

Abstract
The c-Jun N-terminal kinases (JNKs) are members of the mitogen-activated protein kinase family and have been implicated in tumorigenesis. One isoform in particular, JNK2α, has been shown to be frequently activated in primary brain tumors, to enhance several tumorigenic phenotypes and to increase tumor formation in mice. As JNK is frequently activated in non-small cell lung carcinoma (NSCLC), we investigated the role of the JNK2α isoform in NSCLC formation by examining its expression in primary tumors and by modulating its expression in cultured cell lines. We discovered that 60% of the tested primary NSCLC tumors had three-fold higher JNK2 protein and two- to three-fold higher JNK2α mRNA expression than normal lung control tissue. To determine the importance of JNK2α in NSCLC progression, we reduced JNK2α expression in multiple NSCLC cell lines using short hairpin RNA. Cell lines deficient in JNK2α had decreased cellular growth and anchorage-independent growth, and the tumors were four-fold smaller in mass. To elucidate the mechanism by which JNK2α induces NSCLC growth, we analyzed the JNK substrate, signal transducer and activator of transcription 3 (STAT3). Our data demonstrates for the first time that JNK2α can regulate the transcriptional activity of STAT3 by phosphorylating the Ser727 residue, thereby regulating the expression of oncogenic genes, such as c-Myc. Furthermore, reintroduction of JNK2α2 or STAT3 restored the tumorigenicity of the NSCLC cells, demonstrating that JNK2α is important for NSCLC progression. Our studies reveal a novel mechanism in which phosphorylation of STAT3 is mediated by a constitutively active JNK2 isoform, JNK2α.
Keywords: NSCLC, JNK, STAT3, MAPK
Introduction
The c-Jun N-terminal kinases (JNK) members of the mitogen-activated protein kinase (MAPK) family can mediate apoptosis or proliferation through the phosphorylation of transcription factors (Yeager et al., 1995; Butterfield et al., 1997; Arbour et al., 2002; Lee et al., 2008). Several lines of evidence indicate that JNK is important in promoting cell growth and tumorigenesis (Bode and Dong, 2007; Bogoyevitch and Arthur, 2008). First, enhanced JNK activity promotes tumorigenesis in cells (Berner et al., 1999). Second, several tumor cell lines have been reported to have constitutive JNK activity and enhanced expression (Xu et al., 1996; Rodrigues et al., 1997). Lastly, inhibition of JNK expression reduces glial tumor formation (Potapova et al., 2000).
There are three distinct JNK genes (JNK1, JNK2 and JNK3) and 10 different splicing isoforms. The JNK signaling pathway is activated by numerous stimuli, resulting in several seemingly contradictory cellular responses (Cheng et al., 2002). For example, JNK1 activity is associated with apoptosis and tumor suppression, whereas JNK2 activity can stimulate cell proliferation and tumor formation (Chen et al., 2001; Yang et al., 2003). Consequently, each JNK gene and individual isoform most likely mediate different functions depending on the stimulus (Bogoyevitch, 2006).
Several findings suggest that one particular JNK2 isoform, JNK2α, has a significant role in tumorigenesis. The JNK2α isoform is unique, possessing the ability to autophosphorylate and consequently autoactivate. It has been shown to have enhanced expression in primary glial tumors (Cui et al., 2005; Tang et al., 2006), and this constitutively active isoform has been found to increase tumorigenic phenotypes and enhance tumor formation in nude mice (Tang et al., 2006; Nitta et al., 2008). Thus far, the JNK2α isoform has only been linked to glial tumorigenesis; however, it may also be involved in non-small cell lung carcinoma (NSCLC) progression. Recent studies show that NSCLC tumors and cell lines have constitutively active JNK and that the inhibition of JNK activity decreased NSCLC cell growth (Khatlani et al., 2007). Furthermore, reduced expression of JNK2, but not JNK1, decreased tumorigenic phenotypes in NSCLC cells (Berner et al., 1999). On the basis of these reports we hypothesized that the constitutively active JNK2α isoform may be an important component of NSCLC tumorigenesis.
In this study we sought to determine whether JNK2α has a role in NSCLC tumorigenesis. We examined the expression of this particular JNK isoform in primary NSCLC tumors and used genetic and biochemical approaches to modulate JNK2α expression and activity in cultured NSCLC cells. We found that the majority of NSCLC tumors have increased JNK2α expression. To determine whether this JNK isoform has a direct role in NSCLC formation, we generated multiple NSCLC cell lines deficient in JNK2α, using retroviruses to introduce short hairpin RNA (shRNA). We discovered that the reducing JNK2α expression decreased tumorigenic phenotypes and tumor formation in mice. To elucidate the mechanism by which JNK2α induces NSCLC tumorigenesis, we studied a well-known oncogene and JNK substrate, STAT3. Reducing JNK2α expression in a NSCLC cell line yielded a significant reduction in STAT3 phosphorylation at the Ser727 residue and a decrease in STAT3 transcriptional activity. Furthermore, reintroducing JNK2α2 or STAT3 rescued the tumorigenicity of NSCLC cells deficient in JNK2α demonstrating that the JNK2α isoform is directly involved in NSCLC progression. Overall our findings show that JNK2α can induce NSCLC tumor formation by regulating the activity of oncogenic downstream substrates, such as STAT3.
Results
The expression of JNK2α is enhanced in NSCLC tumors
We initially analyzed 10 primary NSCLC tumors for JNK expression. 60% of these samples had two- to three-fold increased levels of JNK2, but not JNK1 or JNK3, as compared with a representative normal lung sample (Figure 1a, Supplementary Figure 1A, Supplementary Table 1). Presently available commercial JNK antibodies can only be used to distinguish the three JNK genes (JNK1, JNK2 and JNK3) and not specific isoforms, so we designed isoform-specific quantitative reverse transcriptase PCR (QRT-PCR) primers to differentiate between the JNK isoforms. Despite having very similar amino acid sequences, the JNK isoforms have sufficiently divergent nucleotide sequences that enabled us to differentiate between these isoforms. To monitor JNK expression, we compared the tumor samples to two representative normal lung samples that were extracted from similar regions of the lung. Using QRT-PCR, we discovered that the JNK2α isoform had two- to three-fold higher mRNA expression in NSCLC tumors, whereas the very similar JNK2β isoform had no significant change (Figure 1b). It is interesting that the QRT-PCR analysis also revealed that the JNK2α-isoform is the predominant JNK2 isoform expressed, having significantly higher mRNA expression compared with JNK2β (data not shown). We extended our QRT-PCR analysis to seven additional primary tumors and discovered that 70% had two- to three-fold higher JNK2α mRNA expression (Supplementary Figure 1B).
Figure 1.
