

Abstract
Despite improvements in treatment strategies, colorectal cancer (CRC) still has high mortality rates. Most CRCs develop from adenopolyps via the adenoma-carcinoma sequence. A mechanism for inhibition of this sequence in individuals with a high risk of developing CRC is urgently needed. Differential studies of mitochondrial (mt) gene expressions in the progressive stages of CRC with villous architecture are warranted to reveal early risk assessments and new targets for chemoprevention of the disease. In the present study, reverse transcription-quantitative PCR (RT-qPCR) was used to determine the relative amount of the transcripts of six mt genes [MT-RNR1, MT-ND1, MT-COI, MT-ATP6, MT-ND6, and MT-CYB (region 648–15887)] which are involved in the normal metabolism of mitochondria. A total of 42 pairs of tissue samples obtained from colorectal adenopolyps, adenocarcinomas, and their corresponding adjacent normal tissues were examined. Additionally, electron transport chain (ETC), complexes I (NADH: ubiquinone oxidoreductase) and III (CoQH2-cytochrome C reductase), and carbonyl protein group contents were analyzed. Results indicate that there were differential expressions of the six mt genes and elevated carbonyl protein contents among the colorectal adenopolyps compared to their paired adjacent normal tissues (p < 0.05). The levels of complexes I and III were higher in tumor tissues relative to adjacent normal tissues. Noticeably, the expression of MT-COI was overexpressed in late colorectal carcinomas among all studied transcripts. Our data suggest that increased expressions in certain mt genes and elevated levels of ROS may potentially play a critical role in the colorectal tumors evolving from adenopolyps to malignant lesions.
Keywords: Colorectal tumors, Mitochondria, Gene expression, Carbonyl content, Cancer progression
Introduction
Despite advances in detection of colorectal cancer (CRC), it remains the third leading cause of cancer-related deaths in both men and women in the USA [1, 2]. Survival rates are related to the stage of cancer at the time of diagnosis [3]. Colorectal tumors represent a wide spectrum of neoplasms, mostly of epithelial origin, and 97 % of CRCs are adenocarcinomas originating from benign lesions of adenomatous polyps (adenopolyps) [4, 5]. Several lines of evidence suggest that this disease is a result of multiple mutations in tumor suppressor genes (P53, APC) and oncogene (K-ras) that are involved in regulation of cell growth [2, 6, 7].
Mitochondrial DNA (mtDNA) mutations have been associated with development and progression of cancers [6, 8–10]. The higher mutation rates for mtDNA relative to nuclear DNA have been attributed to the production of reactive oxygen species (ROS) during oxidative phosphorylation [11]. A recent study observed higher carbonyl protein group content, a characteristic of oxidative stress, in the progression of serous ovarian carcinoma tumors [12]. Given these associations, mt genes and elevated ROS production have been proposed as targets for early diagnosis and intervention for therapeutic treatments for certain cancers [9, 10, 12]. A past study found alterations in the expression of mitochondrial-encoded respiratory chain subunits among patient tissue samples obtained from both primary CRC and malignant familial adenomatous polyposis (FAP) coli syndrome [13]. In that study, levels of ND2 messenger RNA (mRNA) were elevated in the CRC metastatic tissues when compared to normal tissues from the same individuals [13]. Moreover, the expression of the β-subunit of F1-ATPase (required for mitochondrial ATP synthesis) has been found to decrease in malignant tissues [14], and overexpressions of 16S/RNR2, ND4, ND4L, CYB, COII, ATP6, and ATP8 have been reported in colorectal adenocarcinoma of HT-29 cells [15]. Also, Yamamoto et al. [16] have suggested changes in the rate of synthesis and in the degradation of mt-RNA in the process of carcinogenesis which indicate that these changes are responsible for differences in gene levels in neoplastic and normal tissues. Additionally, decreased expressions of 12S/RNR1, COI, COII, and ATP6 transcripts compared to their paired adjacent normal tissues had been previously observed in prostate cancer tissues [17]. However, studies that evaluate mt-RNA transcripts expression levels in primary tissues of colorectal adenopolyps and invasive adenocarcinomas relative to their adjacent normal tissues are currently lacking.
Here, we compared the relative RNA amount of the transcripts of six mt genes (ND1, ND6, CYB, CO1, ATP6, and 12S/MT-RNR1) and complexes I (NADH: ubiquinone oxidoreductase) and III (CoQH2-cytochrome C reductase) in colorectal adenopolyps and adenocarcinomas, to their paired adjacent normal tissues to determine if colorectal tumoral changes leads to differential changes in mt genes expression. We also evaluated whether carbonyl protein content levels were progressively proportional in tumoral stages from colorectal adenopolyps (tubular (TA), tubulovillous (TV), villous (V) to invasive adenocarcinomas.
Materials and methods
Study samples
Forty-two pairs of primary CRC tissues, from patients who underwent surgical resections, were obtained from the Southern Regional Cooperative Human Tissue Network. All studies were implemented under protocols approved by the Institutional Review Boards of Morehouse School of Medicine and the University of Alabama at Birmingham. Tissue samples from patients with familial adenomatous polyposis (FAP), hereditary nonpolyposis syndrome, or inflammatory bowel disease were excluded. The selected tumors including their adjacent normal tissues were from adenopolyps which are comprised of tubular, tubulovillous, villous adenomas, and the invasive carcinomas that were defined as adenocarcinoma stages I–IV not arranged in a tubular, tubulovillous, and/or villous architecture. Of the 42 paired tissue samples, 11 were from African-Americans, 29 were from Caucasians, and two were demographic unknowns. Fourteen of the samples were from men and 28 from women, all with a mean age of 66 years.
