

Abstract
The relationship between mutated proteins and the cancer stem cell population is unclear. Glioblastoma tumors frequently express EGFRvIII, an EGFR variant that arises via gene rearrangement and amplification. However, expression of EGFRvIII is restricted despite the prevalence of the alteration. Here we show that EGFRvIII is highly co-expressed with CD133 and that EGFRvIII+/CD133+ defines the population of cancer stem cells with the highest degree of self-renewal and tumor initiating ability. EGFRvIII+ cells are associated with other stem/progenitor markers while markers of differentiation are found in EGFRvIII− cells. EGFRvIII expression is lost in standard cell culture but its expression is maintained in tumor sphere culture, and cultured cells also retain the EGFRvIII+/CD133+ co-expression and self-renewal and tumor initiating abilities. Elimination of the EGFRvIII+/CD133+ population using a bispecific antibody reduced tumorigenicity of implanted tumor cells better than any reagent directed against a single epitope. This work demonstrates that a mutated oncogene can have CSC specific expression and be used to specifically target this population.
Keywords: Glioblastoma, Cancer Stem Cells, EGF Receptor Variant III, EGFRvIII-CD133 bi-specific antibody
INTRODUCTION
The cancer stem cell (CSC) hypothesis has been a useful concept for explaining several biologic properties of tumors, including the observation that stem-like cells are tumorigenic while most of a tumor consists of differentiated cells with low tumor forming ability (1). Whether this principle can be exploited to enhance therapy is not clear. The CSC population may not necessarily represent the original tumor initiating cell (2) and there is plasticity in the general tumor mass such that cells can reacquire a CSC-like phenotype. The repertoire of markers available for identification of CSC also complicates therapy. Stem cell markers and other cellular proteins have been shown to enrich for CSCs but these otherwise normal proteins lack tumor specificity and no marker has precisely selected for these cells (3–5).
Mutations or rearrangements in genes that potentially drive neoplasia can be identified in many cancers. In vitro work has shown that the resulting oncogenic proteins can contribute to CSC related pathways (6). It stands to reason that the products of such altered genes could be used to identify and potentially target CSCs. In practice this has been difficult to establish because driver mutations are present in cells throughout the mass and typically are not specific to any subpopulation. Thus, mutant proteins may not have any direct role in CSCs and perhaps only generally potentiate tumor growth (7). In addition, most altered proteins are intracellular.
While not all tumors follow a CSC model, glioblastoma (GBM) has been strongly associated with the presence of CSCs (3, 8). Amplification of the EGFR gene is common in this tumor, and 20–40% of GBMs express EGFRvIII, an altered form of the EGFR gene which arises via gene rearrangement and amplification (9). Some studies have seen EGFRvIII expression as high as 70% in GBM (10). In addition to GBM, EGFRvIII has been found in a high percentage of breast (11, 12), lung (13), head and neck, ovarian, and prostate cancers. Importantly, it is rarely found in normal tissue (11) and this almost exclusive expression in tumors makes it an intriguing target for therapy (14). The presence of EGFRvIII correlates with a worse prognosis for both glioblastoma and anaplastic astrocytoma patients (15, 16). EGFRvIII expression is strongly associated with the “classical” molecular subtype of glioblastoma where it is found in conjunction with PTEN mutations but is mutually exclusive with P53 or IDH1 mutations (17). Other laboratories and ours have shown that a peptide vaccine targeting the EGFRvIII antigen can effectively reduce tumor progression in preclinical models (18). Human clinical trials have demonstrated improved overall survival and an EGFRvIII specific immune response in patients treated with the vaccine in several Phase II trials (14, 19).
Despite this improvement in patient survival, a paradoxical observation is that the typical expression pattern for EGFRvIII in positive tumors is either sporadic cells or focal areas of positive cells, unlike wildtype (wt) EGFR which is usually broadly seen across the same tumor (20, 21) despite prevalence of the gene rearrangement/amplification (22). Interestingly, gene amplification in GBM is a clonal event (23) where only one gene rearrangement is seen in EGFRvIII+ tumors (9, 24). These observations point to EGFRvIII being an early development in tumorigenesis. Thus, the restricted expression of EGFRvIII may reflect its association with the CSC population. CSCs show enhanced resistance to radiation therapy and increased DNA repair mechanisms (25) and interestingly, EGFRvIII+ cells are also highly resistant to ionizing radiation due to increased DNA repair mechanisms (26). On the other hand, EGFRvIII expression may only promote growth or have a less specific paracrine function via expression of cytokines (7). Because EGFRvIII is the result of an early genetic alteration and is a transmembrane receptor, it provides a unique opportunity to test if mutated oncogenes can indeed play a role in CSCs.
Materials and Methods
Dissociation of primary human brain tumors and culture
Freshly resected human glioblastoma tumor samples were obtained from the Stanford University tissue and brain bank under IRB approved protocols. Dissociated tissue samples were cultured on non-adherent plates using defined media containing EGF, bFGF, and heparin. For neurospheres from non-neoplastic tissue, recombinant human LIF was also added. For experiments in which tumor spheres were induced to differentiate, cells were cultured in the same media without EGF and FGF plus the addition of either 5% Fetal Bovine Serum and 5% Horse Serum, or by a cocktail of CNTF, BDNF and retinoic acid.
Flow cytometry
Freshly dissociated cells were co-stained with a monoclonal anti-EGFRvIII antibody (G100) (13) or rabbit anti-EGFRvIII and CD133/1-APC and CD133/2-APC. Cells from the primary tumor itself were used for compensation using an anti-MHC I biotin antibody. Appropriate isotype controls were used to control for non-specific isotype background. Sorted cells were collected in tumor stem media and used for orthotopic intracranial transplantation or in vitro assays.
Limiting dilution and tumor sphere formation analysis
Limiting dilution analysis (LDA) was done as described previously. An extreme LDA algorithm was used to determine the frequency of renewing cells (27). To estimate the ability to form tumor spheres after ADCC, NK cells were separated from GBM cells using the MACS lineage depletion kit (Miltenyi Biotech). For the secondary sphere assay, spheres generated during the LDA were re-dissociated and plated 10 cells per well on a 96 well plate and the number of spheres counted after 8 weeks.
Intracranial transplantation and tumor formation
6–8 week NOD-SCID mice were intracranially injected and monitored for 22–26 weeks or until neurological defects were observed. For experiments to test for an anti-tumor effect of the BsAb, human GBM tumor cells were mixed with either BsAb, human IgG1-Fc, di-EGFRvIII or di-CD133 at a final concentration of 0.01ug/100,000 cells prior to injection without pre-incubation.
