 enzymatic function on lipid metabolism likely contributes to risk for coronary artery disease and RA, it is not clear that this is the case for IBD. In fact, IBD risk is associated with the ancestral haplotype, whereas RA and coronary artery disease are associated with the derived haplotype. Moreover, we report that the IBD risk allele marked by rs174537 has a significant eQTL effect associated with increased expression of TMEM28. In contrast, the RA risk allele from this locus (rs968567) is associated with an eQTL effect resulting in decreased expression of FADS1. Collectively, our findings support the interpretation that IBD risk is conferred by TMEM258, while RA and lipid phenotypes are controlled by effects on FADS.
Given that no clear function has yet been attributed to TMEM258 in vivo, its association with IBD was not immediately evident. Here we identify TMEM258 as a central regulator of intestinal homeostasis and demonstrate its requirement for efficient N-linked protein glycosylation and protection from ER stress-induced apoptosis. During the course of generating the Tmem258 knockout mouse and preparation of this manuscript, another report found that TMEM258 interacts with the OST complex and controls glycosylation (Blomen et al., 2015). Our results extend these findings and map the interaction between TMEM258 and the essential OST subunit RPN1. Moreover, we have generated a mouse knockout model to link TMEM258 function with intestinal homeostasis. Further analysis of human genetic data indicates that the IBD risk haplotype in the chromosome 11 (61.5-61.65) locus is associated with increased expression of TMEM258. In this context, ectopic expression of TMEM258 impairs OST-mediated glycosylation and secretion. As a subunit of the large multi-subunit OST complex, TMEM258 abundance above or below optimal levels may alter the stoichiometry of the complex and impair its catalytic assembly or activity. Consequently, our data implicate perturbation in protein glycosylation in the induction of pathological ER stress and intestinal inflammation.
The ER stress response is mediated by three interconnected networks of sensors and effectors. Accumulation of unfolded proteins in the ER is sensed by the chaperone Bip/Grp78 (HSPA5), which then dissociates from and activates ATF6 (ATF6), IRE1 (ERN1 and 2), and PERK (EIF2AK3). In turn, ATF6 elicits transcriptional activation of the cytoprotective unfolded protein response. Similarly, IRE1 controls transcription of a subset of unfolded protein response genes by initiating splicing and activation of the transcription factor XPB1. In contrast, PERK activation triggers cell death in response to unresolved ER stress by activating ATF4 and the executioner CHOP (DDIT3). In the context of pathological ER stress, GWASs and fine mapping studies have associated IBD with several genes involved in the ER stress response, such as ATF4 and ERN2 (IRE1-ß) (Jostins et al., 2012). Moreover, hypomorphic variants of XBP1 are susceptibility factors in Crohn’s disease and ulcerative colitis (Kaser et al., 2008). In mouse models, conditional deletion of Xbp1 or Ire1α (Ern1) leads to colitis resulting from excessive ER stress and apoptosis induced by CHOP (DDIT3) (Kaser et al., 2008; Zhang et al., 2015). Thus, mounting evidence highlights genes and pathways involved in ER homeostasis as critical genetic factors imparting risk to IBD.
From a broader perspective, the ER stress response has been proposed to engage in pathway-level interactions with additional processes underlying IBD pathophysiology. Specifically, a Crohn’s disease risk variant of the autophagy gene ATG16L1 is associated with elevated ER stress in Paneth cells and poor prognosis in patients (Deuring et al., 2014). Moreover, ER stress in goblet cells can be ameliorated by stimulation of IL10, another IBD-associated gene (Hasnain et al., 2013). With mounting evidence implicating the ER stress response in several immunological diseases including arthritis and interstitial lung disease (Bettigole and Glimcher, 2015; Watkin et al., 2015), approaches aimed at alleviating pathological ER stress may represent a therapeutic opportunity in a range of pathological contexts (Das et al., 2013; Fu et al., 2015).
