

Abstract
Conformational flexibility allows proteins to adopt multiple functionally important conformations, but can also lead to non-functional structures. We analyzed the dynamic behavior of the enzyme Guanylate Kinase as it evolved into the GK protein interaction domain (GKPID) to investigate the role of flexibility in the evolution of new protein functions. We found that the ancestral enzyme is very flexible, allowing it to adopt open conformations that can bind nucleotide and closed ones that enable catalysis of phosphotransfer from ATP to GMP. Historical mutations that converted the GK from an enzyme to a protein interaction domain dramatically reduce flexibility, predominantly by inhibiting rotations of the protein backbone that are coupled to the global closing motion. Removing flexibility prevents adoption of conformations that cannot fit the protein partner in the binding site. Our results highlight the importance of mutations that optimize protein conformational flexibility with function during evolution.
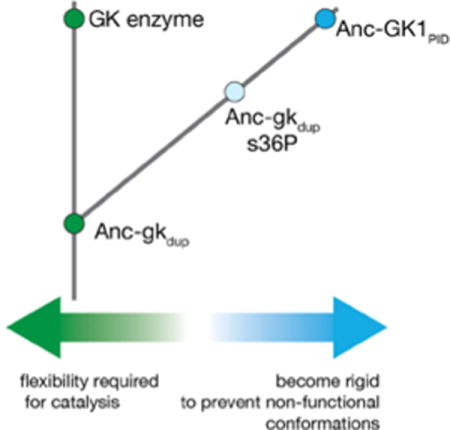
Graphical abstract
Background and Rationale
Proteins are sometimes thought of as “molecular machines”, in large part because they undergo conformational rearrangements in a manner that is coordinated with their function1. These coordinated movements are possible because protein structures are flexible enough to assume multiple functionally relevant conformations. Because flexibility underlies the ability to assume distinct conformations, it is essential to processes as diverse as translation by the ribosome2, cellular energy production by ATP synthase3, and regulation by allostery4. Although proteins need to be flexible for many of the functions they perform in biology, flexible structures can also be buffeted by thermal energy into non-functional conformations5. Evolutionary processes likely “tune” proteins to be flexible enough to undergo the specific movements that are necessary for function, while at the same time preventing the adoption of non-functional conformations.
Conformational flexibility may have been optimized during the evolution of the GK family of protein interaction domains (GKPID). These domains are found in animals and choanoflagellates, and are part of proteins (known as Membrane Associate Guanylate Kinases, or MAGUKs) that were likely important for the evolution of organized multicellularity in animals6–8. The GKPID family shares a common ancestor with extant Guanylate Kinase enzymes, which play a key role in nucleotide metabolism by catalyzing the formation of GDP using GMP and ATP (Figure 1A, 1B). Members of the GKPID family lack catalytic activity but instead function as phosphoprotein recognition modules, binding selectively to the phosphorylated form of their target proteins9–11. The common ancestor of these functionally diverse proteins, Anc-gkdup (dup = duplicated gene), has been analyzed using ancestral protein reconstruction6 – phylogenetic inference of ancestral sequences followed by gene synthesis, genetic manipulation, and experimental characterization12,13. Anc-gkdup has nucleotide kinase but not PID activity, whereas the ancestor to the PID family, Anc-GK1PID, binds the protein ligand Partner of Inscuteable (Pins) with high affinity but lacks nucleotide kinase activity (Figure 1A). Thus, the transition from Anc-gkdup to Anc-GK1PID defines the evolutionary interval over which the GKPID family evolved from its enzyme ancestor.
Figure 1.