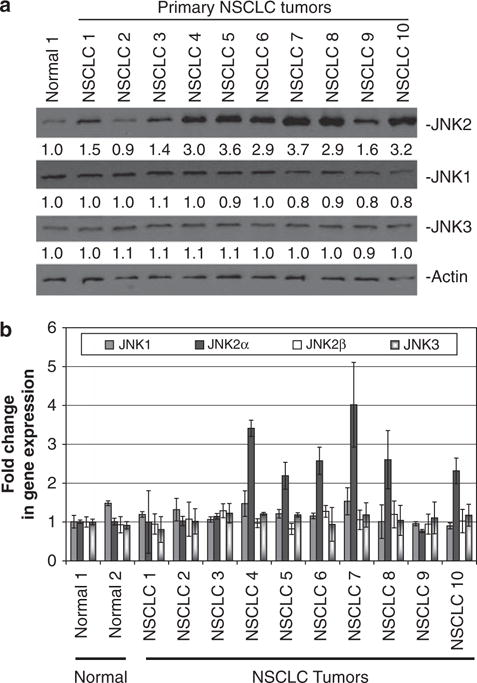
Expression of JNK2α in human non-small cell lung carcinoma (NSCLC). Expression of JNK isoforms in normal lung and in NSCLC tumors. (a) Protein samples were separated using 4–20% sodium dodecyl sulphate–poly acrylamide gel electrophoresis (SDS–PAGE). The numbers under each blot corresponds to the fold change in protein levels relative to normal lung. One representative normal lung sample of five samples is shown. (b) The JNK isoform mRNA levels were determined using quantitative reverse transcriptase PCR (RT–PCR).
Reduction of endogenous JNK2α in NSCLC cell lines using shRNAs
To determine whether JNK2α is directly involved in lung tumorigenesis, we studied the tumorigenic effects of JNK2α in two NSCLC cell lines, NCI-H2009 and HCC-827. Previous reports indicated that these two cell lines have constitutively active JNK that enhances cell proliferation, but did not specify which isoform was responsible (Khatlani et al., 2007). We analyzed the significance of the constitutively active JNK2α in these cell lines, hypothesizing that JNK2α is responsible for inducing lung tumorigenesis and, therefore, reducing endogenous JNK2α should ameliorate the tumorigenic potential of these cells. To test this hypothesis, we utilized the divergent nucleotide sequences in the JNK isoforms to introduce shRNA specific for a nucleotide sequence unique to the JNK2α gene (shJNK2α). To control for the retroviral infections, we infected cells with a nonspecific scrambled sequence (shScrambled) and analyzed uninfected cells (uninfected). Western analysis revealed that the HCC-827 shJNK2α cells had a six-fold decrease in JNK2 compared with the uninfected or shScrambled control cells, whereas the NCI-H2009 shJNK2α cells had a seven-fold reduction (Figure 2a). JNK1 and JNK3 levels were similar in all the tested cell lines indicating that the knockdown was specific to the JNK2 gene. As a further control, we also analyzed another MAPK protein, ERK1/2 and determined that our shRNA specifically targeted the JNK pathway. QRT-PCR analysis verified that the JNK2α isoform was being specifically knocked down as the HCC-827 shJNK2α cells had a 3.5-fold reduction in JNK2α expression, whereas the H2009 shJNK2α cells had a four-fold decrease (Figure 2b). Consistently, the expression of JNK1, JNK3 and the highly similar JNK2β isoform were not altered.
Figure 2.
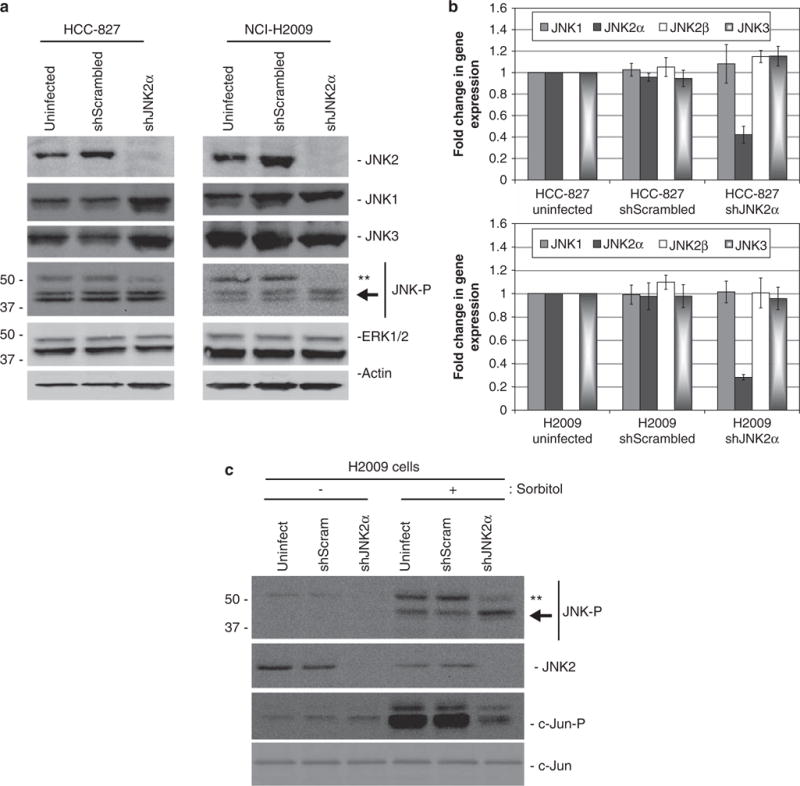
Reduction of endogenous JNK2α in NSCLC cell lines. Retroviral infection of shRNAs targeted to JNK2α in HCC-827 or NCI-H2009 cells (shJNK2α). Uninfected cells and shRNAs targeted to a scrambled sequence (shScrambled) were used as controls. (a) Protein samples were separated using 4–20% SDS–PAGE. (b) Relative expression of JNK isoforms using QRT–PCR analysis for HCC-827-infected cells (top) and NCI-H2009-infected cells (bottom). (c) H2009 cells were serum starved for 24h and then stimulated with 500 mM sorbitol. The protein samples were separated using 4–20% SDS–PAGE and detected with the specified antibodies. **-represents the JNK2 isoform. The arrow indicates the JNK1 isoforms.
We subsequently examined whether JNK activity was reduced in our shJNK2α cell lines. Western analysis showed that the cells deficient in JNK2α had reduced JNK2 phosphorylation compared with the control cell lines, whereas the phosphorylation levels of the other JNK proteins were not altered (Figure 2a). In addition, when we stimulated JNK activity with sorbitol, we discovered that the H2009 shJNK2α cells had reduced JNK2 and c-Jun phosphorylation compared with the uninfected control cells (Figure 2c). Together these results suggest that the reducing JNK2α expression in H2009 cells not only reduced the overall levels of JNK phosphorylation but also reduced the ability of JNK to phosphorylate downstream substrates, such as c-Jun.
JNK2α is necessary for tumorigenic phenotypes in NSCLC cells in vitro
On the basis of the previous studies showing that a constitutively active JNK2α isoform is expressed in human gliomas, we determined whether decreasing JNK2α expression could decrease or prevent NSCLC tumor formation (Tsuiki et al., 2003). We discovered that the shJNK2α-treated cells had a significantly slower growth rate compared with the control cells and a reduced ability to grow in soft agar (Figures 3a–c). To verify these findings, we extended our analysis to two additional NSCLC cell lines, NCI-H1650 and HCI-H1975. These cell lines were not shown to have constitutive JNK activity, but JNK inhibitors decreased their cell growth (Khatlani et al., 2007). Consistent with our previous findings, we were able to specifically reduce JNK2α expression five-fold (Supplementary Figure 2A & B), which correlated with a significant reduction in cell growth and colony formation in soft agar (Supplementary Figure 2C & D). To determine whether JNK2α has a similar role in small cell lung carcinomas tumorigenesis, we reduced JNK2α expression in two SCLC cell lines, NCI-H82 and NCI-H62. We discovered that reducing expression of JNK2α did not alter cell growth or anchorage-independent growth in SCLC cells, suggesting that JNK2α may not be important for SCLC formation (Supplementary Figures 3A–D).