Immunohistochemistry (IHC) and immunofluorescence (IF)
IHC was performed according to standard protocols. For the anti-CD31 and anti-EGFRvIII double labeling, the MACH 2 double stain kit was employed using the Vulcan Fast Red chromagen to detect the anti-CD31 antibody. For staining of cultured tumor spheres, spheres were fixed with paraformaldehyde, permeabilized with Triton X-100, and subjected to a standard PBS-based immunofluorescence protocol. For immunofluorescence samples were visualized using a Leica SP2 confocal microscope with a 63x oil immersion objective.
Creation, expression and purification of antibody constructs
Anti-CD133 scFv was obtained by PCR amplification of the variable regions of the light and heavy chains from AC133.1 hybridoma (ATCC No. HB-12346). Assembly of the scFv was done by splicing using overlap extension PCR and the purified PCR product cloned into phagemid pAK100 (kind gift from Prof. Andreas Pluckthun). Anti-EGFRvIII scFv was artificially synthesized based on the published sequence available from Genbank (accession no.U76382). A pBudCE4:Her2:CD16 bispecific minibody vector was used as the base vector and was a kind gift from Dr. Louis Weiner. This construct has scFv against Her2/neu and CD16 in a bi-cistronic vector and the CH3 region has a “knob-into-hole” configuration to enhance heterologous dimer formation. We replaced the scFv’s for Her2/neu with the scFv of anti-EGFRvIII Ab and anti-CD16 scFv with the scFv of anti-CD133. V5 and His6 tag sequences are present at the 3′-end of the anti-EGFRvIII binding arm, and Myc and His6 tag sequences are present at the 3′-end of the anti-CD133 binding arm. As controls, antibodies containing either two scFv chains against AC133 or EGFRvIII were also created. The constructs were stably transfected into HEK 293 cells. Antibodies were purified from prepared supernatant using a His-Trap column protocol (Amersham Biosciences).
Plasma membrane preparation and ELISA for BsAb Analysis
Plasma membrane protein was quantitated and adsorbed onto 96 well ELISA plates. Wells were blocked with human γ-globulin and incubated with different concentrations of antibody. The secondary antibody used to detect bound antibody was THE™ Anti-c-Myc-tag [HRP] mAb (Genscript). Plates were developed using the SureBlue TMB Microwell Substrate (KPL).
Live cell immunoprecipitation
Cell lines expressing EGFRvIII, CD133 or both were dissociated, blocked with human γ-globin and counted. Viable cells were incubated with various concentrations of antibodies. For a qualitative analysis of affinity, the cells were lysed and processed for immunoblot analysis with anti-CD133, anti-EGFRvIII or anti-V5 antibodies. For quantitative analysis of surface bound antibody, live cells were eluted with glycine stripping buffer. Eluted antibody was neutralized and adsorbed on 96 well plates and detected using HRP labeled anti-myc antibody.
Competitive sandwich ELISA assay
U87 cells lines expressing either or both epitopes (104 cells to 109 cells) were incubated with 400nM BsAb and then added to 96 well plates coated with the membrane fraction from U87 cells transfected with both EGFRvIII and CD133 (~105 cells). The cells were then washed off and membrane fraction bound BsAb was quantitated.
NK cell purification and culture
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll density gradient centrifugation. NK cells were purified using anti-CD56-coated microbeads, followed by two rounds of positive selection using autoMACS. CD3+ cells were subsequently depleted using anti-CD3 coated magnetic beads.
Cytotoxicity assay
The antibodies were tested for their ability to induce antibody dependent cellular cytotoxicity (ADCC) using NK cells as the effectors against target cells. The aCella-Tox kit was used to quantitate ADCC (Cell Technology, Inc.). The protocol was carried out according to the manufacturers’ recommendation and luciferase activity was measured using a luminometer.
Cell lines and expression plasmids utilized
U87 cells expressing EGFRvIII (U87vIII) were a kind gift of Dr. Donald O’Rourke. U87-EGFRvIII/CD133 double expressing cells lines were generated by stable transfection of EGFRvIII and CD133. The CD133 clone was obtained from Plasmid Information Database (PlasmID) maintained by Dana-Farber/Harvard Cancer Center DNA Resource Core. The CD133 gene was PCR amplified and subcloned into the pCR3.1 vector. EGFRvIII was also subcloned into the pCR3.1 vector (28).
Statistical analysis
To validate the significance of the observed differences we analyzed variables using two-sided t tests or two-way ANOVA. Differences in survival estimates were calculated using the Log Rank test.
RESULTS
EGFRvIII is found in primary human GBM CSCs
CD133 has been widely used for the identification of GBM CSCs (3). While some studies have found it to be problematic (29–31), it still remains one of the most generally used markers and has now been used to identify CSCs from colon, lung, ovary and prostate tumors among many others. We reasoned that if EGFRvIII is found in CSCs, then it may show an association with CD133. We first analyzed 28 freshly resected GBM samples by flow cytometry to assess EGFRvIII and CD133 expression. Figure 1A shows representative flow cytometry plots from three tumors, two showing co-expression of EGFRvIII and CD133 (GBM10 & GBM13) and one EGFRvIII−/CD133+ tumor (GBM15). The gating strategy for these results is demonstrated in Figure S1. We found that 57% (16/28) of tumors were CD133+ (as well as two normal brain (NB) samples from adult resections of epileptic seizure foci), 71% (20/28) were EGFRvIII+, and 81% (13/16) of the CD133+ tumors also expressed EGFRvIII (Fig. 1B). Patient time-to-recurrence, survival, EGFRvIII/CD133 status, tumor location, and treatment data for available samples are shown in Table S1, and EGFRvIII/CD133 expression in all samples is shown in Table S2. We consistently observed a significant fold increase in the actual percentage of cells which express both proteins as compared to the co-expression expected through random co-incidence (Fig. 1C, p<0.001 for average of entire set), demonstrating that EGFRvIII is preferentially expressed with CD133 in a subset of tumor cells.
Figure 1. EGFRvIII expression and functional characteristics.