Specimens were micro-dissected and adjacent normal tissues were defined as tissues excised from an area at least 5 cm away from the tumors. Tumor samples used in this study were diagnosed and histopathologically staged by pathologists based on the criteria of the American Joint Committee on Cancer. The samples were stabilized by snap-freezing immediately in cryovials and immersed in liquid nitrogen after excision and dissection. All samples were transferred to −80 °C for long-term storage, as recommended for measurement of RNA and proteins as previously described [18].
RNA extraction and quantitative-PCR analysis
Total RNA extraction of tissue samples were performed with RNeasy Mini kits (Qiagen). Briefly, frozen samples were cut on ice and washed with PBS twice before following the manufacturer’s protocol. Following tissue homogenization, samples were placed in QIAshredder (Qiagen) columns to reduce viscosity. Purity of the RNAwas assessed by 1 % agarose gel electrophoresis and quantified with a Nanodrop ND-2000 spectrophotometer (Nanodrop Technologies, Wilmington, DE). For complementary DNA (cDNA) polymerization, RNA concentrations were adjusted to equal concentrations prior to analysis. Reverse transcription (RT) PCR was performed with iscript cDNA synthesis kits (Bio-Rad) following the manufacturer’s instructions, with 500 μg of total RNA as a template and random hexamers as primers. Quantitative PCR was performed in a 480 Light cycler detector (Roche Diagnostics) with a SYBR green I master mix, 10 ng of cDNA, and specific primers designed for the mt transcripts of ND1, ND6, CYB, COI, ATP6, and 12S/RNR1 (Table 1). Agarose gel electrophoresis was used to confirm primer specificity by checking for a single product of the expected length for each RT-qPCR transcript. The standard curves were generated for each primer set using serial dilutions of pooled cDNA to standardize variations in PCR reactions. In addition, after each run, melting curves were used to verify the melting temperature of the amplicon. All qPCR reactions were performed in triplicates. Second-derivative maximum analysis was used to calculate the crossing point (Cp) values and the mRNA concentrations. For each gene (Table 1), the value used for further calculations was the mean of the three replicates for each sample. To normalize against nonspecific variations in real-time PCR, the expression levels of three endogenous reference genes, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), ubiquitin C (UBC), and β-actin (ATCB), in adjacent normal and tumor tissue samples were analyzed. The levels of RNA expression ratios in tumor tissue were based on comparisons of tumors and their adjacent normal tissues.
Table 1.
Oligonucleotides used for qPCR
| Gene | Primer sequence |
|---|---|
| Mitochondria-encoded genes | |
| rRNA | |
| 12S/MT-RNR1 | Forward: 5′-TAGAGGAGCCTGTTCTGTAATCGAT |
| Reverse: 5′-CGACCCTTAAGTTTCATAAGGGCTA | |
| MRC-complex I | |
| MT-ND1 | Forward: 5′-CCACCTCTAGCCTAGCCGTTTA |
| Reverse: 5′-GGGTCATGATGGCAGGAGTAAT | |
| MT-ND6 | Forward: 5′-CAAACAATGTTCAACCAGTAACCACTAC |
| Reverse: 5′-ATATACTACAGCGATGGCTATTGAGGA | |
| MRC-complex III | |
| MT-CYB | Forward: 5′-ATCACTCGAGACGTAAATTATGGCT |
| Reverse: 5′-TGAACTAGGTCTGTCCCAATGTATG | |
| MRC-complex IV | |
| MT-COI | Forward: 5′-GACGTAGACACACGAGCATATTTCA |
| Reverse: 5′-AGGACATAGTGGAAGTGAGCTACAAC | |
| MRC-complex V | |
| MT-ATP6 | Forward: 5′-TAGCCATACACAACACTAAAGGACGA |
| Reverse: 5′-GGGCATTTTTAATCTTAGAGCGAAA | |
| Housekeeping Genes | |
| ACTB | Forward: 5′-TGCGTTACACCCTTTCTTGACA |
| Reverse: 5′-GCAAGGACTTCCTGTAACAATG | |
| GAPDH | Forward: 5′-GAAGGTGAAGGTCGGAGTC |
| Reverse: 5′-GAAGATGGTGATGGGATTTC | |
| UBC | Forward: 5′-ATTTGGGTCGCGGTTCTTG |
| Reverse: 5′-TGCCTTGACATTCTCGATGG | |
rRNA ribosomal RNA, ATCB β-actin, GAPDH glyceraldehyde-3-phosphate dehydrogenase, UBC ubiquitin C, MT mitochondrial, MT-ATP6 ATP synthase 6, MT-COI mitochondrial encoded cytochrome oxidase I, MT-CYB mitochondrial encoded cytochrome b, MT-ND1 mitochondrial encoded NADH dehydrogenase 1, MT-ND6 mitochondrial encoded NADH dehydrogenase 6
Western blot analysis
The expression of mitochondria-encoded polypeptides, and complexes I and III was analyzed by SDS-PAGE and Western blot immunoassays. Total protein was isolated with the Mammalian Protein Extraction Reagent (Pierce). Protein samples were resolved on 10 % stain-free SDS-polyacrylamide gels (Bio-Rad). Proteins were transferred to Nitrocellulose membranes, blotted with a complex I and III antibody (Invitrogen), and detected with a Super-Signal® West Pico chemiluminescent substrate (Pierce). Glyceraldehyde-3-phosphate dehydrogenase (Invitrogen) was used as a loading control, and blots were quantified by the relative optical density on the level of band intensity using the NIH ImageJ image analysis program.