Figure 3.
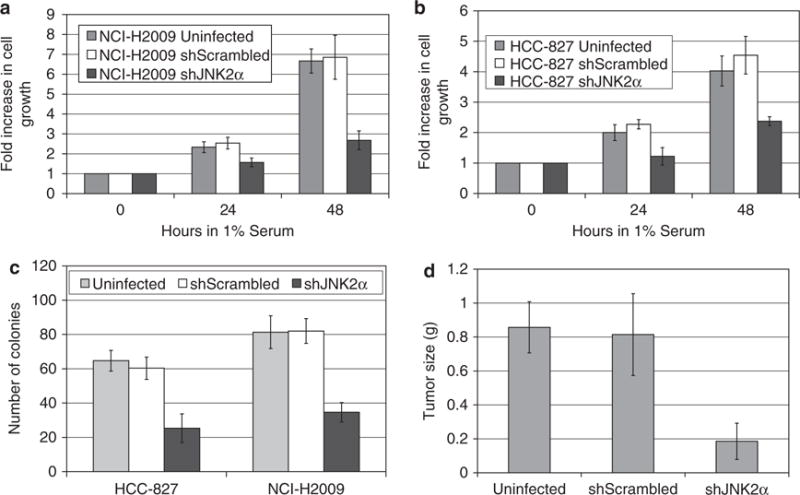
JNK2α promotes tumorigenesis in NSCLC in vitro and in vivo. (a) Cell growth analysis of NCI-H2009-infected cells. The cell lines were cultured in 1% fetal bovine serum and viable cells were counted daily. Results are from two separate experiments, each done in triplicate. (b) Cell growth analysis of HCC-827. (c) Anchorage-independent growth of each cell lines in soft agar. Results are from two separate experiments, each done in triplicate. (d) NOD/SCID mice were injected subcutaneously with the specified NCI-H2009-infected cell lines (n = 10 for each group). The tumors were removed after 7 weeks and were subsequently weighed.
JNK2α is important for NSCLC tumorigenicity in NOD/SCID mice
We extended our study by testing the effects of reducing JNK2α on tumor formation, predicting that reducing endogenous JNK2α expression would reduce the tumorigenicity of the NSCLC cells. To this end, we subcutaneously injected the H2009 Uninfected, shScrambled, or shJNK2α cells into the hind limb of athmyic mouse. After 7 weeks, we removed the tumors and discovered the mice injected with the control cells formed tumors that were ∼0.8g, whereas the shJNK2α cells yielded tumors that were significantly smaller in mass, only ∼0.2g (Figure 3d). We verified that the smaller tumors had reduced JNK2α expression through western and QRT-PCR analysis (data not shown).
JNK2α forms a complex with STAT3
One of the primary functions of JNK is to phosphorylate and activate transcription factors. One intriguing substrate of JNK is STAT3 (Lim and Cao, 1999). STAT3 is a well-established oncogene that is known to regulate pro-survival and pro-proliferative signaling in NSCLC cells (Alvarez et al., 2006). To elucidate the mechanism by which JNK2α achieves its tumorigenic effects, we determined whether JNK2α could form a complex with STAT3. 3xFlag tagged JNK2α2 (a JNK2α isoform) was transfected into H2009 cells and co-immunoprecipitation experiments were performed. We discovered that the 3xFlag JNK2α2 was able to co-immunoprecipitate with endogenous STAT3 (Figure 4a). We extended our analysis by determining whether the formation of the JNK2—STAT3 complex was decreased in cells lacking JNK2α. To this end, we conducted co-immunoprecipitation experiments using antibodies specific for endogenous STAT3 or JNK2 (as there are no commercially available JNK2α antibodies) in the H2009 shJNK2α cells. We discovered that the absence of JNK2α significantly decreased the amount of STAT3 that co-immunoprecipitated with JNK2 suggesting that JNK2α is an important component of the JNK–STAT3 complex (Figure 4b).
Figure 4.
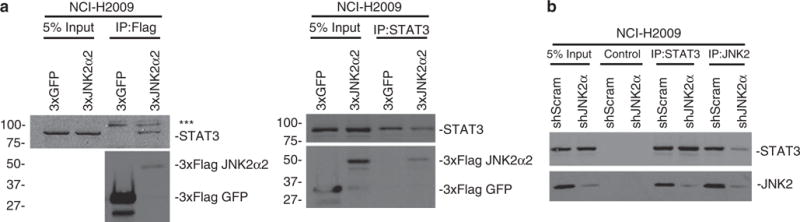
JNK2α can form a complex with STAT3 in NSCLC cells. Co-immunoprecipitation reactions indicate that JNK2α can interact with STAT3 in cells. (a) 3xFlag-JNK2α2 was transfected in NCI-H2009 cells and the immunoprecipitates obtained with anti-Flag (left) or anti-STAT3 (right) were separated by SDS–PAGE and detected by immunoblotting with anti-STAT3, JNK2 or Flag. ***-represents a non-specific band in the immunoprecipitation due to a well-known contaminant (IgG chain). (b) Co-immunoprecipitation for endogenous STAT3 and JNK2 using H2009 shJNK2α or shScrambled (shScram) cells. An IgG control antibody was used as the control.
JNK2α can regulate the phosphorylation and activity of STAT3 in NSCLC cells
To ascertain whether JNK2α is involved in regulating STAT3 activity, we looked at the ability of JNK2α2 to phosphorylate serine 727 of STAT3. Phosphorylation of this residue has been shown to enhance cell proliferation and tumorigenesis (Qin et al., 2008; Yeh et al., 2009). We initially conducted in vitro kinase assays using bacterially expressed and purified JNK2α2 and STAT3. Wild-type JNK2α2 enhanced the phosphorylation of Ser727 three-fold compared with STAT3 alone (Figure 5a). As JNK can phosphorylate serine/threonie and tyrosine residues, we tested whether the JNK2α2 phosphorylation of STAT3 was non-specific by analyzing the phosphorylation of tyrosine 705. As Tyr705 phosphorylation was not altered, we concluded that JNK2α2 can only phosphorylate Ser727 (Figure 5a). To verify that JNK2α2 is directly responsible for Ser727 phosphorylation, we also analyzed a kinase dead mutation, K55R. Consistent with previous findings, we discovered that the inactive K55R mutant did not autophosphorylate, and did not alter the phosphorylation of Ser727 (Nitta et al., 2008).
Figure 5.