Experimental Procedures
DSS-Induced Acute Colitis
Mice were given 2.5% (w/v) dextran sulfate sodium (DSS, MP Biomedicals; MW = 36,000-50,000) dissolved in drinking water for 7 days followed by one day of regular drinking water. Mice were euthanized at day 8 and colons were assessed for length and histology. Clinical assessment of DSS-treated mice included daily evaluation of weight loss, stool consistency, blood in stool, posture, ruffled fur, and mobility. Each parameter was individually scored (ranging from 0-3) and disease severity (0-15) was calculated by sum of individual scores.
Histology
The colon was extracted from DSS-treated mice and divided into proximal, middle, and distal sections, and fixed in 10% formalin. Sections were stained with hematoxylin and eosin (H&E) and histologic examination was performed in a blinded fashion using a modified validated scoring system (Krieglstein et al., 2007). Three independent parameters were measured: (a) severity of inflammation, (b) depth of injury/inflammation, and (c) crypt damage. Each score was multiplied by a factor reflecting a percentage of the tissue involved, generating a maximum possible score of 40. The histopathology score was the sum of the scores obtained from the three segments of the colon, yielding a maximum possible score of 120 (Kabashima et al., 2002). To detect epithelial cell proliferation, tissues sections were immunostained for Ki-67 and the frequency of Ki-67-positive epithelial cells was quantitated.
RNA-seq on Colon Samples
Mice were administered DSS as described above, and RNA was purified from colon tissue samples by Trizol extraction (Life Technologies). Purified RNA was treated with Turbo DNase (Thermo Fisher Scientific) prior to first-strand cDNA synthesis and template switching with Maxima reverse transcriptase (Thermo Fisher Scientific). PolydT oligos were appended on the 5′ end with sample barcodes, unique molecular identifier sequences, and a truncated Illumina adapter. Template switch oligos were comprised of a truncated Illumina adapter and three modified ribocytosines at the 3′ end. Following reverse transcription, samples were treated with RNaseH (Thermo Fisher Scientific) and subjected to second-strand cDNA synthesis with NEBnext (NEB). Double-stranded cDNA (1 ng) was then tagmented with the Nextera XT kit (Illumina) for 15 min, followed by 12-cycle PCR to generate 3′ DGE libraries. Samples were pooled to a final concentration of 4 nM and sequenced with a 50-cycle MiSeq kit (Illumina) by paired-end reads of 21 and 40 cycles followed by 1 adapter read.
shRNA Knockdown
HeLa cells (ATCC) were seeded in 24-well plates at 3.4 x 104/300 μl media. After 24 h, cells were transduced with 150 μl lentiviral supernatant packaged in HEK293T cells. The following constructs were used to produce lentiviral particles: TMEM258-TRCN0000188656, TMEM258-TRCN0000160130, Luc-TRCN0000072264, and LacZ-TRCN0000072228 (from the Genetic Perturbation Platform, Broad Institute). Three days after transduction, cells were passaged into 6-well plates and media was supplemented with puromycin (InvivoGen) at a concentration of 3 μg/ml. After two days in selection, cells were harvested for analysis.
Immunofluorescence
HeLa cells (ATCC) were transduced with lentivirus for ectopic expression of TMEM258 appended with an N-terminal V5 tag. Cells were seeded onto coverslips and allowed to adhere for 30 min at 37°C. Subsequently, cells were fixed in 4% paraformaldehyde for 15 min at 37°C and stained with mouse monoclonal anti-V5 (Invitrogen) and rabbit anti-Calnexin (Cell Signaling Technology) diluted 1:250 in staining buffer (3% BSA, 0.1% saponin, PBS). The following secondary antibodies were used at a 1:1000 dilution in staining buffer: Alexa Fluor488 goat anti-mouse IgG (Life Technologies R37121) and Alexa Fluor594 goat anti-rabbit IgG (Life Technologies A27034). Cells were mounted in Vectashield medium containing DAPI (Vector Laboratories) and imaged with a Zeiss Axio A1 microscope equipped with 63x/1.25 objective. Image acquisition was performed with the AxioVision (Rel.4.8) software package.