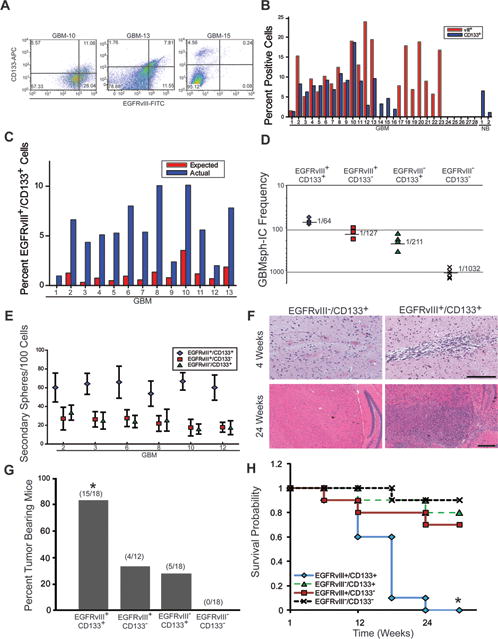
Primary GBM and normal brain samples were dissociated and analyzed. A) Flow cytometry plots of EGFRvIII (x axis) and CD133 (y axis) surface expression in live dissociated GBM cells. Representative plots are shown from 28 samples analyzed. B) Percentage of EGFRvIII and CD133 expressing cells in all samples tested, including two normal brain samples (NB) from adult seizure focus resection of epileptic brain. C) Comparison of actual versus expected co-expression. D) An extreme limiting dilution analysis algorithm was used to determine the frequency of GBMsp forming ( ) EGFRvIII+/CD133+ cells, ( ) EGFRvIII+/CD133− cells, ( ) EGFRvIII−/CD133+ cells, and (X) EGFRvIII−/CD133− cells. E) Neurospheres from (D) above were re-dissociated without sorting to assay for self-renewal of ( ) EGFRvIII+/CD133+ cells, ( ) EGFRvIII+/CD133− cells, and ( ) EGFRvIII−/CD133+ cells. Plotted is the mean ± SEM. F) Sorted primary GBM samples were intracranially injected into NOD-SCID mice. Shown is representative H&E staining of brains injected with 1000 EGFRvIII+/CD133+ or EGFRvIII−/CD133+ cells at 4 weeks and 24 weeks post injection. Scale bars = 500μm. G) Tumor incidence from injection of sorted cells into the brains of NOD-SCID mice. *= statistical significance (p<0.001). H) Kaplan-Meier survival probability estimates of mice injected with sorted cells. *= statistical significance (p<0.001). See also Figures S1 and 2, and Tables S1 and 2.
Tumor derived neurospheres, or tumor spheres are widely used as an in vitro model for studying CSCs, and EGFRvIII has been shown to be expressed in these cells (32, 33). We hypothesized that the EGFRvIII+ cells should show enhanced tumor sphere forming ability. Using limiting dilution analysis of the four sorted populations from six primary GBM samples, we observed that EGFRvIII+/CD133+ and EGFRvIII+/CD133− cells had the highest frequency of GBM sphere initiating cells (1/64 and 1/127 respectively) as compared to EGFRvIII−/CD133+ (1/211), and EGFRvIII−/CD133− cells (1/1032) (Figure 1D). Re-dissociated tumor spheres from EGFRvIII+/CD133+ and EGFRvIII+/CD133− cells consistently showed the highest potential for sphere self-renewal compared to the EGFRvIII−/CD133+ populations (Figure 1E). Additionally, we observed in EGFRvIII+/CD133− tumors that selection for EGFRvIII+ resulted in a population capable of self-renewal whereas the EGFRvIII− population showed no self-renewal (4/4 tumors analyzed).
An essential property of CSCs is tumor forming ability in mice. CD133+ cells from primary GBM tumors show enrichment for tumor formation when implanted intracranially into NOD-SCID mice (3). Figure 1F shows representative tumors formed 4 weeks and 24 weeks after injection with EGFRvIII+/CD133+ versus EGFRvIII−/CD133+ primary GBM cells. Review of the histopathology of these tumors shows several classic features of glioblastoma including increased vascularity, pseudopalisades, increased mitoses, and varied cellular morphology, but there did not appear to be any consistent difference in tumor histology among the immunophenotypic groups. Also, it has been reported that glioblastoma CSCs exist in a perivascular niche (34) and can transdifferentiate (35, 36) into endothelial cells. Interestingly, when both primary human GBM samples and mouse xenografts from cultured human GBM tumor spheres were stained for EGFRvIII alone or EGFRvIII in combination with the endothelial marker CD31, they occasionally revealed the presence of EGFRvIII around and in vessels (Fig. S2). In primary human GBM samples, this staining pattern was observed in 23% of human GBM tumors analyzed (5/21), demonstrating another property that EGFRvIII+ cells have in common with CSCs. In agreement with previous reports (20, 21), overall EGFRvIII staining appeared to be either sporadic or focal with no strong association with the edges of necrotic areas.
Using six GBM samples, we found that 83% (15/18) of mice receiving EGFRvIII+/CD133+ cells, and 33% (4/12) of mice receiving EGFRvIII+/CD133− cells developed tumors, whereas only 28% (5/18) of mice receiving EGFRvIII−/CD133+ cells showed tumors (Fig. 1G). None of the mice (0/18) injected with EGFRvIII−/CD133− cells developed tumors even with doses as high as 100,000 cells. The survival probability of EGFRvIII+/CD133+ cell injected mice was also significantly decreased as compared to mice injected with the EGFRvIII+/CD133− or EGFRvIII−/CD133+ population (Fig. 1H).
EGFRvIII+ tumor spheres recapitulate properties observed with primary tumor cells
EGFRvIII amplification and expression is rapidly lost when GBM tumors are grown under standard serum based culture conditions (37). However, because tumor spheres represent the CSC population in cell culture if EGFRvIII expression marks a CSC population, then its expression should be retained in spheres grown under stem cell culture conditions. EGFRvIII expression was found in all EGFRvIII+/CD133+ cultured tumor spheres (GBMsp) derived from EGFRvIII+/CD133+ primary GBMs when examined by immunoblot, RT-PCR, flow cytometry, and immunofluorescence (Fig. 2 A–C, Fig. 3 and 4). Expression was not detected in cultured spheres derived from normal tissue or primary GBM tumors that were EGFRvIII− by flow cytometry. These data demonstrate that EGFRvIII does not arise spontaneously during culture, and also suggest that it is the selection for CSC properties that preserves EGFRvIII expression in tumor spheres. EGFRvIII+ tumor sphere cells also co-express CD133 (Fig. 2C), maintain greater than expected co-expression of both receptors (Fig. 2D), and are the highest self-renewing population of cells (Fig. 2E), just as in the original tumors. Lastly, EGFRvIII+ cultured tumor spheres are also tumorigenic, as orthotopic transplantation of 104 cells into NOD-SCID mice cortex formed high grade glioma tumors (Fig. 2F). This data extends the results of others showing EGFRvIII expression in spheres (38, 39) by suggesting that it is the EGFRvIII+ cells that are the key cells within GBM tumor spheres for self-renewal and tumorigenicity. Overall, the data from figures 1 and 2 support our hypothesis that EGFRvIII identifies a putative CSC population. The fact that the EGFRvIII+/CD133+ population showed the highest tumor formation and self-renewal capacity suggests that the use of these two markers further refines detection of the CSC population.
