

Abstract
The synthesis of unsymmetrical cyclobutanes by controlled heterodimerization of olefins remains a substantial challenge, particularly in an enantiocontrolled fashion. Here we show that chiral Lewis acid catalyzed triplet sensitization enables the synthesis of highly enantioenriched diarylcyclobutanes by photocycloaddition of structurally varied 2′-hydroxychalcones with a range of styrene coupling partners. We demonstrate the utility of this reaction through a direct synthesis of a representative norlignan cyclobutane natural product.
Keywords: asymmetric catalysis, cycloaddition, cyclobutane, photocatalysis, triplet sensitization
Cross Products
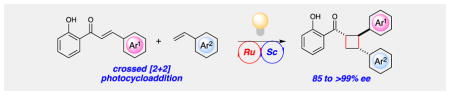
Lewis acid catalyzed triplet sensitization enables the highly enantioselective crossed cycloaddition of chalcones and styrenes to forge the cyclobutane cores of multiple bioactive natural products.
Asymmetric catalytic [2+2] cycloadditions1 have long been underdeveloped compared to analogous [4+2], [3+2], and [2+1] cycloadditions.2 Consequently, the enantiocontrolled synthesis of four-membered carbocycles remains more challenging than the preparation of structurally complex cyclohexanes, cyclopentanes, and cyclopropanes. One reason for this discrepancy may be the unique challenge of controlling crossed vs homodimerization in [2+2] cycloadditions, which introduces an additional complication beyond the issues of regio- and stereoselectivity common to all cycloaddition methods. Among the most successful enantioselective catalytic methods for crossed [2+2] cycloaddition of alkenes are Lewis acid catalyzed3 and organocatalytic processes.4 The polar nature of these thermal reactions, however, limits their scope to highly electronically biased substrates and thus restricts their general applicability in complex target-oriented synthesis. A handful of enantioselective photochemical methods for intermolecular [2+2] cycloaddition have also been recently developed.5,6,7 Most of the catalytic methods for asymmetric [2+2] photocycloaddition, however, require relatively complex cyclic substrates for efficient intermolecular cycloaddition and are generally limited to electron-deficient coupling partners.8
Thus, many cyclobutane-containing natural products (Figure 1) and similarly complex bioactive cyclobutane compounds are challenging to prepare using existing cycloaddition methods. Recent syntheses have circumvented the problem of crossed [2+2] cycloaddition by utilizing complementary strategies. Baran pioneered one approach involving the directed C–H arylation of a simplified cyclobutane scaffold, a strategy that has been utilized in three elegant but racemic total syntheses.9,10 Tang and Fox, meanwhile, have offered enantioselective routes to cyclobutane alkaloids involving ring-expansions of cyclopropane intermediates.11 Notably, the connectivity and stereochemistry of cyclobutane natural products can be difficult to determine spectroscopically, and the proposed structures of several cyclobutane natural products have required reassignment through total synthesis.9ab,11a Thus, controlled chemical synthesis remains an important tool for the study of this class of natural products.
Figure 1.
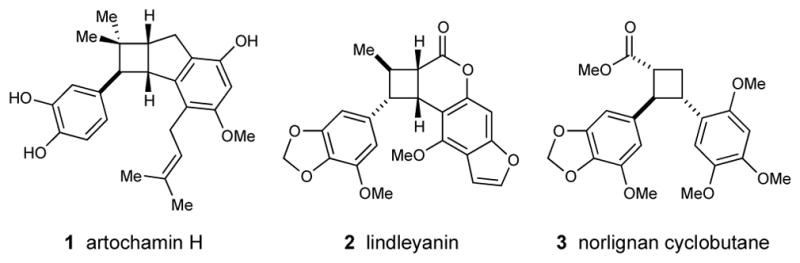
Representative cyclobutane natural products.