RNA-seq on HeLa Cells
TMEM258 was knocked down in HeLa cells as described above. Cells were lysed, and mRNA was purified with polydT microbeads (Dynabeads mRNA Direct Kit, Invitrogen). First-strand cDNA synthesis with template switching was performed as described above. Second-strand cDNA was generated by NEBnext kit (NEB). Double-stranded cDNA (0.5 ng) was then tagmented with the Nextera XT kit (Illumina) for 12 min, followed by a 12-cycle PCR to amplify the whole transcriptome. Samples were then pooled to a final concentration of 4 nM and sequenced with a 50-cycle MiSeq kit (Illumina) by paired-end reads of 30 and 29 cycles followed by 2 adapter reads.
Tunicamycin Stress Test
TMEM258 was knocked down in HeLa cells as described above. Cells were treated with escalating doses of tunicamycin (Sigma Aldrich) for 4 days. Cell viability was then monitored by Alamar blue reagent (Invitrogen) according to the manufacturer’s recommendations.
Glycosylation Assays
TMEM258 knockdown was performed in HeLa cells as described above. Cells were then gently scraped off the plate and washed in serum-free DMEM prior to lectin staining with 1 μg FITC-concanavalin A (Sigma). All samples were run on an Accuri C6 flow cytometer (Becton Dickinson) and analyzed with FlowJo software. In addition, HeLa cells grown to confluency in 6-well plates were subjected to western blot for the glycoprotein reporter basigen (BSG). Designated samples were deglycosylated with PNGaseF according to the manufacturer’s protocol (NEB). Samples were then resolved by PAGE, and western blot was performed to detect Bsg (anti-CD147, clone EPR4052, Abcam).
Immunoprecipitation
293T cells (ATCC) were transfected with Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer. The indicated cDNA constructs were cloned as PCR products into pCMV (Invitrogen) by Gibson assembly. Transfected cells were lysed (1% Triton-X 100, PBS pH 7.4), and V5-TMEM258 was immunoprecipitated with anti-V5 magnetic agarose beads (MBL International) with rotation at 4°C for 4 hours. Samples were then resolved by PAGE, and western blot was performed with HRP-conjugated anti-Flag (anti-DYKDDDDK, Cell Signaling Technologies) to detect RPN1-Flag. In secretion studies, 293T cells were transfected with the indicated cDNA constructs as described above. Secreted alpha 1 anti-trypsin (SerpinA1-V5) was immunoprecipitated from culture supernatant as described above. Samples were then resolved by PAGE, and western blot was performed with HRP-conjugated anti-V5 (Sigma Aldrich).
Immunofluorescence on Colon Samples
Colon samples were fixed in 10% formalin, embedded in paraffin, and sectioned. Rehydration was performed by serial wash steps with Xylene (10 min), 100% ethanol (twice, 20 sec), 90% ethanol (20 sec), 70% ethanol (20 sec), and water (twice, 20 sec). Sections were then bathed in antigen retrieval buffer (BD Pharmingen, Retrievagen A, 550524, used per manufacturer instructions). After blocking in IFF (PBS 1% BSA, 2% FCS, filtered) for 30 min, sections were stained with the following primary antibodies diluted in IFF: anti-cleaved caspase 3, anti-Ki67, anti-BiP (all from Cell Signaling Technology). After staining for 60 min at room temperature in a humidified chamber, sections were washed in PBS and stained as above with Alexa Fluor 488 goat anti-rabbit IgG (Life Technologies). Sections were mounted in Vectashield medium containing DAPI (Vector Laboratories) and imaged with a Zeiss Axio A1 microscope equipped with 63x/1.25 objective. Image acquisition was performed with the AxioVision (Rel.4.8) software package.