Characterization of ancestral guanylate kinase conformational equilibria. A. Reduced phylogeny of the protein family containing gk enzymes (green) and protein-binding GKPIDs (blue). Mutation of the ancestral serine 36 to proline (Anc-gkdup s36P) abolishes catalytic activity and confers moderate protein binding affinity. The affinities for Pins are shown (ND = not detected)6. B. Extant GK enzymes (green) are primarily in an open conformation until nucleotides bind, at which point they close to position ATP and GMP phosphates for catalysis. The conformational change involves rotation of the GMP binding domain (GBD) towards the “CORE” and ATP-binding “LID”. A “hinge” composed of residues that connect the core and GBD mediates rotation. The extant GK domain (blue) from Discs large (Dlg) binds phosphorylated Partner of Inscuteable (Pins) during cortically-directed mitotic spindle orientation in metazoa. The PDB IDs for the structures used throughout are as follows: GK enzyme open conformation (1EX6, chain A), GK enzyme closed conformation (1EX7), s36P GK enzyme open conformation (4F4J), apo GK PID (1KJW), Pins-bound GK PID (3TVT). C. 1H-15N HSQC NMR experiment. Signals are observed for nitrogen atoms bonded to hydrogens, primarily peptide bonds (except for proline which does not have a hydrogen atom at this position). The position of the signal is dependent on the chemical shift of both atoms, which is in turn sensitive to structure. D. GMP binding induces linear changes in the chemical shifts of GK residues consistent with GK closing. The 1H-15N HSQC NMR signals for several GK residues that don’t directly contact GMP are shown when GMP concentrations are gradually increased (concentration key shown at right). The full spectra are shown in Supporting Information. E. Origin of GK 1H-15N HSQC NMR signals and the effect of GMP. An example signal that is influenced by GMP binding is shown. The GK exists as a structural ensemble in the absence of nucleotide (see Figure 3) so the HSQC signals from many GK residues are the average of many conformations. GMP-binding shifts the conformation to a more homogenous closed conformation. HSQC signals can be influenced directly by GMP (for residues near the binding site) and/or through the indirect effects on GK conformation. The signal at intermediate, non-saturating concentrations of GMP results from averaging of the relatively few closed and many open conformations.
Several observations are consistent with a role for flexibility in the evolution of the GKPID family. First, GK enzymes are flexible enough to allow them to transition from an open to a closed conformation in response to substrate binding, a key step in the catalytic mechanism (Figure 1B)14,15. In contrast, the GKPID remains in an open conformation even when bound to its protein ligand10,11, suggesting that the flexibility required for catalysis is not necessary for mediating protein interactions. Second, the genetic changes responsible for the transition from enzyme to PID function suggest that conformational flexibility may have been an important variable in the evolutionary process. Remarkably, a single mutation that occurred along the trajectory from Anc-gkdup to Anc-GK1PID, and is not in the protein-binding site, explains much of their functional difference. Placing this large-effect mutation – s36P (ancestral state, lowercase; derived state, capitalized) – into Anc-gkdup results in a complete loss of the ancestral nucleotide kinase function and gain of the derived Pins binding function (Figure 1A)6. In the context of the extant yeast GK enzyme, this mutation causes it to lose the ability to close in response to binding of its nucleotide substrate16, pointing to a role for flexibility in the transition between the two functions.
The differences in the response of the GK enzyme and PID to ligand binding (closing versus remaining open), and the effect of the s36P mutation on the yeast enzyme (preventing it from closing in response to guanylate binding), suggest that changes in flexibility might have been important in the evolution of PID activity. However, it is also possible that flexibility was unaltered, or did not play a significant role in this process. For example, function switching mutations such as s36P could simply shift the population of open and closed GK, or alter the mechanisms that couple ligand binding to closing, without affecting flexibility. Furthermore, even if flexibility was important in the functional transition from enzyme to PID, we do not know how it was altered, or how doing so could lead to such a dramatic change in function. We have addressed these fundamental questions in molecular evolution by analyzing the transition from Anc-gkdup to Anc-GK1PID using a combination of Nuclear Magnetic Resonance (NMR) spectroscopy and Molecular Dynamics (MD) simulation.
Results
Distinct conformational effects of GMP binding and a PID mutation
To determine how PID activity evolved in the GK family, we first tested a simple two-state model, in which the s36P mutation shifts the GK equilibrium towards the open state, which could preferentially bind proteins because of its larger binding groove10. We measured the conformational equilibrium between open and closed states of the GK using 1H-15N HSQC NMR, which resolves signals throughout the protein (Figure 1C). We used GMP binding to define the effect of shifting the equilibrium on the GK HSQC signals. In extant GK enzymes, addition of GMP alters signals in the GMP binding site, but also at distant sites that are influenced by the closing conformational change16,17. In a similar manner, GMP binding to Anc-gkdup influenced the chemical shift of residues both near to and distant from the GMP binding site (Figure 1D, Supporting Information). In the absence of GMP, signals that are sensitive to GK conformation are averaged due to the mixture of open and closed states (Figure 1E)18–20. GMP binding shifts the equilibrium to the closed state, and HSQC signals that are influenced by GK conformation shift position based on the fraction of GK that is in the open or closed conformation.