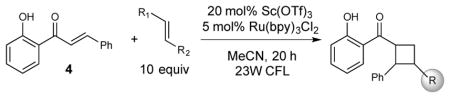
Recently, we reported a conceptually novel method for chiral Lewis acid catalyzed crossed [2+2] photocycloaddition that results in the highly enantioselective synthesis of a range of vinylcyclobutanes (Scheme 1).12 If this reaction could be generalized, it might offer a powerful method for the synthesis of diverse cyclobutane-containing structures. The enabling mechanistic feature of this reaction is a dramatic lowering of the singlet–triplet gap of 2′-hydroxychalcone 4 (ET = 54 kcal/mol) upon Lewis acid coordination. This effect renders triplet energy transfer from a Ru(bpy)32+ photosensitizer (ET = 45 kcal/mol) thermodynamically feasible only to the Lewis-acid-bound substrate (5, ET = 32 kcal/mol) and provides an opportunity for highly enantioselective asymmetric catalysis when a chiral Lewis acid is used. Triplet [2+2] cycloadditions generally proceed in a stepwise fashion; we surmised from the regioselectivity of this reaction that the benzylic–allylic 1,4-diradical 6 is a key intermediate. We hypothesized, therefore, that styrenes and other alkene coupling partners that would produce similarly stabilized diradicals might also participate in this cycloaddition. To test this hypothesis, we examined the reaction of hydroxychalcone 4 with a range of alkenes upon irradiation with a compact fluorescent light bulb (CFL) in the presence of Sc(OTf)3 and Ru(bpy)32+ (Table 1). Simple aliphatic alkenes (7 and 8) that would give relatively unstabilized 1,4-diradical intermediates failed to produce cyclobutane cycloadducts. Electron-rich vinyl ethers (9) are rapidly polymerized in the presence of the Lewis acid co-catalyst. However, less strongly activated electron-rich alkenes such as vinyl sulfides (10) and styrenes (11) underwent smooth cycloadditions to afford the corresponding cyclobutanes.
Scheme 1.
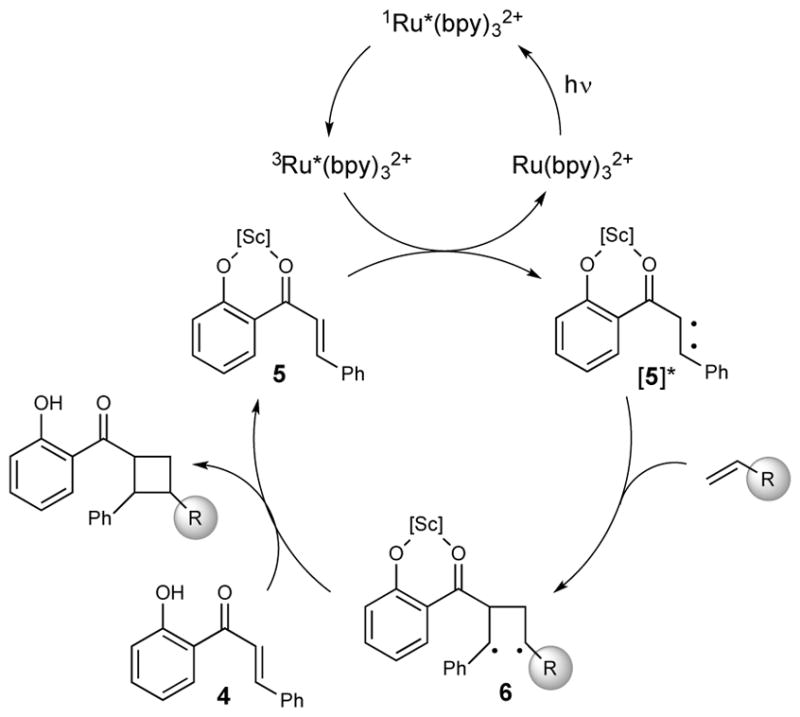
Proposed mechanism of cycloadditions featuring Lewis acid catalysed triplet sensitization.