Figure 2. Expression of EGFRvIII in Cultured GBM Neurospheres (GBMsp).
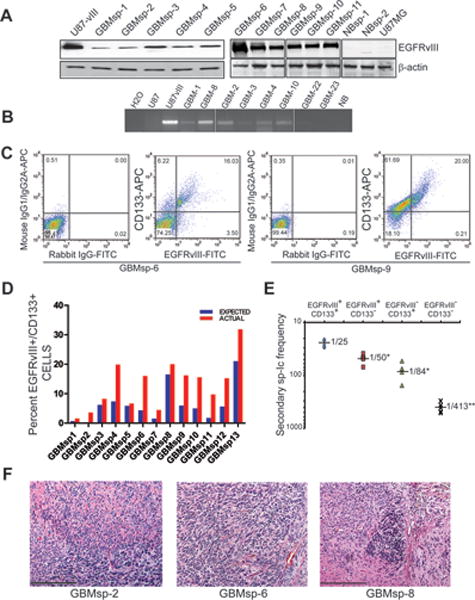
Tumor spheres derived from dissociated primary human GBMs were cultured in stem cell media prior to analysis. A) Immunoblot analysis of lysates from cultured GBMsp (GBMsp), normal brain spheres (NBsp) from epileptic patients, and control U87MG and U87-vIII for EGFRvIII expression and β- actin. B) RT-PCR for EGFRvIII transcription from total RNA of cultured GBMsp and normal brain (NB) and U87MG and U87-vIII samples as controls. C) Flow cytometry analysis of co-expression of EGFRvIII (x axis) and CD133 (y axis) in dissociated GBMsp. The left panels of each sample show isotype control background (mouse IgG1/IgG2B for CD133 and rabbit IgG for EGFRvIII), and the right panels show signal for anti-CD133 and anti-EGFRvIII. D) Comparison of actual versus expected CD133/EGFRvIII co-expression in all cultured GBMsp samples tested. E) Secondary sphere initiating (sp-Ic) frequency of 6 tumor sphere lines from cells sorted into 4 populations ( ) EGFRvIII+/CD133+, ( ) EGFRvIII+/CD133−, ( ) EGFRvIII−/CD133+, (×) EGFRvIII−/CD133−. *= statistical significance (p<0.01), **= statistical significance (p<0.001). F) Cultured GBMsp cells were intracranially injected into NOD-SCID mice and assayed for tumor formation. Shown are micrographs of H & E staining of three sphere lines (Scale bar = 500 μm).
Figure 3. Effects of Differentiation on EGFRvIII and Marker Expression.
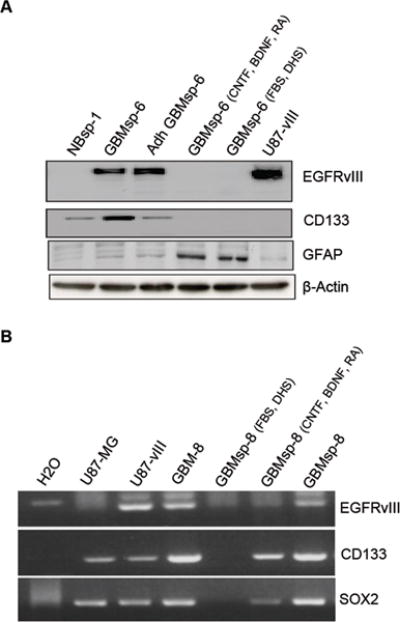
GBMsp were grown either as a suspension culture or adherent culture in tumor stem media or differentiated by withdrawing EGF and FGF and exposing to two separate differentiating conditions; CNTF, BDNF and retinoic acid (CNTF, BDNF, RA) or 5%FBS + 5% donor horse serum (FBS, DHS). A) Immunoblot analysis with anti-EGFRvIII, anti-CD133, anti-GFAP, and anti β-Actin as control. Four separate cell lines were tested; representative blots of GBMsp-6 are shown. B) Whole cell RNA was utilized in RT-PCR analysis with primers specific for EGFRvIII, CD133, and SOX2 gene products. Each experiment was performed in two different GBM sphere lines. Representative gels of GBMsp-8are shown.
Figure 4. EGFRvIII is not co-localized with markers of differentiation in cultured GBMsp.
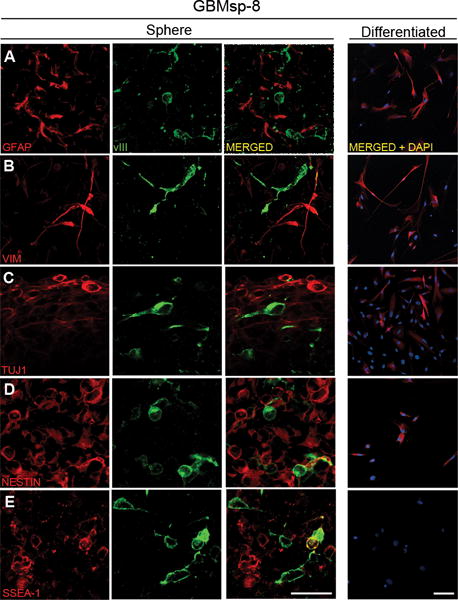
EGFRvIII+ neurospheres were maintained in stem cell media (Sphere) or dissociated and differentiated with media containing 10% serum (Differentiated). Cells were immunostained with antibodies against EGFRvIII (vIII) and: A) Glial Fibrillary Acidic Protein (GFAP), B) Vimentin (VIM), C) β-III Tubulin (TUJ1), D) Nestin (NESTIN), and E) Stage Specific Embryonic Antigen 1(SSEA-1), and imaged by confocal microscopy. Each staining combination was performed on three separate GBM neurosphere lines with similar results. Images are from GBMsp-8 (0.16μm sections). Scales = 37.5μm in A–E (Sphere), and 75μm in all Differentiated micrographs. See also Table S3.
EGFRvIII association with stem cell markers
Much like neural stem cells, during differentiation GBM CSCs lose expression of stem cell markers and express markers for neural and astrocytic lineages (40, 41). Immunoblot and RT-PCR analysis comparing undifferentiated vs. differentiated tumor sphere samples showed that loss of EGFRvIII expression upon differentiation correlates with the loss or reduction of expression of the stem/progenitor cell markers CD133 and SOX2. Conversely, differentiation resulted in the expression of the glial lineage marker GFAP (Fig. 3 A, B). To better understand on a cell-by-cell basis the effects of differentiation on EGFRvIII and other markers, we performed immunofluorescence microscopy. In cultured tumor spheres, EGFRvIII showed no co-expression with vimentin, and only very low co-expression with GFAP or Tuj1, but showed a ~ 4–17 fold greater co-expression with nestin and SSEA-1 (Fig. 4 and Table S3). GBM tumor sphere (GBMsp) cells were differentiated by culture in 10% serum, and similar to Figure 3 all sphere lines examined lost all EGFRvIII expression, as well as SSEA-1expression, yet maintained GFAP, Vimentin and TuJ1 (Fig. 4 Differentiated). Of note, wtEGFR expression was maintained upon differentiation (data not shown). These findings strongly support the notion that EGFRvIII expression can define a subpopulation of stem/progenitor-like cells in GBM by showing that EGFRvIII is associated with markers of undifferentiated cells that is lost upon differentiation while markers of differentiation are maintained or gained.