The perturbations of Anc-gkdup NMR signals in response to GMP binding provides a background for understanding how mutations that induce PID function, such as s36P, influence GK conformation. To understand how the s36P mutation affects the equilibrium between the Anc-gkdup open and closed states, we compared HSQC signals from Anc-gkdup s36P (without GMP) to the Anc-gkdup GMP titration series (Figure 2A, Supporting Information). To avoid chemical shift perturbations not directly related to changes in GK conformation, we restricted our analysis to signals from residues distant from both the mutation site and GMP binding pocket. If the s36P mutation simply increased the open Anc-gkdup state at the expense of the closed one, the mutation would shift HSQC signals along the same trajectory as GMP, but in the opposite direction. In general, however, the s36P mutation induces signal shifts that are much different than those caused by GMP binding (Figures 2, Supporting Information). The response of the Anc-gkdup HSQC spectrum to the s36P mutation is complex, with an average chemical shift perturbation that is not along the trajectory (either in the same or opposite direction) from GMP binding-induced perturbations (Figure 2B). The distinct effects of GMP-binding and the s36P mutation on Anc-gkdup chemical shifts indicate that the mutation does not simply alter the equilibrium between pre-existing open and closed states.
Figure 2.

Diverse conformational effects of GMP and the s36P PID mutation. A. The s36P mutation induces complex chemical shift perturbations. GMP binding and s36P induced chemical shift perturbations are shown mapped onto the GK structure. Assigned signals from Anc-gkdup and Anc-gkdup s36P residues distant from the GMP binding and mutation sites were taken from the HSQC spectra, rotated so that the Anc-gkdup signal without GMP is on the left and the GMP saturated signal is on the right, and placed on their position on the structure of GMP-bound yeast GK enzyme (PDB 1EX7). The HSQC signals from Anc-gkdup s36P are shown in blue. The full spectra with Anc-gkdup s36P overlaid are shown in Supporting Information. B. Comparison of chemical shift perturbations induced by the s36P mutation and GMP binding. Each vector represents the chemical shift perturbation of a single residue induced by the s36P mutation, relative to the shift induced by saturating concentrations of GMP. Signals from each residue shown in (A) were converted into vectors with angle defined by the angle between the s36P- and GMP-induced shifts (0° represents a shift in the same direction as GMP), and length defined by the ratio of the s36P- and GMP-shift magnitudes. Because the s36P induced shift magnitudes are normalized, residues with smaller absolute shifts may yield vectors of greater magnitude than those with larger absolute shifts. A vector representing the average shift is shown in blue.
A loss of flexibility along the trajectory from enzyme to PID
To determine if conformational flexibility changes along the evolutionary interval from Anc-gkdup to Anc-GK1PID, we monitored the internal dynamics of the peptide backbone from proteins along this trajectory using CPMG-based R2 relaxation dispersion NMR21,22. This technique is sensitive to motions on the μs-ms timescale, which is appropriate for large subdomain movements like GK closing23. At 25°C, Anc-gkdup did not exhibit large, clear individual dispersions (Figures 3A, Supporting Information). Small R2 dispersions indicate that backbone motions in apo Anc-gkdup deriving from the open-closed reaction are either too slow or too fast to detect with this technique20,23. A related enzyme, Adenylate Kinase, undergoes a open-close transition that is similarly too fast to observe significant dispersions at 25°C but yields large dispersions at 10°C20. Reducing the temperature to 10°C resulted in a large increase in dispersions, consistent with slowing of a process occurring at a rate faster than approximately 0.5 ms−1 (Figures 3A, Supporting Information). Residues exhibiting dispersions were found throughout the protein (Figure 3B, Supporting Information). The global nature of this dynamic process and the known open-closed GK conformational transition14, led us to conclude that the dispersions in Anc-gkdup at 10°C arise from interconversion between the open and closed GK conformations.
Figure 3.