Table 1.
Screen of alkene coupling partners for [2+2] photocycloaddition.
| |||||
|---|---|---|---|---|---|
|
7 |
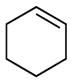 8 |
9 |
10 |
11 |
|
| Yields:a | 0% | 0% | 0% | 54% | 70% |
Yields determined by 1H NMR analysis using phenanthrene as a calibrated internal standard.
Moreover, the [2+2] photocycloaddition with styrene was easily rendered enantioselective under conditions similar to those utilized for chalcone–diene cycloadditions. Optimal conditions utilized 10 mol% of Sc(OTf)3, 15 mol% of t-BuPybox, and 2.5 mol% of Ru(bpy)32+ as a triplet photosensitizer. Upon irradiation of 7 and styrene with a 23W CFL, cycloadduct 12 was isolated in excellent yield, and the major diastereomer was formed with very high enantioselectivity (Table 2).13 Styrenes bearing substituents at all positions of the aromatic ring react smoothly and with high ee (13–23). A variety of electron-withdrawing moieties on the styrene ring are readily tolerated. These included an ester group (16) whose Lewis basicity did not interfere with the action of the chiral Sc Lewis acid, and potentially UV-sensitive halide substituents (13, 14, 20, 22) that survived irradiation without homolytic degradation. A boronate ester moiety was also readily tolerated (17), providing a versatile handle for subsequent derivatization of the cycloadduct. A variety of electron-donating substituents could also be incorporated on all positions of the styrene ring (19, 23, 24). Substituents on the styryl double bond were also tolerated (25 and 26). Finally, we also observed excellent ee in a reaction with phenyl vinyl sulfide (27), suggesting that a modified version of this enantioselective method may be extended to other electron-rich alkene partners.
Table 2.
Reaction scope with respect to styrenes.[a]

|
Isolated yields are the averaged results of two reproducible experiments. Diastereomer ratios were determined by 1H NMR analysis of the unpurified reaction mixtures. Enantiomeric excesses for the major diastereomer were determined by chiral SFC or HPLC. See Supporting Information for details. (S,S)-t-BuPyBox = 2,6-bis[4′-(S)-(tert-butyl)oxazolin-2′-yl]pyridine.
The scope of this reaction with respect to the hydroxychalcone is summarized in Table 3. Chalcones bearing electronically varied β-aryl groups are readily tolerated and react with high ee (28–36). As we observed in our study of cycloadditions with dienes, electron-rich 2′-hydroxychalcones can exhibit modest levels of background cycloaddition with near-UV irradiation, a feature that can lead to diminished ee’s using a broad-spectrum CFL light source. Thus, while β-p-methoxyphenyl cycloadduct 34 was formed with relatively modest ee using the standard protocol (70% ee), the selectivity could be improved to 85% ee by irradiating with a monochromatic blue LED. The 2-acylphenol moiety of the substrate was also readily tolerated substitution, including electron-donating and –withdrawing groups as well as multiple substitutions (37–42). However, the chelating phenolyl moiety was required for optimal results. Unsubstituted chalcone itself provided low yields and low ee (43), suggestive of a diminished propensity to bind to the Lewis acid and poorer organization of this monodentate substrate around the Sc(III) center. Finally, replacement of the β-aryl moiety with an aliphatic group affords an unreactive substrate (44), consistent with the hypothesis that the less extensive conjugation of this substrate shifts the triplet energy of its Lewis acid complex above a range accessible using Ru(bpy)32+ as a triplet sensitizer.
Table 3.
Reaction scope with respect to chalcones.a

|
Isolated yields are the averaged results of two reproducible experiments. Diastereomer ratios were determined by 1H NMR analysis of the unpurified reaction mixtures. Enantiomeric excesses for the major diastereomer were determined by chiral SFC. See Supporting Information for details.
Reaction was irradiated with monochromatic high-intensity blue LED.