Protein carbonyl assay
To determine if the levels of oxidized proteins correlated with the levels of disease-specific mitochondrial gene expression patterns in colorectal adenopolyps, spectrophotometric techniques were used to measure reactive protein carbonyl groups in the samples. Generally, reactive oxygen species (ROS) react with histidine, arginine, lysine, and proline groups in proteins to produce carbonyl groups that react with 2,4-dinitrophenylhydrazine (DNPH), leading to formation of stable dinitrophenylhydrazone adducts [19, 20]. DNPH was used to determine the amounts of reactive protein carbonyl contents in colorectal tissues using a protein carbonyl content assay kits (Sigma). Briefly, DNA-free homogenates of samples (100 μl) were placed in micro-centrifuge tubes, 100 μl of 10 mM DNPH in 2 M HCl was added, and the preparations were incubated at room temperature for 10 min. The hydrazone derivatives were precipitated with 30 μl of 100 % of trichloroacetic acid. The preparations were vortexed, incubated on ice for 5 min, and centrifuged at 13,000g for 2 min. The supernatants were removed, and 500 μl of ice-cold acetone were added to the pellets, which were incubated at −20 °C for 5 min and centrifuged for 2 min at 13,000g. The pellets were dissolved in 6 M guanidine hydrochloride and read at optical densities of 370 nm. The absorptions of DNPH (nmol) incorporated as per milligram of proteins were calculated using the Lambert equation and an extinction coefficient 22,000.
Statistical and data analysis
Each experiment was repeated at least three times. Data were shown as means ± s.e.m. and analyzed using Sigma Plot 12.3. Statistical comparisons between groups were analyzed using Student’s t test and Kruskal-Wallis one-way analysis test. A value of p < 0.05 was considered to indicate statistical significance. Calculations were made with Microsoft Excel/Access 2010. Charts and graphs were drawn with Microsoft Excel.
Results
Housekeeping gene determination
Three housekeeping genes (GAPDH, ATCB, and UBC) were evaluated to determine the best candidate to normalize each sample category. The three genes have been used as internal normalizers for analysis of gene expression in cancer samples [21, 22] (Fig. 1). Andersen et al. ranked UBC and GAPDH among the top three normalization genes based on the least amount of systematic error both introduced [23]. Additionally, ATCB was found to have relatively stable expression in CRC samples [22]. Here, we evaluated the RNA levels of all three genes in each of the paired samples. The levels in adenopolyps (TA, TV, and V) and invasive adenocarcinoma tissues were compared to those in adjacent normal tissues. Based on a <5 % difference in median Cp values between the normal and tumor tissues of within the studied groups, GAPDH was determined as the most consistent housekeeping gene in this study. Additionally, the Cp medians of each group were inside 3 % differences of averaged Cp values for all groups.
Fig. 1.

Reference gene identification. Box and whisker plots detailing the minimum, first quartile, median, third quartile and maximum Cp values of the indicated genes (glyceraldehyde-3-phosphate dehydrogenase (GAPDH), actin, ubiquitin C (UBC)) obtained by RT-qPCR. Five groups of samples were tested: tubular adenoma (TA), tubulovillous adenoma (TV), villous adenoma (V), early adenocarcinomas (ECA, stages I and II), and late adenocarcinomas (LCA, stages III and IV)
Mitochondrial gene expression
Expression of mitochondrial genes (ND1, ND6, CYB, COI, 12S/RNR1, and ATP6) was analyzed by quantitative PCR. Each gene was normalized against GAPDH, creating expression ratios as shown in Fig. 2. The values for each sample were compared with their paired adjacent normal tissue. The expression levels of the mt genes were higher in adenocarcinomas relative to their adjacent normal tissues. The levels of expression in the studied mt gene were lower in tubular adenomas, while the levels of expression were higher in the adenocarcinomas (Fig. 2a–f). In tubulovillous adenomas when compared with their paired adjacent normal tissues the levels of expression mirrored that of tubular adenomas, except for the mt-encoded ND6 transcripts as shown in Fig. 2b. Colorectal adenoma samples with the villous architecture showed overexpressions of all mt transcripts, with the exception of mt-encoded ND1 (Fig. 2a). Of the studied mt transcripts, ATP6, COI, and 12S/RNR1 had the highest values of expression when compared with their paired adjacent normal tissues (Fig. 2d–f). Notably, levels of expression were higher in all the studied mt genes in the late stages of adenocarcinoma (stages III and IV) when compared with early stages of adenopolyp (tubular, tubulovillous, and villous) (Fig. 3). To assess the level of expression of mitochondria-encoded peptides in complexes I and III, Western blot analyses were used (Fig. 4a). In comparison to corresponding adjacent normal tissues, the level of complexes I and III was relatively equal in TA but higher in TV, V, and adenocarcinoma stages. Further, analysis revealed that complex I expression was significantly higher in the villous samples when compared to TA, TV, and CA samples (Fig. 4b).
Fig. 2.

Differential mitochondria gene expression in paired colorectal adenoma and carcinoma samples. Gene expression profiles of mitochondria encoded ND1 (a), mitochondrial-encoded ND6 (b), mitochondrial-encoded CYB (c), mitochondrial-encoded COI (d), mitochondrial-encoded ATP6 (e), and 12S ribosomal RNA/RNR1 (f) genes analyzed using RT-qPCR. Glyceraldehyde-3-phosphate dehydrogenase was used as a housekeeping gene for all experiments. Relative gene expression is expressed as the ratio of tumor tissues as compared to their paired adjacent normal tissue. TA tubular adenoma, TV tubulovillous adenoma, V villous adenoma, CA adenocarcinoma. Significant differences between groups compared to cancer with a p value (<0.05) is denoted with an asterisk. Data are presented as mean ± s.e.m.
Fig. 3.