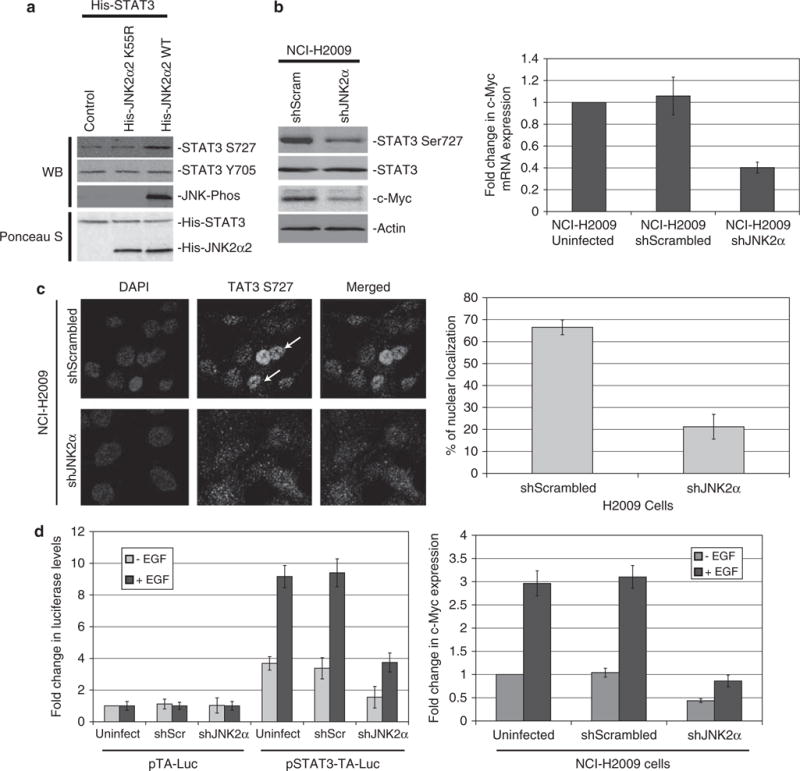
JNK2α is important for STAT Ser727 phosphorylation and STAT3 transcriptional activity in NSCLC cells. (a) In vitro kinase reactions with recombinant His-STAT3 and His-JNK2α2 wild-type (WT) or a kinase dead JNK2α2 mutant (K55R). (b) Left. Western analysis of H2009-infected cells. Right. QRT—PCR analysis of NCI-H2009-infected cells for c-MYC mRNA expression. (c) Left. Immunofluorescence of phosphorylated S727 on STAT3 in H2009-infected cells. Arrow indicates cell with a strong nuclear localization. Right. Percent of infected cells that possessed a nuclear localization. (d) Left. NCI-H2009 cells were transiently co-transfected with a STAT3 luciferase reporter construct (pSTAT3-TA-Luc) or the vector control (pTA-Luc) and a Renilla reporter construct (pRL-CMV), serum starved for 24 h and then stimulated with EGF. Data represent ratios of firefly-Luc activity derived from pSTAT3-TA-Luc over Renilla-Luc activity. The relative fold change of luciferase was calculated by normalizing to the serum-starved (-EGF) H2009-uninfected cells. Data shown represent the mean ± s.e. from three separate experiments performed in triplicate. Right. QRT–PCR analysis of serum-starved NCI-H2009-infected cells stimulated with EGF for c-MYC expression.
We extended our analysis to H2009 shJNK2α cells and discovered that Ser727 phosphorylation was decreased four-fold compared with the shScrambled cells (Figure 5b). It is interesting that the phosphorylated Ser727 produced a strong nuclear signal in the shScrambled cells, yet this nuclear accumulation was significantly impaired in the H2009 shJNK2α cells (Figure 5c). To be transcriptionally active, STAT3 must translocate to the nucleus and recent reports suggest that phosphorylation of Ser727 has an important role in this translocation (Bhattacharya et al., 2005; Qin et al., 2008). To determine whether the decrease in STAT3 Ser727 phosphorylation and nuclear accumulation significantly altered STAT3 transcriptional activity, we analyzed the expression of a STAT3 regulated gene, c-MYC. c-MYC expression is enhanced in NSCLC, is necessary for NSCLC metastasis and has been shown to be transcriptionally upregulated upon Ser727 phosphorylation (Broers et al., 1993; Gazzeri et al., 1994; Yakut et al., 2003; Qin et al., 2008; Rapp et al., 2009). H2009 shJNK2α cells had three-fold less c-MYC protein compared with the control cells and 2.5-fold less mRNA demonstrating that the loss of JNK2α significantly reduced expression of a STAT3-regulated oncogene (Figure 5b).
To determine whether JNK2α is important for STAT3 transcriptional activity, we performed reporter gene assays that measured STAT3-dependent transcriptional activity in the H2009 shJNK2α cells. A firefly-luciferase-based reporter plasmid that contains four copies of the STAT3 enhancer element (pSTAT3-TA-Luc) or the empty vector (pTA-Luc) was co-transfected into the NSCLC cells with a Renilla-luciferase reporter plasmid (pRL-CMV). The Renilla-luciferase plasmid was used as an internal control for the normalization of STAT3 reporter activity. The cells were serum starved for 24h after which both firefly and Renilla luciferase levels were measured. We discovered that the Uninfected and shScrambled cells transfected with pSTAT3-TA-Luc had a two- to three-fold increase in luciferase activity compared with the pTA-Luc transfected cells, whereas the H2009 shJNK2α cells had no significant change suggesting that cells deficient in JNK2α had reduced basal activity of STAT3. To verify that pSTAT3-TA-Luc was specific for STAT3 activity, we stimulated the cells with epidermal growth factor (EGF) that was shown to enhance STAT3 transcriptional activity through phosphorylation of Ser727 (Lufei et al., 2007; Onishi et al., 2008). We found that the pSTAT3-TA-Luc transfected cells had a 2.5-fold increase in luciferase activity, whereas the cells transfected with pTA-Luc control vector had no change (Figure 5d). It is interesting that, even with EGF stimulation the H2009 shJNK2α cells had 2.5-fold less luciferase activity compared with the control cells showing that the absence of endogenous JNK2α significantly decreased, but did not completely abolish, STAT3 transcriptional activity (Figure 5d). To verify that cells deficient in JNK2α had reduced expression of STAT3-regulated genes under EGF stimulation, we analyzed c-MYC mRNA levels and determined that EGF stimulation increased c-MYC expression three-fold in the control cells. The H2009 shJNK2α cells had two-fold less expression under serum deprivation conditions and a three-fold decrease with EGF stimulation (Figure 5d). Together these findings show that JNK2α is important for STAT3 transcriptional activity and regulation of STAT3-dependent genes through the phosphorylation of Ser727.
Reintroduction of JNK2α2 or STAT3 rescues tumorigenic phenotypes in NSCLC cells
To directly establish the significance of JNK2α in NSCLC tumorigenesis we stably introduced a JNK2α2 mutant that is resistant to the shJNK2α knockdown (JNK2α2res) into the H2009 shJNK2α cells. It is surprising that the introduction of the yellow fluorescent protein (YFP)-tagged JNK2α2res increased untagged JNK2 expression. This is most likely due to either proteolytic cleavage of the YFP tag or competition with shRNA. Western analysis showed that YFP–JNK2α2res restored phosphorylation of STAT3 Ser727 and the expression of c-Myc (Figure 6a). In addition, JNK2α2res restored cell proliferation and anchorage independence confirming the importance of JNK2α in NSCLC tumor progression (Figures 6b and c). To determine whether JNK2α induces tumorigenic phenotypes occurs through phosphorylation of Ser727, we stably overexpressed wild-type STAT3, a phospho-deficient mutant (S727A) or a phospho-mimetic mutant (S727E) in H2009 shJNK2α cells. Expression of the exogenous wild-type STAT3 and S727E mutant restored S727 phosphorylation along with c-MYC expression, whereas there were no changes with the expression of the phospho-deficient mutant S727A (Figure 6a). It is interesting that the wild-type STAT3 restored cell proliferation 80% and anchorage-independent growth 85%, whereas the S727A mutant phenocopied the shJNK2α vector control cells and the S727E phenocopied the shScrambled control cells (Figures 6b and c). This finding suggests that, although the JNK2α-STAT3 complex has an important role in NSCLC progression, there are other downstream substrates of JNK2α that may also partially contribute to NSCLC tumorigenesis.