Reaction was irradiated for 40 h.
Yields determined by 1H NMR analysis using phenanthrene as a calibrated internal standard.
This method should provide a direct approach to the synthesis of diarylcyclobutane natural products. As an initial demonstration of utility, we next developed an expeditious synthesis of norlignan 3 (Scheme 2),14 a representative member of a small class of cyclobutane compounds obtained from medicinal plants in the Piperacaeae family.15 The fully elaborated styrene appropriate for synthesis of 3 unfortunately suffered from competitive polymerization under the strongly Lewis acidic conditions of the enantioselective photocycloaddition. The brominated analogue 46, however, undergoes cycloaddition with chalcone 45 smoothly in 94% ee. The o-hydroxyaryl ketone moiety of cycloadduct 47 proved resistant to Baeyer–Villiger oxidation. In order to install the required cyclobutyl carboxylate, we adapted a one-pot method recently reported by Chen and coworkers16 for the conversion of 2′-hydroxylaryl ketimines into benzoxazoles. Condensation of 47 with ammonia followed by exposure of the resulting imine to NaOCl triggered Beckmann rearrangement to afford 48. At this point, the diastereomers could easily be separated by chromatography. Hydrolysis of the benzoxazole ring and methylation of the cyclobutane carboxylate are readily accomplished under standard basic conditions, affording the corresponding methyl ester (49) without loss of stereochemical integrity. Elaboration to the reported structure of the natural product was accomplished by a Pd-catalyzed Buchwald etherification,17 and the spectral data of the resulting compound 3 matched all reported values except for the optical rotation, which differed in sign and suggests that the absolute stereochemistry is the opposite of that arbitrarily depicted in the original isolation report.14
Scheme 2.
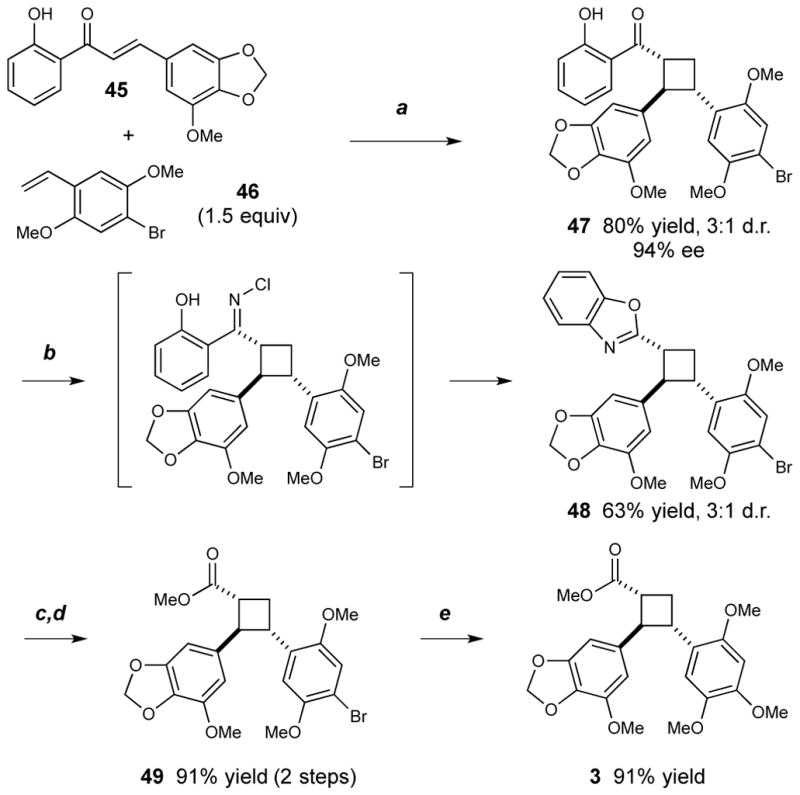
Synthesis of norlignan cyclobutane 3: a. 2.5 mol% Ru(bpy)3(PF6)2, 32 mol% Sc(OTf)3, 38 mol% (S,S)-tBuPybox, 12 h, 23 W CFL. b. NH3, MeOH/THF, 12 h; then NaOCl, THF, 30 min. c. NaOH, H2O/EtOH, 95 °C, 36 h. d. MeI, K2CO3, DMF, 1 h. e. 10 mol% RockPhos G3 precatalyst, 2.0 equiv Cs2CO3, 5.0 equiv MeOH, PhMe, 90 °C, 24 h.