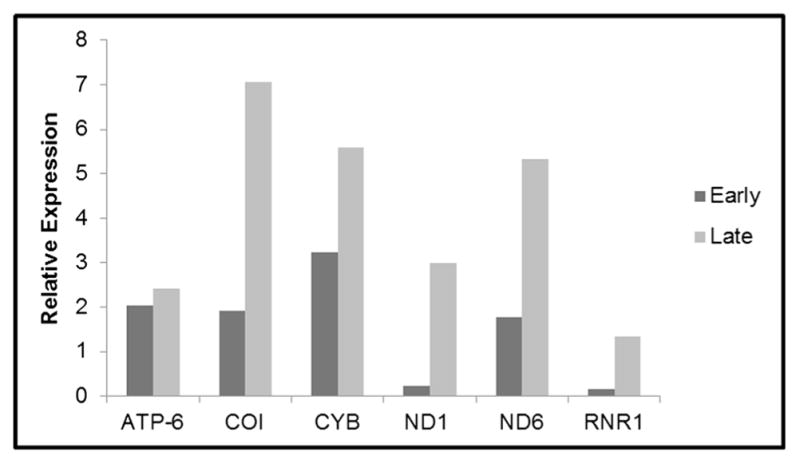
Comparison of gene expression in early- and late-stage paired colorectal carcinomas. Gene expression of mitochondria encoded genes in early-stage adenocarcinomas comprised of stages I and II, and late-stage adenocarcinomas comprised of stages III and IV. Staging of tumors was based on the criteria of the American Joint Committee on Cancer. Gene expression profiles were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using RT- quantitative PCR. Gene name abbreviations are as follows: ATP synthase 6 (ATP-6), cytochrome oxidase I (COI), cytochrome b (CYB), 12S/MT-RNR1 12S ribosomal RNA (12S/MT-RNR1), NADH dehydrogenase 1 (ND1), NADH dehydrogenase 6 (ND6)
Fig. 4.

Expression of mitochondrial complexes I and III in colorectal tumors and paired adjacent normal tissues. a Lysates were analyzed by Western blot to examine the levels of complex proteins in tubular adenoma (TA), tubulovillous tissue (TV), villous tissue (V), and adenocarcinoma tissue (CA) compared to paired adjacent normal tissues (ANT). Complex proteins were normalized against GAPDH. b Protein bands were quantified using relative optical density calculated using NIH ImageJ software. Expression profiles of complexes I (CI) and III (CIII) were normalized against their adjacent normal tissues. Significant differences between tested groups with a p value (<0.05) is denoted with an asterisk when compared to V and Δ when compared to CA. Data are presented as mean ± s.e.m.
Carbonyl protein expression
Although this effort focused on the differences between mitochondrial RNA expression and colorectal adenoma tumor stage, the levels of carbonyl protein were also determined using the DNPH spectrophotometer method. Reactive carbonyl proteins formed through reaction with ROS, which are a characteristic of oxidative stress. Carbonyl protein levels were progressively higher from the early colorectal tumor stage of tubular to adenocarcinoma (Fig. 5), similar to the level of gene expression. Also, the expression patterns were consistent with our previous work using Western blot analysis [24].
Fig. 5.

Levels of carbonyl reactive proteins in colorectal tissue as measured by spectrophotometry. a Ratios of carbonyl protein in tissue samples normalized against adjacent normal tissues. b Protein carbonyl content comparison in individual patients paired adjacent normal and tumor tissue comprised of tubular adenoma (TA), tubulovillous adenoma (TV), villous adenoma (V), and adenocarcinoma (CA). Significant differences between groups compared to cancer were calculated using ANOVA *p < 0.05. Data are presented as mean ± s.e.m.
Discussion
Mitochondria are involved in the generation of free radicals, in cell metabolism, and in apoptosis. Alterations in the integrity of mitochondria have been examined in colorectal cell lines and in primary tissues [25]. Sequencing of mtDNA has led to the discovery of mutations in all genes encoded by the mitochondrial genome. Thus, these mutations could be sources of varied expression at both the transcriptional and posttranscriptional levels [17]. Each mt gene we studied has exhibited differential expression in prostate, ovarian, head and neck, thyroid, and breast carcinomas [17, 21, 26, 27]. Here, we evaluated the expression levels of electron transport chain (ETC) subunits in colorectal adenopolyps and invasive adenocarcinomas compared to their adjacent normal tissues. There were differential expression levels of each subunit as colorectal lesions progressed from polyps to adenocarcinoma. Also, carbonyl proteins were elevated progressively in lesions of tubular adenomas to adenocarcinomas. This suggests that alterations in gene expression could be a product of mtDNA mutations and/or posttranscriptional modification that could influence the progressive stages of colorectal tumors.
Previous studies have indicated that deficient Complex I activity leads to the production of ROS [25, 28–30]; hence, most subunits of Complex I are encoded by the mitochondria, causing it to become susceptible to defects, such as mtDNA mutations [25, 29]. We observed elevated expression levels of ND1 and ND6 transcripts in villous adenomas and colorectal adenocarcinomas compared to tubular adenoma counterparts. These results are similar to those previously reported by Yamamoto et al. [16] using tissue samples of familial adenomatous polyposis. They found elevated levels of ND1 in CRC metastatic samples and suggested that these levels were due to changing energy requirements of rapidly growing cells. Additionally, a study analyzing the role of ND1 in CRCs found that multiple somatic mutations were present in many of their samples. The authors suggested that these mutations were caused by ROS and also inferred that these mutations could interrupt the oxidative phosphorylation pathway, creating more ROS [29]. Mitochondrial damage mediated by ROS may lead to the inactivation of ETC complexes and thereby alter normal mitochondrial function. In fact, a study had shown that components of the ETC and ATPase are damaged by ROS [31]. The increase in the expression levels of ND1 in the prelesion of colorectal adenopolyps in this study may indicate an association of the accumulation of mutations found in mtDNA as we previously suggested [9].