Organoids
Procedures for establishing and maintaining mouse organoids/spheroids were based on a previously published method (Miyoshi and Stappenbeck, 2013). The only difference was the addition of Primocin antibiotic (InvivoGen 1:500) to all organoid media. In brief, crypts were isolated from mouse distal colon and suspended in Matrigel (BD Biosciences; 15 μl/well). Matrigel mixture was plated into 48-well tissue culture plates at room temperature. To prevent crypts from attaching to the plastic, plates were flipped and incubated at 37°C to polymerize the Matrigel. 500 μl of 50% L-WRN CM (Miyoshi et al., 2012) and fresh primary culture media (Advanced DMEM/F-12 [Invitrogen] supplemented with 20% fetal bovine serum, 2 mM glutaMAX, 100 units/mL penicillin, and 0.1 mg/mL streptomycin), supplemented with 10 μM Y-27632 (ROCK inhibitor; Tocris Bioscience, R&D Systems) and 10 μM SB 431542 (TGFBR1 inhibitor; Cayman Chemical Company) was added to each well. Organoids were passaged (1:3) every 1 to 3 days for two weeks up to transduction and every 2-3 days (1:2 – 1:3) following transduction. We followed the following steps to obtain single-cell suspensions of organoid cells for flow cytometry, FACS, and lentivirus transduction: Culture media was removed from organoid/Matrigel well and 500 μl PBS EDTA (0.5 mM EDTA in PBS) was added to each well. We scratched and suspended the Matrigel with a 1000 μl pipette. Organoids were spun at 200 x g for 5 min and PBS was removed to the level of the Matrigel containing organoids. Organoids were dissociated by incubation in 0.25% (wt/vol) trypsin for 1 min followed by vigorous pipetting. Trypsin was washed with wash media and cells were then either transduced or stained for flow cytometry.
Single-cell suspensions of organoids were obtained from two wells of Matrigel organoids and plated as one well in infection media (50% L-WRN media in DMEM/F12 + 10 μM Y-27632 + 10 μM SB 431542). 3 μl of each virus (concentrated by ultracentrifugation) was added per well (with 4 wells/virus/mouse) for a total volume of 250 μl/well. Cells were left to be transduced for 6 h at 37°C. Organoid suspensions were then washed and replated in Matrigel (two wells/well transduced).
For FACS sorting, single-cell suspensions were obtained and filtered (40 μm). DAPI was added just before the sort as a live/dead marker. Cells were sorted on GFP-positive DAPI-negative into TRIzol (Ambion).
For imaging, organoids were plated on chamber wells (Nunc Lab-Tek II Chamber slide system) with 50% L-WRN for one day and differentiation media (primary culture media and 5 μM DAPT) for two days. Organoids were then fixed in PFA for 1 h at 37°C. All steps that followed were at room temperature. The tissue was permeabilized for 30 min in 1% Triton X-100 in PBS and then blocked for 1 h at room temperature (2% BSA, 3% goat serum, 0.2% Triton in PBS). Primary antibodies (rabbit anti-GFP AlexaFluor647 (1:400), Life Technologies) were added to stain overnight in working buffer (0.2% BSA 0.2% Triton, 0.3% goat serum). Fixed organoids were washed all day in working buffer and stained overnight in secondary antibodies plus Hoechst. Organoids were washed 4 times and the chamber was removed. Coverslips were mounted with ProLong Gold Antifade Reagent (Life Technologies). Organoids were imaged by confocal microscopy using a 40x objective.