Finally, diarylcyclobutanes are key structural features of several families of bioactive natural products, and the ability to access this motif in enantiopure form should facilitate the exploration of the biological properties of these otherwise difficult-to-access architectures. For instance, the cycloaddition of chalcone 4 with indene affords a tricyclic core (50) common to the cyclobutane-containing artochamin natural products18 with excellent ee. Similarly, cycloaddition of 4 with 2H-chromene affords a cyclobutane (51) that represents the core ring system of lindleyanin19 in highly enantiomerically enriched form.
In summary, Lewis acid catalyzed sensitization of chalcones enables the highly enantioselective crossed [2+2] cycloaddition of styrenic olefins. This method provides a controlled method to directly synthesize 1,2-diarylcyclobutane structures that are present in the cores of a variety of natural products. These results thus demonstrate that the Lewis acid catalyzed triplet-lowering effect recently discovered in our laboratory can be applied to solve a challenging synthetic problem that we hope will prove enabling in the synthesis of structurally unique bioactive cyclobutane compounds.
Supplementary Material
Figure 2.
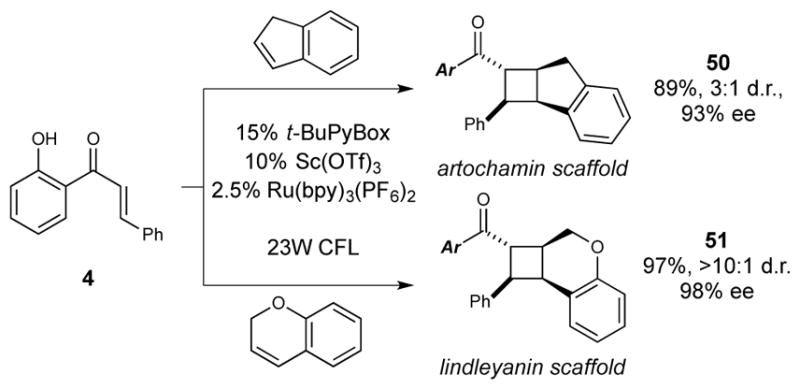
Representative cyclobutane scaffolds accessible by enantioselective [2+2] cycloaddition.
Acknowledgments
We thank Dr. Ilia A. Guzei for determining absolute configurations of the cycloadducts by X-ray crystallography and Dr. Travis R. Blum for insightful discussions. This work was funded by the NIH (GM098886). The mass spectroscopy facilities at UW–Madison are funded in part by the NIH (S10 OD020022), as are the NMR facilities (S10 OD012245).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.For recent reviews, see: Xu Y, Conner ML, Brown MK. Angew Chem Int Ed. 2015;54:11918–11928. doi: 10.1002/anie.201502815.Poplata S, Tröster A, Zou Y, Bach T. Chem Rev. 2016;116:9748–9815. doi: 10.1021/acs.chemrev.5b00723.
- 2.For recent reviews, see: Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew Chemie Int Ed. 2002;41:1668–1698. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z.Lebel H, Marcoux JF, Molinaro C, Charette AB. Chem Rev. 2003;103:977–1050. doi: 10.1021/cr010007e.Grover HK, Emmett MR, Kerr MA. Org Biomol Chem. 2015;13:655–671. doi: 10.1039/c4ob02117g.Klier L, Tur F, Poulsen PH, Jørgensen KA. Chem Soc Rev. 2017;46:1080–1102. doi: 10.1039/c6cs00713a.