Development of a bispecific EGFRvIII/CD133 antibody
If EGFRvIII defines a CSC population, then depleting EGFRvIII+ and more specifically EGFRvIII+/CD133+ cells should prevent tumor formation. A bispecific antibody that recognizes EGFRvIII and CD133 would have the highest affinity towards a cell expressing both proteins but lower affinity towards cells expressing either antigen alone. Single chain variable fragments (scFv) against CD133 and EGFRvIII were cloned. The scFv against EGFRvIII has been previously described (42) and the scFv against CD133 was generated as described in the Methods. These scFv were combined to create the anti-EGFRvIII/CD133 chimera named BsAb (bi-specific antibody), and bi-valent constructs for EGFRvIII (di-EGFRvIII) and CD133 (di-CD133). Each construct contains a human IgG1-Fc domain for engaging natural killer (NK) cells (Fig. 5A). We confirmed that the BsAb has the highest binding for cells expressing both epitopes, whereas di-EGFRvIII and di-CD133 have high affinity for cells expressing only high levels of each protein respectively (Fig. 5B, S3A and B). EGFRvIII+ /CD133+ cells bound BsAb at concentrations as low as 200 nM, whereas mixtures of cells expressing each antigen independently showed low affinity towards the BsAb even at 2000 nM (Fig. 5B). The BsAb does not cross-link cells independently expressing each protein as pools of U87-EGFRvIII and U87-CD133 cells do not bind (Fig. 5B and S3C). To be effective the BsAb must be able to identify a rare stem cell population amongst the more abundant differentiated cell population. Comparison of an EGFRvIII specific antibody to the BsAb by IHC demonstrates that the BsAb recognizes only a subset of cells recognized by the EGFRvIII antibody in GBM samples (Fig. S3D), and a competitive sandwich ELISA using EGFRvIII+/CD133+ cells showed that the BsAb bound to dual epitope expressing cells even in the presence of a vast excess of cells expressing individual proteins (Figure 5C). Furthermore, using four different neurosphere lines (3 tumor, 1 non-neoplastic) we found specific binding of the BsAb correlated with EGFRvIII+/CD133+ expression (Figure 5D). These results demonstrate that the BsAb preferentially binds to individual cells co-expressing both proteins.
Figure 5. Characterization of the anti-CD133/anti-EGFRvIII bi-specific antibody (BsAb).
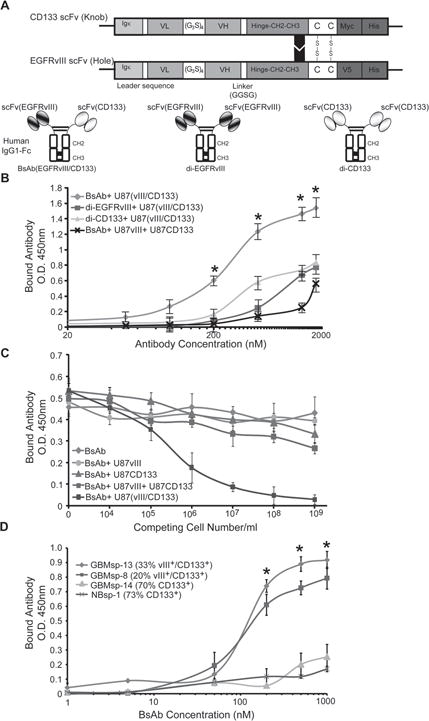
A) Schematic of the BsAb showing scFv chains of anti-CD133 and anti-EGFRvIII. Igκ = leader sequence from kappa chain of human IgG1, VL = variable light chain, (G3S)4 = hinge peptide, VH = variable heavy chain, CH2-CH3 = Fc domain from human IgG1, Myc, V5 and His = tags for purification and identification. Di-valent anti-CD133 (di-CD133) and anti-EGFRvIII (di-EGFRvIII) antibodies were also created. B) ELISA dose-response analysis of the BsAb ( ), di-EGFRvIII ( ), and di-CD133 ( ), reagents on U87 cells co-transfected with EGFRvIII and CD133, and analysis of the BsAb reagent on an equal mix of cells transfected with either EGFRvIII or CD133 (x). * = statistical significance (p < 0.001). C) Competitive sandwich ELISA to identify specificity of BsAb towards EGFRvIII+/CD133+ cells. BsAb was not pre-incubated ( ) or pre-incubated with U87 cells expressing either ( ) CD133, ( ) EGFRvIII, or both proteins expressed in the same cell (EGFRvIII/CD133) ( ), or a pool of cells expressing CD133 or EGFRvIII ( ) prior to incubation with membrane fractions from U87-EGFRvIII/CD133 cells (105cells/well). D) Live cell immunoprecipitation and ELISA analysis of BsAb binding to GBM tumor spheres in cultures with variable EGFRvIII and CD133 expression. ( ) GBMsp-13, ( ) GBMsp-8, ( ) GBMsp-14, (x) NBsp-1. * = statistical significance (p<0.001). Data points are represented as mean ± SEM of three individual experiments. See also Figure S3.
The BsAb is cytotoxic to human GBM cells
We established that di-EGFRvIII and BsAb exhibited significant NK cell induced toxicity towards U87-EGFRvIII/CD133 cells (Fig. S4A). Then, using a effector to target (E:T) ratio of 10:1, we found as little as 0.1ug/ml (8.3nM) of BsAb was sufficient to induce cytotoxicity in EGFRvIII+/CD133+ cells (Fig. S4B). Mirroring the binding results, the BsAb induced the greatest cytotoxicity in EGFRvIII+/CD133+ cells while the di-valent constructs worked best in cells expressing only the respective epitope (Figure S4C). Finally we tested freshly resected GBM tumor (GBM-8) or dissociated GBMsp to assay cytotoxicity towards cells from human patients. In GBM-8 and GBMsp lines that had high EGFRvIII+/CD133+ cells (GBMsp-4 and GBMsp-6), the BsAb induced higher cytotoxicity compared to GBMsp lines which expressed either CD133 (GBMsp-14, GBMsp-15) or EGFRvIII (GBMsp-17) alone (Figure 6A). Importantly we also found low toxicity towards normal neural stem cells (NBsp-1, NBsp-2).