Cytochrome b is the only subunit of complex III that is encoded by mtDNA and, like complex I, is an important component of the ETC, which is responsible for the generation of ROS [25, 32]. Dasgupta et al. showed that a 21-bp deletion mutation in mitochondria-encoded cytochrome b genes led to overexpression of the protein and increased generation of ROS that induced significant tumor growth in vitro and in vivo by triggering rapid cell cycle progression through upregulation of the nuclear factor-kappa B2 signaling pathway, using a murine xenograft and human model of bladder cancer [33]. Upregulation of gene expression associated with cytochrome b was found in HT-29 cells of the villous subtype as well as in carcinomas [14, 15]. A study on genomic instability in colorectal adenomas and adenocarcinomas found that non-synonymous mutations in complex III were more frequent in adenomas and complex 1 mutations were more frequent in adenocarcinomas. This implies that the higher expression levels of mt-cytochrome b in tumors, compared to adjacent normal tissue, are due to mutations that lead to an increase in cell growth [36]. Therefore, the elevated expression of mt-cytochrome b that we observed in the villous adenoma could be driving its induction to invasive adenocarcinoma.
Given that MT-ATP6 and MT-COI are genes that code for proteins composing the heavy strand of the mt-genome [17], differential expression of these two subunits may be a result of posttranscriptional mechanisms, such as the upregulation or downregulation of miRNA expression. In this study, overexpression of MT-ATP6 transcripts that were observed in the villous adenoma and adenocarcinoma samples may be attributed to the higher energy required in rapidly growing colorectal cells transforming from adenopolyps to malignant cells. It has been demonstrated that subunits COI, II, and III of complex IV are frequently mutated in CRC metastases [35, 36] and the expression of each of these subunits varies in adenomas and adenocarcinomas in respect to the terms of well differentiated vs. moderate or poor cell differentiation [35]. The COI expressions were elevated in well-differentiated tumors and in HT-29 colorectal cells. In contrast, low expression levels of COX were observed in biopsies of human colonic adenocarcinomas relative to adjacent normal tissue [37]. The ATP6 subunit of complex V varied in the expression levels within different tumor types as previously observed in glioblastomas, prostate, and thyroid cancers [31, 35, 38]. Additionally, it has been suggested that mutations in this gene resulted in overexpression of ND2 protein that enhanced ROS production in prostate and head and neck cancers. Thus, changes in these genes via a mutation or by a posttranslational modification could lead to an increase in cell proliferation and/or to a decrease in apoptosis, as seen in other functional studies [37, 39].
Interestingly, our Western blot results reveal that the levels of complexes I and III appear to progressively change in tumors and their associated normal adjacent tissues when compared (Fig. 4). Normally, tissue biochemical transformation (tumor biochemical margins) would be expected to precede the tumor anatomical margins. This usually occurred as a result of molecular features in the histologically normal-appearing tissue adjacent to the histological tumor border which may have an increased or decreased protein expression relative to the tumor. The progressive change in the levels of transcripts in mt-Complexes, although not entirely mirrored in normal adjacent associated with tumor samples in this study, may be an indicative of the aberrant cells outside of the tumor margin or infiltrative tumor cells that go undetected by conventional histology[40]. Alternatively, these cells could be benign tumor cells that have already undergone malignant transformation at the molecular level yet to show no phenotypic characteristics of tumor cells. On the other hand, the possibility of cross-talk or secretory interactions between the tumor and the normal adjacent tissue should not be ruled out which could cause signaling pathway of cells outside of the histological margin to express some molecular features characteristic of the tumor [40].
Additionally, data from this study showed that there were many proteins overexpressed and under-expressed in the intermediate stage of tubulovillous adenomas relative to villous and adenocarcinoma samples. However, those of particular interest are the proteins that showed abnormal patterns outside of the histologically defined tumor margin. For example, mitochondrial proteins in this study were consistently overexpressed in the precancerous stages of tubular and tubulovillous tumors as well as in the histologically normal tissue adjacent to the tumor in complexes I and III (Fig. 4). Complexes I, II, and III are considered as some of the main sources of ROS production within the mitochondria [39, 41–43], and they have subunits that are coded by both mitochondrial and nuclear DNA [34]. Previous studies have indicated that there is an existence of a cross-talk mechanism between nucleus and mitochondria in regulating oxidative phosphorylation [17, 44, 45]. Although, the intermediate stage of tubulovillous adenomas showed more expression of these proteins relative to villous and carcinoma samples by Western blot analysis, there was an inverse relationship between these stages relative to reactive carbonyl protein content levels. A reported study that examined triple-negative breast cancers revealed a similar low expression of complex I and complex III relative to receptor-positive cell lines [34]. Also, a past study with K-ras in the transformed cells of colorectal showed a reduction in the expression of mitochondrial complex I [46] which appeared to occur in one cell of a preexisting small adenoma through clonal expansion, to produce a large and more dysplastic tumor [47] due to impaired assembly in the lack of posttranslational events or alteration of assembly factors [46, 48].
Since carbonyl protein content may reflect the amount of ROS in cells, complex I and III results in this study suggest an alteration in the ETC chain components that may lead to the disruption of electron transport, causing electron leakage that could result in the generation of ROS. The actual consequence of these deficiencies of complex I- and III-altered expression in the colorectal adenopolyps observed in the present study is unknown. However, larger scale expression profiling studies have identified genes that are differentially expressed between histological subtypes of tumors and premalignant samples [49–52]. For gastric cancer, this deficiency correlated with the induction of HIF-1α, which in turn leads to ROS production and impairment of chain components [49, 50, 52].
Prakash and Chandran found that differences in gene expression were much greater in tumors than in asymptomatic benign prostatic hyperplastic tissues adjacent to tumors which is consistent with our colorectal adenopolyps study [53, 54]. These results suggest that normal adjacent appearing tissues around tumors may also undergo tumor-related changes commonly found in tumors themselves and thought to drive their growth. The spread of clonal cell populations over large areas of epithelium appears to be an early part of the developmental pathway of some cancer types [55]. The observed significant changes in the expression levels of some mitochondrial proteins in colorectal tumors relative to adjacent normal tissues, in this study, may be considered to be driver rather than the neutrality of the passenger protein expression changes in that it predicts future tumor development and/or progression [55].