CRISPR Design
All sgRNAs were designed for efficacy using sgRNA Designer (Doench et al., 2014) and for specificity with CCTop (Stemmer et al., 2015). sgRNA sequences were as follows: exon 2 GAAGTACCACAGTCAGGTGA, exon 3 GTAGAGGTGACCTCGTAACT, intronic 1 AGGAGGCTGGTGCTGCCCAA, intronic 2 GGTGGTTCATTGGTATTTGG. Guides were cloned as oligonucleotides into lentiCRISPR v2 plasmid (Sanjana et al., 2014) in which spCas9-2A-PAC was replaced with cDNA encoding Cre recombinase. Genomic sequencing primers were as follows: forward i5adapter-TTCCTTAGGAGCTCGAAGCC and reverse i7adapter-GTCTCAATCCCACCTCAAAGGA, forward i5adapter-CCCCATGAAAGGTAGTCAGAGAC and reverse i7adapter-CCTGTTGATATGGCTCTACGAGG, forward i5adapter-TTTCCTTTGAGGTGGGATTGAGA and reverse i7adapter-CAGCTGGGACATGAGCACTT, forward i5adapter-TCCAACTTCAAATTTGCTCAGTTTA and reverse i7adapter-TCGCCTCTAGCCTCTAGTCA. Sequencing was performed with a 150-cycle MiSeq kit (Illumina).
Mice
All animal studies were conducted under protocols approved by the Subcommittee on Research Animal Care (SRAC) at Massachusetts General Hospital. Tmem258tm1(KOMP)Vlcg knockout mice (KOMP, UC Davis) were generated by targeting in VGB6 ES cells derived from C57BL/6NTac mice. Blastocyst injections were performed by KOMP, UC Davis. Where indicated, Tmem258+/- mice were bred to Cas9 transgenic knock-in mice (Gt(ROSA)26Sortm1(CAG-cas9*,-EGFP)Fezh) (Platt et al., 2014) obtained from Jackson Laboratories.
Statistics
Statistical procedures specific for each technology are described in their respective sections above. For comparisons between two groups, unpaired Student’s t tests (two-tailed) or non-parametric tests were employed. See figure legends for additional details.
Supplementary Material
Acknowledgments
This work was supported by funding from The Leona M. and Harry B. Helmsley Charitable Trust, the Crohn’s and Colitis Foundation of America, and National Institutes of Health grants DK043351and DK097485 to R.J.X. We thank Richard Cummings, Isabel Latorre, Elizabeth Creasey, Natalia Nedelsky, and Bihua Li for scientific input and technical expertise.
Footnotes
The authors have no competing financial interests to declare.
Author Contributions
Conceptualization, D.B.G., and R.J.X.; Methodology, D.B.G., S.A.C., and M.S.; Formal Analysis, P.D., M.V., A.N., N.K., and A.K.B.; Investigation, D.B.G., A.L., A.B., A.N.D., C.P.P., and G.G.; Resources, C.W., M.J.D., M.A.R., and S.A.C., Writing – Original Draft, D.B.G., and R.J.X.; Supervision, D.B.G. and R.J.X.; Funding Acquisition, R.J.X.