Quenching of GK open-close interconversion dynamics during GKPID evolution. A. Rex in Anc-gkdup increases dramatically at 10°C compared to 25°C. The average Rex is shown for all assigned residues in Anc-gkdup at 25°C and 10°C. Error bars represent standard error and asterisks represent p < 0.001 (t-test). A residue by residue comparison of Rex at both temperatures is shown in Supporting Information. B. Rex in Anc-gkdup at 10°C monitors a global process. Anc-gkdup residues with significant Rex at 10°C (spheres) are shown on the structure of the yeast GK enzyme (PDB 1EX6). For comparison, Anc-gkdup chemical shift perturbations induced by GMP are also shown. Residues shifting 0.15–0.59 ppm are colored light orange, while those with a greater shift magnitude are dark orange. C. The evolution of PID function was accompanied by slowed GK dynamics. The mean exchange rate constants (kex) for Anc-gkdup, Anc-gkdup s36P, and Anc-GK1PID (all from measurements at 25°C) are shown. Error bars represent standard error and asterisks represent p < 0.001 (t-test). D, E. Anc-gkdup s36P (D) and Anc-GK1PID (E) residues used to determine kex (i.e. those with significant Rex).
Next, we determined if the rapid interconversion exhibited by Anc-gkdup persisted along the evolutionary trajectory from enzyme to PID by measuring relaxation dispersions in both Anc-gkdup s36P and Anc-GK1PID at 15 and 25 °C, and at two magnetic field strengths. Although Anc-gkdup s36P is identical to Anc-gkdup except at a single position, its dynamics are dramatically different – unlike Anc-gkdup, R2 dispersions for Anc-gkdup s36P are readily observable at 25°C (Figures 3A, B, Supporting Information). Indeed, while the exchange rate constant (kex) for interconversion in Anc-gkdup can only be approximately determined at 25°C because the process is so rapid (Figure 3C) (2505 s−1, Standard Error range: 1608–2877 SE), kex for Anc-gkdup s36P can be precisely determined and is much slower (930 s−1, 881–962 SE). Remarkably, open-close interconversion dynamics are further quenched in Anc-GK1PID (kex = 571 s−1, 436–679 SE) (Figures 3C–3E, Supporting Information) indicating that there is a strong correlation between activity (from enzyme to PID) and loss of GK flexibility. The effect on dynamics was not correlated with differences in overall stability of the proteins, however, which were not significant (Supporting Information).
The mutations that change GK function from enzyme to PID also change how it responds to GMP-binding. Guanylate-binding shifts Anc-gkdup HSQC signals from residues both close to and distant from the GMP binding site, indicating that binding is coupled to GK closing. In contrast, the nucleotide only shifts signals from residues that directly contact GMP in Anc-GK1PID (Figure 1D, Supporting Information). This suggests that while GMP binding is coupled to GK closing in Anc-gkdup, the s36P mutation uncouples nucleotide binding from the conformational change, an effect also observed in an extant GK enzyme16. The uncoupling of substrate binding and GK closing is likely responsible for the loss of nucleotide kinase activity that occurs along the evolutionary trajectory from Anc-gkdup to Anc-GK1PID, and why catalytic function is mutually exclusive with PID activity.
E. Valine 35 backbone conformation is influenced by open-close interconversion and conformations corresponding to closed GK are restricted by the s36P mutation. In the GK enzyme (s35; upper panels), the backbone conformations sampled by the V35 backbone depend on the GK hinge angle. Note that only the upper left quadrant of the plot in (C) is shown. Shaded area represents disallowed angles (regular valine in the top row and pre-proline valine in the bottom row). The dashed line shows the pre-proline disallowed angle boundary for comparison. Backbone conformations corresponding to partially and fully open GKs overlap with steric clash introduced by s36P mutation (shaded area).
PID mutations restrict GK backbone rotations that are coupled to global motions
Our findings demonstrate that mutations that occurred along the evolutionary trajectory from Anc-gkdup to Anc-GK1PID, in particular s36P, not only changed GK function, but also altered its flexibility. We sought to understand how this mutation affects GK flexibility. The s36P mutation is located in the GK’s “hinge”, a short segment that mediates interconversion between open and closed conformations (Figure 1B)14. Closing involves movement of the GBD towards the “CORE” and ATP-binding “LID” subdomains with rotations in the peptide backbones of hinge residues (Figure 4A). Proline backbone dihedral angles are severely limited compared to serine24, which could affect the coupling between local hinge residue backbone movements and the global closing conformational transition. To examine the hinge angles adopted by the GK during interconversion, we performed 1.5 μs molecular dynamic simulations of apo WT (s36) and s36P GKs (PDB IDs 1EX6 and 4F4J, respectively). During these long simulations, both proteins sampled the continuum of conformations between fully open and closed (85°–110° hinge angle; Figure 4B, C), although the s36P protein tended to remain in more open conformations (Figure 4C, bottom panel). The s36 protein underwent numerous transitions between open and closed during the simulation, as did s36P, although less frequently (5340 versus 424 transitions, respectively; Figure 4B).