Conclusion
In summary, we have shown a progressive increase of MT-COI, MT-CYB, MT-ND1, and MT-RNR1 gene expression in colorectal adenomas and adenocarcinomas when compared to their adjacent normal tissues. Additionally, protein analysis using complex I and III antibodies showed similar results of increased expression in tumors compared to adjacent normal tissues. These data suggest that certain mitochondrial gene expression changes could be involved in the progression to malignancy of precursor lesions of colorectal adenopolyps to invasive adenocarcinoma as a result of their high metabolism during the mitotic division given the progressive increase in carbonyl protein contents among the colorectal adenopolyps observed in this study and that carbonyl protein is used as a measurement of oxidative stress in cells; it is tempting to speculate that the increase in ROS production may be critical in the induction of colorectal adenomas to invasive adenocarcinomas. Of course, the results in this study are not sufficient to support a definitive statement that higher ROS production directly correlates with colorectal adenopolyps progression. Nevertheless, we believe that the current study provides essential clues that link functional changes in mitochondrial RNA expression and higher ROS production in colorectal tumors during evolution from benign to malignant lesions.
Acknowledgments
We acknowledge the RCMI G12 MBRC Program from the National Institute of Minority Health and Health Disparities, Grant Number 8G12MD007602. To those investigators whose meritorious works could not be cited due to space limitations, we honestly apologize. This work was supported by grant NIH-NIGMS GM099663 awarded to Dr. Felix O Aikhionbare. We would also like to acknowledge William Roth and Saravanakumar Muthusamy for their technical and editing support. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH-NIMHD and NIGMS.
Abbreviations
- 12S/MT-RNR1
Mitochondrial encoded 12S ribosomal RNA
- ATCB
Beta actin
- CA
Adenocarcinoma
- Cp
Crossing point
- CRC
Colorectal cancer
- DNPH
2,4-Dinitrophenylhydrazine
- ETC
Electron transport chain
- FAP
Familial adenomatous polyposis
- GAPDH
Glyceraldehyde 3 phosphate dehydrogenase
- HILF1α
Hypoxia inducible factor 1-alpha
- MT
Mitochondrial
- MT-ATP6
ATP synthase F0 subunit 6
- MT-COI
Mitochondrial encoded cytochrome oxidase I
- MT-CYB
Mitochondrial encoded cytochrome b
- MT-DNA
Mitochondria DNA
- MT-ND1
Mitochondrial encoded NADH dehydrogenase 1
- MT-ND6
Mitochondrial encoded NADH dehydrogenase 6
- ROS
Reactive oxygen species
- RT-qPCR
Reverse transcription quantitative polymerase chain reaction
- TA
Tubular adenoma
- TV
Tubulovillous
- UBC
Ubiquitin
- V
Villous adenoma
Footnotes
Author contributions Conception and design: L. Wallace, F.O. Aikhionbare, and S. Mehrabi; development of methodology: L. Wallace, S. Mehrabi, and F.O. Aikhionbare; acquisition of data: L. Wallace, S. Mehrabi, and M. Bacanamwo; analysis and interpretation of data: L. Wallace, S. Mehrabi, X. Yao, and F.O. Aikhionbare; writing: L. Wallace, S. Mehrabi, and F.O. Aikhionbare.
Compliance with ethical standard
Conflicts of interest The authors declare no conflicts of interest.
Contributor Information
LaShanale Wallace, Email: lwallace@msm.edu.
Sharifeh Mehrabi, Email: msharifeh@msm.edu.
Methode Bacanamwo, Email: mbacanamwo@msm.edu.
Xuebiao Yao, Email: xyao@msm.edu.
Felix O. Aikhionbare, Email: faikhionbare@msm.edu.
References
- 1.Duarte FV, Palmeira CM, Rolo AP. The Role of microRNAs in mitochondria: small players acting wide. Genes (Basel) 2014;5:865–86. doi: 10.3390/genes5040865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol Mech Dis. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 3.Winawer SJ, Zauber AG, Ho MN, O’Brien MJ, Gottlieb LS, Sternberg SS, et al. Prevention of colorectal cancer by colonoscopic polypectomy. N Engl J Med. 1993;329:1977–81. doi: 10.1056/NEJM199312303292701. [DOI] [PubMed] [Google Scholar]
- 4.Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends in Genetics. 1993;9:138–41. doi: 10.1016/0168-9525(93)90209-z. [DOI] [PubMed] [Google Scholar]
- 5.Czarnecka A, Golik P, Bartnik E. Mitochondrial DNA mutations in human neoplasia. J Appl Genet. 2006;47:67–78. doi: 10.1007/BF03194602. [DOI] [PubMed] [Google Scholar]
- 6.Weren RDA, Ligtenberg MJL, Kets CM, de Voer RM, Verwiel ETP, Spruijt L, et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet. 2015;47:668–71. doi: 10.1038/ng.3287. [DOI] [PubMed] [Google Scholar]
- 7.Bettington M, Walker N, Clouston A, Brown I, Leggett B, Whitehall V. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology. 2013;62:367–86. doi: 10.1111/his.12055. [DOI] [PubMed] [Google Scholar]