Accession Numbers
RNA-Seq data from HeLa cells is accessible at GEO: GSE89542.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ameur A, Enroth S, Johansson A, Zaboli G, Igl W, Johansson AC, Rivas MA, Daly MJ, Schmitz G, Hicks AA, et al. Genetic adaptation of fatty-acid metabolism: a human-specific haplotype increasing the biosynthesis of long-chain omega-3 and omega-6 fatty acids. Am J Hum Genet. 2012;90:809–820. doi: 10.1016/j.ajhg.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthakrishnan AN, Khalili H, Konijeti GG, Higuchi LM, de Silva P, Fuchs CS, Willett WC, Richter JM, Chan AT. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn’s disease. Gut. 2014;63:776–784. doi: 10.1136/gutjnl-2013-305304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudoin M, Goyette P, Boucher G, Lo KS, Rivas MA, Stevens C, Alikashani A, Ladouceur M, Ellinghaus D, Torkvist L, et al. Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet. 2013;9:e1003723. doi: 10.1371/journal.pgen.1003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettigole SE, Glimcher LH. Endoplasmic reticulum stress in immunity. Annu Rev Immunol. 2015;33:107–138. doi: 10.1146/annurev-immunol-032414-112116. [DOI] [PubMed] [Google Scholar]
- Blomen VA, Majek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A, et al. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015;350:1092–1096. doi: 10.1126/science.aac7557. [DOI] [PubMed] [Google Scholar]
- Costea I, Mack DR, Lemaitre RN, Israel D, Marcil V, Ahmad A, Amre DK. Interactions between the dietary polyunsaturated fatty acid ratio and genetic factors determine susceptibility to pediatric Crohn’s disease. Gastroenterology. 2014;146:929–931. doi: 10.1053/j.gastro.2013.12.034. [DOI] [PubMed] [Google Scholar]
- Das I, Png CW, Oancea I, Hasnain SZ, Lourie R, Proctor M, Eri RD, Sheng Y, Crane DI, Florin TH, et al. Glucocorticoids alleviate intestinal ER stress by enhancing protein folding and degradation of misfolded proteins. J Exp Med. 2013;210:1201–1216. doi: 10.1084/jem.20121268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuring JJ, Fuhler GM, Konstantinov SR, Peppelenbosch MP, Kuipers EJ, de Haar C, van der Woude CJ. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut. 2014;63:1081–1091. doi: 10.1136/gutjnl-2012-303527. [DOI] [PubMed] [Google Scholar]
- Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJ, Shishkin AA, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Yalcin A, Lee GY, Li P, Fan J, Arruda AP, Pers BM, Yilmaz M, Eguchi K, Hotamisligil GS. Phenotypic assays identify azoramide as a small-molecule modulator of the unfolded protein response with antidiabetic activity. Sci Transl Med. 2015;7:292ra298. doi: 10.1126/scitranslmed.aaa9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DB, Xavier RJ. From genetics of inflammatory bowel disease towards mechanistic insights. Trends Immunol. 2013;34:371–378. doi: 10.1016/j.it.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grun D, Muraro MJ, Boisset JC, Wiebrands K, Lyubimova A, Dharmadhikari G, van den Born M, van Es J, Jansen E, Clevers H, et al. De Novo Prediction of Stem Cell Identity using Single-Cell Transcriptome Data. Cell Stem Cell. 2016;19:266–277. doi: 10.1016/j.stem.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W, Steffen BT, Lemaitre RN, Wu JH, Tanaka T, Manichaikul A, Foy M, Rich SS, Wang L, Nettleton JA, et al. Genome-wide association study of plasma N6 polyunsaturated fatty acids within the cohorts for heart and aging research in genomic epidemiology consortium. Circ Cardiovasc Genet. 2014;7:321–331. doi: 10.1161/CIRCGENETICS.113.000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberman Y, Tickle TL, Dexheimer PJ, Kim MO, Tang D, Karns R, Baldassano RN, Noe JD, Rosh J, Markowitz J, et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest. 2014;124:3617–3633. doi: 10.1172/JCI75436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasnain SZ, Tauro S, Das I, Tong H, Chen AC, Jeffery PL, McDonald V, Florin TH, McGuckin MA. IL-10 promotes production of intestinal mucus by suppressing protein misfolding and endoplasmic reticulum stress in goblet cells. Gastroenterology. 2013;144:357–368. e359. doi: 10.1053/j.gastro.2012.10.043. [DOI] [PubMed] [Google Scholar]