Figure 4.

The s36P mutation preferentially destabilizes V35 peptide backbone conformations associated with the closed GK. A. Interconversion between closed and open GK. The three GK subdomains are shown: LID, CORE, and guanosine binding domain (GBD). The GK hinge angle is shown connecting crosses specifying the center-of-mass of the GBD, hinge residues, and CORE subdomains. Structures are from PDB 1EX7 (closed) and 1EX6 (chain A; open). An alignment of the hinge regions from the ancestral GKs are shown with those from the yeast GK enzyme (GUK1) and the extant GKPID from Discs large (Dlg GKPID). The s36P mutation is boxed. B. GK hinge angle as a function of molecular dynamics simulation time for GK s36 and s36P. Simulations were performed with structures from PDBs 1EX6 chain A (s36) and 4F4J (s36P). The s36 protein sampled hinge angles ranging from 78° – 123° during the simulation while s36P sampled angles from 83° – 126°. Closed (80° – 93°), partially open (93° – 108°), and open (108° – 120°) conformations are highlighted. C. Probability distribution functions for s36 and s36P GK hinge angles during the 1.5 μs molecular dynamics simulations shown in panel (B). D. Comparison of the backbone dihedral angles sampled by residue 36 (serine, left panel; proline, right panel) during 1.5 μs molecular dynamics simulations. The s36P mutation does not affect the conformations sampled by residue 36. Shaded areas represent disallowed angles for serine in the left panel and for proline in the right panel. Inset: protein structure near the s36P mutation. Interactions between the proline (s36P) δ carbon, valine 35’s (V35) α hydrogen, and serine 34’s (S34) carbonyl oxygen are shown. Structures are from PDB 1EX6 (chain A) and 4F4J (chain A).
To determine how the s36P mutation influences the configuration of the peptide backbone during open-closed interconversion, we compared the distribution of backbone angles for both proteins over the simulation. Surprisingly, the backbone dihedral angles sampled by residue 36 are only minimally affected by the s36P mutation – the backbone remains in configurations that are allowed for serine and the much more restrictive proline, regardless of the GK hinge angle (Figure 4D). However, the backbone configuration of the residue preceding the mutation site, valine 35 (V35), is strongly affected by the mutation (Figure 4E). When the GK is in open conformations, the V35 backbone is compatible with both serine and proline at position 36 (Figure 4E, right panels). However, as the GK assumes partially open and fully closed conformations (Figure 4E, middle and left panels, respectively), the backbone moves into configurations that become unfavorable for residues preceding proline (Figure 4E, shaded area) because of steric clashes with proline 36 sidechain’s δ carbon (Figure 4D, inset)25,26. Because the unfavorable interactions only occur in closed and partially open GKs, these conformations are destabilized relative to open ones. Thus, rotations about the V35 backbone that are coupled to the global closing motion become restricted by the s36P mutation, significantly reducing GK flexibility.
Taken together, the NMR and simulation results indicate that the function switching s36P mutation dramatically alters GK flexibility, and correspondingly the ensemble of conformations accessed by the protein. The mutation destabilizes the closed conformation, but also reshapes the set of open conformations occupied by the protein. The complex change in the landscape of Anc-gkdup conformations explains the confounding effects of the mutation on Anc-gkdup’s HSQC spectrum compared to the effects of GMP binding (Figures 2A, Supporting Information). Rather than purely shifting populations between open and closed states, the mutation causes a redistribution among the open conformations sampled by the GK. The redistribution of open conformations shifts HSQC signals from residues that are sensitive to GK conformation much differently compared to when GMP binds. We conclude that the reduction in GK flexibility that occurred during the transition from enzyme to PID resulted from steric restrictions in backbone residues whose movements are coupled to the global open-close interconversion. This change in flexibility correspondingly redefined the “open state” of the GK by dramatically changing which open conformations are accessed in the absence of nucleotide substrate or protein ligand.