Figure 6.
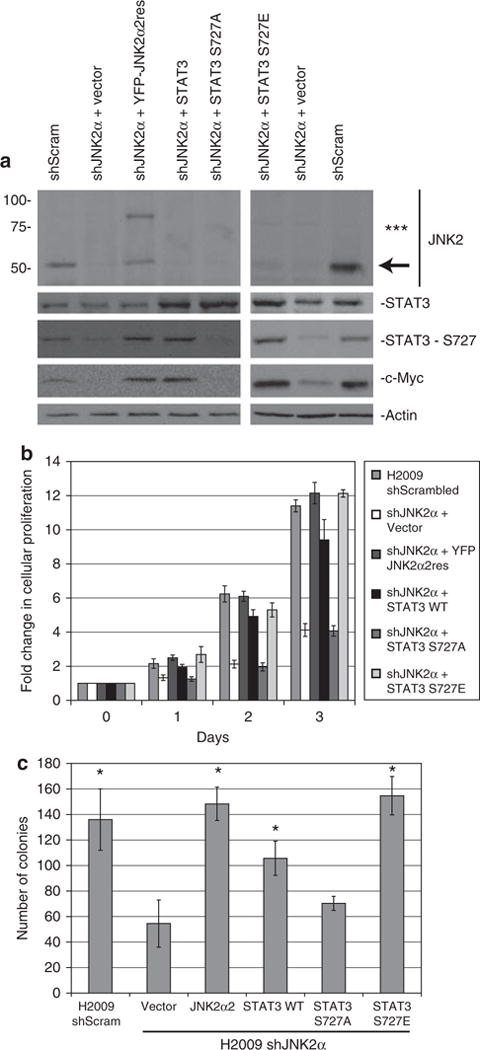
Reintroduction of JNK2α2 or STAT3 rescues the tumorigenic phenotypes in H2009 shJNK2α cells. Retroviral infection of JNK2α2 that is resistant to shJNK2α (JNK2α2res) or STAT3 in NCI-H2009 shJNK2α cells. (a) Protein samples were separated using 4–20% SDS–AGE and detected by immunoblot. ***-represents the YFP-JNK2α2res and the arrow depicts untagged JNK2. (b) Cell growth analysis of H2009 shJNK2α-infected cells. The cell lines were cultured with 1% fetal bovine serum and viable cells were counted daily. Results are from two separate experiments, each done in triplicate. (c) Anchorage-independent growth of each cell lines in soft agar. Results are from two separate experiments, each done in triplicate. Statistically significant differences compared with H2009 shJNK2α cells are indicated (*P values < 0.02).
Discussion
In this study, we investigated the role of JNK2α in NSCLC tumorigenesis by examining its expression in primary NSCLC tumors and by modulating its expression in cultured NSCLC cells. We showed for the first time that a constitutively active JNK isoform has increased expression in the majority of primary NSCLC tumors tested. To directly determine the significance of JNK2α in NSCLC progression, we used retroviruses to specifically reduce endogenous JNK2α in multiple NSCLC cell lines. Each cell line with decreased levels of JNK2α had significantly reduced JNK activity that resulted in the decreased tumorigenic phenotypes, such as decreased cellular growth, anchorage-independent growth and tumor formation in NOD/SCID mice. To elucidate the mechanism by which JNK2α induces NSCLC formation, we analyzed a well-known oncogene and JNK substrate STAT3. Our data show that JNK2α isoforms can interact with and regulate STAT3 transcriptional activity by phosphorylating Ser727. In addition, we demonstrated that the reintroduction of JNK2α2 or STAT3 restored the tumorigenic phenotypes in cells deficient in JNK2α, demonstrating the importance of JNK2α in NSCLC progression.
JNK2α regulation of oncoproteins or tumor suppressors
Our study suggests that JNK2α has a major role in NSCLC tumorigenesis by regulating the activity of the oncoprotein STAT3. Although the role of JNK2α in other tumors is still unknown, our preliminary work suggests that JNK2α has a direct role in glioblastoma multiforme (GBM) tumorigenesis (Tang et al., 2006). We have discovered that reducing endogenous JNK2α expression in two GBM cell lines, U87-MG and U251, decreased tumorigenic phenotypes and tumor formation in mice (data not shown). Yet, JNK2α is not a universal oncogene as reducing JNK2α expression in SCLC cell lines did not alter cell growth or anchorage-independent growth (Supplementary Figures 3A–D). Consequently, to fully understand the role of JNK2α in various cancers it is important to study the relationship between JNK2α and its substrates. Previous research has already shown that JNK2α can phosphorylate oncoproteins such as eIF4E, Akt (Tang et al., 2006), and β-catenin (Wu et al., 2008). Other oncogenic JNK2α substrates, such as Sirt1 (Ford et al., 2008) and Ras (Shair et al., 2007), have been shown to be phosphorylated by JNK2 specifically. The JNK2 isoforms have also been associated with phosphorylation and regulation of the activity of tumor suppressors. Recently, p53 was shown to directly interact with and be phosphorylated by JNK2, whereas the well-known JNK substrate, c-Jun, was discovered to inhibit p53 activity (Maeda and Karin, 2003; Oleinik et al., 2007). Our study, in conjunction with previous reports, suggests that the JNK2α has a key role in the regulation of oncoproteins and tumor suppressors. Further research should be conducted to identify additional JNK2α substrates that may regulate tumorigenesis.
Phosphorylation of STAT3 at S727 in NSCLC tumorigenesis
Activation of the STAT3 transcription factor has been implicated in cellular transformation and tumor progression. The oncogenic activities of STAT3 were first observed when an overexpressed, constitutively active STAT3-induced tumor formation in nude mice (Bromberg et al., 1999). Subsequent biochemical analysis revealed that STAT3 could promote cell growth and cell survival in tumor cells by transcriptionally upregulating genes, such as c-MYC, BCL-2, cyclin D1 and VEGF (Gao and Bromberg, 2006). STAT3 has also been shown to be constitutively active in a variety of solid and hematological tumors, including breast, prostate, ovarian and lung cancers (Kortylewski et al., 2005). In particular, STAT3 activity has been closely linked to NSCLC tumorigenesis. For example, two independent reports observed increased expression or enhanced phosphorylation of STAT3 in the majority of NSCLC tumors (Haura et al., 2005; Achcar Rde et al., 2007). Moreover, the inhibition of STAT3 activity using G-quartet oligodeoxynucleotides decreased cell growth and reduced tumor formation in NSCLC bearing nude mice (Weerasinghe et al., 2007; Zhang et al., 2007).