- Hester AG, Murphy RC, Uhlson CJ, Ivester P, Lee TC, Sergeant S, Miller LR, Howard TD, Mathias RA, Chilton FH. Relationship between a common variant in the fatty acid desaturase (FADS) cluster and eicosanoid generation in humans. J Biol Chem. 2014;289:22482–22489. doi: 10.1074/jbc.M114.579557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Hea. Association mapping of inflammatory bowel disease loci to single variant resolution. bioRxiv. 2015 doi: 10.1038/nature22969. http://dx.doi.org/10.1101/028688. [DOI] [PMC free article] [PubMed]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashima K, Saji T, Murata T, Nagamachi M, Matsuoka T, Segi E, Tsuboi K, Sugimoto Y, Kobayashi T, Miyachi Y, et al. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest. 2002;109:883–893. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieglstein CF, Anthoni C, Cerwinka WH, Stokes KY, Russell J, Grisham MB, Granger DN. Role of blood- and tissue-associated inducible nitric-oxide synthase in colonic inflammation. Am J Pathol. 2007;170:490–496. doi: 10.2353/ajpath.2007.060594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak JH, Paik JK, Kim OY, Jang Y, Lee SH, Ordovas JM, Lee JH. FADS gene polymorphisms in Koreans: association with omega6 polyunsaturated fatty acids in serum phospholipids, lipid peroxides, and coronary artery disease. Atherosclerosis. 2011;214:94–100. doi: 10.1016/j.atherosclerosis.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedlander MR, t Hoen PA, Monlong J, Rivas MA, Gonzalez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SW, Lin K, Ma P, Zhang ZL, Zhou YD, Lu SY, Zhou X, Liu SM. FADS gene polymorphisms confer the risk of coronary artery disease in a Chinese Han population through the altered desaturase activities: based on high-resolution melting analysis. PLoS ONE. 2013;8:e55869. doi: 10.1371/journal.pone.0055869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979–986. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli N, Girelli D, Malerba G, Guarini P, Illig T, Trabetti E, Sandri M, Friso S, Pizzolo F, Schaeffer L, et al. FADS genotypes and desaturase activity estimated by the ratio of arachidonic acid to linoleic acid are associated with inflammation and coronary artery disease. Am J Clin Nutr. 2008;88:941–949. doi: 10.1093/ajcn/88.4.941. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, Stappenbeck TS. Wnt5a potentiates TGF-beta signaling to promote colonic crypt regeneration after tissue injury. Science. 2012;338:108–113. doi: 10.1126/science.1223821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc. 2013;8:2471–2482. doi: 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin SY, Hu D, Matsumoto K, Takeda K, Matsumoto N, Yamaguchi Y, Yamamoto K. Malectin forms a complex with ribophorin I for enhanced association with misfolded glycoproteins. J Biol Chem. 2012;287:38080–38089. doi: 10.1074/jbc.M112.394288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, Boucher G, Ripke S, Ellinghaus D, Burtt N, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer M, Thumberger T, Del Sol Keyer M, Wittbrodt J, Mateo JL. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS ONE. 2015;10:e0124633. doi: 10.1371/journal.pone.0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Shen J, Abecasis GR, Kisialiou A, Ordovas JM, Guralnik JM, Singleton A, Bandinelli S, Cherubini A, Arnett D, et al. Genome-wide association study of plasma polyunsaturated fatty acids in the InCHIANTI Study. PLoS Genet. 2009;5:e1000338. doi: 10.1371/journal.pgen.1000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintle NL, Pottala JV, Lacey S, Ramachandran V, Westra J, Rogers A, Clark J, Olthoff B, Larson M, Harris W, et al. A genome-wide association study of saturated, mono- and polyunsaturated red blood cell fatty acids in the Framingham Heart Offspring Study. Prostaglandins Leukot. Essent Fatty Acids. 2015;94:65–72. doi: 10.1016/j.plefa.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, Sha Y, Thamsen M, Santos-Cortez RL, Lee K, Gambin T, et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet. 2015;47:654–660. doi: 10.1038/ng.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HS, Chen Y, Fan L, Xi QL, Wu GH, Li XX, Yuan TL, He SQ, Yu Y, Shao ML, et al. The Endoplasmic Reticulum Stress Sensor IRE1alpha in Intestinal Epithelial Cells Is Essential for Protecting against Colitis. J Biol Chem. 2015;290:15327–15336. doi: 10.1074/jbc.M114.633560. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.