PID mutations prevent the adoption of non-functional GK conformations
The reduction in flexibility that occurred along the evolutionary trajectory to PID function suggests that flexibility may have been detrimental to protein binding activity, but why? Flexibility, combined with the random effects of thermal energy, can lead to the adoption of non-functional conformations5, so we sought to determine which GK conformations are capable of binding proteins. When the GK enzyme closes, it forms a tight fitting pocket around guanylate14,15, but because the Pins protein ligand is much larger than the nucleotide it is unlikely to fit into this pocket10. Thus, we suspected that once the GK closes beyond a certain threshold, it can no longer function as a PID. To determine the degree to which the GK can close with Pins still bound, we performed molecular dynamics simulations with Pins docked into the GK enzyme’s latent site and followed the distribution of hinge angles that it was able to access (Figure 5A). As we expected, Pins binding prevents the GK from accessing closed and partially open conformations that would sterically clash with the large protein ligand. The incompatibility of these conformations with protein binding activity indicates that the flexibility of the GK allows it to access a significant number of conformational substates that are unable to bind protein. Thus, GK enzymes may be too flexible to function as PIDs, because flexibility allows for the adoption of conformations whose binding groove is not large enough to engage the protein ligand.
Figure 5.

The role of conformational flexibility in GK protein interaction domain evolution. A. Probability distribution function of s36 GK hinge angles when Pins is docked into its latent protein binding site during a 1.5 μs molecular dynamics simulation. The protein sampled hinge angles ranging from 90° – 119°. The distribution functions for s36 (without Pins) and s36P from Figure 4B are shown for comparison. B. Probability distribution function of the hinge angles from the extant GKPID from PSD-9527 during a 1.5 μs molecular dynamics simulation. The distribution function for the s36 GK enzyme with Pins docked (from panel A) is shown for comparison. C. Conformational flexibility model for the birth of the GK protein interaction domain family. The apo enzyme is able to access both open and closed conformations because of flexibility required for catalysis. The enzyme harbors a protein-binding site, but protein can only bind to a subset of conformations–thereby introducing an entropic penalty to binding. D. Diverging flexibility in GK enzyme and protein interaction domains. The ancestral enzyme (Anc-gkdup) is flexible, which allowed it to function catalytically, but prevented Pins binding. Extant enzymes retain these characteristics. A single mutation (s36P) significantly reduced GK flexiblity, abolishing catalytic activity but allowing the Pins binding site to become active by reducing the entropic barrier to binding. Binding was enhanced by mutations that further reduced the flexibility of AncGK1PID.
If GK enzymes were too flexible to support PID activity, then one path to PID function would have been via mutations that reduced flexibility. However, these mutations would have to limit flexibility in a very specific way–preventing the adoption of conformations that lack protein binding activity, while still allowing those that can bind protein. The reduction in flexibility caused by the s36P mutation limits the adoption of GK conformations that aren’t accessed in the GK-Pins complex (Figure 5A). However, the distribution of GK conformations accessed in the s36P variant still contains many conformations that are not able to bind protein. We tested how the distribution of hinge angles evolved by analyzing an extant GKPID27. Remarkably, the hinge angle distribution accessed by this protein nearly exactly overlays the distribution of the docked GK enzyme-Pins complex (Figure 5B) suggesting that a key factor in the evolution of protein binding activity of the GKPID family was reducing flexibility to prevent the adoption of conformations that aren’t able to bind protein, while still allowing those that are able to engage the protein ligand.
Discussion
The GKPID family resulted from a large functional leap–from nucleotide kinase to phosphoprotein recognition module–that occurred around the time that animals appeared on Earth6,8. A key difference between the ancestral and derived functions is the type of molecular recognition events required for each, GK-nucleotide versus GK-protein10. In particular, the GK-protein interaction requires much more recognition surface than its nucleotide counterpart. A simple model for the functional transition is that the new activity evolved by the construction of a new active site that would encompass the larger protein interaction surface. However, in our previous work we were surprised to find that the GK enzyme ancestor has an intact, but non-functional protein binding site that overlaps with the GMP binding pocket6,10,16. In the present work, we have identified a key role for protein flexibility in activating the enzyme’s latent site to give rise to the GKPID family.
GK enzyme and PID functions have very different conformational flexibility requirements. The ancestral enzymatic function requires flexibility for the catalytic mechanism–the open conformation mediates substrate binding and the closed conformation enables phosphotransfer14. However, the flexibility that is required for nucleotide binding to be coupled to GK closing, also allows thermal energy to push the nucleotide-free enzyme into the closed conformation5. This gives rise to the dynamics that we observe for Anc-gkdup (Figure 3). During these conformational transitions, the GK crosses a threshold where it can no longer fit the protein ligand into its binding groove. Thus, the flexibility that is necessary for catalysis allows the GK to access conformational substates that cannot bind protein (Figure 5). Mutations that promote protein interaction domain function reduce GK flexibility, preventing the corresponding thermally-induced motions that the enzyme undergoes and the GMP coupled conformational transition that is required for catalytic activity. We have found that one large effect mutation, s36P, reduces GK flexibility by restricting backbone movements in the protein’s hinge that are coupled to the global conformational change (Figure 4).