STAT3 activation and the subsequent increase in transcriptional activities have been linked to the phosphorylation of two different STAT3 residues: tyrosine 705 and serine 727. The function of Tyr705 phosphorylation has been carefully studied and reports show that it leads to STAT3 homodimerization through its SH2 domain, which enhances translocation to the nucleus and activation of STAT3 target genes (Chung et al., 1997). The role of Ser727 phosphorylation is less clear. Phosphorylation of Ser727 was shown to enhance STAT3 homodimerization and DNA binding or to solely affect transcriptional activity (Wen et al., 1995; Zhang et al., 1995). Although the mechanism by which Ser272 phosphorylation regulates STAT3 activity remains unknown, Ser727 was found to induce prostate and lung tumorigenesis independent of Tyr705 phosphorylation by enhancing the transcription of pro-proliferative genes, such as c-MYC (Qin et al., 2008; Yeh et al., 2009). In addition, Ser727 has been implicated with the ability of STAT3 to transform cells in a Ras-dependent manner by targeting STAT3 to the mitochondria (Gough et al., 2009). Subsequently, many researchers have tried to identify the STAT3 serine kinase(s) hoping to identify new therapeutic targets.
JNK phosphorylation of STAT3
The STAT3 carboxyl-terminal Ser727 residue is located within a consensus site of MAPK phosphorylation. MAPK proteins are serine- and threonine-specific kinases and have been shown to phosphorylate Ser727. For example, one report showed that ERK, but not JNK1, could phosphorylate Ser727 in vitro and in vivo (Chung et al., 1997). It is interesting that, a subsequent study demonstrated that JNK1 has the ability to phosphorylate Ser727 when activated by ultraviolet light or by an upstream kinase, MEKK7 (Lim and Cao, 1999). These seemingly contradictory results could be due to the differential activation of JNK1. Unlike JNK2α, JNK1 is not constitutively active and, therefore, stimuli must be used to activate this JNK protein (Tsuiki et al., 2003). As JNK2α has the unique ability to autophosphorylate, and consequently autoactivate, we studied the relationship between this JNK isoform and STAT3 phosphorylation. Our discovery that JNK2α can regulate STAT3 activity by directly phosphorylating STAT3 at Ser727 is critical in understanding the therapeutic potential of targeting this pathway. Significant research has been conducted to identify potent serine kinases for STAT3. By identifying a serine kinase that enhances tumorigenicity in NSCLC cells, we have discovered a novel therapeutic target for NSCLC tumors.
Materials and methods
Cell culture and transfection
The NSCLC cell lines NCI-H2009, HCC-827, NCI-H1650 and NCI-H1975 (from American Type Culture Collection, Manassas, VA, USA), were cultured in RPMI-1640 media supplemented with 10% fetal bovine serum and 2mM-L-glutamine. Cells were transfected using TransIT LT1 (Mirus, Madison, WI, USA) according to the manufacturer’s protocol.
Protein analysis and immunoprecipitations
Protein extracts from cells were harvested and immunoblotted as previously described (Nitta et al., 2007). The following antibodies were used for immunoblotting or immuoprecipitations: green fluorescent protein (Roche, Palo Alto, CA, USA), phosphorylated JNK (Cell Signaling Technology, Danvers, MA, USA), JNK2 (Cell Signaling), c-Myc (Santa Cruz Biotechnlogy, Santa Cruz, CA, USA), JNK1 (Santa Cruz Biotechnlogy), JNK3 (Upstate, Billerica, MA, USA), actin (Chemicon, Billerica, MA, USA), STAT3 (B&D Bioscience, San Jose, CA, USA), phospho STAT3 S727 (Cell Signaling Technology), phospho STAT3 Y705 (Cell Signaling Technology) and Flag (Sigma, St Louis, MO, USA). Chemiluminescence signals were quantitated using NIH Image J (National Institutes of Health, Bethesda, MD, USA). Immunoprecipitations were conducted as previously described (Nitta et al., 2006).
Retroviral infections
Retroviral small inhibitory RNA (shRNA) constructs were generated by ligating annealed oligonucleotides (JNK2α 5′-AAGGTTGTGTGATATTCCA-3′ or scrambled 5′-GTAAACAAAGCAATGTATA-3′) into pSuper.retro-Puro (Oligoengine, Seattle, WA, USA). Retroviral infections were carried out as previously described (Xie et al., 1997). 36 h after infection, the cells were selected using 5 μg/ml of puromyocin for 3 days. JNK2α2res and STAT3 were reintroduced into H2009 shJNK2α cells using the pMXIH retroviral vector as previously described (Xie et al., 1997). The JNK2α2 mutant resistant to shJNK2α was generated by mutating the following nucleotides: T666C and G669C.
Cell growth analysis
NSCLC-infected cells were plated in six-well plates (5 × 105 cells per well) and cultured in RPMI-1640 media supplemented with 1% fetal bovine serum, 2mM L-glutamine, and 100 units per ml penicillin/streptomycin. The numbers of live cells were counted daily by means of trypan blue exclusion assay. Experiments were done in triplicate and results are expressed as mean ± s.d.
Soft agar assay
NSCLC cells were plated in six-well plates (3 × 105 cells per well), suspended in RPMI-1640 as previously described (Tang et al., 2006). The presence of colonies was scored after 10 days using Genetools software (Syngene, Frederick, MA, USA). Experiments were done in triplicate and results are expressed as mean± s.d.
In vivo tumor formation in NOD/SCID mice
2 × 106 cells of NCI-H2009 cells were treated and subcutaneously injected into the hind limbs of six to eight-week old mice as previously described (Tang et al., 2006). Around 7 weeks after injection the tumors were weighed.
QRT-PCR analysis
Total RNA from each cell line was extracted using Trizol reagent (Promega, Madison, WI, USA) according to the manufacturer’s protocol. The reverse transcription and quantitative PCR (qPCR) were carried out using the Brilliant II Syber Green QRT-PCR Master Mix Kit according to the manufacturer’s protocol (Stratagene, La Jolla, CA, USA). Primers were designed specifically for the JNK2α, JNK2β, JNK1 or JNK3 genes and the specificity was verified by sequencing. Their sequences were: JNK2α (forward), 5′-GCGTGCACTAACTTCATGATGAC-3′; JNK2α (reverse), 5′-GCAACCCACTGACCAGATATCAAC-3′; JNK2β (forward), 5′-GCACCCTGAAGATCCTTGAC-3′; JNK2β (reverse), 5′-CGGGAACAGGACTTTATGGA-3′; JNK1 (forward), 5′-GGCTCAGGAGCTCAAGGAATAG-3′; JNK1 (reverse), 5′-GATTCTGAAATGGTCGGCTTAG-3′; and JNK3 (forward), 5′-CATAGTTTGTGCCGCGTATG-3′; JNK3 (reverse), 5′-GGCATCCATCAGTTCCATTAC-3′; c-Myc (forward) 5′-TGAGGAGACACCGCCCA-3′; c-Myc (reverse) 5′-AACATCGATTTCTTCCTCA-3′; glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (forward) 5′-GTCCACCACCCTGTTGCTGTA-3′; GAPDH (reverse) 5′-ACCACAGTCCATGCCATC-3′. All samples were normalized to GAPDH. Fresh-frozen NSCLC tumors and adjacent normal tissues were obtained from the Biosample Repository at Fox Chase Cancer Center (Philadelphia, PA, USA). All specimens were accrued and evaluated under institutional review board (IRB)-approved protocols.