- 8.Adams G, Mehrabi S, Vatcharapijarn Y, Iyamu OI, Akwe JA, Grizzle WE, et al. Frequencies of mtDNA mutations in primary tissue of colorectal adenopolyps. Front Biosci (Elite Ed) 2013;5:809–13. doi: 10.2741/e661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aikhionbare F, Khan M, Carey D, Okoli J, Go R. Is cumulative frequency of mitochondrial DNA variants a biomarker for colorectal tumor progression? Mol Cancer. 2004;3:30. doi: 10.1186/1476-4598-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polyak K, Li Y, Zhu H, Lengauer C, Willson JKV, Markowitz SD, et al. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet. 1998;20:291–3. doi: 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- 11.Sun C, Reimers LL, Burk RD. Methylation of HPV16 genome CpG sites is associated with cervix precancer and cancer. Gynecol Oncol. 2011;121:59–63. doi: 10.1016/j.ygyno.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mehrabi S, Partridge EE, Seffens W, Yao X, Aikhionbare FO. Oxidatively modified proteins in the serous subtype of ovarian carcinoma. Biomed Res Int. 2014;2014:585083. doi: 10.1155/2014/585083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chester KA, Robson L, Begent RHJ, Pringle H, Primrose L, Talbot IC, et al. In situ and slot hybridization analysis of RNA in colorectal tumours and normal colon shows distinct distributions of mitochondrial sequences. J Pathol. 1990;162:309–15. doi: 10.1002/path.1711620406. [DOI] [PubMed] [Google Scholar]
- 14.Lee HC, Yin PH, Lin JC, Wu CC, Chen CY, Wu CW, et al. Mitochondrial Genome Instability and mtDNA Depletion in Human Cancers. Annals of the New York Academy of Sciences. 2005;1042:109–22. doi: 10.1196/annals.1338.011. [DOI] [PubMed] [Google Scholar]
- 15.Lu X, Walker T, MacManus JP, Seligy VL. Differentiation of HT-29 human colonic adenocarcinoma cells correlates with increased expression of mitochondrial RNA: effects of trehalose on cell growth and maturation. Cancer Research. 1992;52:3718–25. [PubMed] [Google Scholar]
- 16.Yamamoto A, Horai S, Yuasa Y. Increased level of mitochondrial gene expression in polyps of familial polyposis coli patients. Biochemical and Biophysical Research Communications. 1989;159:1100–6. doi: 10.1016/0006-291x(89)92222-5. [DOI] [PubMed] [Google Scholar]
- 17.Abril J, De Heredia ML, González L, Cléries R, Nadal M, Condom E, et al. Altered expression of 12S/MT-RNR1, MT-CO2/COX2, and MT-ATP6 mitochondrial genes in prostate cancer. Prostate. 2008;68:1086–96. doi: 10.1002/pros.20771. [DOI] [PubMed] [Google Scholar]
- 18.Evans P, Lyras L, Halliwell B. Methods in Enzymology Oxidants and Antioxidants Part B. Academic Press; 1999. Measurement of protein carbonyls in human brain tissue; pp. 145–56. [DOI] [PubMed] [Google Scholar]
- 19.Beal MF. Oxidatively modified proteins in aging and disease1,2. Free Radical Biology and Medicine. 2002;32:797–803. doi: 10.1016/s0891-5849(02)00780-3. [DOI] [PubMed] [Google Scholar]
- 20.Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG, et al. Methods in enzymology oxygen radicals in biological systems part B: oxygen radicals and antioxidants. Academic Press; 1990. Determination of carbonyl content in oxidatively modified proteins; pp. 464–78. [DOI] [PubMed] [Google Scholar]
- 21.Bragoszewski P, Kupryjanczyk J, Bartnik E, Rachinger A, Ostrowski J. Limited clinical relevance of mitochondrial DNA mutation and gene expression analyses in ovarian cancer. BMC Cancer. 2008;8:292. doi: 10.1186/1471-2407-8-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubie C, Kempf K, Hans J, Su T, Tilton B, Georg T, et al. Housekeeping gene variability in normal and cancerous colorectal, pancreatic, esophageal, gastric and hepatic tissues. Molecular and Cellular Probes. 2005;19:101–9. doi: 10.1016/j.mcp.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 23.Andersen CL, Jensen JL, Ørntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Research. 2004;64:5245–50. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- 24.Mehrabi S, Wallace L, Cohen S, Yao X, Aikhionbare FO. Differential measurements of oxidatively modified proteins in colorectal adenopolyps. Int J Clin Med. 2015;6:288–99. doi: 10.4236/ijcm.2015.64037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carew JS, Huang P. Mitochondrial defects in cancer. Mol Cancer. 2002;1:9. doi: 10.1186/1476-4598-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grzybowska-Szatkowska L, Slaska B, Rzymowska J, Brzozowska A, Floriańczyk B. Novel mitochondrial mutations in the ATP6 and ATP8 genes in patients with breast cancer. Mol Med Rep. 2014;10:1772–8. doi: 10.3892/mmr.2014.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun AS, Cederbaum AI. Oxidoreductase activities in normal rat liver, tumor-bearing rat liver, and hepatoma HC-252. Cancer Research. 1980;40:4677–81. [PubMed] [Google Scholar]
- 28.Chandra D, Singh KK. Genetic insights into OXPHOS defect and its role in cancer. Biochimica et Biophysica Acta (BBA) -Bioenergetics. 2011;1807:620–5. doi: 10.1016/j.bbabio.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akouchekian M, Houshmand M, Akbari MHH, Kamalidehghan B, Dehghan M. Analysis of mitochondrial ND1 gene in human colorectal cancer. J Res Med Sci. 2011;16:50–5. [PMC free article] [PubMed] [Google Scholar]
- 30.Rigoulet M, Yoboue ED, Devin A. Mitochondrial ROS generation and its regulation: mechanisms involved in H2O2 signaling. Antioxidants & Redox Signaling. 2010;14:459–68. doi: 10.1089/ars.2010.3363. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Marcillat O, Giulivi C, Ernster L, Davies KJ. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. Journal of Biological Chemistry. 1990;265:16330–6. [PubMed] [Google Scholar]
- 32.Saybaşili H, Yϋksel M, Haklar G, Yal in AS. Effect of mitochondrial electron transport chain inhibitors on superoxide radical generation in rat hippocampal and striatal slices. Antioxid Redox Signal. 2001;3(6):1099–104. doi: 10.1089/152308601317203602. [DOI] [PubMed] [Google Scholar]