Figure 6. BsAb eliminates EGFRvIII+/CD133+cells from culture, enhances cytotoxicity, impairs GBMsp tumorigenicity, and enhances survival.
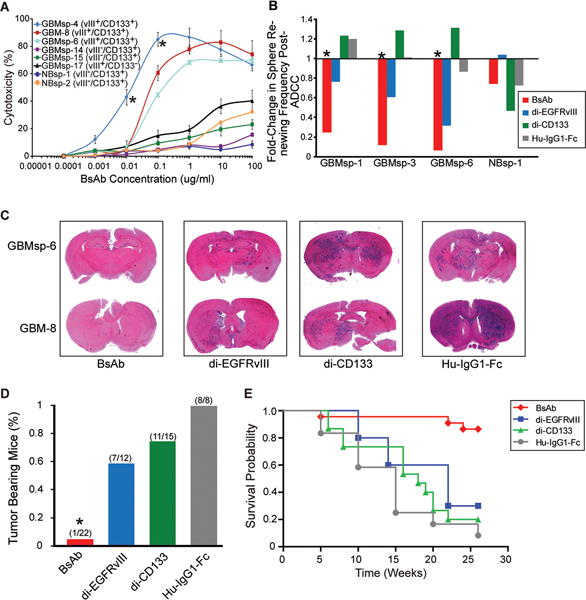
A) BsAb Antibody-Dependent Cell Mediated Cytotoxicity (ADCC) on primary GBM cells (GBM-8), GBM tumor spheres (GBMsp), and normal neurospheres from epileptic patients (NBsp). All experiments were carried out in triplicate. Error bars represent ± SEM. *= statistical significance (p<0.001). B) Fold-change in the frequency of NBsp and GBMsp initiating cells pre and post-ADCC. Data represented as change in sphere renewing frequency, with and without the presence of BsAb (red) di-EGFRvIII (blue), di-CD133 (green) and human-IgG1-Fc (grey). C) Antibodies were mixed with unsorted cells (100,000 total) from EGFRvIII+/CD133+ tumors, injected into the brains of NOD-SCID mice, and assayed after 26 weeks. Coronal sections from mouse brain were stained with H&E. D) Percent tumor bearing mice from four human glioma samples (GBMsp-4, GBMsp-6, GBM8 and GBM12) co-injected with the various antibodies. Mice were analyzed after 26 weeks. E) Kaplan Meier survival probability shows the BsAb significantly enhances survival (p<0.0001) as compared to the mono-specific antibodies. See also Figures S4 and S5.
The BsAb decreases self-renewal and tumorigenicity
Limiting dilution analysis of GBMsp cells pre and post cytotoxicity assay showed a significantly decreased renewal capability from BsAb treatment, while di-EGFRvIII was effective to a lesser degree, and a human IgG1-Fc fragment had no effect. No reagent significantly affected normal brain neurospheres (Figs. 6B, and S5). For tumor prevention analysis, the antibody constructs (all at 0.01ug/105 cells) were combined with GBM or GBMsp cells and injected into the cortex of NOD-SCID mice, which were sacrificed after 26 weeks and analyzed for tumor formation (Figure 6C and 6D). The BsAb severely inhibited tumor formation (26 total mice, 1/22 mice showed tumor formation) (p<0.001) as compared to Hu-IgG1-Fc which showed no reduction (12 injected, 8/8 analyzed showed tumors). Di-EGFRvIII and di-CD133 did not statistically significantly reduce tumor formation (Figure 6D). Furthermore, the survival probability of mice receiving the BsAb is significantly increased compared to the mono-specific antibody recipients, strongly suggesting that targeting of the EGFRvIII+/CD133+ CSC population effectively prevents GBM formation (Figure 6E).
DISCUSSION
Several studies have shown that EGFRvIII is expressed in tumor spheres derived from GBM samples, but whether this was incidental or significant to CSC biology has not been investigated (38, 39). We have found that EGFRvIII is associated with markers used to identify stem cells or CSCs including CD133, nestin, SOX2, and SSEA-1, but shows little to no association with markers of differentiation including vimentin, Tuj-1, and GFAP. EGFRvIII expression, which is difficult to maintain under standard conditions, is preserved using tumor sphere conditions but the differentiation of tumor spheres results in the loss of EGFRvIII. Cell populations containing EGFRvIII have the highest degree of self-renewal and tumorigenicity. Collectively, our data suggest that the EGFRvIII oncogene can be used to define the CSC population.
Antibodies and small molecule inhibitors against wtEGFR also bind to EGFRvIII, but are less potent against this variant (43–45). Thus, if EGFRvIII+ cells represent the tumor initiating population there may be a CSC basis for why anti-EGFR therapy has not been effective against tumors expressing EGFRvIII (46). Interestingly, a peptide vaccine against EGFRvIII has demonstrated improved overall survival for GBM patients in Phase II clinical trials (19). Our data also provides an explanation for why therapy directed against a minority of cells can be effective in increasing survival: it is the targeting of CSCs and not the bulk of the tumor that can indeed be therapeutically effective (19). The vaccine is administered after total resection and radiation/chemotherapy and serves to inhibit tumor recurrence. The finding that the anti-EGFRvIII and BsAb can effectively prevent intracranial tumor formation suggests that it can be used in a similar manner where patients are treated immediately post-resection to prevent tumor recurrence. Because these antibodies are passive immunotherapy it would have specificity against the cells responsible for recurrence and complement the effects of the vaccine if used in combination. Recent preliminary experiments show that the BsAb can be used to treat existing intracranial tumors in mice indicating that this agent can penetrate the blood brain barrier. Whether the BsAb studied here can ultimately be used in clinical trials awaits further preclinical work to confirm that it can induce the regression of pre-existing tumors in animal models.
While CD133 has been widely used to isolate CSC in many solid tumors, its use has been debated (29–31). Clearly this marker can enrich for CSCs, but our work and other studies show that the true CSC may comprise only a fraction of the marker positive pool. We have found that EGFRvIII+/CD133+ primary tumor cells are the most highly enriched population for self-renewal and tumorigenicity, strongly suggesting that the addition of EGFRvIII expression to CD133 expression more accurately defines a CSC population in GBM. An important corollary of the CSC hypothesis is that elimination of the cancer stem cell population is necessary and sufficient to eliminate a tumor. We tested this notion by developing the BsAb to precisely recognize EGFRvIII+/CD133+ cells. The specific lysis of the EGFRvIII+/CD133+ population significantly reduces the implantation of primary glioblastoma tumors in mice and prolongs survival. Because these mice were observed for 26 weeks, this data also suggest that the spontaneous acquisition of tumor initiating properties by non-EGFRvIII+/CD133+ cells is likely to be very low. Thus, our findings provide support for the hypothesis that elimination of cancer stem cells is sufficient to eradicate a tumor in vivo.