In vitro kinase assay
1 μg of the fusion protein was incubated in kinase buffer at 30 °C for 30 min as previously described (Tang et al., 2006). Reactions were terminated by adding protein loading buffer and boiling for 5 min.
Luciferase assay
Using TransIT LT1 (Mirus), NCI-H2009-infected cells were transiently co-transfected with pSTAT3-TA-Luc vector (Clontech, Mountain View, CA, USA) and pRL-CMV (Promega). 24 h after transfection the cells were serum starved for 24 h, and subsequently stimulated with EGF (250 ng/ml). The firefly and Renilla luciferase activities of the stimulated cells were measured using the Dual-Luciferase Reporter Assay System (Promega). The values obtained from the NCI-H2009-uninfected cells were used to normalize the relative STAT3 activity.
Indirect immunofluorescence
Immunofluorescence was performed on formaldehyde-fixed cells followed by a methanol permeabilization as previously described (Lee et al., 2004). The phospho STAT3 S727 antibody (Cell Signaling Technology) was used. Images were taken using Leica SP2 AOBS confocal microscope. Images were collected using equal exposure times and processed similarly.
Protein expression and purification
pET28 and pET42 constructs were expressed in the BL21(DE3)pLysS Escherichia coli strain (Novagen, Gibbs-town, NJ, USA) and induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 4h at 37°C. The tagged proteins were purified according to the manufacturer’s protocol using Ni-NTA agarose beads (Qiagen, Valencia, CA, USA) or Glutathione Sepharose 4B beads (GE Scientific, Piscataway, NJ, USA).
Supplementary Material
Acknowledgments
We are grateful to Emily Piccione, Shawn Badal, Tiffany Lieu and Marina Holgado-Madruga for their useful discussions and critical review of the paper. This work was supported by the Mark Linder/American Brain Tumor Association Fellowship, NIH grants CA69495 and CA124832 and a research grant from the National Brain Tumor Foundation.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- Achcar Rde O, Cagle PT, Jagirdar J. Expression of activated and latent signal transducer and activator of transcription 3 in 303 non-small cell lung carcinomas and 44 malignant mesotheliomas: possible role for chemotherapeutic intervention. Arch Pathol Lab Med. 2007;131:1350–1360. doi: 10.5858/2007-131-1350-EOAALS. [DOI] [PubMed] [Google Scholar]
- Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006;66:3162–3168. doi: 10.1158/0008-5472.CAN-05-3757. [DOI] [PubMed] [Google Scholar]
- Arbour N, Naniche D, Homann D, Davis RJ, Flavell RA, Oldstone MB. c-Jun NH(2)-terminal kinase (JNK)1 and JNK2 signaling pathways have divergent roles in CD8(+) T cell-mediated antiviral immunity. J Exp Med. 2002;195:801–810. doi: 10.1084/jem.20011481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berner JM, Sorlie T, Mertens F, Henriksen J, Saeter G, Mandahl N, et al. Chromosome band 9p21 is frequently altered in malignant peripheral nerve sheath tumors: studies of CDKN2A and other genes of the pRB pathway. Genes Chromosomes Cancer. 1999;26:151–160. [PubMed] [Google Scholar]
- Bhattacharya S, Ray RM, Johnson LR. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem J. 2005;392:335–344. doi: 10.1042/BJ20050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode AM, Dong Z. The functional contrariety of JNK. Mol Carcinog. 2007;46:591–598. doi: 10.1002/mc.20348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoyevitch MA. The isoform-specific functions of the c-Jun N-terminal Kinases (JNKs): differences revealed by gene targeting. Bioessays. 2006;28:923–934. doi: 10.1002/bies.20458. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch MA, Arthur PG. Inhibitors of c-Jun N-terminal kinases: JuNK no more? Biochim Biophys Acta. 2008;1784:76–93. doi: 10.1016/j.bbapap.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broers JL, Viallet J, Jensen SM, Pass H, Travis WD, Minna JD, et al. Expression of c-myc in progenitor cells of the bronchopul-monary epithelium and in a large number of non-small cell lung cancers. Am J Respir Cell Mol Biol. 1993;9:33–43. doi: 10.1165/ajrcmb/9.1.33. [DOI] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- Butterfield L, Storey B, Maas L, Heasley LE. c-Jun NH2-terminal kinase regulation of the apoptotic response of small cell lung cancer cells to ultraviolet radiation. J Biol Chem. 1997;272:10110–10116. doi: 10.1074/jbc.272.15.10110. [DOI] [PubMed] [Google Scholar]
- Chen N, Nomura M, She QB, Ma WY, Bode AM, Wang L, et al. Suppression of skin tumorigenesis in c-Jun NH(2)-terminal kinase-2-deficient mice. Cancer Res. 2001;61:3908–3912. [PubMed] [Google Scholar]
- Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ, et al. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell. 2002;2:497–503. doi: 10.1016/s1534-5807(02)00149-1. [DOI] [PubMed] [Google Scholar]
- Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997;17:6508–6516. doi: 10.1128/mcb.17.11.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Holgado-Madruga M, Su W, Tsuiki H, Wedegaertner P, Wong AJ. Identification of a specific domain responsible for JNK2alpha2 autophosphorylation. J Biol Chem. 2005;280:9913–9920. doi: 10.1074/jbc.M412165200. [DOI] [PubMed] [Google Scholar]
- Ford J, Ahmed S, Allison S, Jiang M, Milner J. JNK2-dependent regulation of SIRT1 protein stability. Cell Cycle. 2008;7:3091–3097. doi: 10.4161/cc.7.19.6799. [DOI] [PubMed] [Google Scholar]
- Gao SP, Bromberg JF. Touched and moved by STAT3. Sci STKE. 2006;2006:pe30. doi: 10.1126/stke.3432006pe30. [DOI] [PubMed] [Google Scholar]
- Gazzeri S, Brambilla E, Caron de Fromentel C, Gouyer V, Moro D, Perron P, et al. p53 genetic abnormalities and myc activation in human lung carcinoma. Int J Cancer. 1994;58:24–32. doi: 10.1002/ijc.2910580106. [DOI] [PubMed] [Google Scholar]
- Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent onco-genic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haura EB, Zheng Z, Song L, Cantor A, Bepler G. Activated epidermal growth factor receptor-Stat-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin Cancer Res. 2005;11:8288–8294. doi: 10.1158/1078-0432.CCR-05-0827. [DOI] [PubMed] [Google Scholar]
- Khatlani TS, Wislez M, Sun M, Srinivas H, Iwanaga K, Ma L, et al. c-Jun N-terminal kinase is activated in non-small-cell lung cancer and promotes neoplastic transformation in human bronchial epithelial cells. Oncogene. 2007;26:2658–2666. doi: 10.1038/sj.onc.1210050. [DOI] [PubMed] [Google Scholar]