- 3.(a) Hayashi Y, Narasaka K. Chem Lett. 1989:793–796. [Google Scholar]; (b) Canales E, Corey EJ. J Am Chem Soc. 2007;129:12686–12687. doi: 10.1021/ja0765262. [DOI] [PubMed] [Google Scholar]; (c) Suárez-Pantiga S, Hernández-Díaz C, Rubio E, González JM. Angew Chem, Int Ed. 2012;51:11552–11555. doi: 10.1002/anie.201206461. [DOI] [PubMed] [Google Scholar]; Conner ML, Xu Y, Brown MK. J Am Chem Soc. 2015;137:3482–3845. doi: 10.1021/jacs.5b00563. [DOI] [PubMed] [Google Scholar]; (e) Hu JL, Feng LW, Wang L, Xie Z, Tang Y, Li X. J Am Chem Soc. 2016;138:13151–13154. doi: 10.1021/jacs.6b08279. [DOI] [PubMed] [Google Scholar]
- 4.(a) Ishihara K, Nakano K. J Am Chem Soc. 2007;129:8930–8931. doi: 10.1021/ja073435w. [DOI] [PubMed] [Google Scholar]; (b) Albrecht L, Dickmeiss G, Acosta FC, Rodríguez-Escrich C, Davis RL, Jørgenson KA. J Am Chem Soc. 2012;134:2543–2546. doi: 10.1021/ja211878x. [DOI] [PubMed] [Google Scholar]; (c) Talavera G, Reyes E, Vicario JL, Carillo L. Angew Chem Int Ed. 2012;51:4104–4107. doi: 10.1002/anie.201200269. [DOI] [PubMed] [Google Scholar]; (d) Duan GJ, Ling JB, Wang WP, Luo YC, Xu PF. Chem Commun. 2013;49:4625–4627. doi: 10.1039/c3cc41785a. [DOI] [PubMed] [Google Scholar]; (e) Qi L, Yang Y, Gui Y, Zhang Y, Chen F, Tian F, Peng L. Org Lett. 2014;16:6436–6439. doi: 10.1021/ol503266q. [DOI] [PubMed] [Google Scholar]; (f) Halskov KS, Kniep F, Lauridsen VH, Iversen EH, Donslund BS, Jørgenson KA. J Am Chem Soc. 2015;137:1685–1691. doi: 10.1021/ja512573q. [1] [DOI] [PubMed] [Google Scholar]; (g) Nielsen AJ, Jenkins HA, McNulty J. Chem Eur J. 2016;22:9111–9115. doi: 10.1002/chem.201601842. [DOI] [PubMed] [Google Scholar]
- 5.(a) Du J, Skubi KL, Schultz DM, Yoon TP. Science. 2014;344:392–396. doi: 10.1126/science.1251511. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maturi MM, Bach T. Angew Chem, Int Ed. 2014;53:7661–7664. doi: 10.1002/anie.201403885. [DOI] [PubMed] [Google Scholar]; (d) Tröster A, Alonso R, Bauer A, Bach T. J Am Chem Soc. 2016;138:7808–7811. doi: 10.1021/jacs.6b03221. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Coote SC, Pöthig A, Bach T. Chem Eur J. 2015;21:6906–6912. doi: 10.1002/chem.201500173. [DOI] [PubMed] [Google Scholar]
- 6.For enantioselective intermolecular [2+2] photocycloadditions using stoichiometric chiral controllers, see: Bach T, Bergmann H, Grosch B, Harms K. J Am Chem Soc. 2002;124:7982–7990. doi: 10.1021/ja0122288.Bach T, Bergmann H. J Am Chem Soc. 2000;122:11525–11526.