- 33.Dasgupta S, Hoque MO, Upadhyay S, Sidransky D. Mitochondrial cytochrome B gene mutation promotes tumor growth in bladder cancer. Cancer Research. 2008;68:700–6. doi: 10.1158/0008-5472.CAN-07-5532. [DOI] [PubMed] [Google Scholar]
- 34.Pelicano H, Zhang W, Liu J, Hammoudi N, Dai J, Xu RH, et al. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: role of mTOR pathway and therapeutic potential. Breast Cancer Research. 2014;16:434. doi: 10.1186/s13058-014-0434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heerdt BG, Halsey HK, Lipkin M, Augenlicht LH. Expression of mitochondrial cytochrome c oxidase in human colonic cell differentiation, transformation, and risk for colonic cancer. Cancer Research. 1990;50:1596–600. [PubMed] [Google Scholar]
- 36.Herrmann PC, Gillespie JW, Charboneau L, Bichsel VE, Paweletz CP, Calvert VS, et al. Mitochondrial proteome: altered cytochrome c oxidase subunit levels in prostate cancer. PROTEOMICS. 2003;3:1801–10. doi: 10.1002/pmic.200300461. [DOI] [PubMed] [Google Scholar]
- 37.Sun AS, Sepkowitz K, Geller SA. A study of some mitochondrial and peroxisomal enzymes in human colonic adenocarcinoma. Laboratory Investigation. 1981;44:13–7. [PubMed] [Google Scholar]
- 38.Dmitrenko V, Shostak K, Boyko O, Khomenko O, Rozumenko V, Malisheva T, et al. Reduction of the transcription level of the mitochondrial genome in human glioblastoma. Cancer Letters. 2005;218:99–107. doi: 10.1016/j.canlet.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 39.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: a mechanism of O2 sensing. Journal of Biological Chemistry. 2000;275(33):25130–8. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 40.Oppenheimer SR, Mi D, Sanders ME, Caprioli RM. A Molecular Analysis of Tumor Margins by MALDI Mass Spectrometry in Renal Carcinoma. J Proteome Res. 2010;9:2182–90. doi: 10.1021/pr900936z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Higuchi M. Roles of Mitochondrial DNA Changes on Cancer Initiation and Progression. Cell Biol (Henderson, NV) 2012;1:109. doi: 10.4172/2324-9293.1000e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 2012;287:27255–64. doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Musatov A, Robinson NC. Susceptibility of mitochondrial electron-transport complexes to oxidative damage. Focus on cytochrome c oxidase. Free Radical Research. 2012;46:1313–26. doi: 10.3109/10715762.2012.717273. [DOI] [PubMed] [Google Scholar]
- 44.Poyton RO, McEwen JE. Crosstalk between nuclear and mitochondrial genomes. Annu Rev Biochem. 1996;65:563–607. doi: 10.1146/annurev.bi.65.070196.003023. [DOI] [PubMed] [Google Scholar]
- 45.Delsite R, Kachhap S, Anbazhagan R, Gabrielson E, Singh K. Nuclear genes involved in mitochondria-to-nucleus communication in breast cancer cells. Mol Cancer. 2002;1:1–10. doi: 10.1186/1476-4598-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baracca A, Chiaradonna F, Sgarbi G, Solaini G, Alberghina L, Lenaz G. Mitochondrial complex I decrease is responsible for bioenergetic dysfunction in K-ras transformed cells. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2010;1797:314–23. doi: 10.1016/j.bbabio.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 47.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 48.De Rasmo D, Panelli D, Sardanelli AM, Papa S. cAMP-dependent protein kinase regulates the mitochondrial import of the nuclear encoded NDUFS4 subunit of complex I. Cellular Signalling. 2008;20:989–97. doi: 10.1016/j.cellsig.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 49.Puurand M, Peet N, Piirsoo A, Peetsalu M, Soplepmann J, Sirotkina M, et al. Deficiency of the complex I of the mitochondrial respiratory chain but improved adenylate control over succinate-dependent respiration are human gastric cancer-specific phenomena. Mol Cell Biochem. 2012;370:69–78. doi: 10.1007/s11010-012-1399-3. [DOI] [PubMed] [Google Scholar]
- 50.Lim HY, Ho QS, Low J, Choolani M, Wong KP. Respiratory competent mitochondria in human ovarian and peritoneal cancer. Mitochondrion. 2011;11:437–43. doi: 10.1016/j.mito.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 51.Simonnet H, Demont J, Pfeiffer K, Guenaneche L, Bouvier R, Brandt U, et al. Mitochondrial complex I is deficient in renal oncocytomas. Carcinogenesis. 2003;24:1461–6. doi: 10.1093/carcin/bgg109. [DOI] [PubMed] [Google Scholar]
- 52.Bonora E, Porcelli AM, Gasparre G, Biondi A, Ghelli A, Carelli V, et al. Defective oxidative phosphorylation in thyroid oncocytic carcinoma is associated with pathogenic mitochondrial dna mutations affecting complexes I and III. Cancer Research. 2006;66:6087–96. doi: 10.1158/0008-5472.CAN-06-0171. [DOI] [PubMed] [Google Scholar]
- 53.Chandran UR, Dhir R, Ma C, Michalopoulos G, Becich M, Gilbertson J. Differences in gene expression in prostate cancer, normal appearing prostate tissue adjacent to cancer and prostate tissue from cancer free organ donors. BMC Cancer. 2005;5:1–11. doi: 10.1186/1471-2407-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prakash K, Pirozzi G, Elashoff M, Munger W, Waga I, Dhir R, et al. Symptomatic and asymptomatic benign prostatic hyperplasia: molecular differentiation by using microarrays. Proceedings of the National Academy of Sciences. 2002;99:7598–603. doi: 10.1073/pnas.112191399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]