- Kortylewski M, Jove R, Yu H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005;24:315–327. doi: 10.1007/s10555-005-1580-1. [DOI] [PubMed] [Google Scholar]
- Lee C, Dhillon J, Wang MY, Gao Y, Hu K, Park E, et al. Targeting YB-1 in HER-2 overexpressing breast cancer cells induces apoptosis via the mTOR/STAT3 pathway and suppresses tumor growth in mice. Cancer Res. 2008;68:8661–8666. doi: 10.1158/0008-5472.CAN-08-1082. [DOI] [PubMed] [Google Scholar]
- Lee SO, Lou W, Johnson CS, Trump DL, Gao AC. Interleukin-6 protects LNCaP cells from apoptosis induced by androgen deprivation through the Stat3 pathway. Prostate. 2004;60:178–186. doi: 10.1002/pros.20045. [DOI] [PubMed] [Google Scholar]
- Lim CP, Cao X. Serine phosphorylation and negative regulation of Stat3 by JNK. J Biol Chem. 1999;274:31055–31061. doi: 10.1074/jbc.274.43.31055. [DOI] [PubMed] [Google Scholar]
- Lufei C, Koh TH, Uchida T, Cao X. Pin1 is required for the Ser727 phosphorylation-dependent Stat3 activity. Oncogene. 2007;26:7656–7664. doi: 10.1038/sj.onc.1210567. [DOI] [PubMed] [Google Scholar]
- Maeda S, Karin M. Oncogene at last–c-Jun promotes liver cancer in mice. Cancer Cell. 2003;3:102–104. doi: 10.1016/s1535-6108(03)00025-4. [DOI] [PubMed] [Google Scholar]
- Nitta R, Okada Y, Hirokawa N. Structural model for strain-dependent microtubule activation of Mg-ADP release from kinesin. Nat Struct Mol Biol. 2008;15:1067–1075. doi: 10.1038/nsmb.1487. [DOI] [PubMed] [Google Scholar]
- Nitta RT, Jameson SA, Kudlow BA, Conlan LA, Kennedy BK. Stabilization of the retinoblastoma protein by A-type nuclear lamins is required for INK4A-mediated cell cycle arrest. Mol Cell Biol. 2006;26:5360–5372. doi: 10.1128/MCB.02464-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta RT, Smith CL, Kennedy BK. Evidence that proteasome-dependent degradation of the retinoblastoma protein in cells lacking A-type lamins occurs independently of gankyrin and MDM2. PLoS One. 2007;2:e963. doi: 10.1371/journal.pone.0000963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleinik NV, Krupenko NI, Krupenko SA. Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene. 2007;26:7222–7230. doi: 10.1038/sj.onc.1210526. [DOI] [PubMed] [Google Scholar]
- Onishi A, Chen Q, Humtsoe JO, Kramer RH. STAT3 signaling is induced by intercellular adhesion in squamous cell carcinoma cells. Exp Cell Res. 2008;314:377–386. doi: 10.1016/j.yexcr.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova O, Gorospe M, Bost F, Dean NM, Gaarde WA, Mercola D, et al. c-Jun N-terminal kinase is essential for growth of human T98G glioblastoma cells. J Biol Chem. 2000;275:24767–24775. doi: 10.1074/jbc.M904591199. [DOI] [PubMed] [Google Scholar]
- Qin HR, Kim HJ, Kim JY, Hurt EM, Klarmann GJ, Kawasaki BT, et al. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphoryla-tion. Cancer Res. 2008;68:7736–7741. doi: 10.1158/0008-5472.CAN-08-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp UR, Korn C, Ceteci F, Karreman C, Luetkenhaus K, Serafin V, et al. MYC is a metastasis gene for non-small-cell lung cancer. PLoS One. 2009;4:e6029. doi: 10.1371/journal.pone.0006029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues GA, Park M, Schlessinger J. Activation of the JNK pathway is essential for transformation by the Met oncogene. Embo J. 1997;16:2634–2645. doi: 10.1093/emboj/16.10.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shair KH, Bendt KM, Edwards RH, Bedford EC, Nielsen JN, Raab-Traub N. EBV latent membrane protein 1 activates Akt, NFkappaB, and Stat3 in B cell lymphomas. PLoS Pathog. 2007;3:e166. doi: 10.1371/journal.ppat.0030166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang GS, Cai JM, Ni J, Xiang YS, Cui JG, Zhu D, et al. Effects of STAT3 antisense oligodeoxynucleotides on apoptosis and proliferation of mouse melanoma cell line B16. Ai Zheng. 2006;25:269–274. [PubMed] [Google Scholar]
- Tsuiki H, Tnani M, Okamoto I, Kenyon LC, Emlet DR, Holgado-Madruga M, et al. Constitutively active forms of c-Jun NH2-terminal kinase are expressed in primary glial tumors. Cancer Res. 2003;63:250–255. [PubMed] [Google Scholar]
- Weerasinghe P, Garcia GE, Zhu Q, Yuan P, Feng L, Mao L, et al. Inhibition of Stat3 activation and tumor growth suppression of non-small cell lung cancer by G-quartet oligonucleotides. Int J Oncol. 2007;31:129–136. [PubMed] [Google Scholar]
- Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F. Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell. 2008;133:340–353. doi: 10.1016/j.cell.2008.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Su K, Wang D, Paterson AJ, Kudlow JE. MDA468 growth inhibition by EGF is associated with the induction of the cyclin-dependent kinase inhibitor p21WAF1. Anticancer Res. 1997;17:2627–2633. [PubMed] [Google Scholar]
- Xu X, Heidenreich O, Kitajima I, McGuire K, Li Q, Su B, et al. Constitutively activated JNK is associated with HTLV-1 mediated tumorigenesis. Oncogene. 1996;13:135–142. [PubMed] [Google Scholar]
- Yakut T, Egeli U, Gebitekin C. Investigation of c-myc and p53 gene alterations in the tumor and surgical borderline tissues of NSCLC and effects on clinicopathologic behavior: by the FISH technique. Lung. 2003;181:245–258. doi: 10.1007/s00408-003-1026-x. [DOI] [PubMed] [Google Scholar]
- Yang YM, Bost F, Charbono W, Dean N, McKay R, Rhim JS, et al. C-Jun NH(2)-terminal kinase mediates proliferation and tumor growth of human prostate carcinoma. Clin Cancer Res. 2003;9:391–401. [PubMed] [Google Scholar]
- Yeager T, Stadler W, Belair C, Puthenveettil J, Olopade O, Reznikoff C. Increased p16 levels correlate with pRb alterations in human urothelial cells. Cancer Res. 1995;55:493–497. [PubMed] [Google Scholar]
- Yeh HH, Giri R, Chang TY, Chou CY, Su WC, Liu HS. Ha-ras oncogene-induced Stat3 phosphorylation enhances oncogenicity of the cell. DNA Cell Biol. 2009;28:131–139. doi: 10.1089/dna.2008.0762. [DOI] [PubMed] [Google Scholar]
- Zhang X, Blenis J, Li HC, Schindler C, Chen-Kiang S. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science. 1995;267:1990–1994. doi: 10.1126/science.7701321. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang J, Wang L, Wei H, Tian Z. Therapeutic effects of STAT3 decoy oligodeoxynucleotide on human lung cancer in xenograft mice. BMC Cancer. 2007;7:149. doi: 10.1186/1471-2407-7-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.