It is important to note that evolution of PID activity required reducing GK flexibility in a very specific manner. GK enzyme flexibility allows the protein access conformations that are both active and inactive for PID activity. Conformations that are inactive because their binding groove is too narrow to bind protein must be selectively destabilized relative to those that are capable of binding proteins (Figure 5). In other words, reducing flexibility alone would not be sufficient to promote PID function, as doing so in a way that promoted non-functional conformations at the expense of functional ones would lead to a more rigid, but inactive protein. Thus, mutations that promote PID activity must selectively destabilize the closed conformations that are unable to bind protein without significantly affecting open conformations. The s36P does this in an imperfect way (Figure 5A), but additional mutations along the trajectory to the extant GKPID lead to a nearly perfect match of conformations that are accessible in the free and bound GK (Figure 5B).
Tuning flexibility to alter function may have advantages over other avenues of neofunctionalization. Because flexibility can be altered by a small number of mutations, evolutionary pathways that manipulate this property may be more probable than those that have more complex requirements, such as the construction of a new active site28. The potential advantages of manipulating flexibility and the resulting dynamics has been recognized by Tawfik and co-workers in the “functional promiscuity” model of neofunctionalizaiton. In this model, the flexibility inherently required for functions such as catalysis allows proteins to access alternate states that may have distinct activities, which can be further selected for following an event such as gene duplication. Our analysis of the GK system extends this model in a number of ways. First, the conformational substate that is promiscuous may be required for the ancestral function. In other words, the GK open conformation promiscuously binds protein but this conformation is also required for nucleotide kinase activity. Second, our results highlight the negative effects of sampling too many conformations by demonstrating that the “promiscuous” activity may not manifest itself until conformations supporting the original function are selectively eliminated.
Energetically, the barrier the GKPID family faced at its inception was an entropic one. The penalty for assuming non-functional conformations in the unbound state is entropic in nature because the protein-bound GK can only bind a subset of the conformations accessed by the enzyme (Figure 5). The mutations that reduced the flexibility of the GK also reduced the entropic penalty to PID activity. Conformational entropy has been demonstrated to be a key energetic component opposing binding29,30, and this appears to be the key challenge that the GKPID faced at its birth.
Protein interaction domains are ubiquitous in cellular signaling proteins31,32. The GKPID represents a unique model system where the ancestral function of the domain family is clearly known. We have now identified the key events that occurred at the birth of the GKPID family, however key questions remain, including the mechanism by which specificity for phosphorylated target proteins evolved. We expect that analysis of the GKPID evolutionary trajectory will continue to yield key insights into the mechanisms by which protein interaction networks evolve.
Supplementary Material
Acknowledgments
The authors thank Mike Harms, Brad Nolen, and Joe Thornton for helpful discussions. Molecular dynamics simulations were performed using the ACISS compute cluster at the University of Oregon.