A dual marker approach may prove more specific than the targeting of a single marker for several reasons. Nearly all studies isolating CSCs have relied on normal stem cell markers, such as CD133 (3) or CD15/SSEA-1 (4, 47, 48), which would present a potential toxicity problem if used as a target. This is especially relevant in the case of brain tumors since the brain has a finite pool of stem cells and limited capacity for regeneration. Many markers have been proposed to define glioblastoma CSCs (3–5) yet no single marker has definitively isolated these cells. The true CSC population may comprise only a fraction of any marker positive pool. The addition of another marker provides an independent selection that enhances the identification of the CSC. This work represents another step towards the goal of precisely identifying the CSC population, which is likely to require a series of studies building upon this and other findings.
The BsAb could be useful in treating other tumor types such as ovarian, prostate, colon and lung, where CD133 (49) and EGFRvIII expression (11, 13) has been reported. However, our bispecific approach demonstrates a proof of principle that could be extended to other cell surface protein and stem cell marker combinations. It is likely there are other oncogenic receptors that show CSC specific expression. Amplification of growth factor receptors is a common event in aggressive malignancies and has been implicated in the genesis of tumors through expansion of the CSC population. For example, the overexpression of HER2 in breast cancer has been linked to an increase in the CSC population (50). As more receptors and CSC markers are identified, this unique approach can be used to develop strategies to identify tumor initiating cancer stem cells in a wide variety of malignancies.
Supplementary Material
Acknowledgments
We thank Navpreet Bains, Supriya Sharma and Fanny Pang for assistance in cell count quantitation, and Beth Hoyte for review and assistance with figure preparation. We also thank the Stanford University tissue bank for acquisition of tissue, Dr. Karen Sachs for help with statistical analysis, Dr. Kalle Söderström for help in developing ADCC assays and NK cell isolation, and Prof. Andreas Pluckthun for his gift of pAK100 phagemid.
A.J.W. is supported by National Institutes of Health grants CA69495, CA124832, 1RC2CA148491, and research grants from the California Institute for Regenerative Medicine, National Brain Tumor Foundation and Lucille Packard Children’s Foundation
Footnotes
Author Contributions: D.R.E. performed differentiation and localization studies, participated in the analysis and interpretation of the data, and writing of the manuscript. P.G. cloned and constructed the di-EGFRvIII and di-CD133 plasmids, and assisted the immunofluorescence. M.H.M. assisted in the design and construct of the BsAb and writing of the manuscript. CDV performed differentiation studies and performed data analysis and manuscript preparation. S.S.M. performed flow cytometry, tumorigenicity, BsAb tumor studies, and participated in the data analysis and writing of the manuscript. S.-Y. H. cloned the scFv fragments, designed and constructed the BsAb plasmid. G.L. assisted with the tumorigenicity experiments. K.J. and H.V. provided glioblastoma specimens and pathology review. L.W.X. retrieved patient data. S.S. studied the co-expression of EGFRvIII and CD133 by flow cytometry. A.J.W. initiated the concepts and designed the study, analyzed the data, supervised the overall project and wrote the manuscript.
Conflicts of interest: none
Reference List
- 1.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–22. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 3.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 4.Son MJ, Woolard K, Nam DH, Lee J, Fine HA. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. 2009;4:440–52. doi: 10.1016/j.stem.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. 2010;6:421–32. doi: 10.1016/j.stem.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez-Climent JA, Andreu EJ, Prosper F. Somatic stem cells and the origin of cancer. Clin Transl Oncol. 2006;8:647–63. doi: 10.1007/s12094-006-0035-7. [DOI] [PubMed] [Google Scholar]
- 7.Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW, Vandenberg S, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010;24:1731–45. doi: 10.1101/gad.1890510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh S, Dirks PB. Brain tumor stem cells: identification and concepts. Neurosurg Clin N Am. 2007;18:31–8. viii. doi: 10.1016/j.nec.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 9.Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A. 1992;89:2965–9. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saikali S, Avril T, Collet B, Hamlat A, Bansard JY, Drenou B, et al. Expression of nine tumour antigens in a series of human glioblastoma multiforme: interest of EGFRvIII, IL-13Ralpha2, gp100 and TRP-2 for immunotherapy. J Neurooncol. 2007;81:139–48. doi: 10.1007/s11060-006-9220-3. [DOI] [PubMed] [Google Scholar]
- 11.Moscatello DK, Holgado-Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW, et al. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 1995;55:5536–9. [PubMed] [Google Scholar]
- 12.Del Vecchio CA, Jensen KC, Nitta RT, Shain AH, Giacomini CP, Wong AJ. Epidermal growth factor receptor variant III contributes to cancer stem cell phenotypes in invasive breast carcinoma. Cancer Res. 2012;72:2657–71. doi: 10.1158/0008-5472.CAN-11-2656. [DOI] [PubMed] [Google Scholar]
- 13.Okamoto I, Kenyon LC, Emlet DR, Mori T, Sasaki J, Hirosako S, et al. Expression of constitutively activated EGFRvIII in non-small cell lung cancer. Cancer Sci. 2003;94:50–6. doi: 10.1111/j.1349-7006.2003.tb01351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Del Vecchio CA, Wong AJ. Rindopepimut, a 14-mer injectable peptide vaccine against EGFRvIII for the potential treatment of glioblastoma multiforme. Curr Opin Mol Ther. 2010;12:741–54. [PubMed] [Google Scholar]
- 15.Heimberger AB, Suki D, Yang D, Shi W, Aldape K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J Transl Med. 2005;3:38. doi: 10.1186/1479-5876-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pelloski CE, Ballman KV, Furth AF, Zhang L, Lin E, Sulman EP, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol. 2007;25:2288–94. doi: 10.1200/JCO.2006.08.0705. [DOI] [PubMed] [Google Scholar]
- 17.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moscatello DK, Ramirez G, Wong AJ. A naturally occurring mutant human epidermal growth factor receptor as a target for peptide vaccine immunotherapy of tumors. Cancer Res. 1997;57:1419–24. [PubMed] [Google Scholar]