- 7.For intramolecular enantioselective catalytic triplet photocycloadditions, see: Müller C, Bauer A, Bach T. Angew Chem Int Ed. 2009;48:6640–6642. doi: 10.1002/anie.200901603.Guo H, Herdtweck E, Bach T. Angew Chem, Int Ed. 2010;49:7782–7785. doi: 10.1002/anie.201003619.Albrecht D, Vogt F, Bach T. Chem Eur J. 2010;16:4284–4296. doi: 10.1002/chem.200902616.Müller C, Bauer A, Maturi MM, Cuquerella MC, Miranda MA, Bach TT. J Am Chem Soc. 2011;133:1668–16697. doi: 10.1021/ja207480q.Brimioulle R, Bach T. Science. 2013;342:840–843. doi: 10.1126/science.1244809.Alonso R, Bach T. Angew Chem Int, Ed. 2014;53:4368–4371. doi: 10.1002/anie.201310997.Vallavoju N, Selvakumar S, Jockusch S, Sibi MP, Sivaguru J. J Angew Chem Int Ed. 2014;53:5604–5608. doi: 10.1002/anie.201310940.Brimioulle R, Bauer A, Bach T. J Am Chem Soc. 2015;137:5170–5176. doi: 10.1021/jacs.5b01740.
- 8.During the preparation of this manuscript, Huang, Wiest, and Meggers published a fascinating report showing that a chiral Rh Lewis acid can catalyze the excited-state photocycloaddition of α,β-unsaturated amides with styrenes and dienes in excellent ee. See: Huang X, Quinn TR, Harms K, Webster RD, Zhang L, Wiest O, Meggers E. J Am Chem Soc. 2017 doi: 10.1021/jacs.7b04363.
- 9.(a) Gutekunst WR, Baran PS. J Am Chem Soc. 2011;133:19076–19079. doi: 10.1021/ja209205x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gutekunst WR, Baran PS. J Org Chem. 2014;79:2430–2452. doi: 10.1021/jo4027148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For a conceptually related, enantioselective cyclobutane arylation strategy via sequential C–H borylation/arylation, see: He J, Shao Q, Wu Q, Yu J-Q. J Am Chem Soc. 2017;139:3344–3347. doi: 10.1021/jacs.6b13389.
- 11.(a) Liu R, Zhang M, Wyche TP, Winston-McPherson GN, Bugni T, Tang W. Angew Chem, Int Ed. 2012;51:7503–7506. doi: 10.1002/anie.201203379. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Panish RA, Chintala SR, Fox JM. Angew Chem, Int Ed. 2016;55:4983–4987. doi: 10.1002/anie.201600766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blum TR, Miller ZD, Bates DM, Guzei IA, Yoon TP. Science. 2016;354:1391–1395. doi: 10.1126/science.aai8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The absolute configuration of the cycloadducts was determined by crystallographic analysis of a derivative of 30. See Supporting Information for experimental details.
- 14.Li Y-Z, Tong A-P, Huang J. Chem Biodivers. 2012;9:769–776. doi: 10.1002/cbdv.201100138. [DOI] [PubMed] [Google Scholar]
- 15.Gutierrez YV, Yamaguchi LF, de Moraes MM, Jeffrey CS, Kato MJ. Phytochem Rev. 2016;15:1009–1033. [Google Scholar]
- 16.Chen C, Andreani T, Li H. Org Lett. 2011;13:6300–6303. doi: 10.1021/ol202844c. [DOI] [PubMed] [Google Scholar]
- 17.Bruno NC, Buchwald SL. Org Lett. 2013;15:2876–2879. doi: 10.1021/ol401208t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y-H, Hou A-J, Chen D-F, Weiller M, Wendel A, Staples RJ. Eur J Org Chem. 2006:3457–3463. [Google Scholar]
- 19.Tan J-J, Tan C-H, Wang Y-Q, Jiang S-H, Zhu D-Y. Helv Chim Acta. 2006;89:117–121. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.