Funding Sources
This work was supported by NIH Grants GM087457 (K.E.P) and AI058072 (B.F.V.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supplementary figures and experimental procedures (PDF)
References
- 1.Alberts B, Miake-Lye R. Cell. 1992;68:415. doi: 10.1016/0092-8674(92)90179-g. [DOI] [PubMed] [Google Scholar]
- 2.Steitz TA. Nat Rev Mol Cell Biol. 2008;9(3):242. doi: 10.1038/nrm2352. [DOI] [PubMed] [Google Scholar]
- 3.Sazanov LA. Nat Rev Mol Cell Biol. 2015;16:375. doi: 10.1038/nrm3997. [DOI] [PubMed] [Google Scholar]
- 4.Motlagh HN, Wrabl JO, Li J, Hilser VJ. Nature. 2014;508:331. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bar-Even A, Milo R, Noor E, Tawfik DS. Biochemistry. 2015;54:4969. doi: 10.1021/acs.biochem.5b00621. [DOI] [PubMed] [Google Scholar]
- 6.Anderson DP, Whitney DS, Hanson-Smith V, Wozni-ca A, Campodonico-Burnett W, Volkman BF, King N, Thornton JW, Prehoda KE. eLife. 2016;5:e10147. doi: 10.7554/eLife.10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Funke L, Dakoji S, Bredt DS. Annu Rev Biochem. 2005;74:219. doi: 10.1146/annurev.biochem.74.082803.133339. [DOI] [PubMed] [Google Scholar]
- 8.de Mendoza A, Suga H, Ruiz-Trillo I. BMC Evol Biol. 2010;10:93. doi: 10.1186/1471-2148-10-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnston CA, Hirono K, Prehoda KE, Doe CQ. Cell. 2009;138:1150. doi: 10.1016/j.cell.2009.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnston CA, Doe CQ, Prehoda KE. PloS One. 2012;7:e36014. doi: 10.1371/journal.pone.0036014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu J, Shang Y, Xia C, Wang W, Wen W, Zhang M. EMBO J. 2011;30:4986. doi: 10.1038/emboj.2011.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harms MJ, Thornton JW. Nat Rev Genet. 2013;14:559. doi: 10.1038/nrg3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harms MJ, Thornton JW. Curr Opin Struct Biol. 2010;20:360. doi: 10.1016/j.sbi.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blaszczyk J, Li Y, Yan H, Ji X. J Mol Biol. 2001;307:247. doi: 10.1006/jmbi.2000.4427. [DOI] [PubMed] [Google Scholar]
- 15.Sekulic N, Shuvalova L, Spangenberg O, Konrad M, Lavie A. J Biol Chem. 2002;277:30236. doi: 10.1074/jbc.M204668200. [DOI] [PubMed] [Google Scholar]
- 16.Johnston CA, Whitney DS, Volkman BF, Doe CQ, Prehoda KE. Proc Natl Acad Sci U S A. 2011;108:E973. doi: 10.1073/pnas.1104365108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Zhang Y, Yan H. J Biol Chem. 1996;271:28038. doi: 10.1074/jbc.271.45.28038. [DOI] [PubMed] [Google Scholar]
- 18.Adén J, Wolf-Watz M. J Am Chem Soc. 2007;129:14003. doi: 10.1021/ja075055g. [DOI] [PubMed] [Google Scholar]
- 19.Hanson JA, Duderstadt K, Watkins LP, Bhattacharyya S, Brokaw J, Chu J-W, Yang H. Proc Natl Acad Sci U S A. 2007;104:18055. doi: 10.1073/pnas.0708600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henzler-Wildman KA, Thai V, Lei M, Ott M, Wolf-Watz M, Fenn T, Pozharski E, Wilson MA, Petsko GA, Karplus M, Hübner CG, Kern D. Nature. 2007;450:838. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- 21.Kempf JG, Loria JP. Cell Biochem Biophys. 2003;37:187. doi: 10.1385/cbb:37:3:187. [DOI] [PubMed] [Google Scholar]
- 22.Loria JP, Berlow RB, Watt ED. Acc Chem Res. 2008;41:214. doi: 10.1021/ar700132n. [DOI] [PubMed] [Google Scholar]
- 23.Kleckner IR, Foster MP. Biochim Biophys Acta. 2011;1814:942. doi: 10.1016/j.bbapap.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho BK, Thomas A, Brasseur R. Protein Sci Publ Protein Soc. 2003;12:2508. doi: 10.1110/ps.03235203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicholson H, Tronrud DE, Becktel WJ, Matthews BW. Biopolymers. 1992;32:1431. doi: 10.1002/bip.360321103. [DOI] [PubMed] [Google Scholar]
- 26.Ho BK, Brasseur R. BMC Struct Biol. 2005;5:14. doi: 10.1186/1472-6807-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGee AW, Dakoji SR, Olsen O, Bredt DS, Lim WA, Prehoda KE. Mol Cell. 2001;8:1291. doi: 10.1016/s1097-2765(01)00411-7. [DOI] [PubMed] [Google Scholar]
- 28.Tokuriki N, Tawfik DS. Science. 2009;324:203. doi: 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- 29.Thorpe IF, Brooks CL. Proc Natl Acad Sci U S A. 2007;104:8821. doi: 10.1073/pnas.0610064104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tzeng S-R, Kalodimos CG. Nature. 2012;488:236. doi: 10.1038/nature11271. [DOI] [PubMed] [Google Scholar]
- 31.Pawson T. Curr Opin Cell Biol. 2007;19:112. doi: 10.1016/j.ceb.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Pawson T, Nash P. Science. 2003;300:445. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.