- 19.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic Escape After Prolonged Progression-Free Survival With Epidermal Growth Factor Receptor Variant III Peptide Vaccination in Patients With Newly Diagnosed Glioblastoma. J Clin Oncol. 2010;28:4722–9. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aldape KD, Ballman K, Furth A, Buckner JC, Giannini C, Burger PC, et al. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J Neuropathol Exp Neurol. 2004;63:700–7. doi: 10.1093/jnen/63.7.700. [DOI] [PubMed] [Google Scholar]
- 21.Nishikawa R, Sugiyama T, Narita Y, Furnari F, Cavenee WK, Matsutani M. Immunohistochemical analysis of the mutant epidermal growth factor, deltaEGFR, in glioblastoma. Brain Tumor Pathol. 2004;21:53–6. doi: 10.1007/BF02484510. [DOI] [PubMed] [Google Scholar]
- 22.Del Vecchio CA, Giacomini CP, Vogel H, Jensen KC, Florio T, Merlo A, et al. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene. 2012 doi: 10.1038/onc.2012.280. [DOI] [PubMed] [Google Scholar]
- 23.Vogt N, Lefevre SH, Apiou F, Dutrillaux AM, Cor A, Leuraud P, et al. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc Natl Acad Sci U S A. 2004;101:11368–73. doi: 10.1073/pnas.0402979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60:1383–7. [PubMed] [Google Scholar]
- 25.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 26.Mukherjee B, McEllin B, Camacho CV, Tomimatsu N, Sirasanagandala S, Nannepaga S, et al. EGFRvIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res. 2009;69:4252–9. doi: 10.1158/0008-5472.CAN-08-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–8. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Moscatello DK, Montgomery RB, Sundareshan P, McDanel H, Wong MY, Wong AJ. Transformation and altered signal transduction by a naturally occurring mutant EGF receptor. Oncogene. 1996;13:85–96. [PubMed] [Google Scholar]
- 29.Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, et al. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–5. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 30.Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L, Hoek M, et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010;70:719–29. doi: 10.1158/0008-5472.CAN-09-1820. [DOI] [PubMed] [Google Scholar]
- 31.Nishide K, Nakatani Y, Kiyonari H, Kondo T. Glioblastoma formation from cell population depleted of Prominin1-expressing cells. PLoS ONE. 2009;4:e6869. doi: 10.1371/journal.pone.0006869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schulte A, Gunther HS, Phillips HS, Kemming D, Martens T, Kharbanda S, et al. A distinct subset of glioma cell lines with stem cell-like properties reflects the transcriptional phenotype of glioblastomas and overexpresses CXCR4 as therapeutic target. Glia. 2011;59:590–602. doi: 10.1002/glia.21127. [DOI] [PubMed] [Google Scholar]
- 33.Witusik-Perkowska M, Rieske P, Hulas-Bigoszewska K, Zakrzewska M, Stawski R, Kulczycka-Wojdala D, et al. Glioblastoma-derived spheroid cultures as an experimental model for analysis of EGFR anomalies. J Neurooncol. 2011;102:395–407. doi: 10.1007/s11060-010-0352-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 35.Soda Y, Marumoto T, Friedmann-Morvinski D, Soda M, Liu F, Michiue H, et al. Feature Article: Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1016030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–8. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- 37.Bigner SH, Humphrey PA, Wong AJ, Vogelstein B, Mark J, Friedman HS, et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 1990;50:8017–22. [PubMed] [Google Scholar]
- 38.Schulte A, Gunther HS, Martens T, Zapf S, Riethdorf S, Wulfing C, et al. Glioblastoma stem-like cell lines with either maintenance or loss of high-level EGFR amplification, generated via modulation of ligand concentration. Clin Cancer Res. 2012;18:1901–13. doi: 10.1158/1078-0432.CCR-11-3084. [DOI] [PubMed] [Google Scholar]
- 39.Witusik-Perkowska M, Rieske P, Hulas-Bigoszewska K, Zakrzewska M, Stawski R, Kulczycka-Wojdala D, et al. Glioblastoma-derived spheroid cultures as an experimental model for analysis of EGFR anomalies. J Neurooncol. 2011;102:395–407. doi: 10.1007/s11060-010-0352-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guan K, Chang H, Rolletschek A, Wobus AM. Embryonic stem cell-derived neurogenesis. Retinoic acid induction and lineage selection of neuronal cells. Cell Tissue Res. 2001;305:171–6. doi: 10.1007/s004410100416. [DOI] [PubMed] [Google Scholar]
- 41.Ying M, Wang S, Sang Y, Sun P, Lal B, Goodwin CR, et al. Regulation of glioblastoma stem cells by retinoic acid: role for Notch pathway inhibition. Oncogene. 2011;30:3454–67. doi: 10.1038/onc.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beers R, Chowdhury P, Bigner D, Pastan I. Immunotoxins with increased activity against epidermal growth factor receptor vIII-expressing cells produced by antibody phage display. Clin Cancer Res. 2000;6:2835–43. [PubMed] [Google Scholar]
- 43.Learn CA, Hartzell TL, Wikstrand CJ, Archer GE, Rich JN, Friedman AH, et al. Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clin Cancer Res. 2004;10:3216–24. doi: 10.1158/1078-0432.ccr-03-0521. [DOI] [PubMed] [Google Scholar]
- 44.Pedersen MW, Pedersen N, Ottesen LH, Poulsen HS. Differential response to gefitinib of cells expressing normal EGFR and the mutant EGFRvIII. Br J Cancer. 2005;93:915–23. doi: 10.1038/sj.bjc.6602793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gajadhar AS, Bogdanovic E, Munoz DM, Guha A. In situ analysis of mutant EGFRs prevalent in glioblastoma multiforme reveals aberrant dimerization, activation, and differential response to anti-EGFR targeted therapy. Mol Cancer Res. 2012;10:428–40. doi: 10.1158/1541-7786.MCR-11-0531. [DOI] [PubMed] [Google Scholar]
- 46.Neyns B, Sadones J, Joosens E, Bouttens F, Verbeke L, Baurain JF, et al. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann Oncol. 2009;20:1596–603. doi: 10.1093/annonc/mdp032. [DOI] [PubMed] [Google Scholar]
- 47.Read TA, Fogarty MP, Markant SL, McLendon RE, Wei Z, Ellison DW, et al. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell. 2009;15:135–47. doi: 10.1016/j.ccr.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ward RJ, Dirks PB. Cancer stem cells: at the headwaters of tumor development. Annu Rev Pathol. 2007;2:175–89. doi: 10.1146/annurev.pathol.2.010506.091847. [DOI] [PubMed] [Google Scholar]
- 49.Schulenburg A, Bramswig K, Herrmann H, Karlic H, Mirkina I, Hubmann R, et al. Neoplastic stem cells: current concepts and clinical perspectives. Crit Rev Oncol Hematol. 2010;76:79–98. doi: 10.1016/j.critrevonc.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 50.Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27:6120–30. doi: 10.1038/onc.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.